
In Silico Investigation of Traditional Chinese Medicine for Potential Lead Compounds as SPG7 Inhibitors against Coronary Artery Disease

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Docking Simulation
2.3. Molecular Dynamics Simulation
3. Results and Discussion
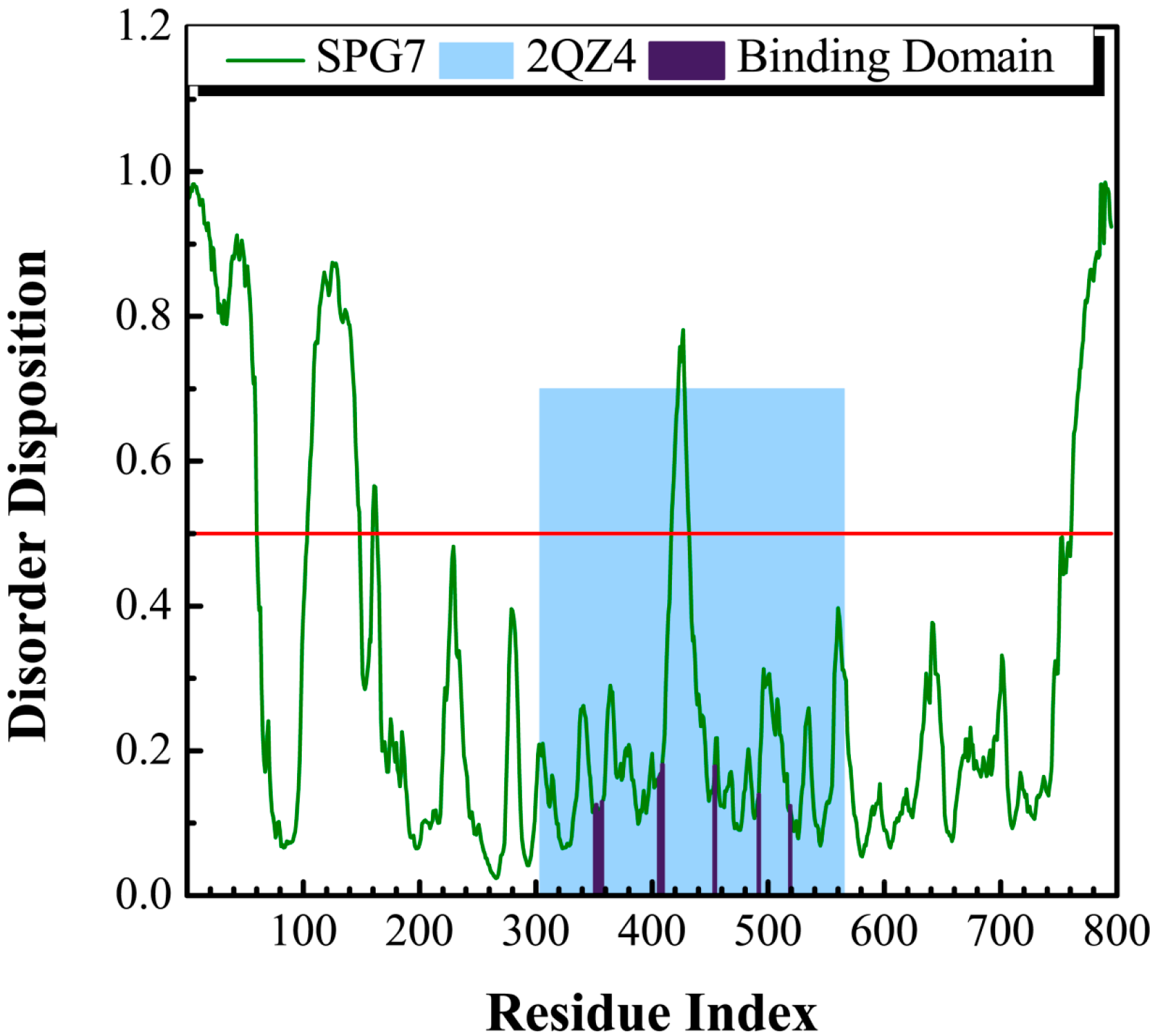
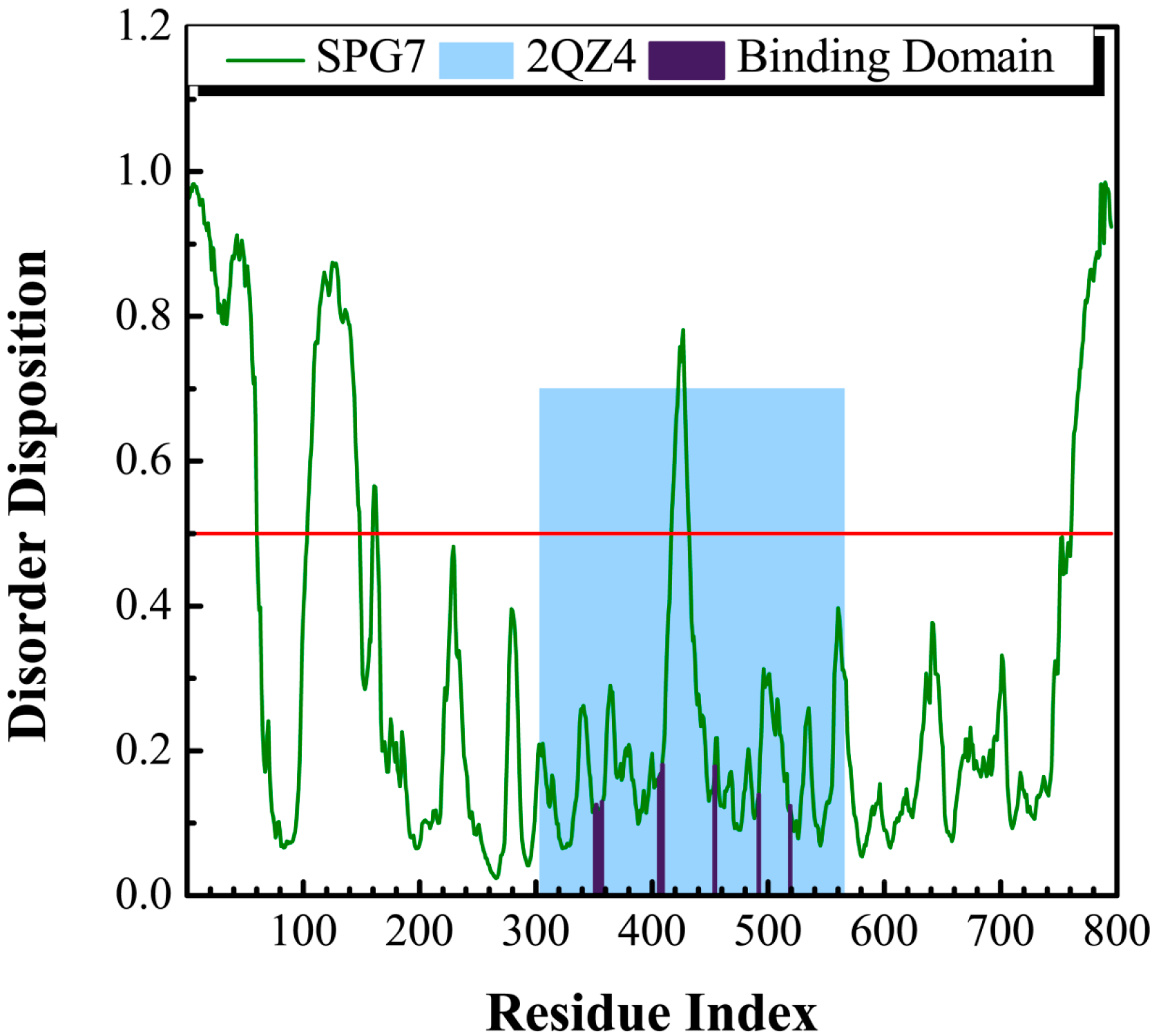
3.1. Disordered Prediction
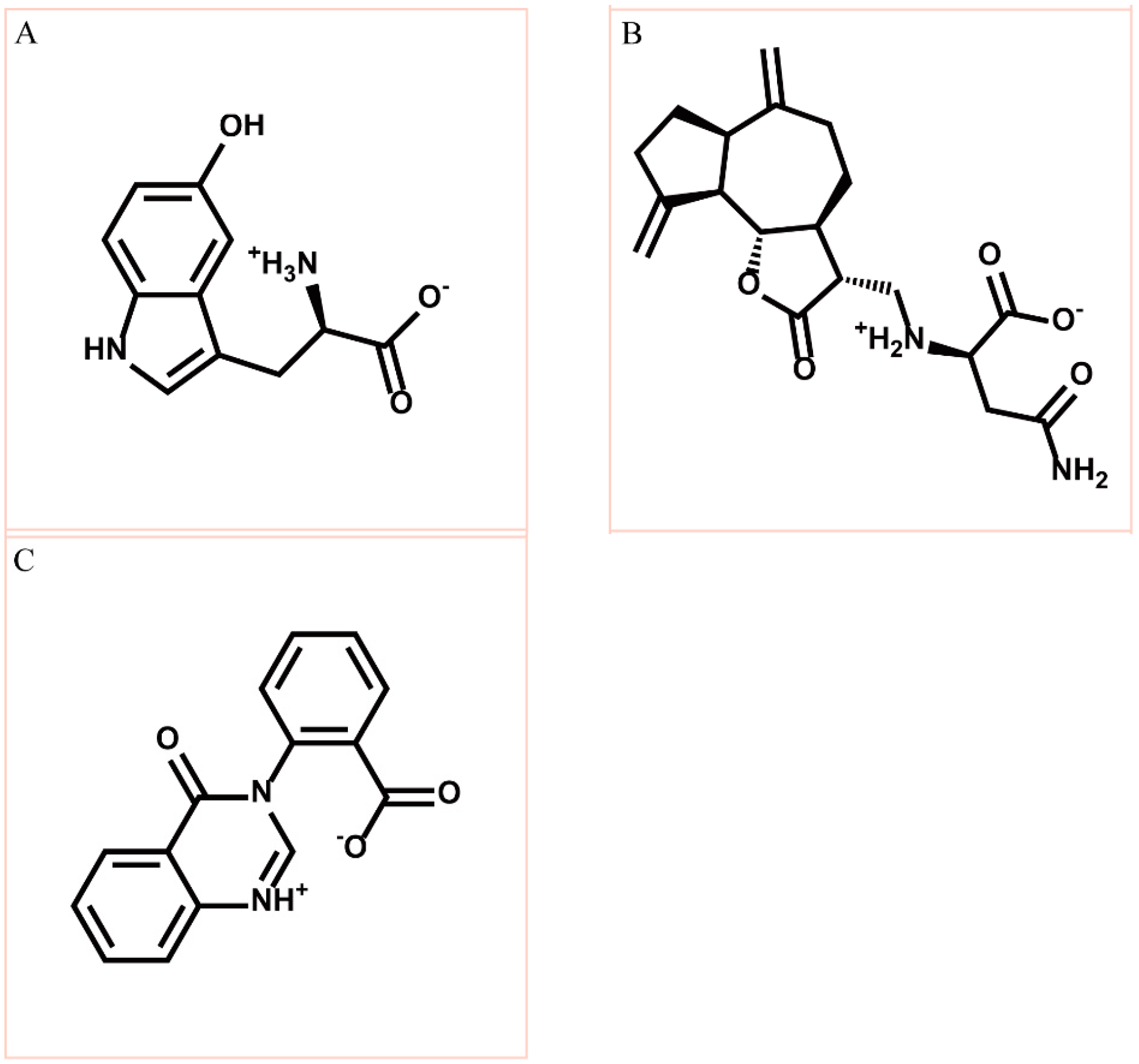
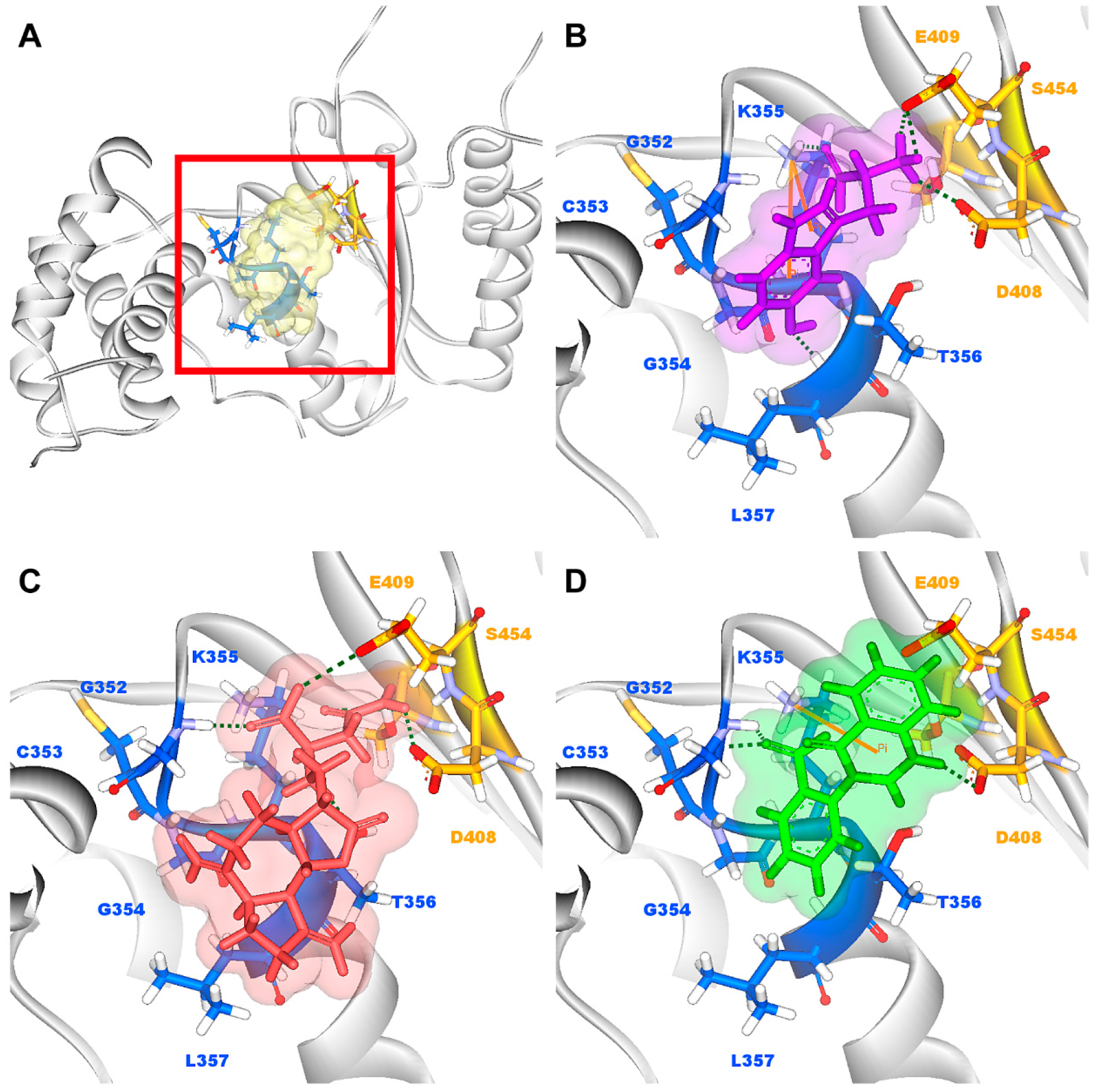
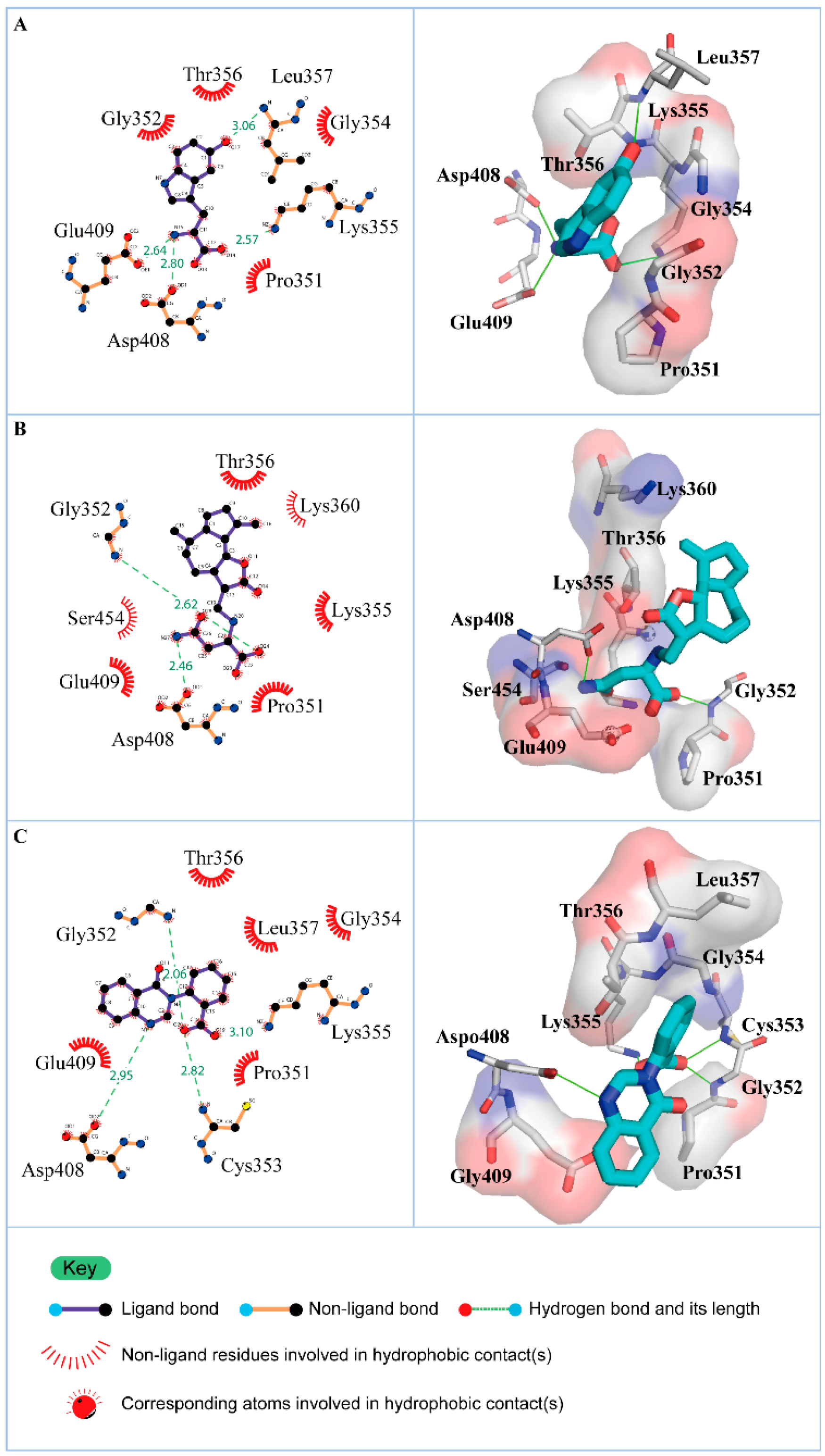
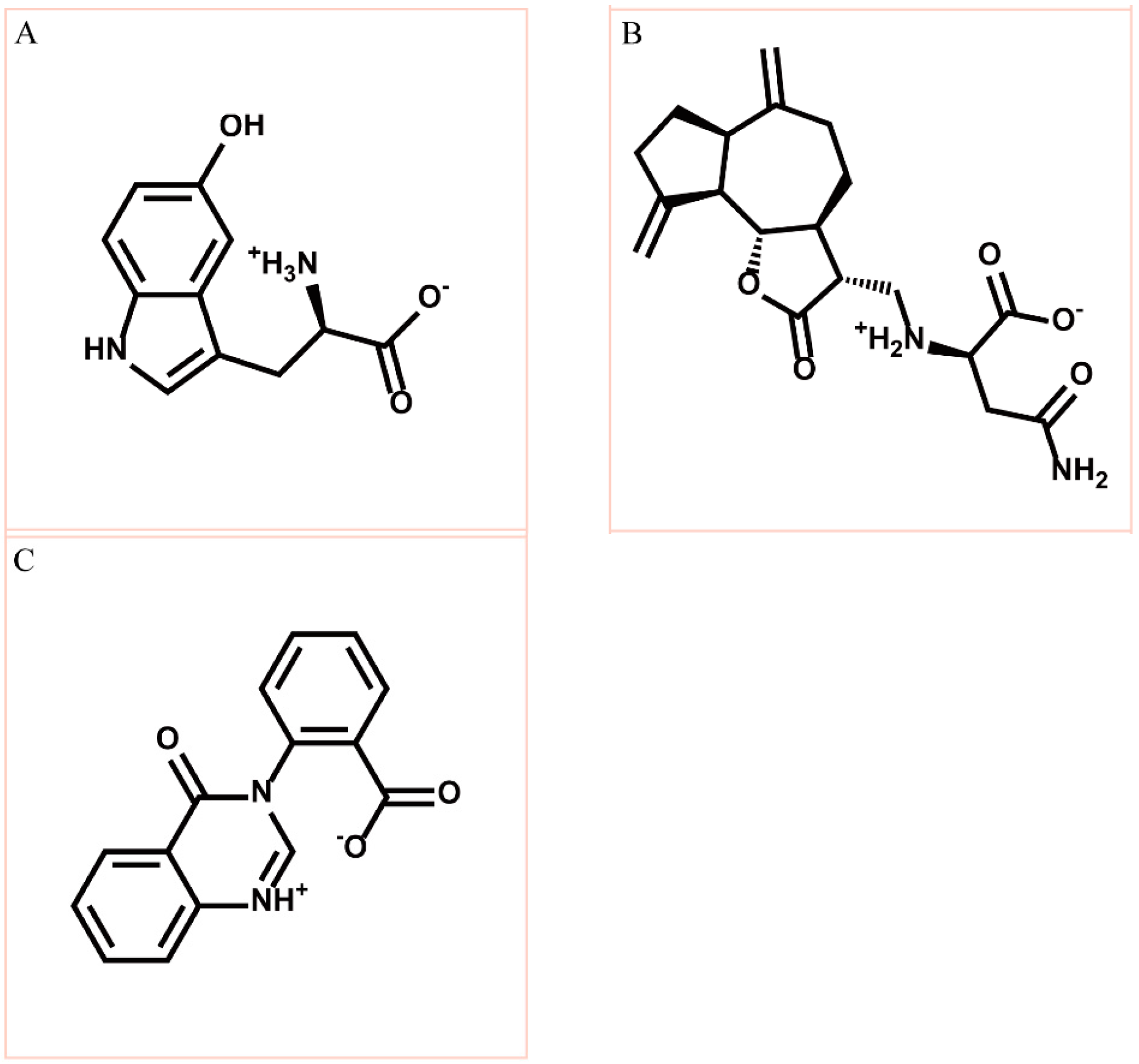
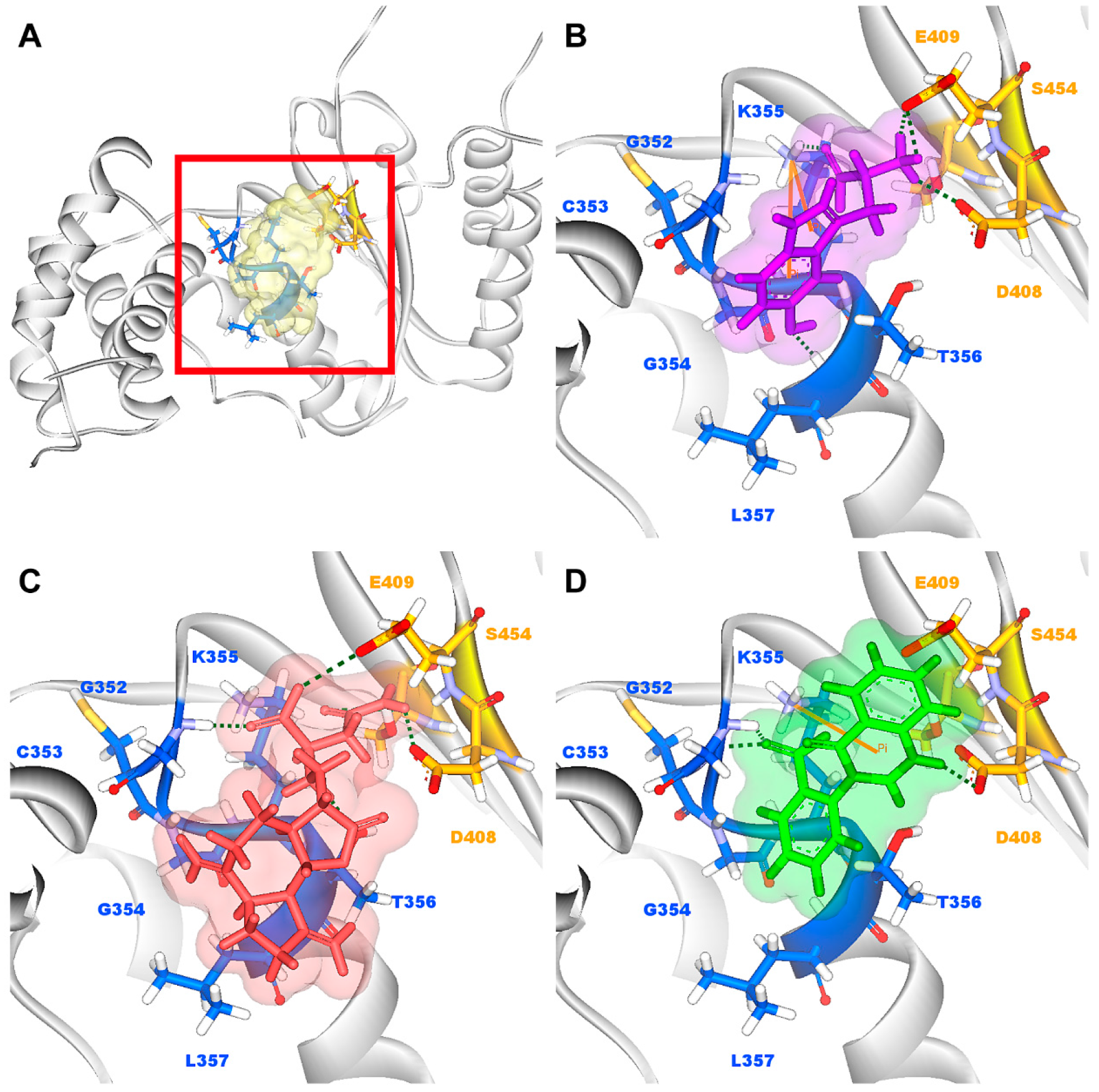
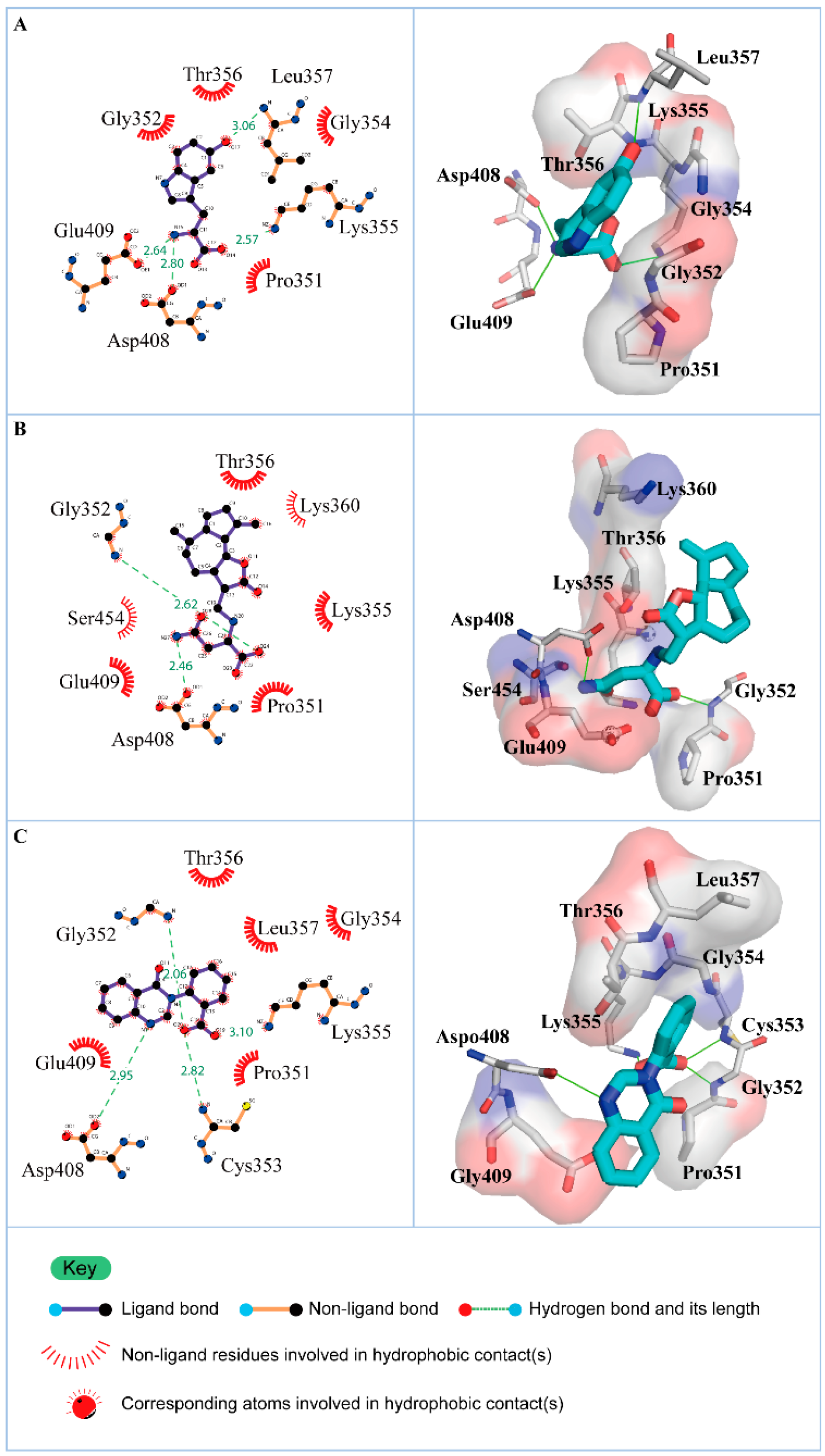
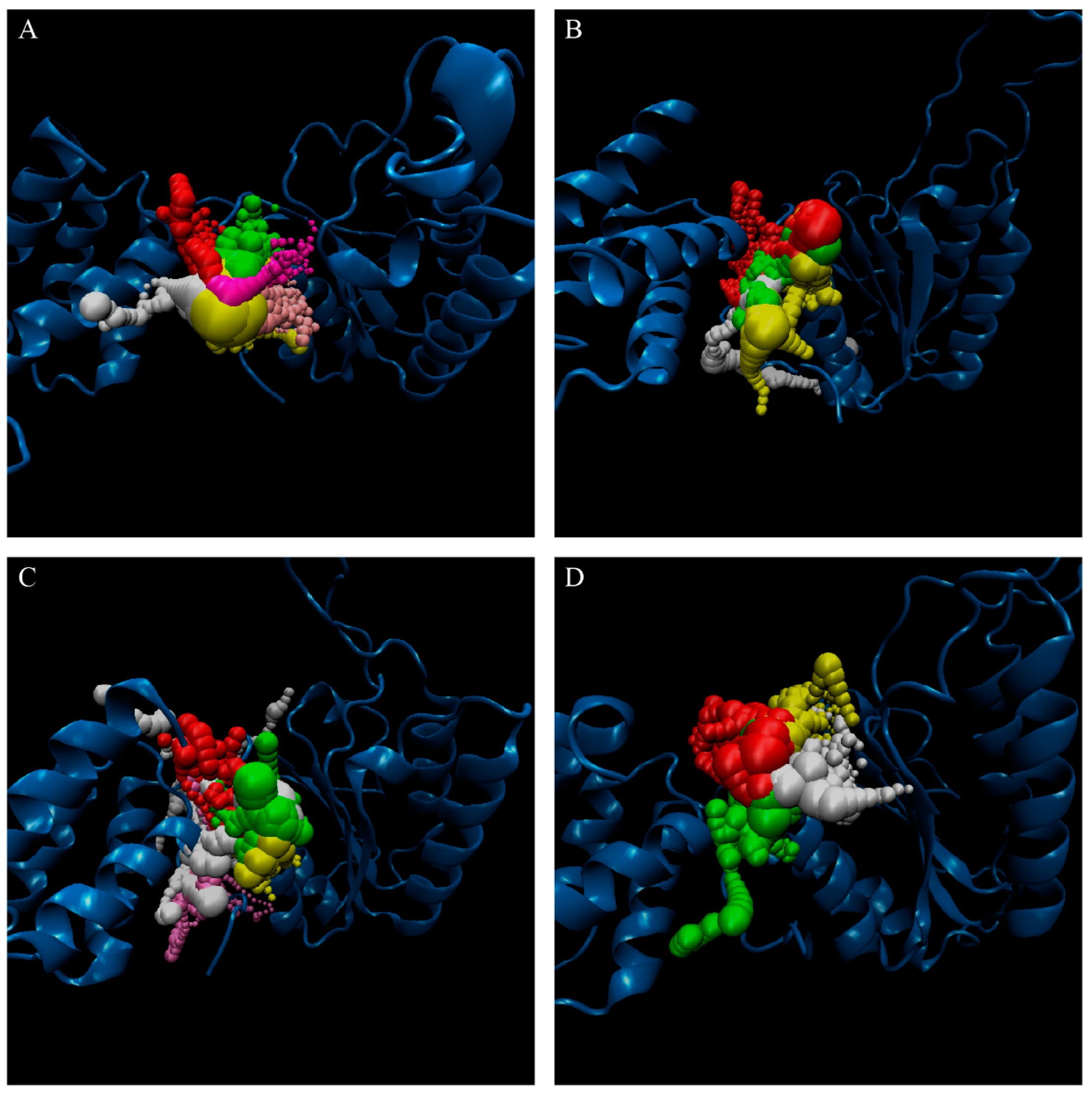
3.2. Docking Simulation
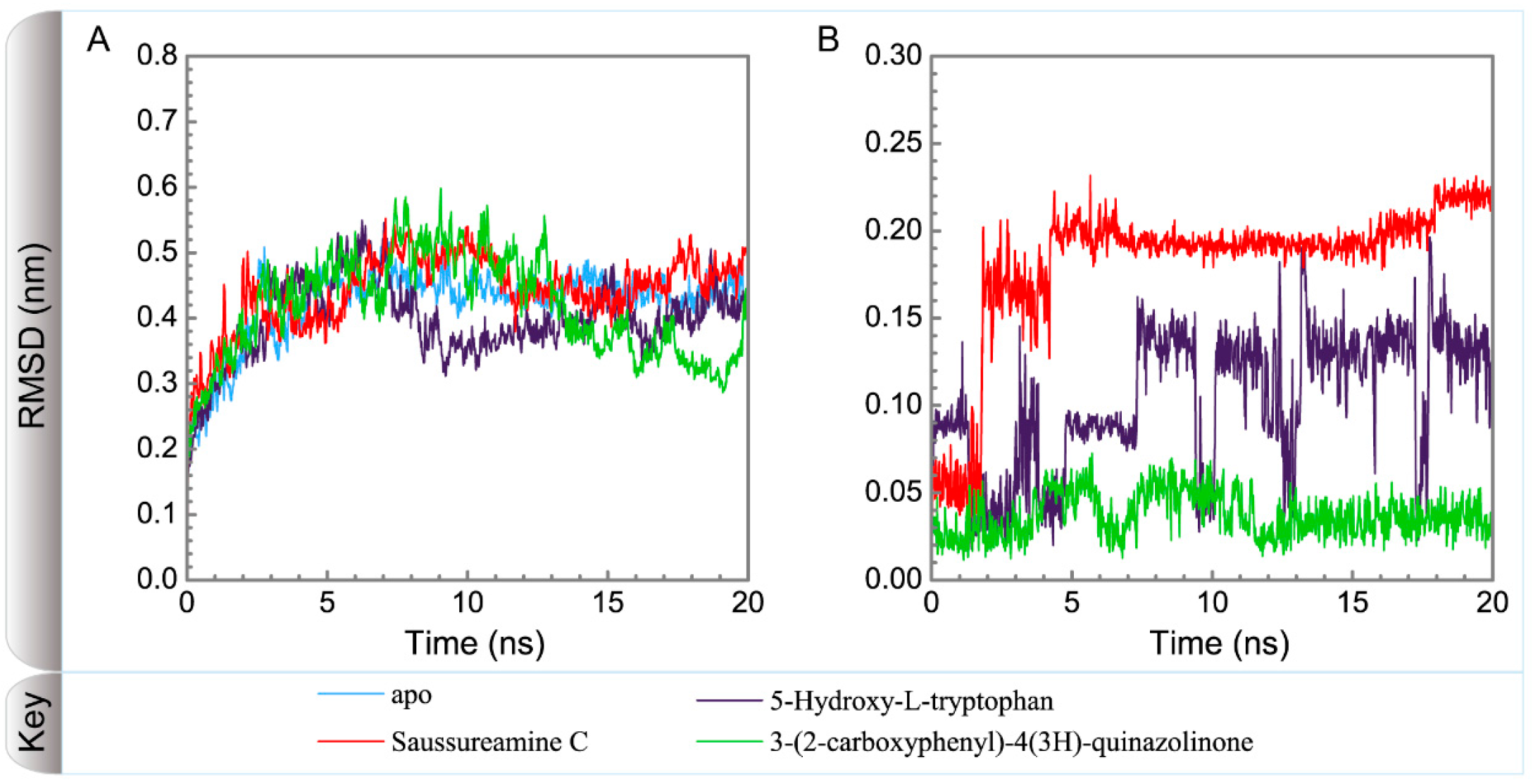
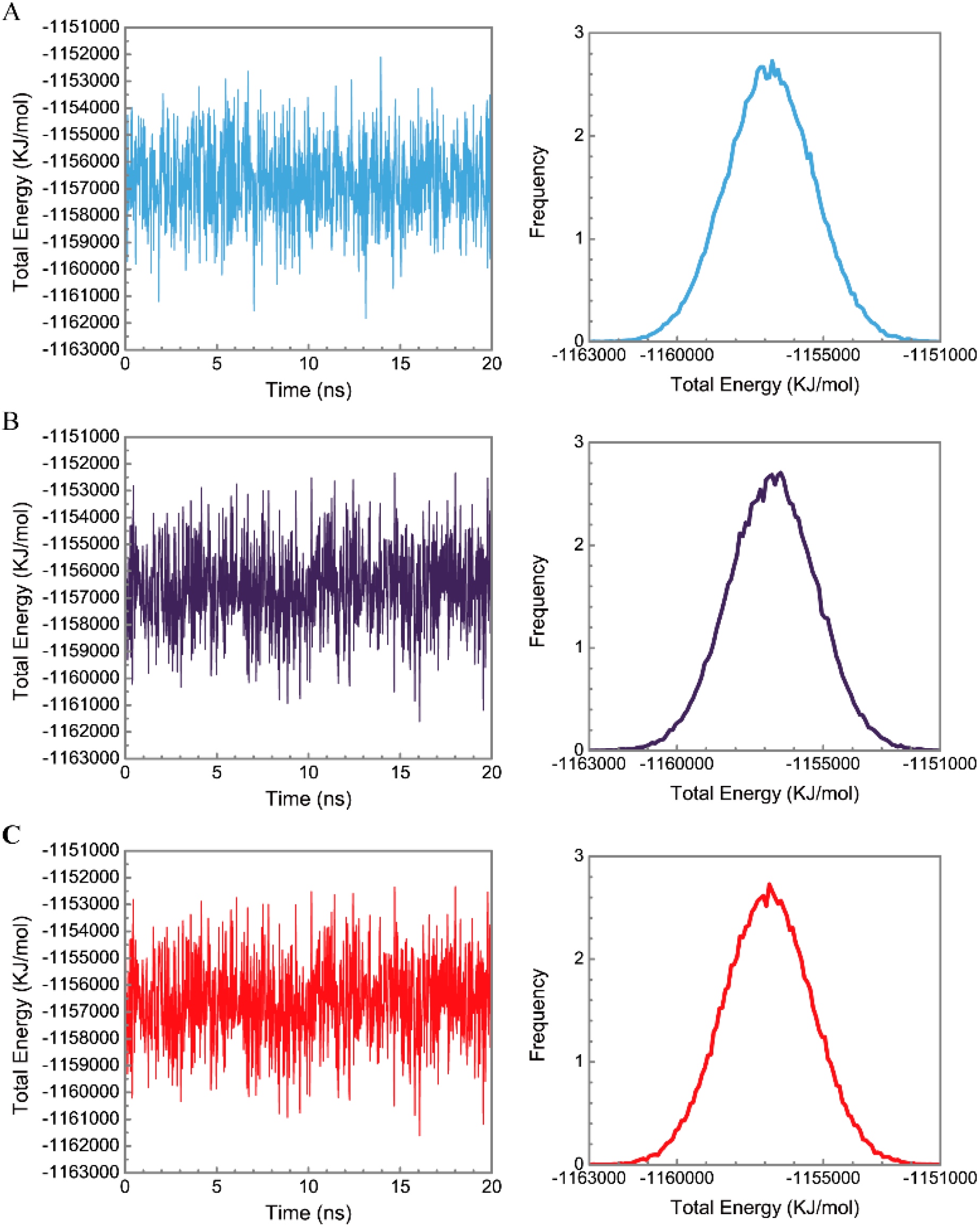
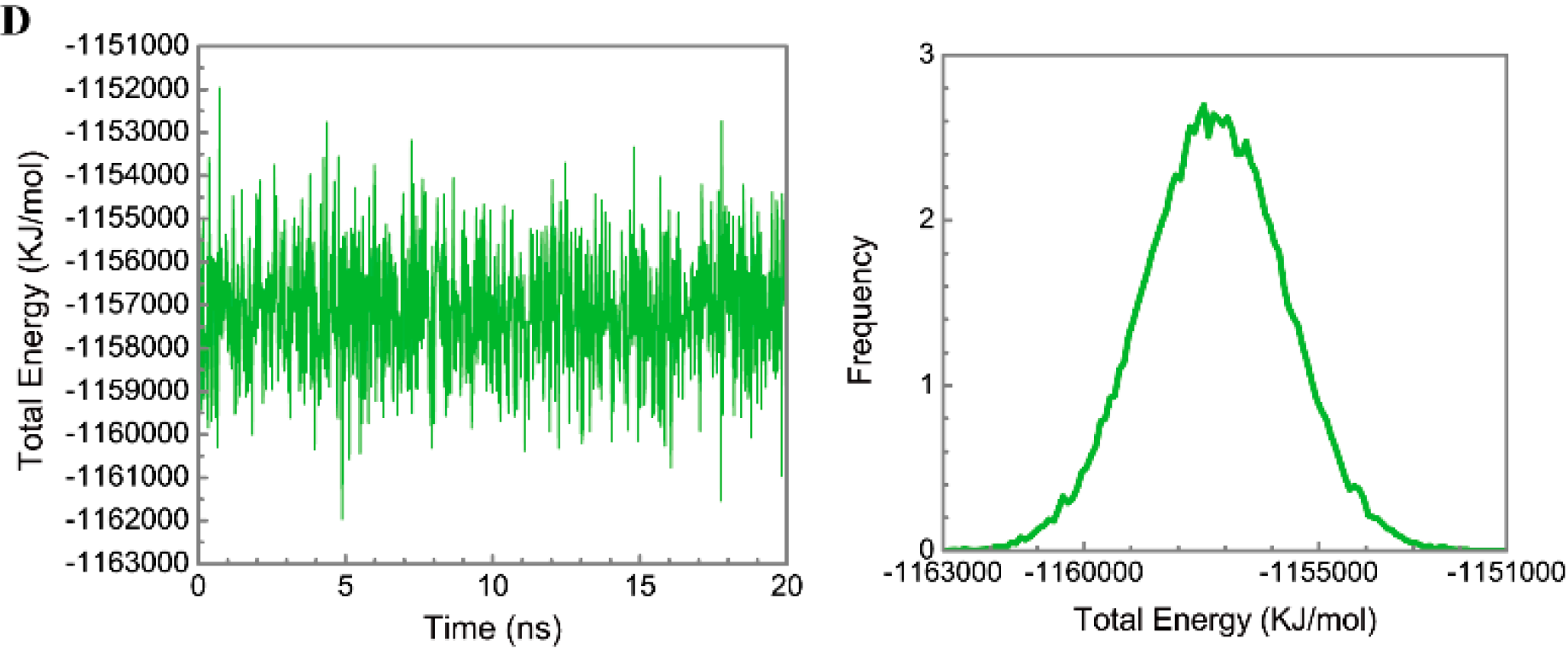
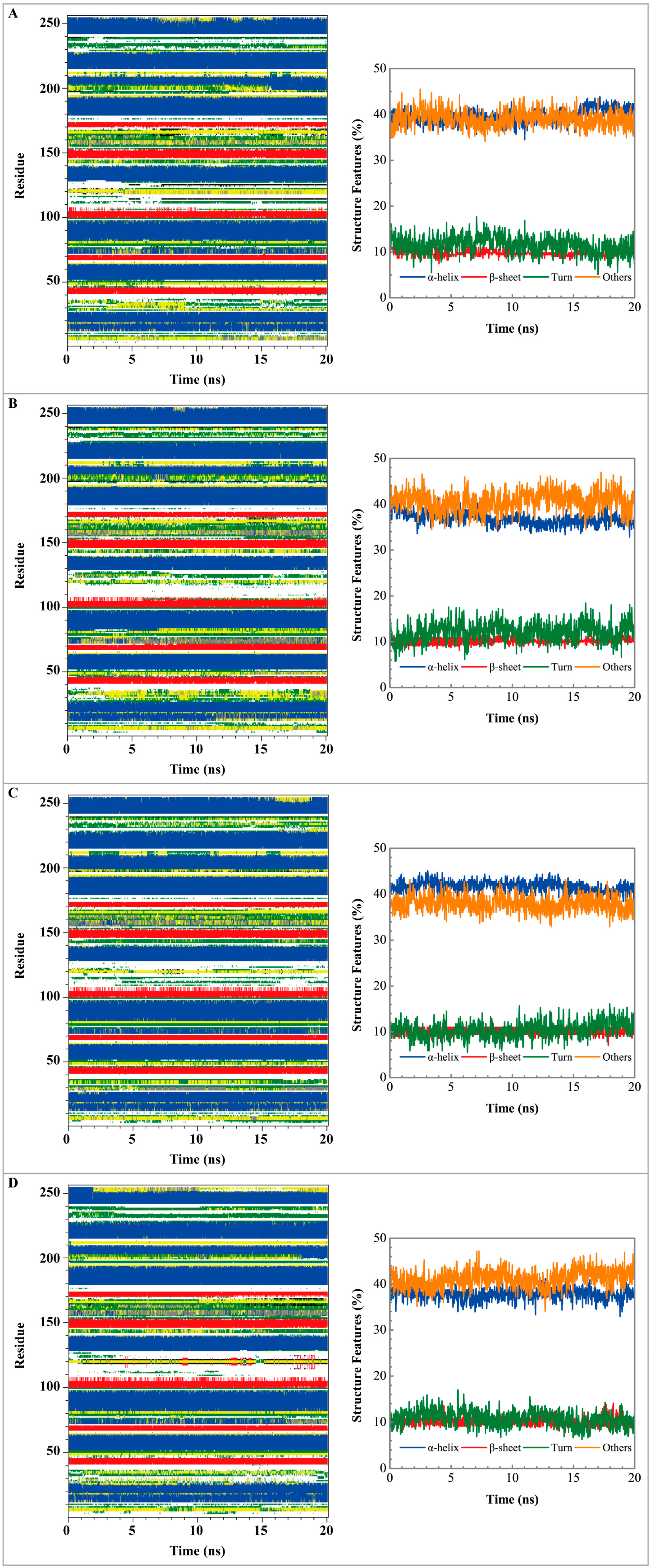
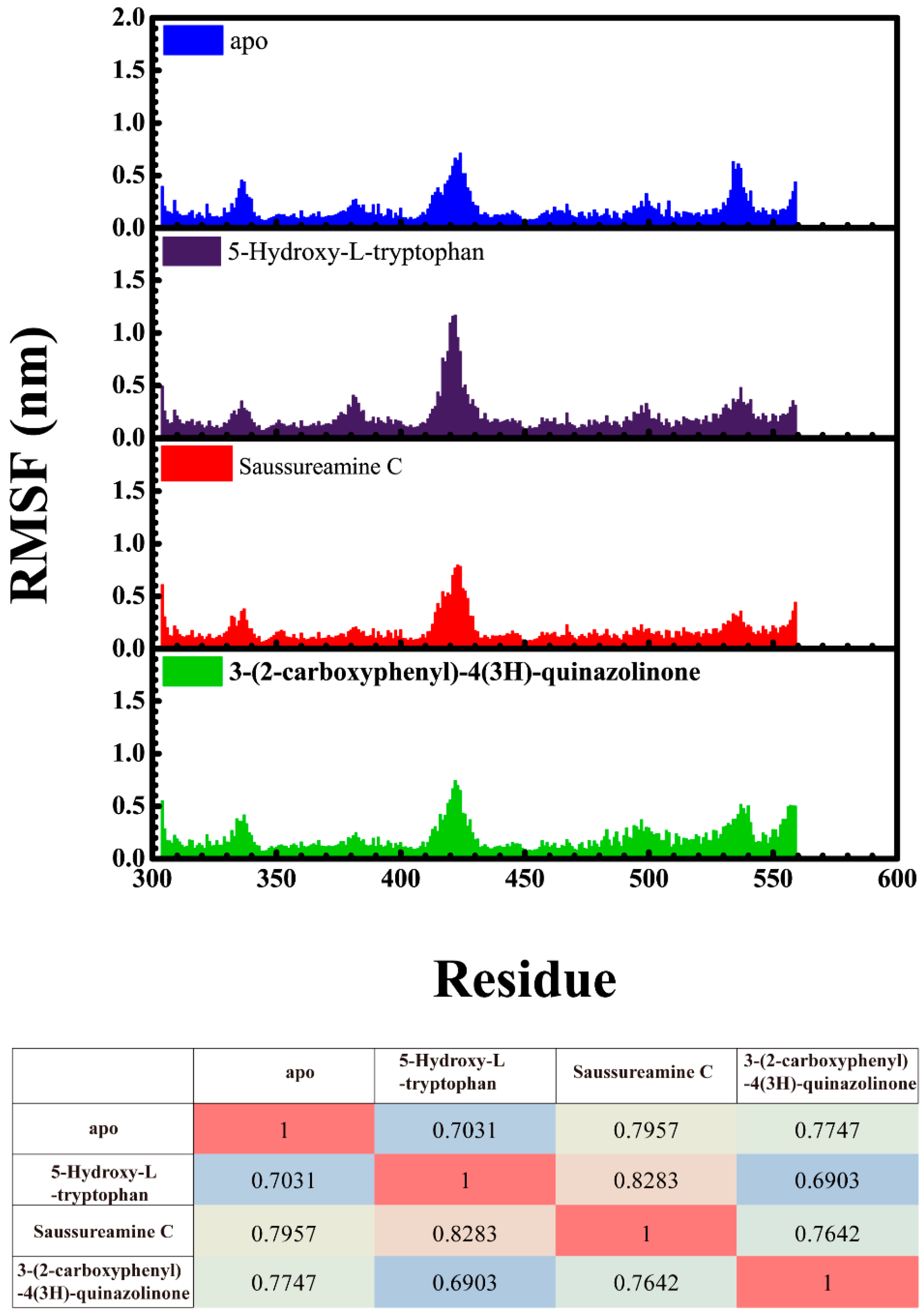
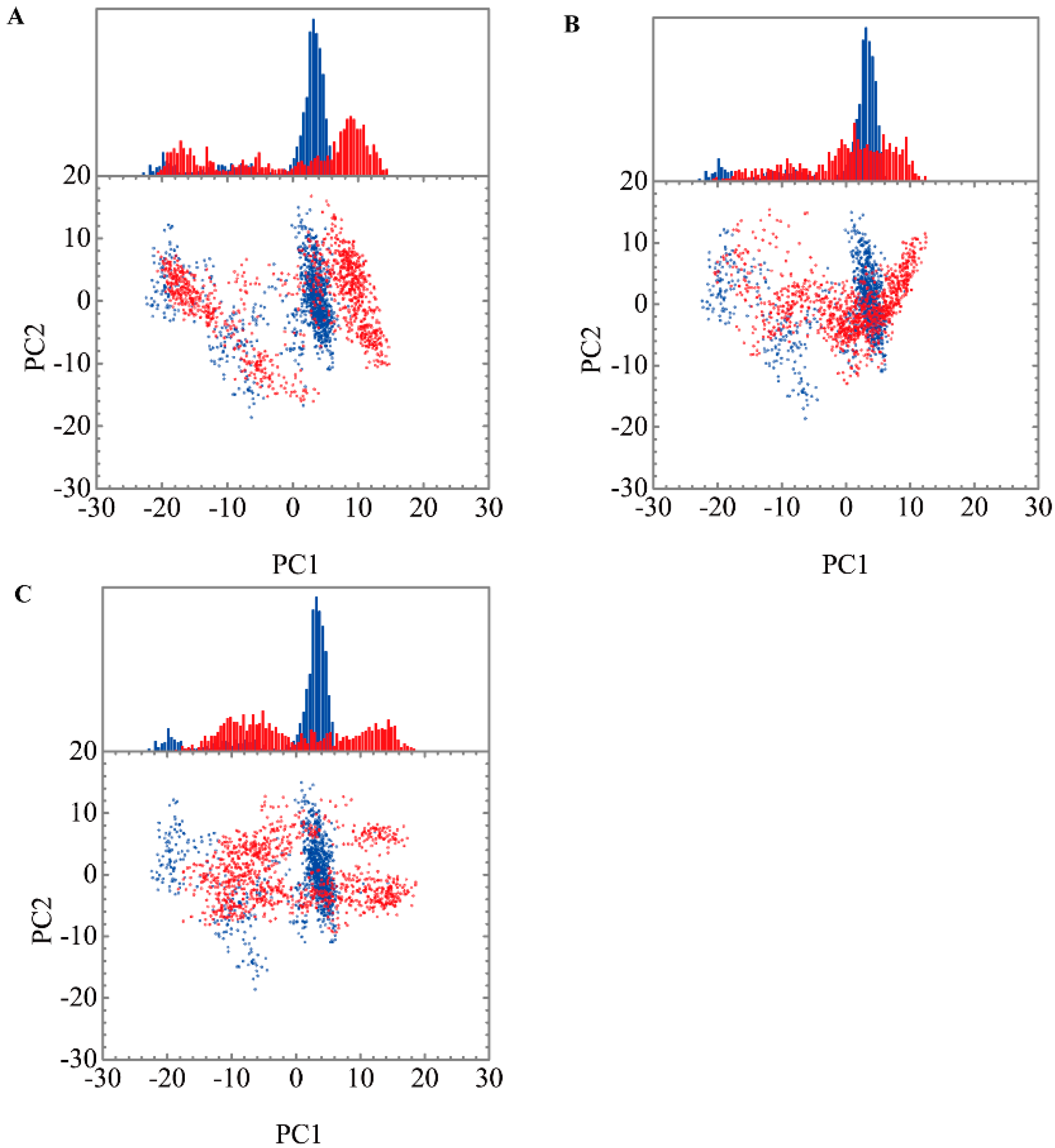
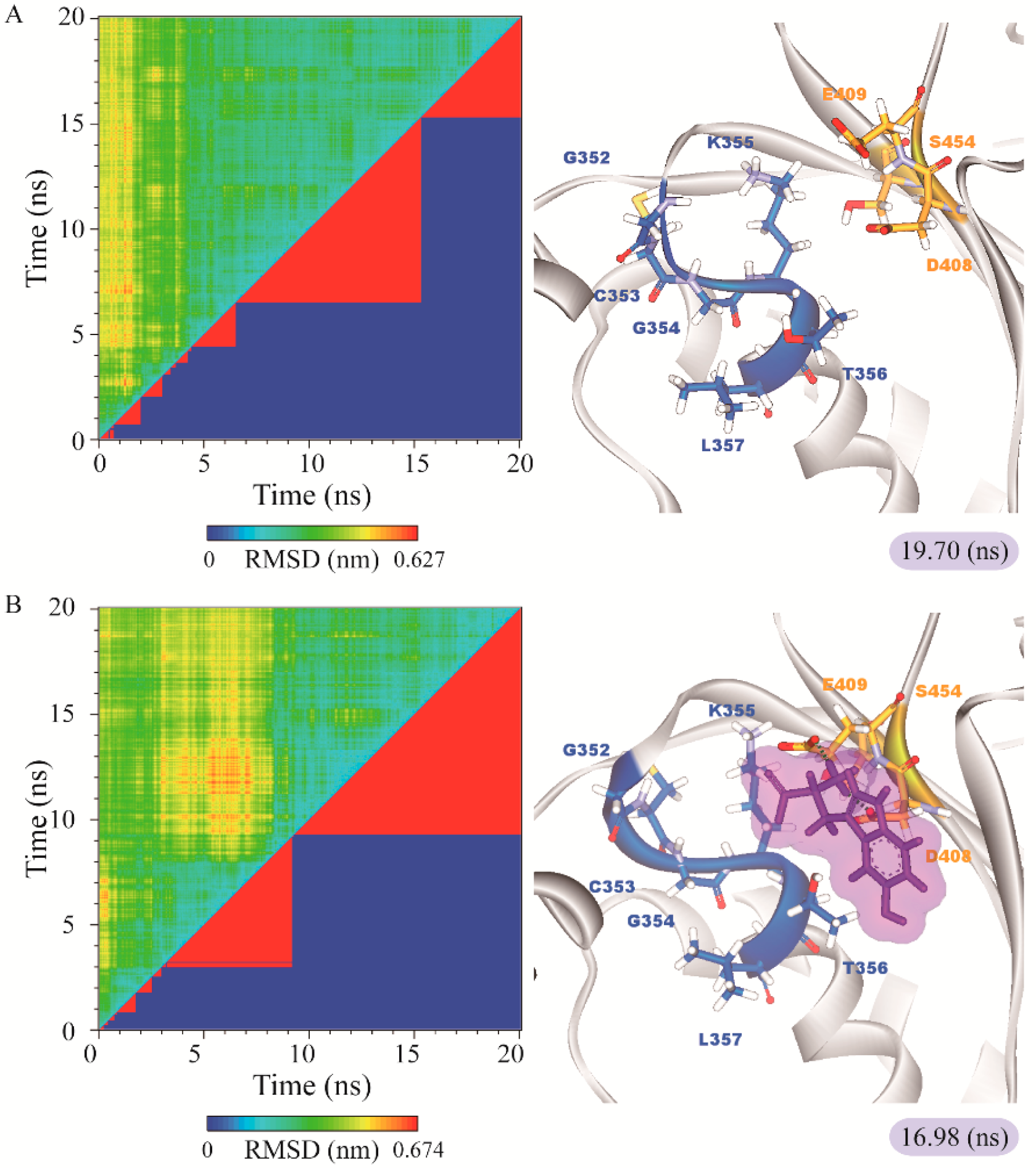
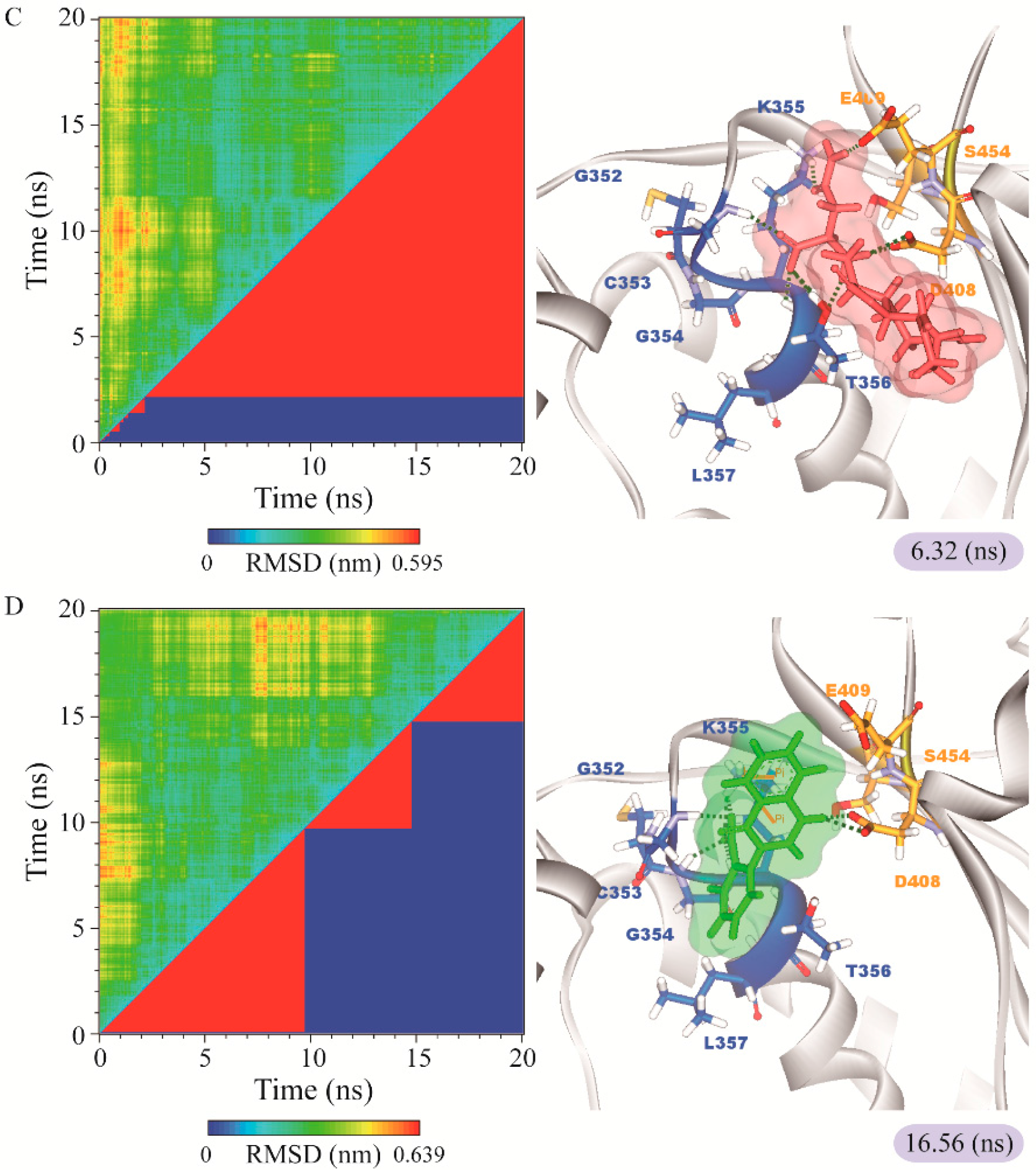
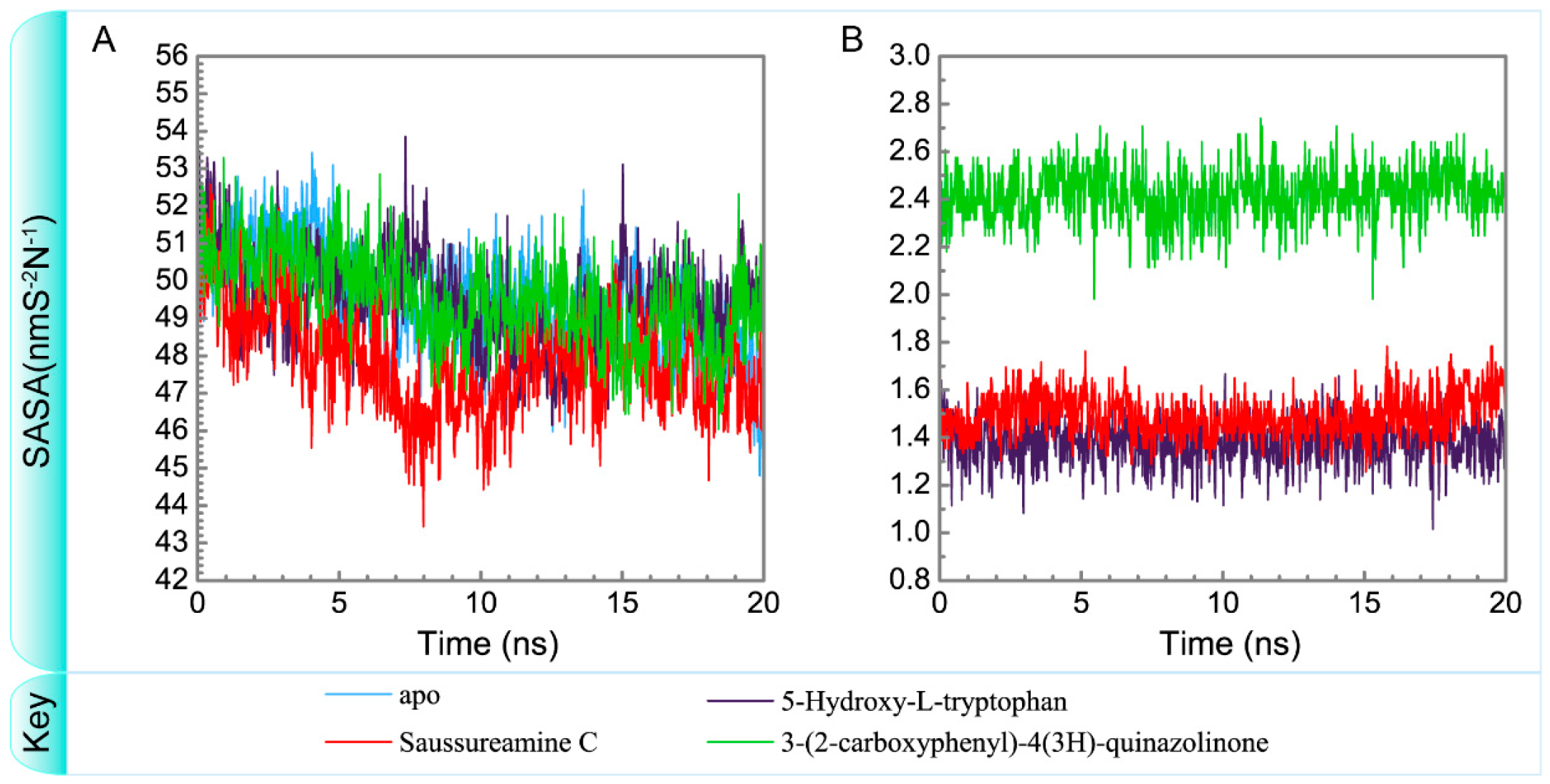
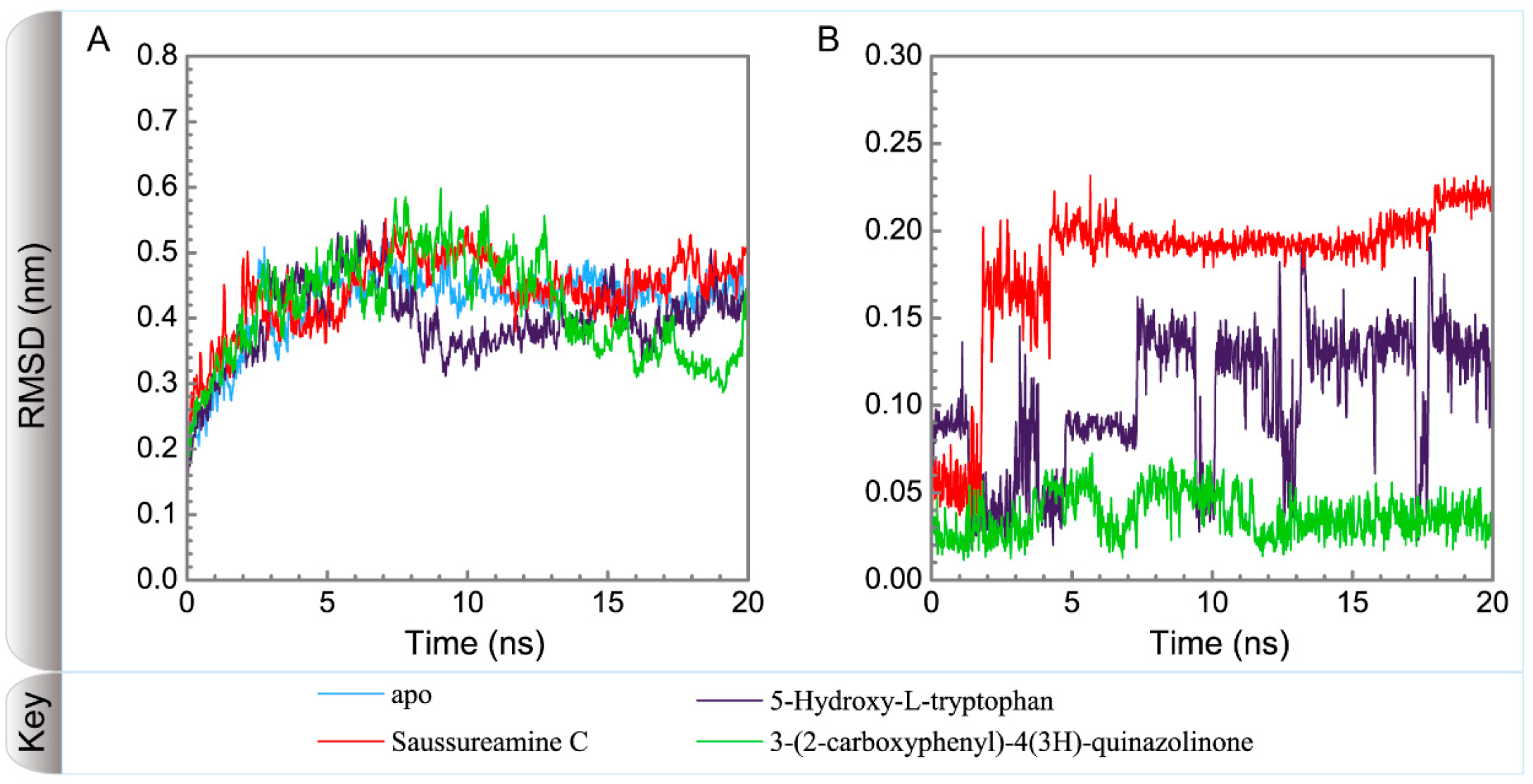
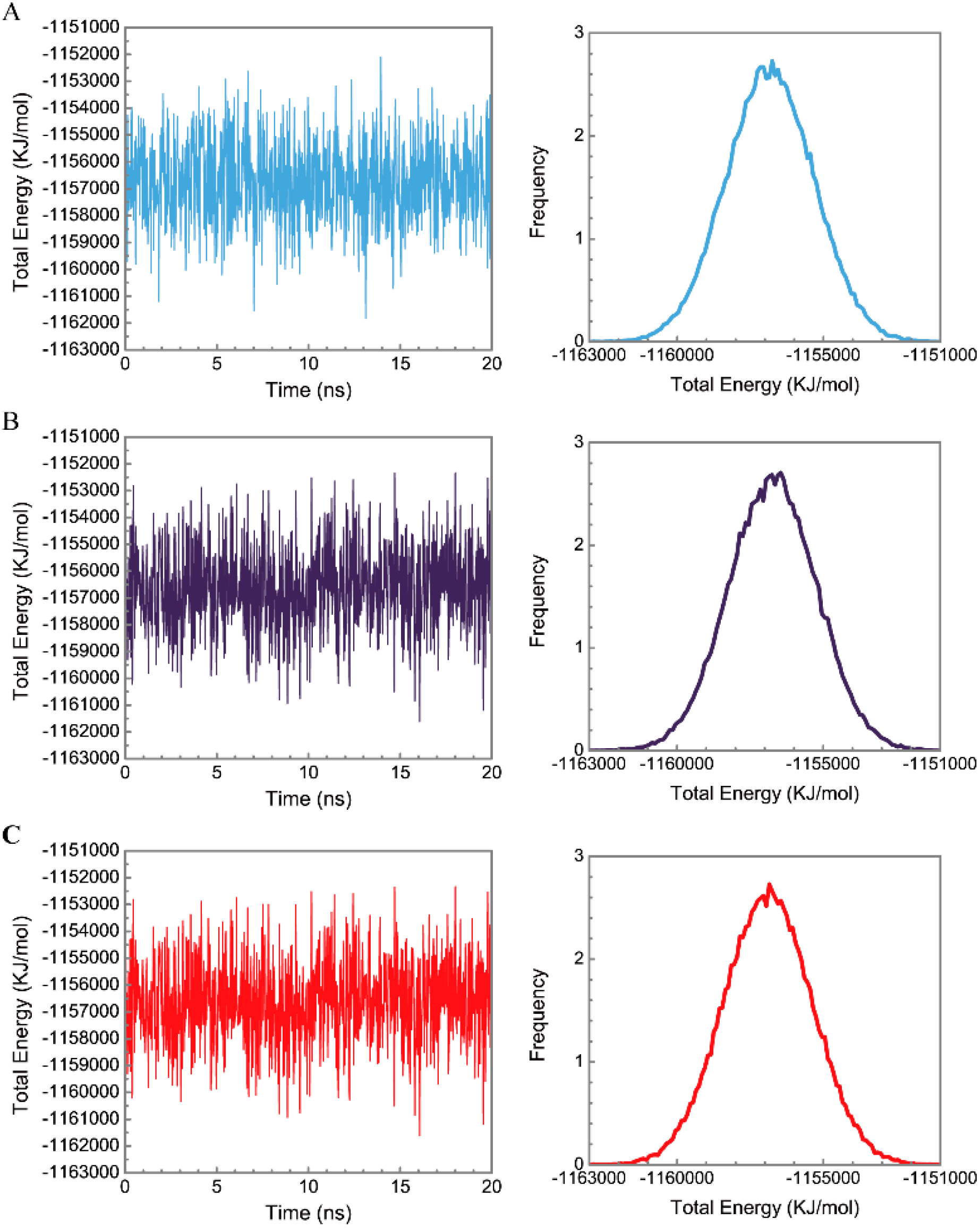
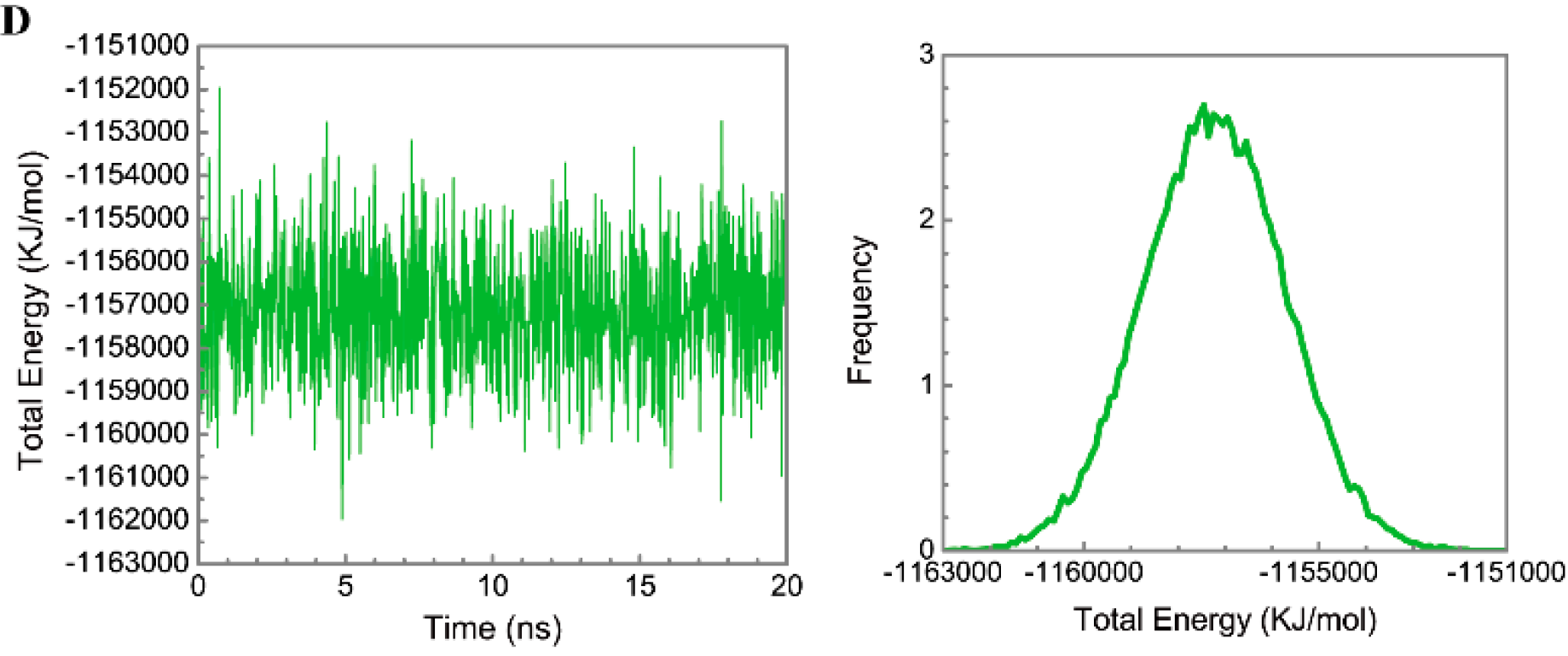
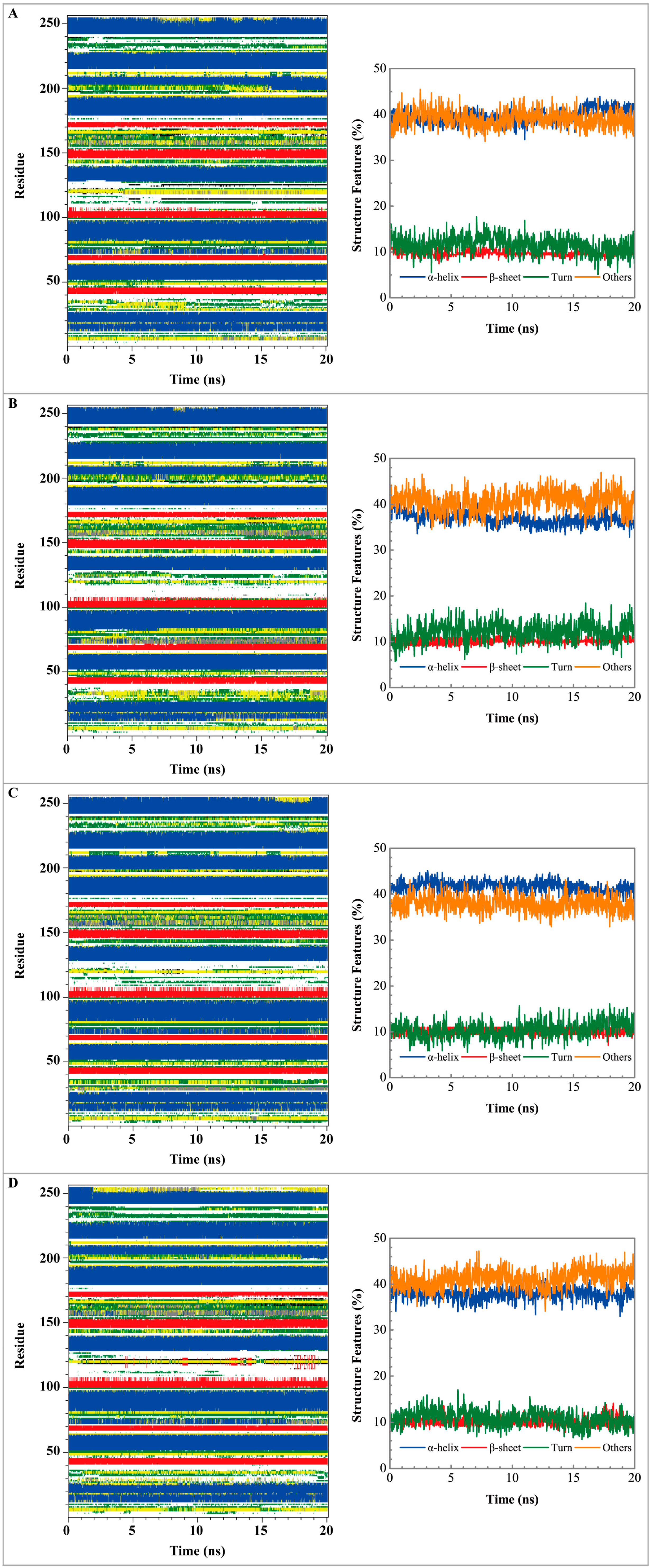
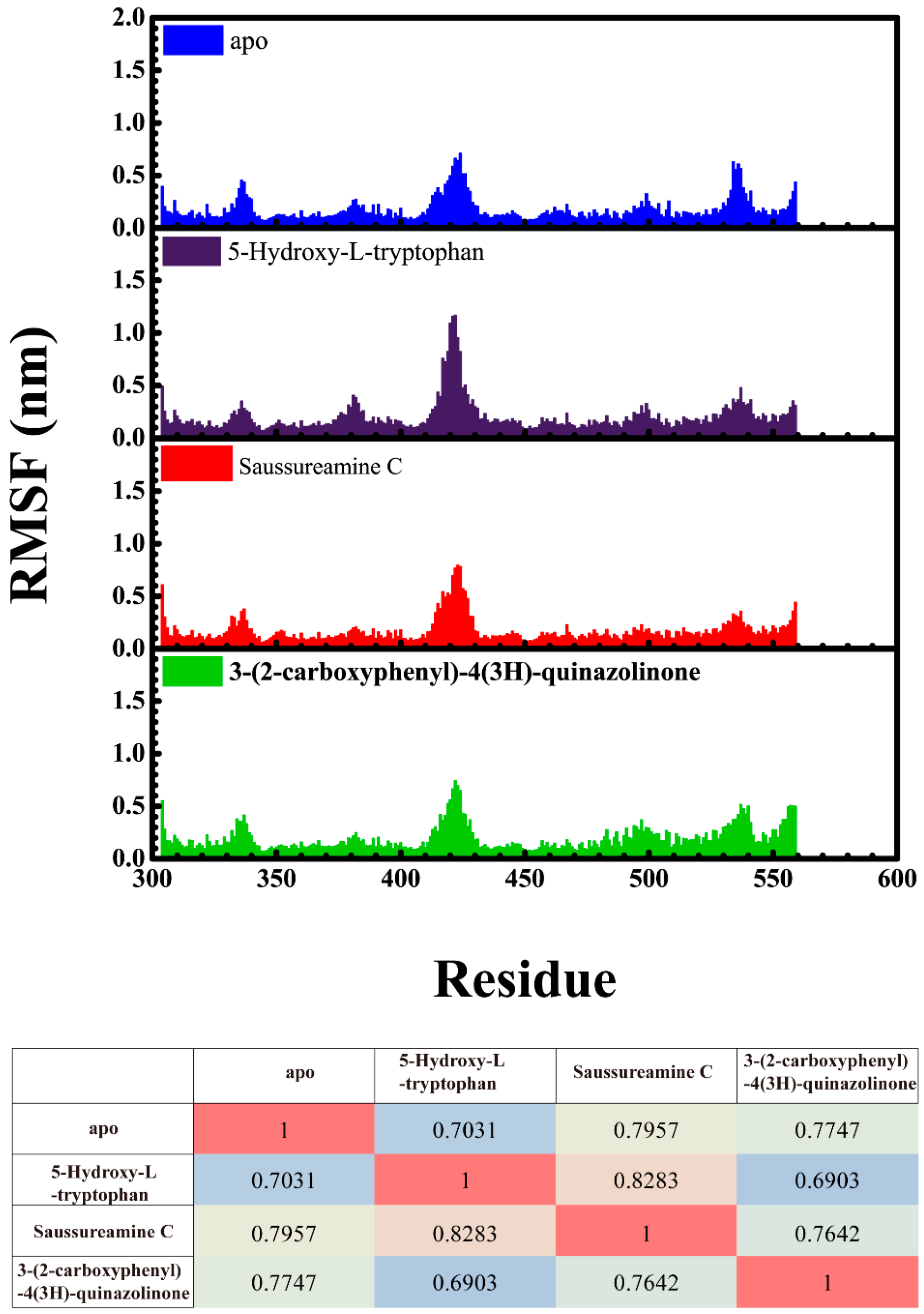
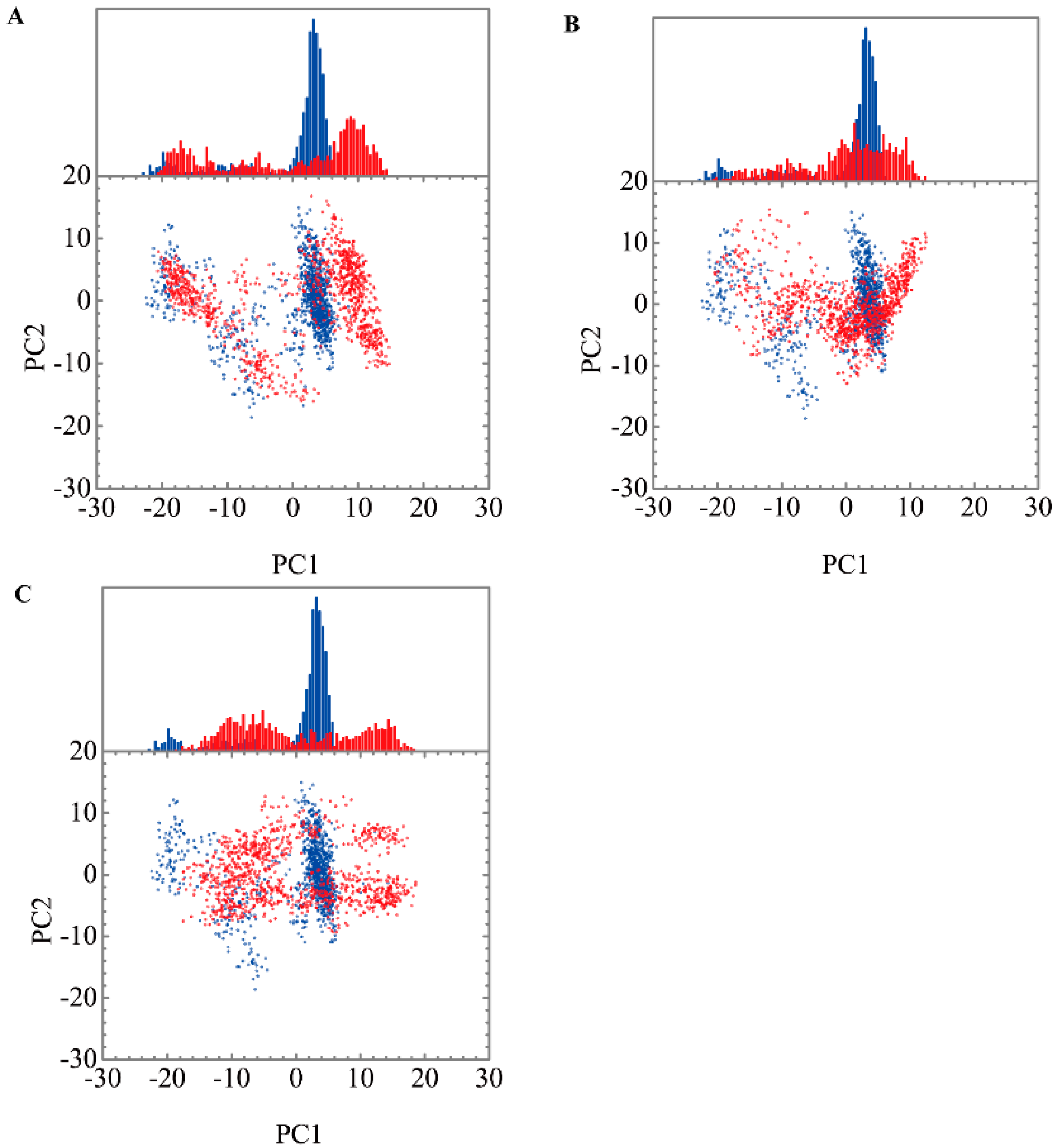
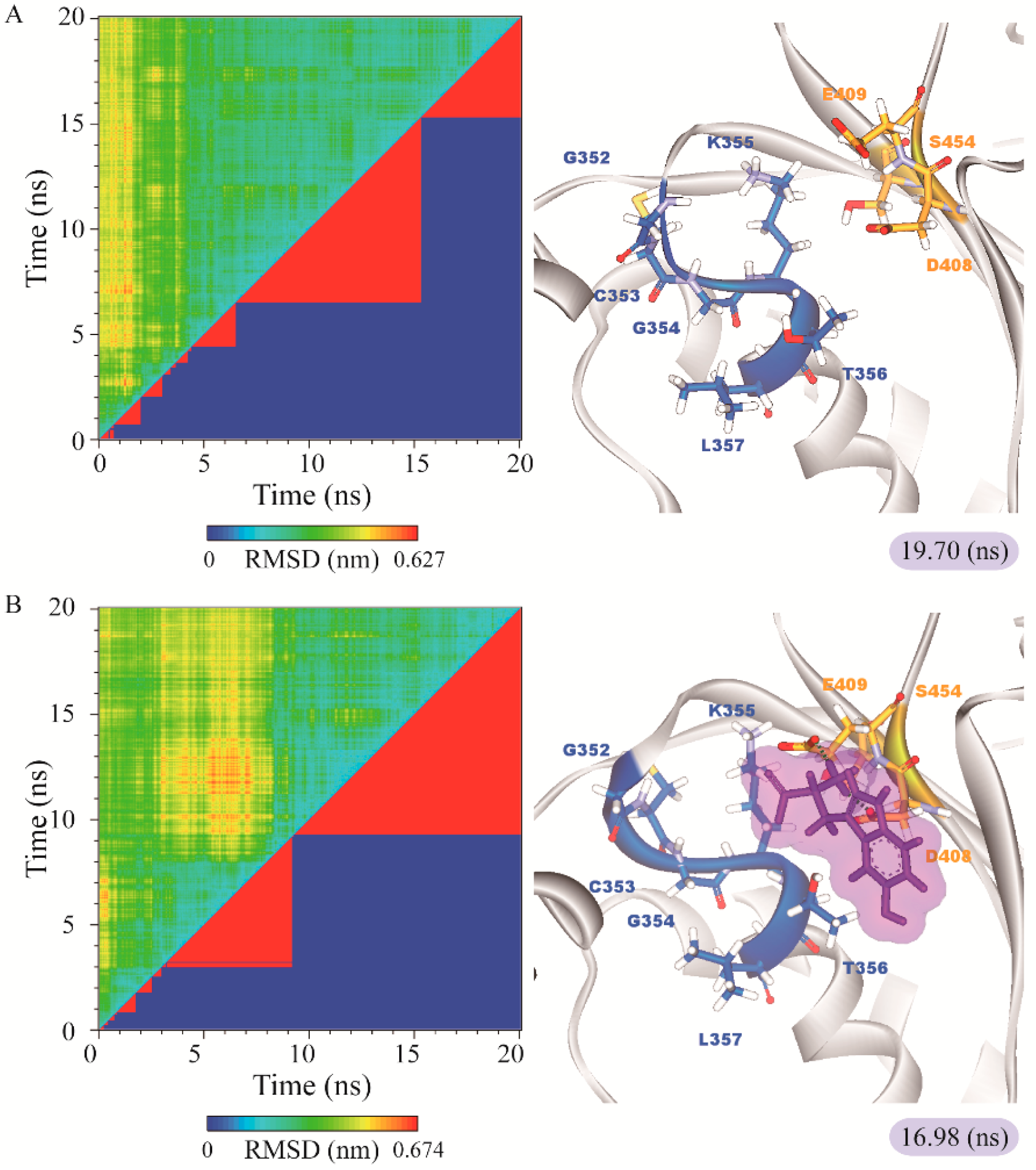
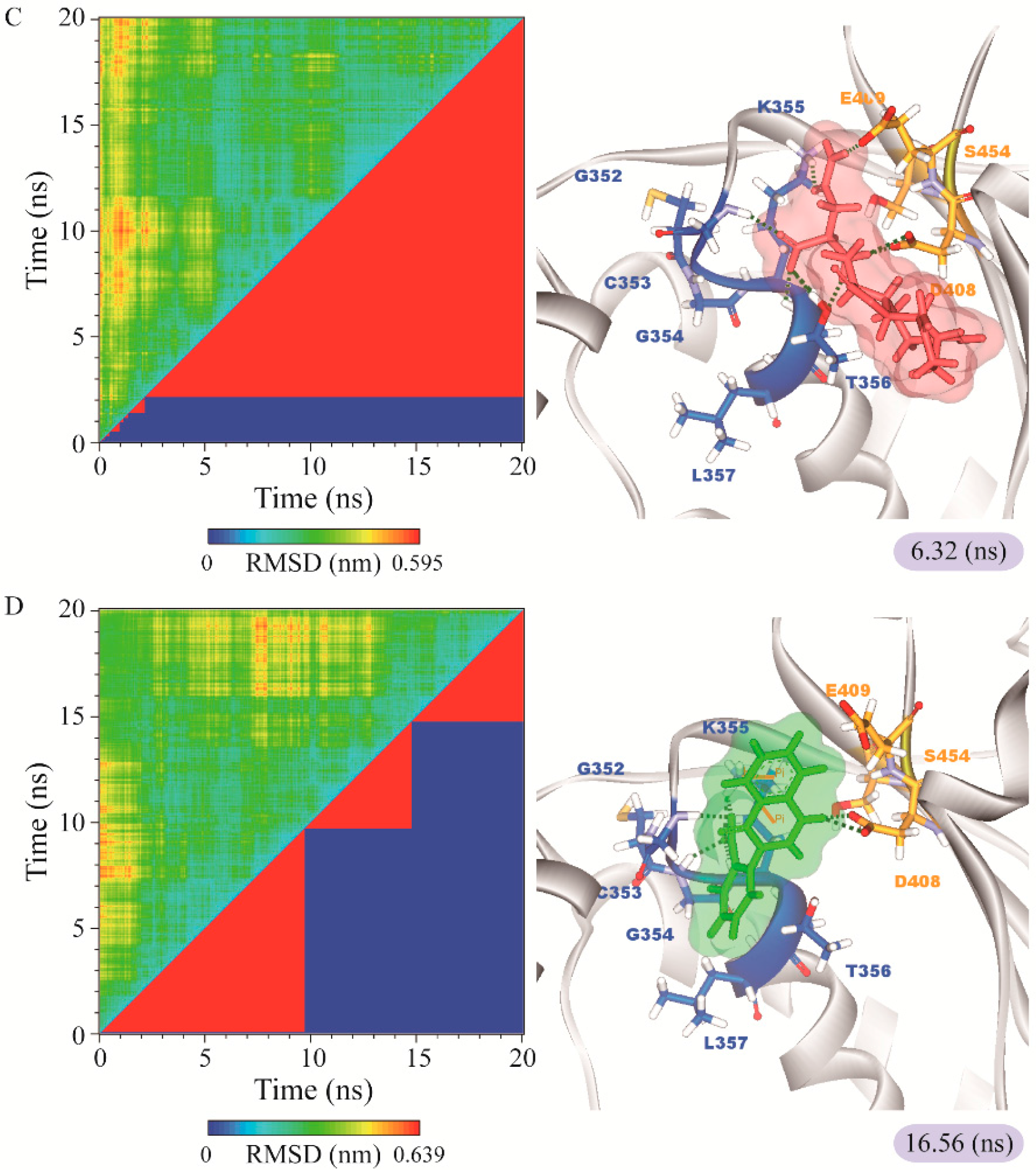
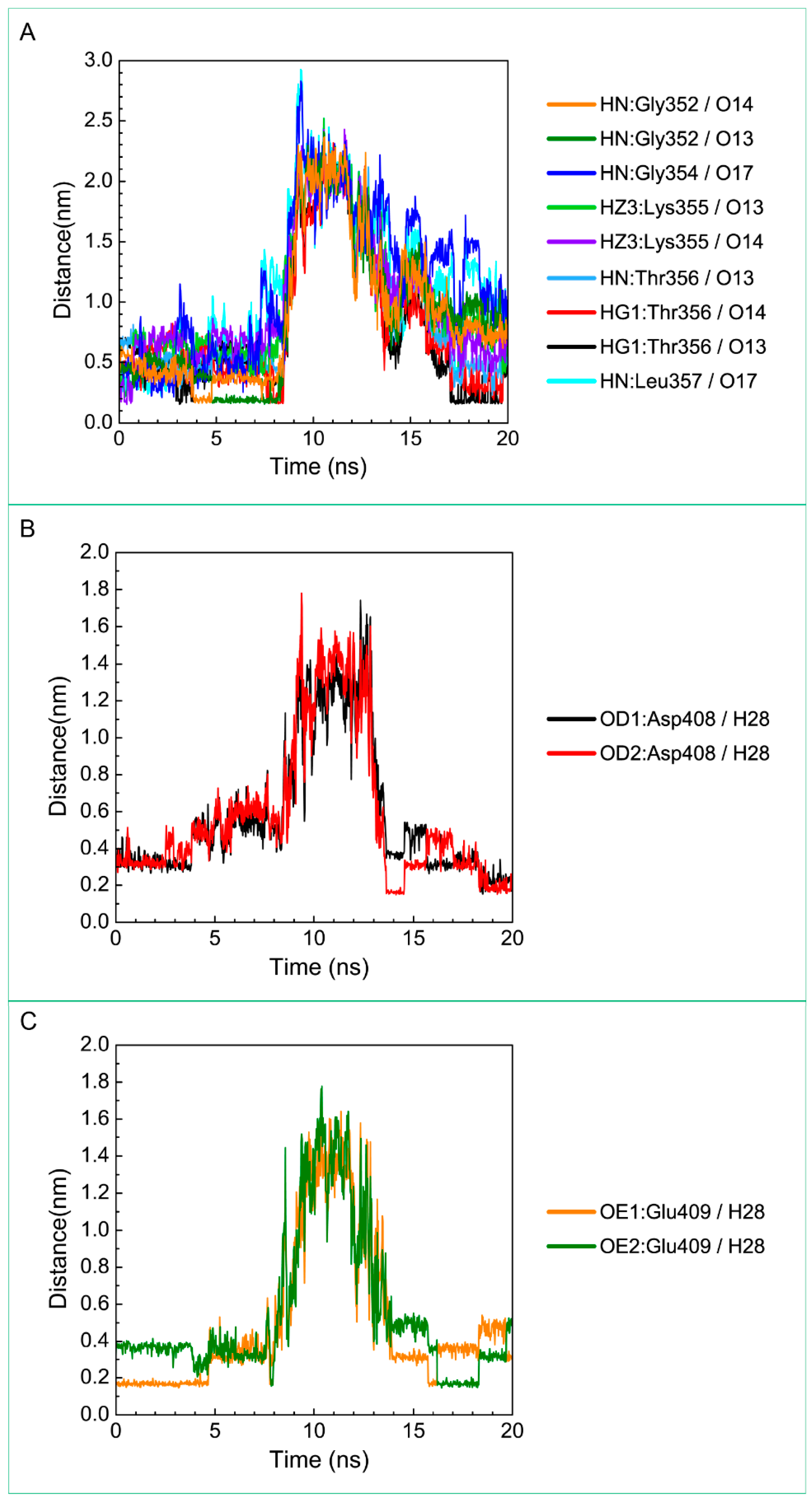
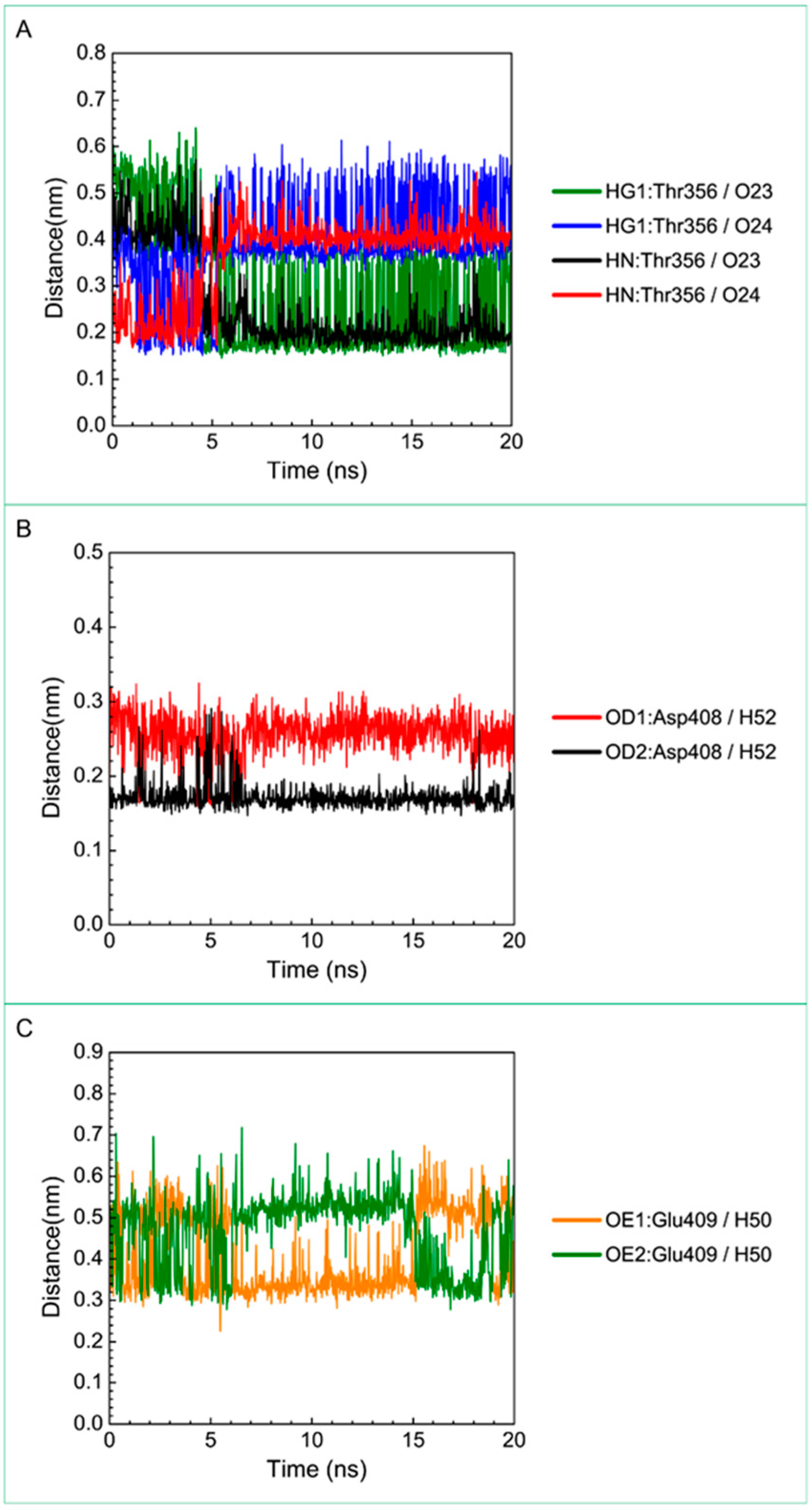
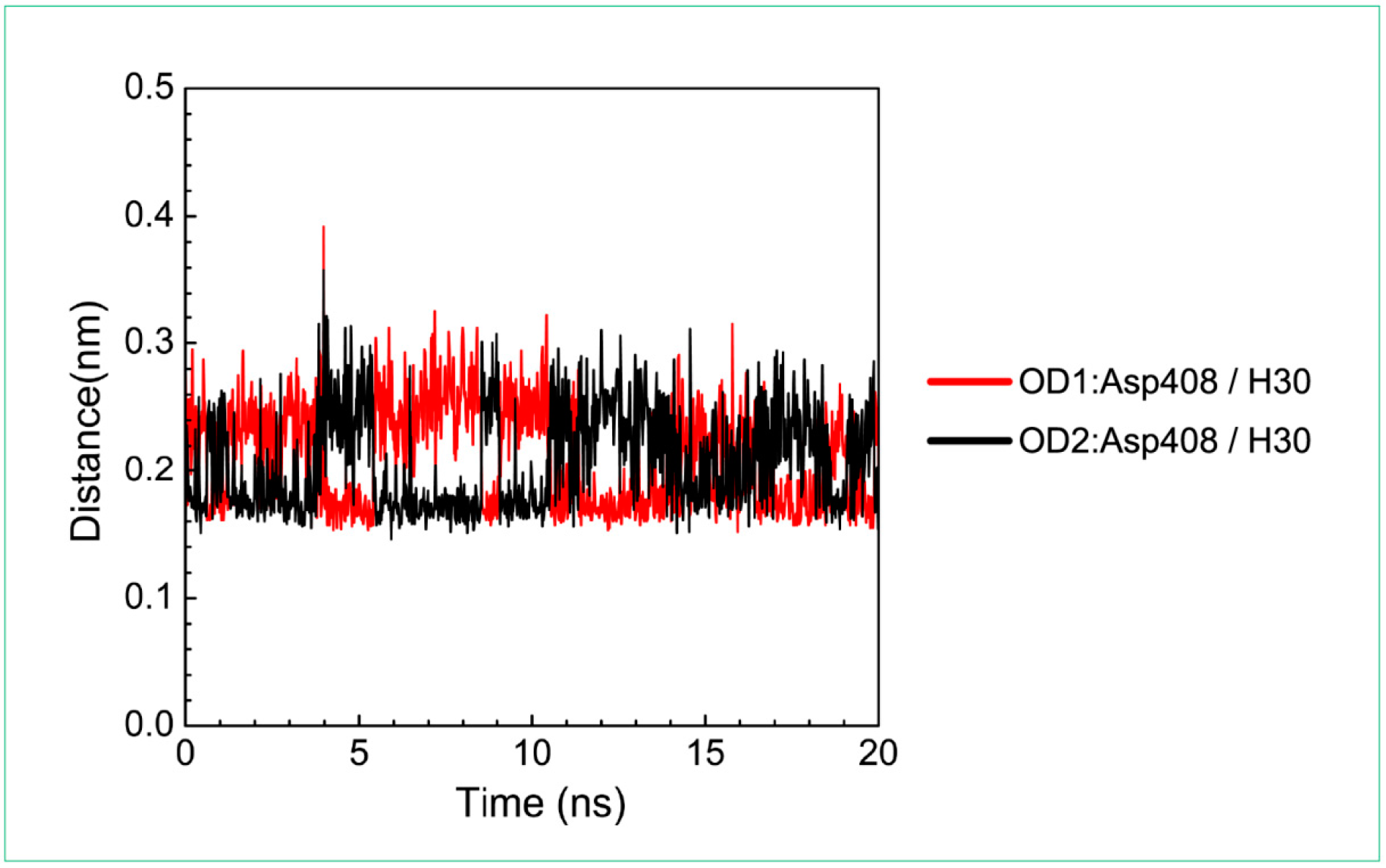
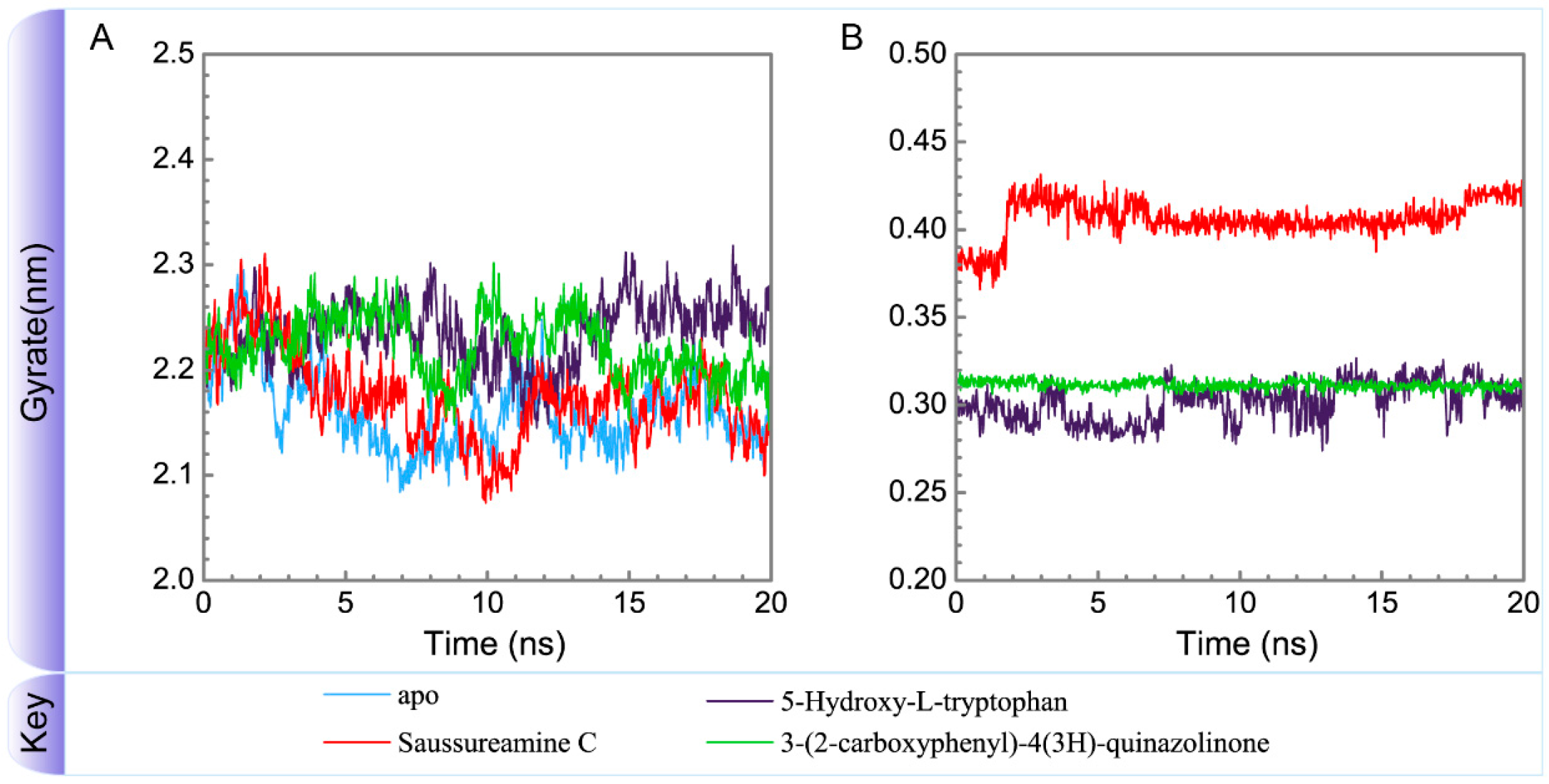
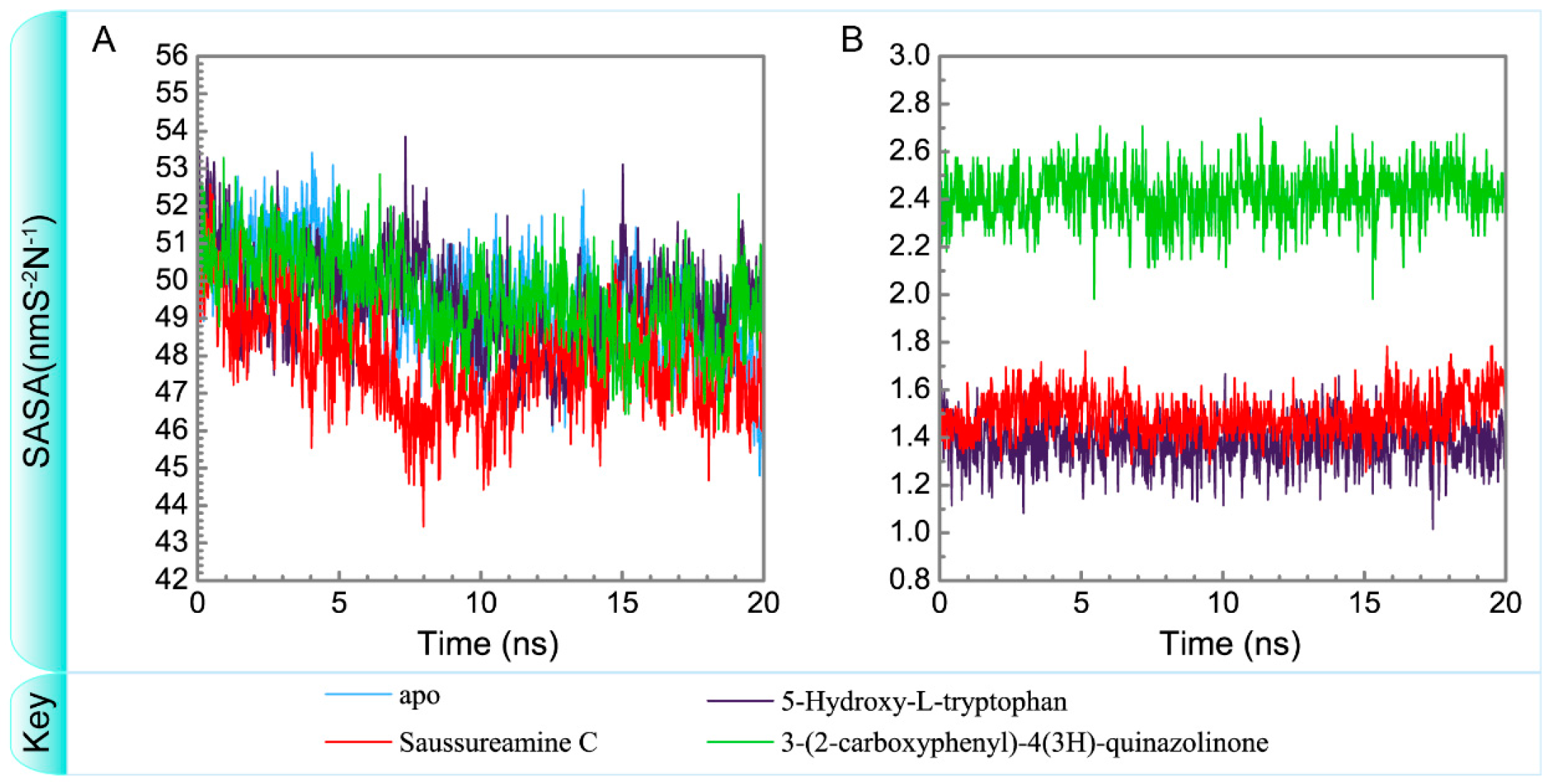
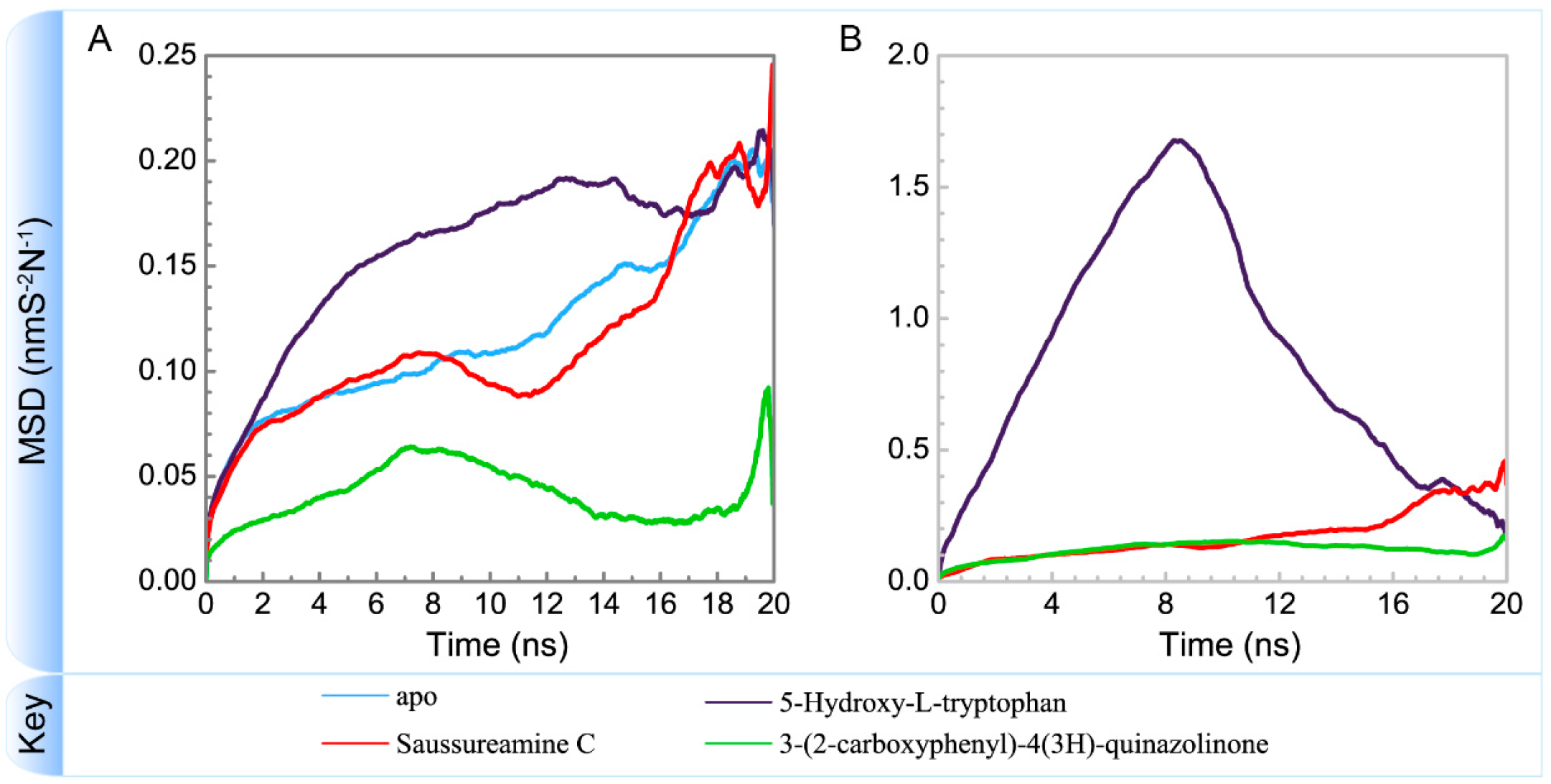
3.3. Molecular Dynamics Simulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Djousse, L.; Gaziano, J.M. Dietary cholesterol and coronary artery disease: A systematic review. Curr. Atheroscler. Rep. 2009, 11, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Faxon, D.P.; Creager, M.A.; Smith, S.C.; Pasternak, R.C.; Olin, J.W.; Bettmann, M.A.; Criqui, M.H.; Milani, R.V.; Loscalzo, J.; Kaufman, J.A.; et al. Atherosclerotic vascular disease conference—Executive summary—Atherosclerotic vascular disease conference proceeding for healthcare professionals from a special writing group of the American Heart Association. Circulation 2004, 109, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Oliver, M.F. Diet and coronary heart disease. Hum. Nutr. Clin. Nutr. 1982, 36, 413–427. [Google Scholar] [PubMed]
- Finegold, J.A.; Asaria, P.; Francis, D.P. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int. J. Cardiol. 2013, 168, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Newman, H.N.; Battino, M. Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: A shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol. 2000 2014, 64, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.R.; Cheng, K.K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010, 107, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Victor, V.M.; Apostolova, N.; Herance, R.; Hernandez-Mijares, A.; Rocha, M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: Mitochondria-targeted antioxidants as potential therapy. Curr. Med. Chem. 2009, 16, 4654–4667. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Harrison, C.M.; Chuang, G.C.; Ballinger, S.W. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat. Res. 2007, 621, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Lin, F.-Y.; Huang, P.-H.; Chen, Y.-L.; Chen, W.-C.; Chen, H.-Y.; Huang, Y.-C.; Liao, W.-L.; Huang, H.-C.; Liu, P.-L.; et al. Endothelial Progenitor Cell Dysfunction in Cardiovascular Diseases: Role of Reactive Oxygen Species and Inflammation. BioMed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Ho, T.J.; Lin, T.H.; Hsu, W.Y.; Huang, S.M.; Liao, C.C.; Lai, C.H.; Liu, X.; Tsang, H.; Lai, C.C.; et al. P-coumaric acid regulates exon 12 splicing of the ATP7B gene by modulating hnRNP A1 protein expressions. Biomedicine 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, X.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; Wei, C.; Guo, F.; Chen, Y.; et al. PKM2 Regulates Chromosome Segregation and Mitosis Progression of Tumor Cells. Mol. Cell 2014, 53, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y. EZH2: Novel therapeutic target for human cancer. Biomedicine 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Almontashiri, N.A.M.; Chen, H.H.; Mailloux, R.J.; Tatsuta, T.; Teng, A.C.T.; Mahmoud, A.B.; Ho, T.; Stewart, N.A.S.; Rippstein, P.; Harper, M.E.; et al. SPG7 Variant Escapes Phosphorylation-Regulated Processing by AFG3L2, Elevates Mitochondrial ROS, and Is Associated with Multiple Clinical Phenotypes. Cell Rep. 2014, 7, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Winsvold, B.S.; Nelson, C.P.; Malik, R.; Gormley, P.; Anttila, V.; Vander Heiden, J.; Elliott, K.S.; Jacobsen, L.M.; Palta, P.; Amin, N.; et al. Genetic analysis for a shared biological basis between migraine and coronary artery disease. Neurol. Genet. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef]
- De Michele, G.; De Fusco, M.; Cavalcanti, F.; Filla, A.; Marconi, R.; Volpe, G.; Monticelli, A.; Ballabio, A.; Casari, G.; Cocozza, S. A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3. Am. J. Hum. Genet. 1998, 63, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Juhola, M.K.; Shah, Z.H.; Grivell, L.A.; Jacobs, H.T. The mitochondrial inner membrane AAA metalloprotease family in metazoans. FEBS Lett. 2000, 481, 91–95. [Google Scholar] [CrossRef]
- Karlberg, T.; van den Berg, S.; Hammarstrom, M.; Sagemark, J.; Johansson, I.; Holmberg-Schiavone, L.; Schuler, H. Crystal structure of the ATPase domain of the human AAA+ protein paraplegin/SPG7. PLoS ONE 2009, 4, e6975. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Liu, S.P.; Lin, H.L.; Chan, M.C.; Chen, Y.C.; Huang, Y.L.; Tsai, M.C.; Fu, R.H. Irisflorentin improves alpha-synuclein accumulation and attenuates 6-OHDA-induced dopaminergic neuron degeneration, implication for Parkinson’s disease therapy. Biomedicine 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.C.; Tung, Y.T.; Kuo, Y.H.; Liao, J.W.; Tsai, H.C.; Chong, K.Y.; Chen, H.L.; Chen, C.M. Anti-inflammatory effects of Antrodia camphorata, a herbal medicine, in a mouse skin ischemia model. J. Ethnopharmacol. 2015, 159, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Tseng, Y.H.; Wu, H.R.; Chu, F.H.; Kuo, Y.H.; Wang, S.Y. Anti-proliferation Effect on Human Breast Cancer Cells via Inhibition of pRb Phosphorylation by Taiwanin E Isolated from Eleutherococcus trifoliatus. Nat. Prod. Commun. 2014, 9, 1303–1306. [Google Scholar] [PubMed]
- Chien, S.C.; Tseng, Y.H.; Hsu, W.N.; Chu, F.H.; Chang, S.T.; Kuo, Y.H.; Wang, S.Y. Anti-inflammatory and Anti-oxidative Activities of Polyacetylene from Dendropanax dentiger. Nat. Prod. Commun. 2014, 9, 1589–1590. [Google Scholar] [PubMed]
- Cichero, E.; Espinoza, S.; Tonelli, M.; Franchini, S.; Gerasimov, A.S.; Sorbi, C.; Gainetdinov, R.R.; Brasili, L.; Fossa, P. A homology modelling-driven study leading to the discovery of the first mouse trace amine-associated receptor 5 (TAAR5) antagonists. Medchemcomm 2016, 7, 353–364. [Google Scholar] [CrossRef]
- Cichero, E.; Espinoza, S.; Franchini, S.; Guariento, S.; Brasili, L.; Gainetdinov, R.R.; Fossa, P. Further Insights Into the Pharmacology of the Human Trace Amine-Associated Receptors: Discovery of Novel Ligands for TAAR1 by a Virtual Screening Approach. Chem. Biol. Drug Des. 2014, 84, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.C. TCM database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in Silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Tou, W.I. How to design a drug for the disordered proteins? Drug Discov. Today 2013, 18, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, R. Optimization; Academic Press: New York, NY, USA; London, UK, 1969. [Google Scholar]
- Maiorov, V.N.; Crippen, G.M. Size-independent comparison of protein three-dimensional structures. Proteins 1995, 22, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Rawat, A.; Keshari, A.K.; Singh, A.K.; Maity, S.; De, A.; Samanta, A.; Saha, S. Antiproliferative effect of isolated isoquinoline alkaloid from Mucuna pruriens seeds in hepatic carcinoma cells. Nat. Prod. Res. 2016, 30, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.K.; Prakash, J.; Chouhan, S.; Westfall, S.; Verma, M.; Singh, T.D.; Singh, S.P. Comparison of the neuroprotective potential of Mucuna pruriens seed extract with estrogen in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mice model. Neurochem. Int. 2014, 65. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.K.; De, A.; Bhadra, S.; Mukherjee, P.K. Angiotensin-converting enzyme (ACE) inhibitory potential of standardized Mucuna pruriens seed extract. Pharm. Biol. 2015, 53, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Hatakeyama, S.; Inoue, Y.; Yamahara, J. Saussureamines A, B, C, D, and E, new anti-ulcer principles from Chinese Saussureae Radix. Chem. Pharm. Bull. 1993, 41, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.M.; Choi, S.I.; Kim, G.H. Anti-oxidant Activity of Saussurea lappa C.B. Clarke Roots. Prev. Nutr. Food Sci. 2012, 17, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Yaeesh, S.; Jamal, Q.; Shah, A.J.; Gilani, A.H. Antihepatotoxic activity of Saussurea lappa extract on d-galactosamine and lipopolysaccharide-induced hepatitis in mice. Phytother. Res. 2010, 24 (Suppl. S2), S229–S232. [Google Scholar] [CrossRef] [PubMed]
- Saleem, T.S.; Lokanath, N.; Prasanthi, A.; Madhavi, M.; Mallika, G.; Vishnu, M.N. Aqueous extract of Saussurea lappa root ameliorate oxidative myocardial injury induced by isoproterenol in rats. J. Adv. Pharm. Technol. Res. 2013, 4, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Sheng, W.; Sun, J.; Qin, G. Chemical constituents of Isatis indigotica. Planta Med. 1997, 63, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Chen, M.H.; Guo, Q.L.; Lin, S.; Xu, C.B.; Jiang, Y.P.; Li, Y.H.; Jiang, J.D.; Shi, J.G. Antiviral glycosidic bisindole alkaloids from the roots of Isatis indigotica. J. Asian Nat. Prod. Res. 2015, 17, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Chang, Y.C.; Lu, K.Z.; Tsou, Y.Y.; Lin, C.W. Antiviral Activity of Isatis indigotica Extract and Its Derived Indirubin against Japanese Encephalitis Virus. Evid. Based Complement. Altern. Med. 2012, 2012. [Google Scholar] [CrossRef]
- Chung, Y.C.; Tang, F.Y.; Liao, J.W.; Chung, C.H.; Jong, T.T.; Chen, S.S.; Tsai, C.H.; Chiang, E.P. Isatis indigotica induces hepatocellular cancer cell death via caspase-independent apoptosis-inducing factor translocation apoptotic pathway in vitro and in vivo. Integr. Cancer Ther. 2011, 10, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, G.; Wang, M.; Jiang, H.; Chen, L.; Zhao, F.; Qiu, F. Indole alkaloids from the roots of Isatis indigotica and their inhibitory effects on nitric oxide production. Fitoterapia 2014, 95, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Dock Score | H-Bond Forming Residues | H-Bond Quantity |
|---|---|---|---|
| 5-Hydroxy-l-tryptophan | 203.573 | Lys355, Leu357, Asp408, Glu409 | 4 |
| Saussureamine C | 188.993 | Lys355, Thr356, Asp408, Glu409, Ser454, Asn456 | 6 |
| 3-(2-Carboxyphenyl)-4(3H)-quinazolinone | 180.181 | Gly352, Lys355, Asp408, Glu409 | 4 |
| Saussureamine B | 164.449 | Lys355, Thr356 | 2 |
| Crotalaburnine | 160.377 | Asp408, Glu409 | 2 |
| Labiatic acid | 154.729 | Gly354, Lys360, His492 | 3 |
| Saussureamine A | 152.977 | Gly352, Lys355, Asp408 | 3 |
| l-Valine-l-valine anhydride | 149.351 | Gly352, Asp408, Glu409 | 3 |
| N-Methyl tyramine-O-alpha-l-rhamnopyranoside | 147.058 | Gly354, Asp408, Glu409 | 3 |
| Riddelline | 146.166 | Gly352, Asp408, Glu409 | 3 |
| Name | H-Bond Interaction | Occupancy |
|---|---|---|
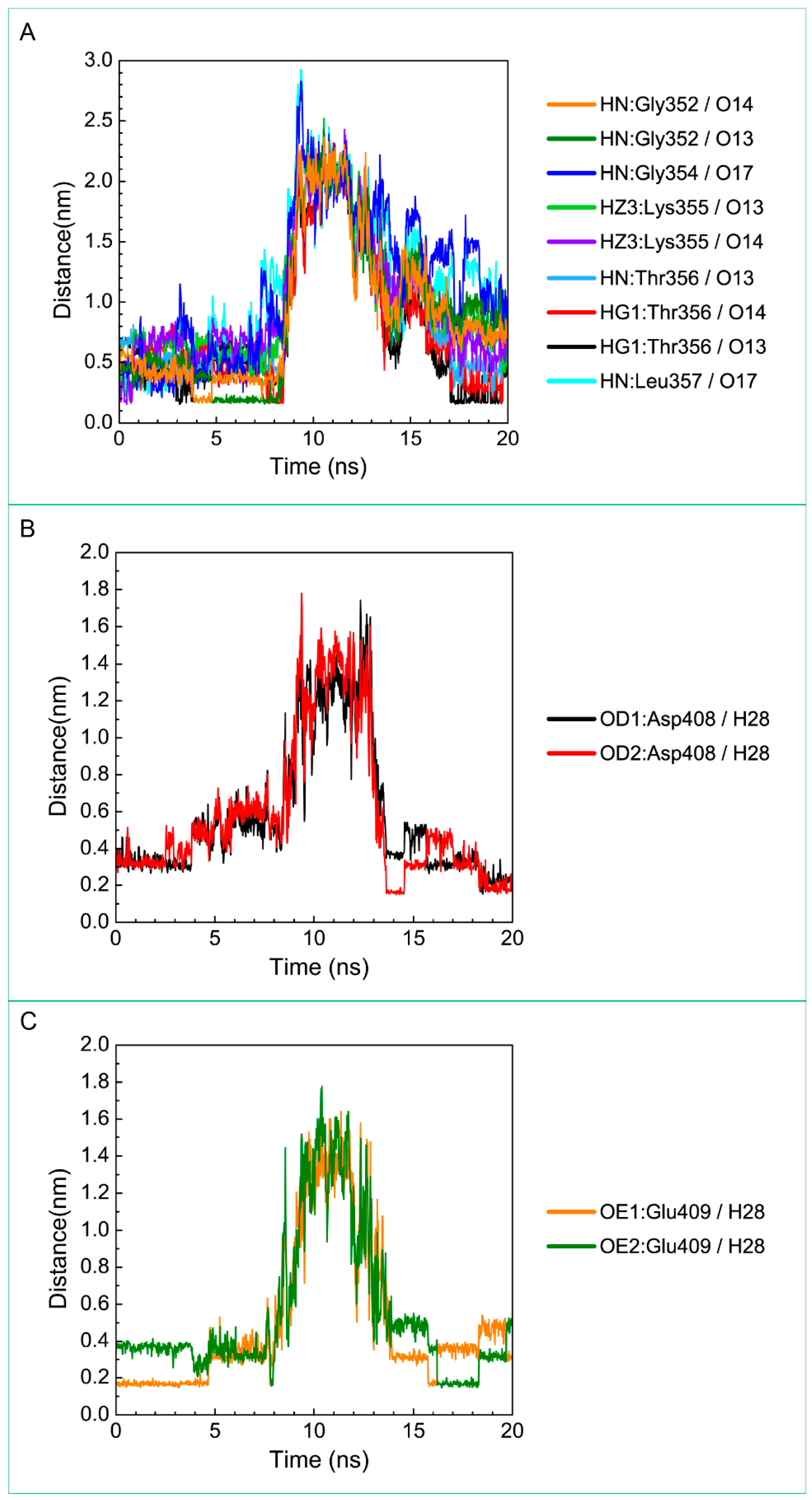
| 5-Hydroxy-l-tryptophan | Gly352: HN/O13 | 19.00% |
| Gly352: HN/O14 | 12.80% | |
| Thr356: HG1/O13 | 18.00% | |
| Thr356: HG1/O14 | 13.30% | |
| Glu377: OE2/H28 | 10.00% | |
| Asp408: OD1/H28 | 36.60% | |
| Asp408: OD2/H28 | 38.00% | |
| Glu409: OE1/H28 | 51.50% | |
| Glu409: OE2/H28 | 37.10% | |
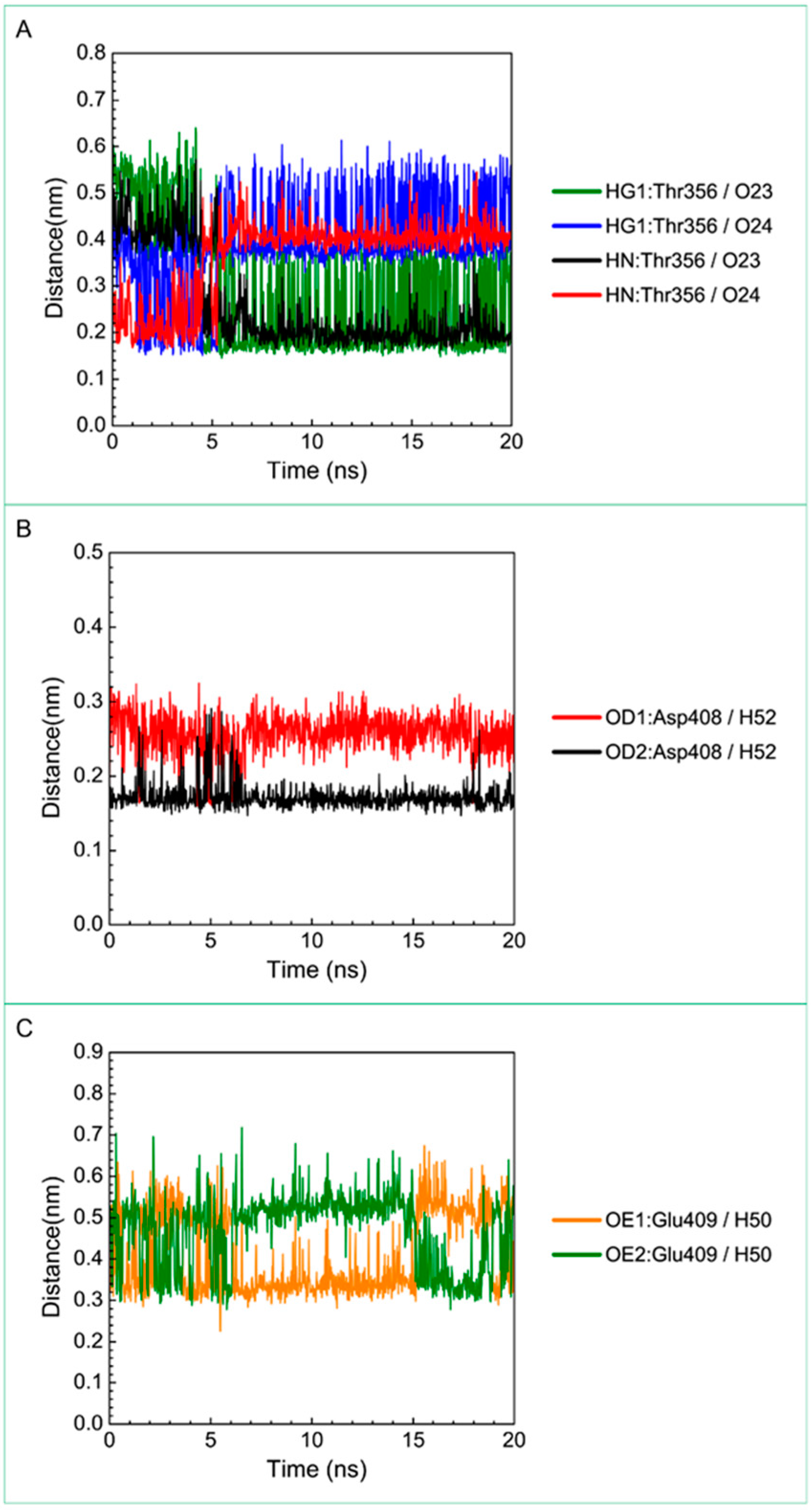
| Saussureamine C | Gly352: HN/O23 | 30.77% |
| Gly352: HN/O24 | 69.83% | |
| Thr356: HG1/O23 | 62.74% | |
| Thr356: HG1/O24 | 15.48% | |
| Thr356: HN/O23 | 75.72% | |
| Thr356:HN/O24 | 23.98% | |
| Thr356: OG1/H52 | 61.64% | |
| Asp408: OD1/H52 | 100.00% | |
| Asp408: OD2/H52 | 100.00% | |
| Glu409: OE1/H50 | 46.15% | |
| Glu409: OE2/H50 | 19.78% | |
| Ser454: HG1/O28 | 76.02% | |
| Asn456: HD22/N27 | 99.70% | |
| Asn456: OD1/H50 | 100.00% | |
| 3-(2-carboxyphenyl)-4(3H)-quinazolinone | Gly352: HN/O11 | 95.50% |
| Gly352: HN/O19 | 81.70% | |
| Gly352: HN/O20 | 88.90% | |
| Cys353: HN/O19 | 34.40% | |
| Cys353: HN/O20 | 97.10% | |
| Gly354: HN/O20 | 96.60% | |
| Lys355: HZ3/O19 | 85.70% | |
| Lys355: HZ3/O20 | 52.40% | |
| Lys355: HN/O20 | 99.40% | |
| Thr356: HN/O20 | 51.30% | |
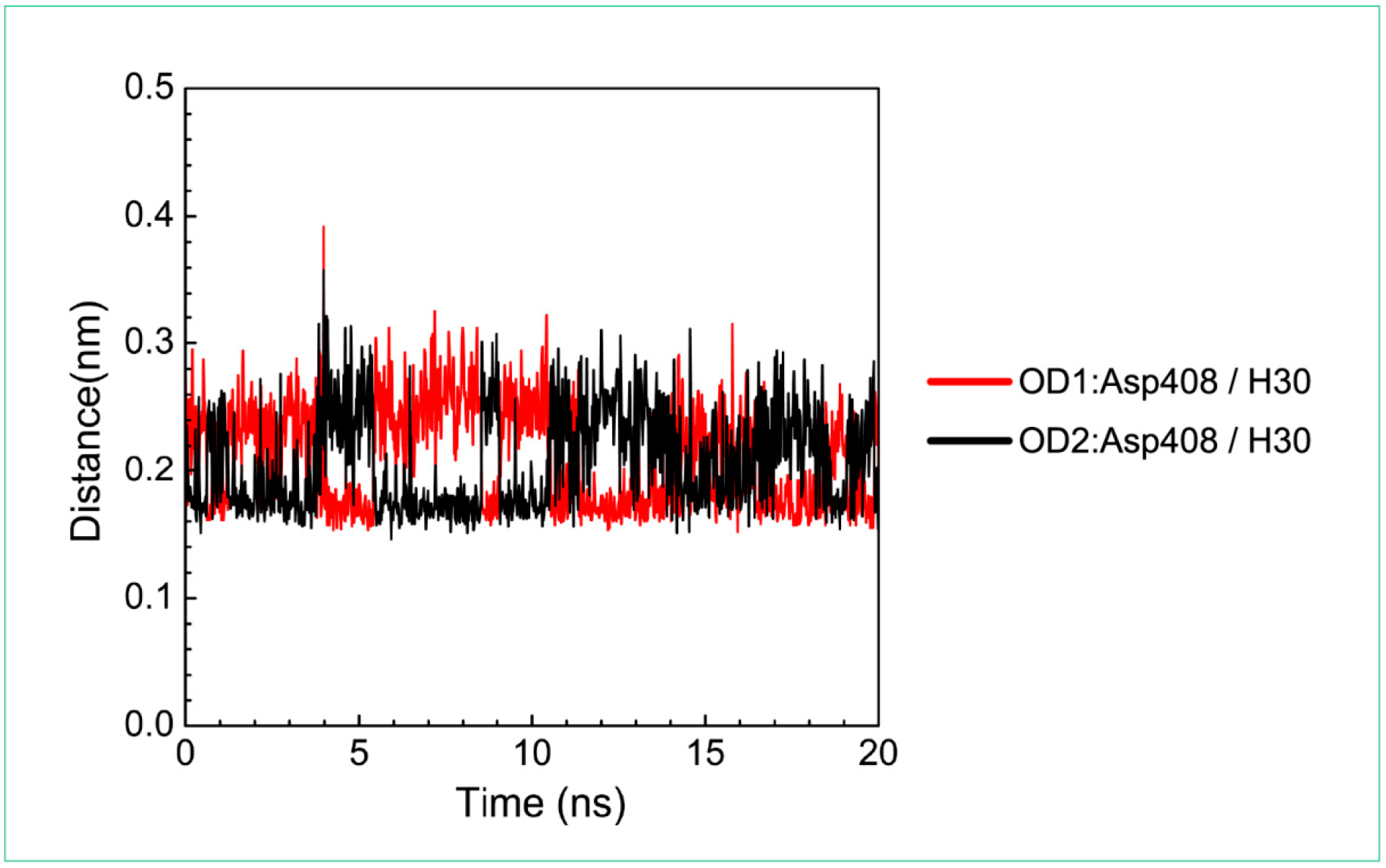
| Asp408: OD1/H30 | 100.00% | |
| Asp408: OD2/H30 | 100.00% |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.-B.; Chen, K.-C.; Chang, Y.-L.; Chang, K.-L.; Chang, P.-C.; Chang, T.-T.; Chen, Y.-C. In Silico Investigation of Traditional Chinese Medicine for Potential Lead Compounds as SPG7 Inhibitors against Coronary Artery Disease. Molecules 2016, 21, 588. https://doi.org/10.3390/molecules21050588
Chen K-B, Chen K-C, Chang Y-L, Chang K-L, Chang P-C, Chang T-T, Chen Y-C. In Silico Investigation of Traditional Chinese Medicine for Potential Lead Compounds as SPG7 Inhibitors against Coronary Artery Disease. Molecules. 2016; 21(5):588. https://doi.org/10.3390/molecules21050588
Chicago/Turabian StyleChen, Kuen-Bao, Kuan-Chung Chen, Ya-Lin Chang, Kun-Lung Chang, Pei-Chun Chang, Tung-Ti Chang, and Yu-Chian Chen. 2016. "In Silico Investigation of Traditional Chinese Medicine for Potential Lead Compounds as SPG7 Inhibitors against Coronary Artery Disease" Molecules 21, no. 5: 588. https://doi.org/10.3390/molecules21050588
APA StyleChen, K.-B., Chen, K.-C., Chang, Y.-L., Chang, K.-L., Chang, P.-C., Chang, T.-T., & Chen, Y.-C. (2016). In Silico Investigation of Traditional Chinese Medicine for Potential Lead Compounds as SPG7 Inhibitors against Coronary Artery Disease. Molecules, 21(5), 588. https://doi.org/10.3390/molecules21050588