Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates


 ,
,
Abstract
:
1. Introduction

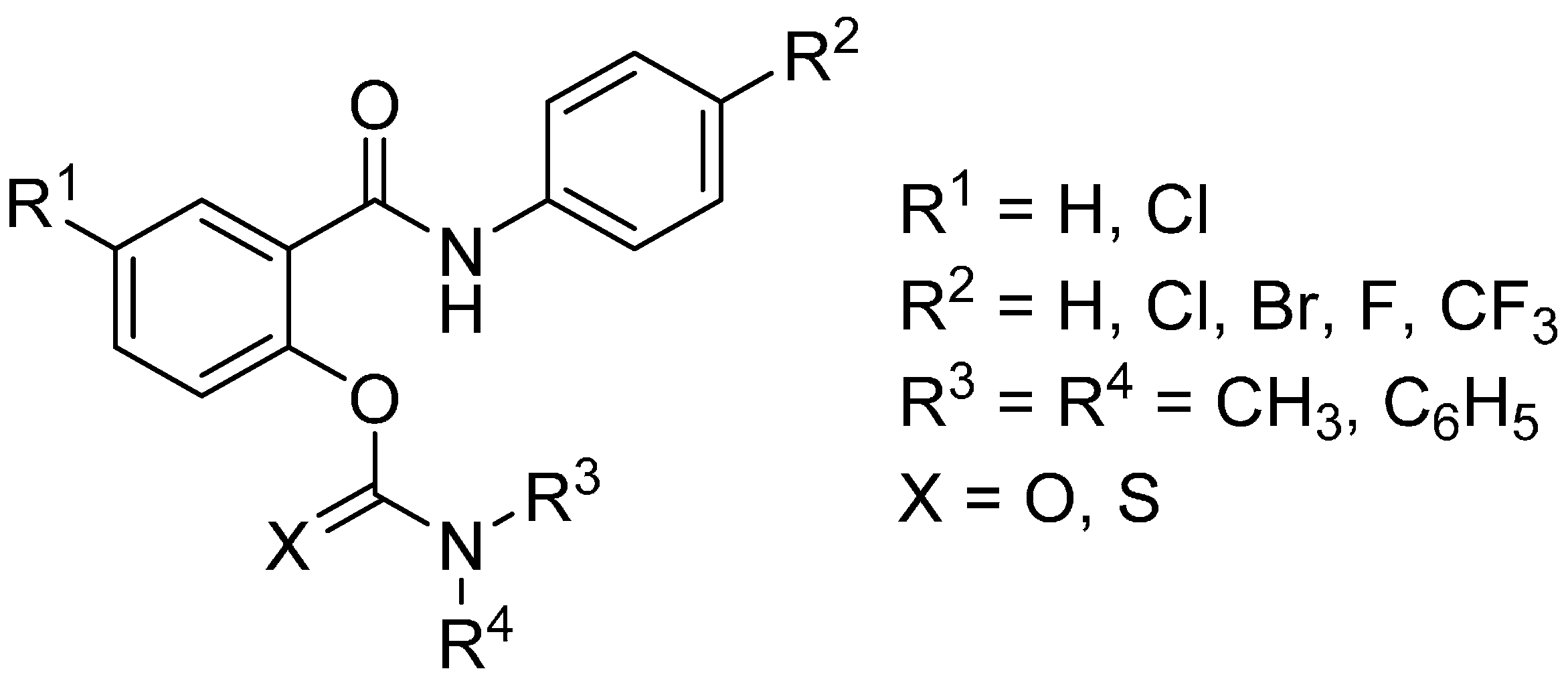
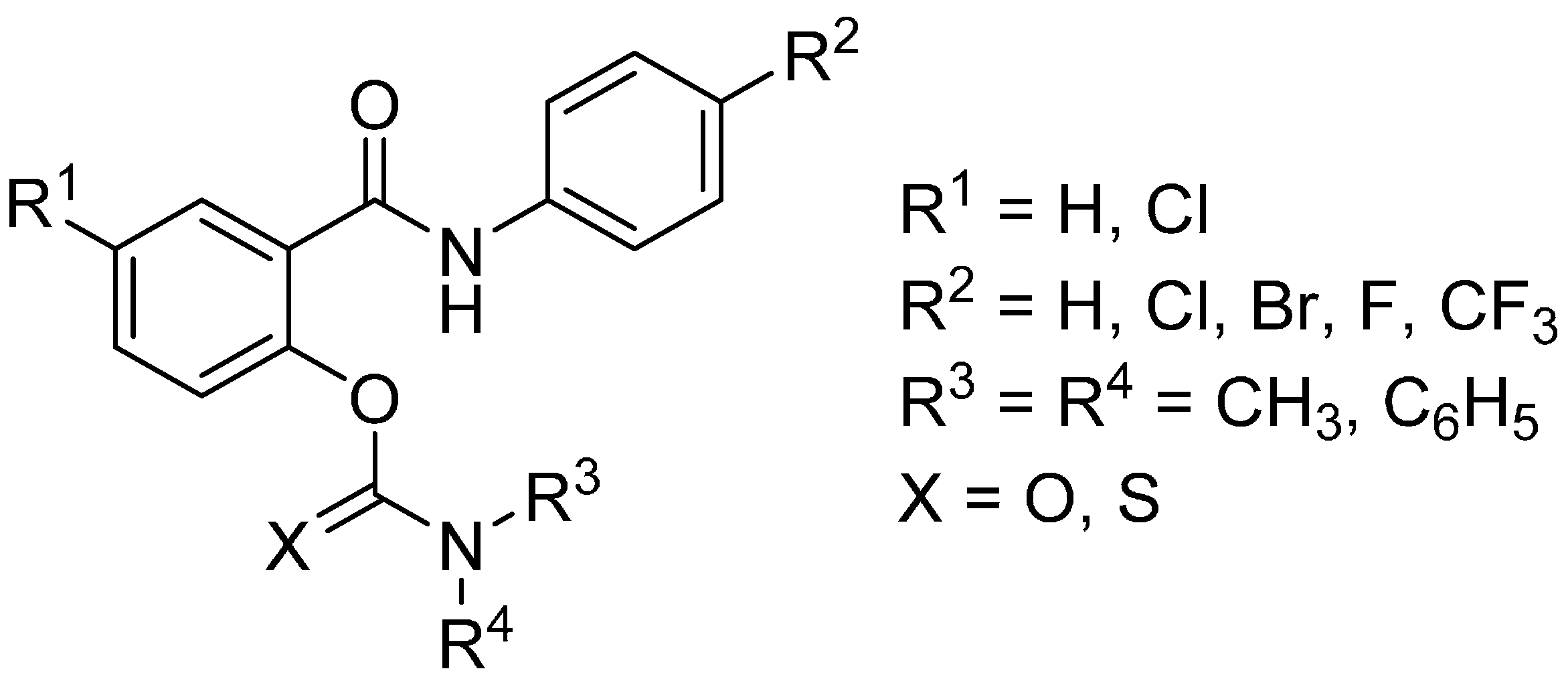
2. Results and Discussion
2.1. In Vitro Inhibition of Acetylcholinesterase and Butyrylcholinesterase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | R1 | R2 | R3 | R4 | X | IC50 for AChE [µM] | IC50 for BChE [µM] | Selectivity to BChE |
|---|---|---|---|---|---|---|---|---|
| 1a | Cl | Cl | CH3 | CH3 | O | 72.87 ± 1.06 | 88.42 ± 1.27 | 0.8 |
| 1b | Cl | Cl | CH3 | CH3 | S | 38.98 ± 1.11 | 51.38 ± 1.65 | 0.8 |
| 1c | Cl | Cl | CH3 | Ph | O | 74.83 ± 2.95 | 4.20 ± 0.18 | 17.8 |
| 1d | Cl | Cl | Ph | Ph | O | 54.81 ± 1.43 | 106.46 ± 3.64 | 0.5 |
| 2a | Cl | Br | CH3 | CH3 | O | 62.38 ± 1.36 | 54.21 ± 0.30 | 1.2 |
| 2b | Cl | Br | CH3 | CH3 | S | 59.05 ± 2.13 | 54.64 ± 0.42 | 1.1 |
| 2c | Cl | Br | CH3 | Ph | O | 81.60 ± 2.92 | 4.30 ± 0.51 | 19.0 |
| 2d | Cl | Br | Ph | Ph | O | 63.29 ± 0.95 | 144.80 ± 29.02 | 0.4 |
| 3a | Cl | F | CH3 | CH3 | O | 76.17 ± 2.87 | 311.00 ± 4.50 | 0.2 |
| 3b | Cl | F | CH3 | CH3 | S | 89.74 ± 4.68 | 91.37 ± 5.45 | 1.0 |
| 3c | Cl | F | CH3 | Ph | O | 62.54 ± 5.03 | 7.34 ± 0.14 | 8.5 |
| 3d | Cl | F | Ph | Ph | O | 49.03 ± 3.88 | 15.63 ± 0.11 | 3.1 |
| 4a | Cl | CF3 | CH3 | CH3 | O | 49.04 ± 0.54 | 28.14 ± 0.86 | 1.7 |
| 4b | Cl | CF3 | CH3 | CH3 | S | 41.98 ± 0.55 | 36.22 ± 0.28 | 1.2 |
| 4c | Cl | CF3 | CH3 | Ph | O | 49.85 ± 2.76 | 1.97 ± 0.02 | 25.3 |
| 4d | Cl | CF3 | Ph | Ph | O | 47.39 ± 2.78 | 49.00 ± 0.17 | 1.0 |
| 5a | H | H | CH3 | CH3 | O | 57.74 ± 1.60 | 83.59 ± 6.84 | 0.7 |
| 5b | H | H | CH3 | CH3 | S | 61.94 ± 6.56 | 26.62 ± 2.78 | 2.3 |
| 5c | H | H | CH3 | Ph | O | 55.01 ± 2.46 | 11.38 ± 0.48 | 4.8 |
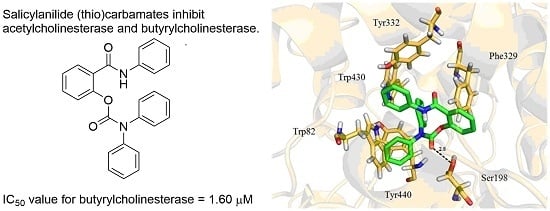
| 5d | H | H | Ph | Ph | O | 53.79 ± 4.26 | 1.60 ± 0.01 | 33.6 |
| Rivastigmine | 501 ± 3.08 | 19.95 ± 0.20 | 25.1 | |||||
| Galantamine | 4 ± 0.13 | 7.96 ± 0.59 | 0.5 | |||||
2.2. Cytotoxicity and Selectivity Indexes of (Thio)Carbamates
| Code | IC50 for HepG2 [µM] [19] | Selectivity Index for AChE | Selectivity Index for BChE |
|---|---|---|---|
| 1b | 83.51 | 2.1 | 1.6 |
| 1c | 289.0 | 3.9 | 68.8 |
| 2c | 945.1 | 11.6 | 219.8 |
| 3c | 512.0 | 8.2 | 69.8 |
| 4a | 6508.0 | 132.7 | 231.3 |
| 4b | 67.29 | 1.6 | 1.9 |
| 4c | ˃100 | ˃2.0 | ˃50.8 |
| 5a | 5985.9 | 103.7 | 71.6 |
| 5d | 211.1 | 3.9 | 131.9 |
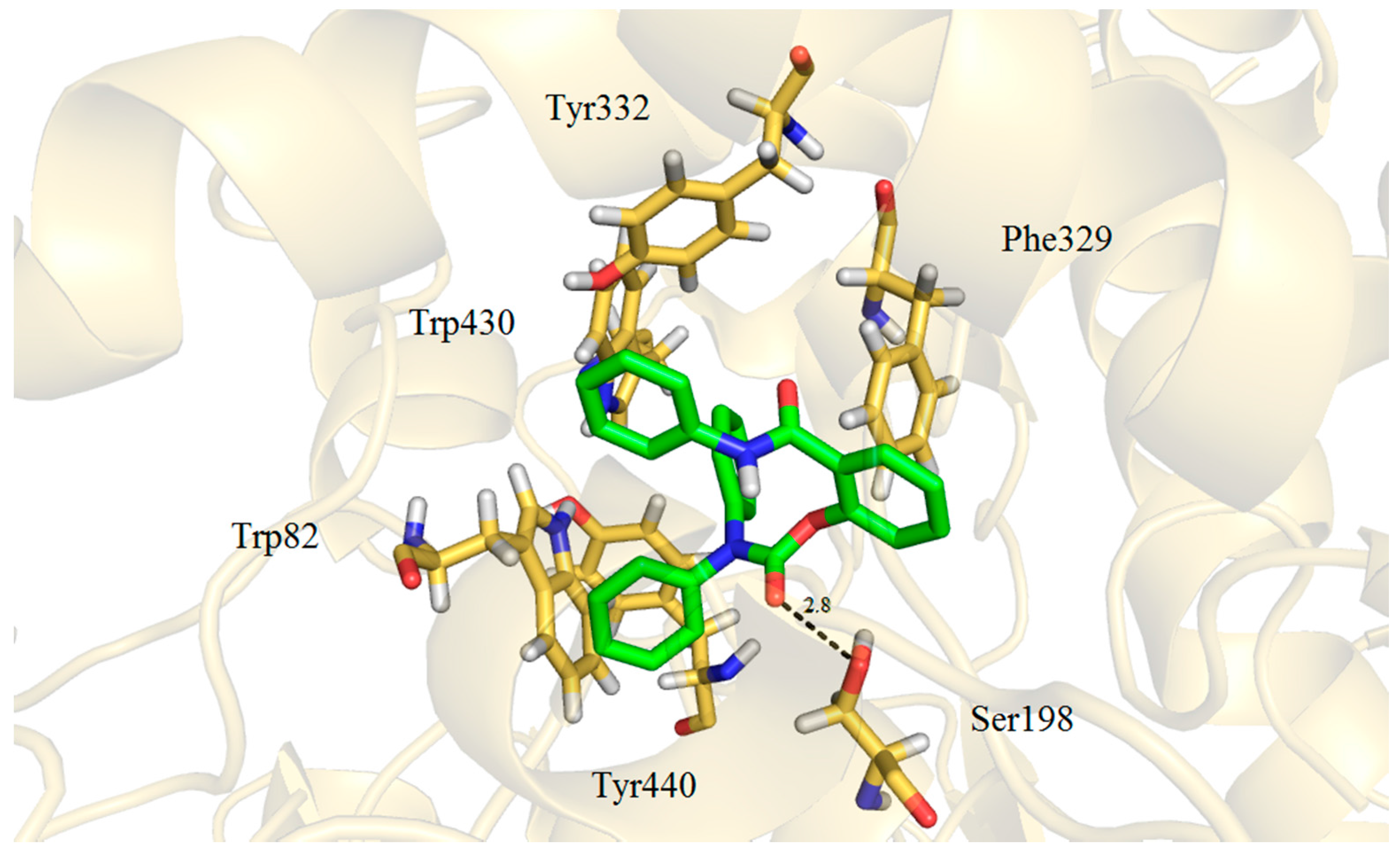
2.3. Molecular Docking
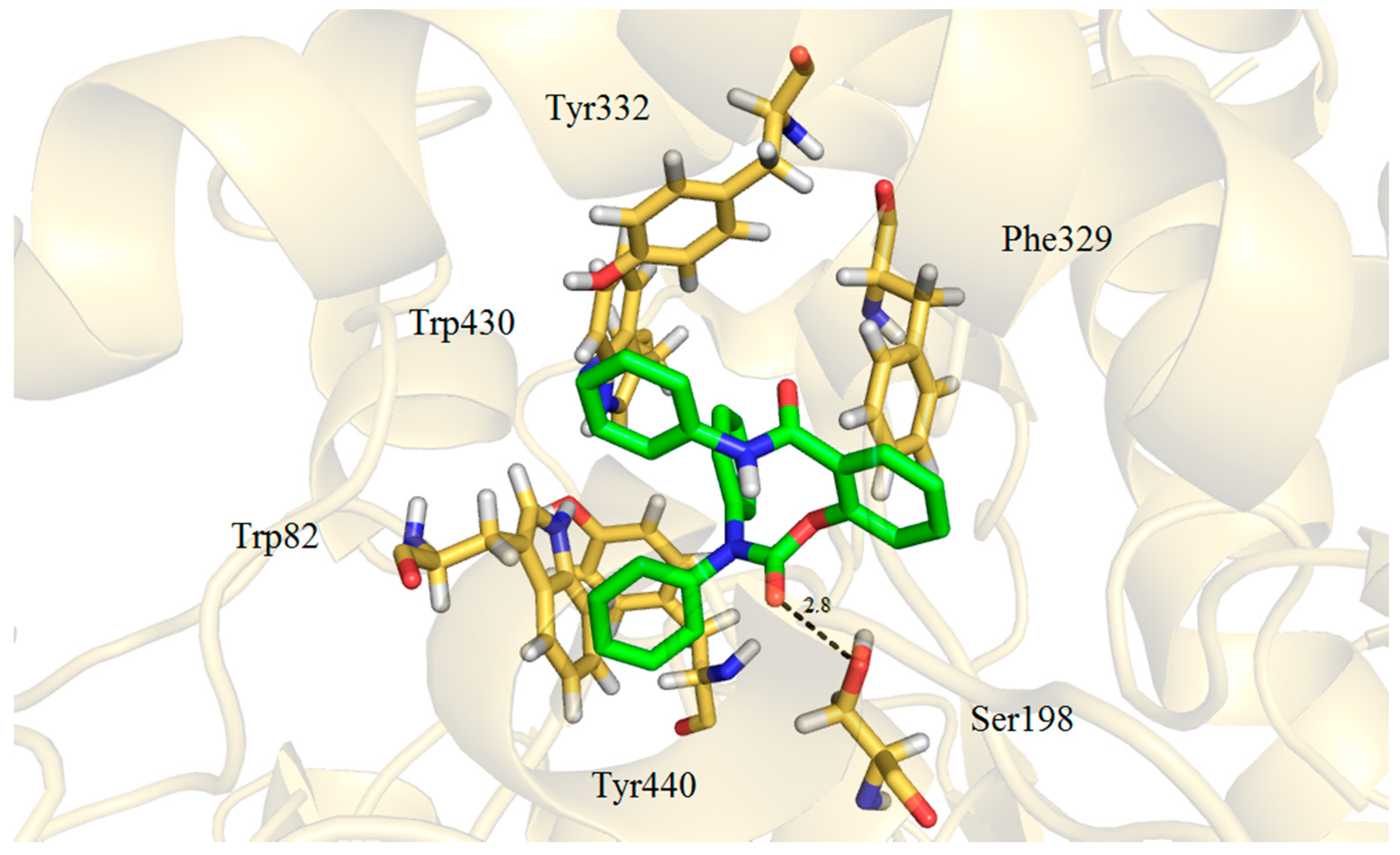
3. Experimental Section
3.1. Determination of Cholinesterase Inhibition
3.2. Molecular Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| ATCh | acetylthiocholine |
| AD | Alzheimer’s disease |
| AChE | acetylcholinesterase |
| BChE | butyrylcholinesterase |
| ChE | cholinesterases |
References
- Schwarz, S.; Lucas, S.D.; Sommerwerk, S.; Csuk, R. Amino derivatives of glycyrrhetinic acid as potential inhibitors of cholinesterases. Bioorg. Med. Chem. 2014, 22, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Mahesar, P.A.; Zaib, S.; Khan, M.S.; Matin, A.; Shahid, M.; Iqbal, J. Synthesis, cytotoxicity and molecular modelling studies of new phenylcinnamide derivatives as potent inhibitors of cholinesterases. Eur. J. Med. Chem. 2014, 78, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Ibach, B.; Haen, E. Acetylcholinesterase inhibition in Alzheimer’s disease. Curr. Pharm. Des. 2004, 10, 231–251. [Google Scholar] [CrossRef] [PubMed]
- Leger, G.C.; Massoud, F. Novel disease-modifying therapeutics for the treatment of Alzheimer’s disease. Expert Rev. Clin. Pharmacol. 2013, 6, 423–442. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Nordberg, A.; Arnold, S.E. A preclinical view of cholinesterase inhibitors in neuroprotection: Do they provide more than symptomatic benefits in Alzheimer’s disease? Trends Pharmacol. Sci. 2005, 26, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Chaudhaery, S.S.; Roy, K.K.; Shakya, N.; Saxena, G.; Sammi, S.R.; Nazir, A.; Nath, C.; Saxena, A.K. Novel Carbamates as Orally Active Acetylcholinesterase Inhibitors Found to Improve Scopolamine-Induced Cognition Impairment: Pharmacophore-Based Virtual Screening, Synthesis, and Pharmacology. J. Med. Chem. 2010, 53, 6490–6505. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Bajda, M.; Guzior, N.; Malawska, B. Novel alkyl- and arylcarbamate derivatives with N-benzylpiperidine and N-benzylpiperazine moieties as cholinesterases inhibitors. Eur. J. Med. Chem. 2010, 45, 5602–5611. [Google Scholar] [CrossRef] [PubMed]
- Imramovský, A.; Pejchal, V.; Štěpánková, Š.; Vorčáková, K.; Jampílek, J.; Vančo, J.; Šimůnek, P.; Královec, K.; Brůčková, L.; Mandíková, J.; et al. Synthesis and in vitro evaluation of new derivatives of 2-substituted-6-fluorobenzo[d]thiazoles as cholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 1735–1748. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Shang, Z.; Qiang, X.; Tan, Z.; Deng, Y. Synthesis of pterostilbene and resveratrol carbamate derivatives as potential dual cholinesterase inhibitors and neuroprotective agents. Res. Chem. Intermed. 2014, 40, 787–800. [Google Scholar] [CrossRef]
- Gocer, H.; Akincioglu, A.; Goksu, S.; Gulcin, I.; Supuran, C.T. Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J. Enzyme Inhib. Med. Chem. 2015, 3, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Cholinesterases, a target of pharmacology and toxicology. Biomed. Pap. 2011, 155, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Stepankova, S.; Vanco, J.; Pauk, K.; Monreal-Ferriz, J.; Vinsova, J.; Jampilek, J. Acetylcholinesterase-Inhibiting Activity of Salicylanilide N-Alkylcarbamates and Their Molecular Docking. Molecules 2012, 17, 10142–10158. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Lai, C.Y.; Liao, W.C. Molecular Recognition by Acetylcholinesterase at the Peripheral Anionic Site: Structure-Activity Relationships for Inhibitions by Aryl Carbamates. Bioorg. Med. Chem. 1999, 7, 2683–2689. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Salicylanilide Ester Prodrugs as Potential Antimicrobial Agents—A Review. Curr. Pharm. Des. 2011, 17, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinšová, J.; Novotná, E.; Wsól, V.; Ulmann, V.; Stolaříková, J.; Fernandes, S.; Bhat, S.; Liu, J.O. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis (Edinb.) 2012, 92, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Trejtnar, F.; Stolaříková, J. Antibacterial Activity of Salicylanilide 4-(Trifluoromethyl)benzoates. Molecules 2013, 18, 3674–3688. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Volková, M.; Novotná, E.; Trejtnar, F.; Stolaříková, J.; Vinšová, J. Synthesis and Biological Activity of New Salicylanilide N,N-Disubstituted Carbamates and Thiocarbamates. Bioorg. Med. Chem. 2014, 22, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinšová, J.; Novotná, E.; Stolaříková, J. Salicylanilide pyrazinoates inhibit in vitro multidrug-resistant Mycobacterium tuberculosis strains, atypical mycobacteria and isocitrate lyase. Eur. J. Pharm. Sci. 2014, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Novotná, E.; Saxena, S.; Yogeeswari, P.; Sriram, D.; Švarcová, M.; Vinšová, J. Salicylanilide Diethyl Phosphates as Potential Inhibitors of Some Mycobacterial Enzymes. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Liu, F.L.; Lee, C.C.; Chen, T.C.; Ali, A.A.A.; Sytwu, H.K.; Chang, D.M.; Huang, H.S. Modified Salicylanilide and 3-Phenyl-2H-benzo[e][1,3]oxazine-2,4(3H)-dione Derivatives as Novel Inhibitors of Osteoclast Differentiation and Bone Resorption. J. Med. Chem. 2014, 57, 8072–8085. [Google Scholar] [CrossRef] [PubMed]
- Vinšová, J.; Krátký, M.; Komlóová, M.; Dadapeer, E.; Štěpánková, Š.; Vorčáková, K.; Stolaříková, J. Diethyl 2-(Phenylcarbamoyl)phenyl Phosphorothioates: Synthesis, Antimycobacterial Activity and Cholinesterase Inhibition. Molecules 2014, 19, 7152–7168. [Google Scholar] [CrossRef] [PubMed]
- Vinšová, J.; Kozic, J.; Krátký, M.; Stolaříková, J.; Mandíková, J.; Trejtnar, F.; Buchta, V. Salicylanilide diethyl phosphates: Synthesis, antimicrobial activity and cytotoxicity. Bioorg. Med. Chem. 2014, 22, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.Y.; Kwok, K.C.; Wang, Y.; Bao, H.Y. An improved method to determine SH and -S-S- group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Naturforsch. C 2004, 59, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, P.H.; Harel, M.; Silman, I.; Sussman, J.L. Structure and dynamics of the active site gorge of acetylcholinesterase: Synergistic use of molecular dynamics simulation and X-ray crystallography. Protein Sci. 1994, 3, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S.W. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar]
- Saxena, A.; Redman, A.M.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in active-site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Chem. Biol. Interact. 1999, 119, 61–69. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1a–5d are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Švarcová, M.; Vinšová, J. Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules 2016, 21, 191. https://doi.org/10.3390/molecules21020191
Krátký M, Štěpánková Š, Vorčáková K, Švarcová M, Vinšová J. Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules. 2016; 21(2):191. https://doi.org/10.3390/molecules21020191
Chicago/Turabian StyleKrátký, Martin, Šárka Štěpánková, Katarína Vorčáková, Markéta Švarcová, and Jarmila Vinšová. 2016. "Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates" Molecules 21, no. 2: 191. https://doi.org/10.3390/molecules21020191
APA StyleKrátký, M., Štěpánková, Š., Vorčáková, K., Švarcová, M., & Vinšová, J. (2016). Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules, 21(2), 191. https://doi.org/10.3390/molecules21020191