2. Results and Discussion
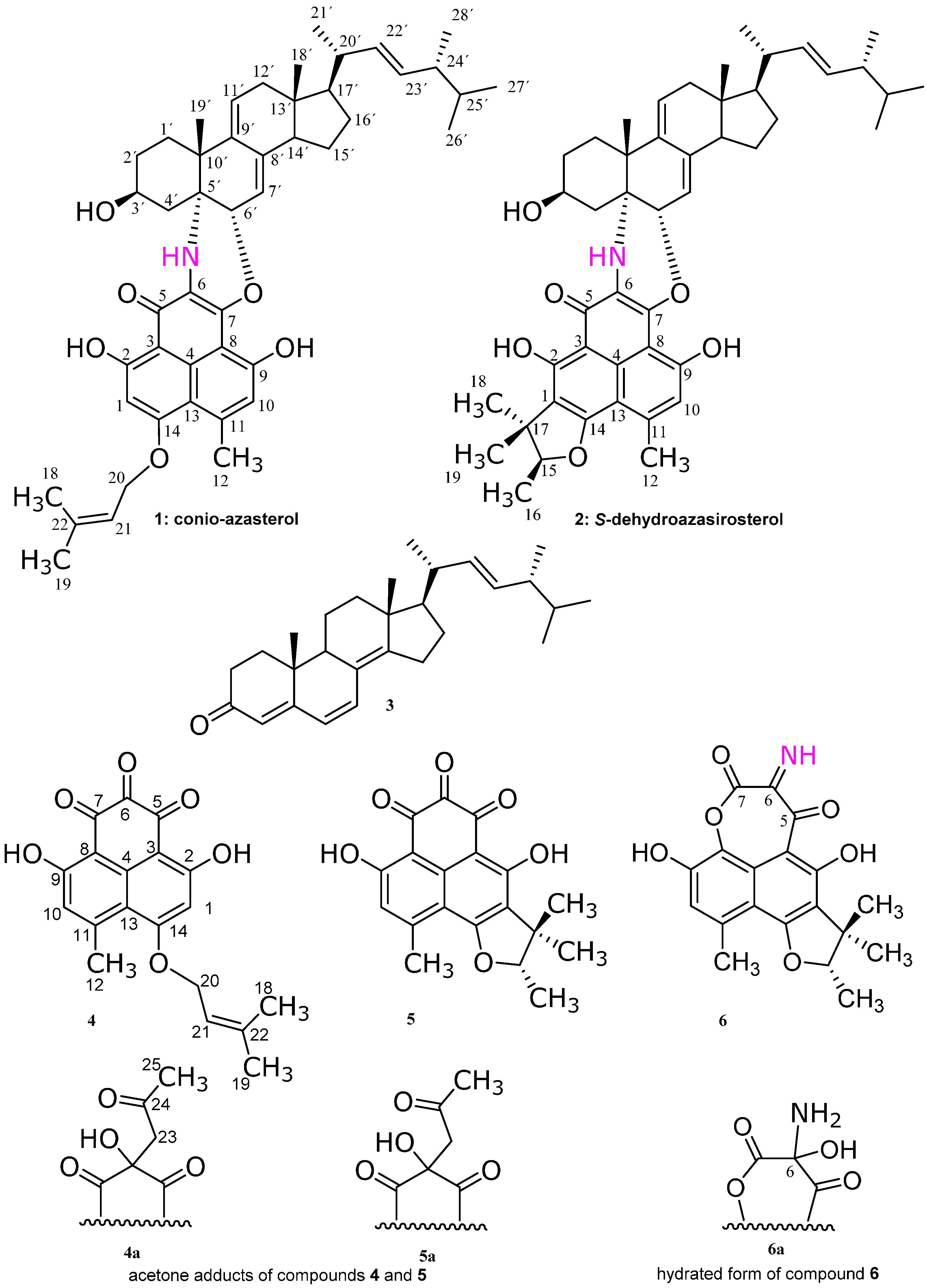
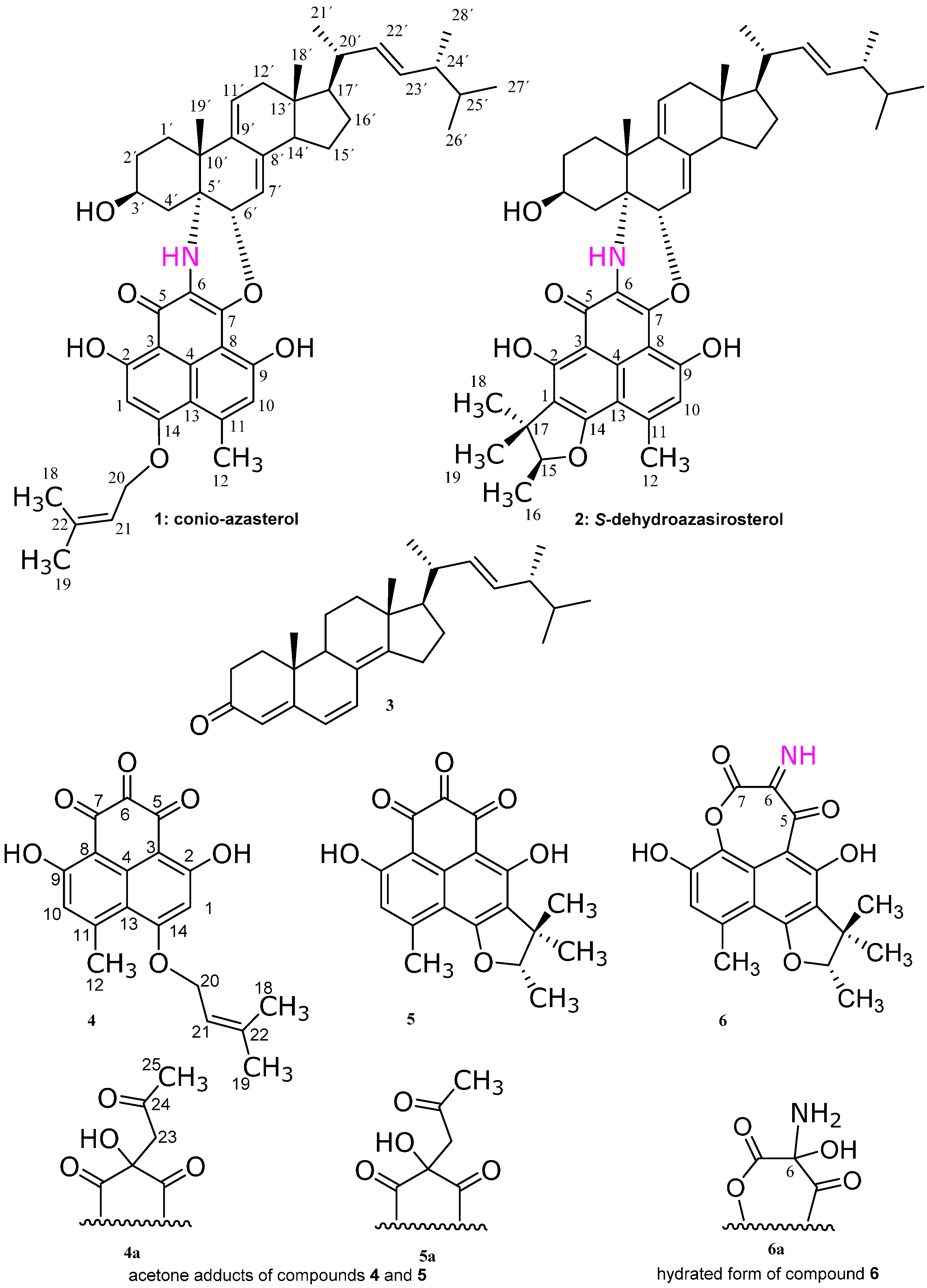
Subfraction 11 of the ethyl acetate extract of the mycelium of C. cereale was subjected to RP-HPLC separation to give the yellowish brown compounds 1 and 2. At first glance, from their 1H- and 13C-NMR spectra, they were suspected to be a mixture of a sterol and a phenalenone like compounds, though the HPLC chromatogram showed only one peak for each compound. Further elucidation of the mass gave a signal at 733 Da correspondent to a molecular formula C47H59NO6, suggesting that compounds 1 and 2 are adducts of a sterol and phenalenone derivatives. Further investigation of the 1D and 2D NMR spectra confirmed the adduct formation as follows.
Compound
1 showed a molecular formula of C
47H
59NO
6 on the basis of the number of signals in both
1H- and
13C-NMR spectra and accurate mass measurement (HRESIMS:
m/
z found = 756.4235 [M + Na]
+,
Figure S2). The
1H-NMR spectrum (
Figure S1) of the phenalenone portion of
1 gave rise to signals for two exchangeable phenolic hydrogens, one is strongly chelated (δ
H 16.91 for 2-OH) with a carbonyl group (IR 1711/3354 cm
−1), and the second is weakly chelated (δ
H 9.69 for 9-OH), in addition to a characteristic NH resonance at δ
H 3.93 which has no correlation in the HSQC (
Figures S3–S6).
The
1H- and
13C-NMR spectra showed an aromatic methyl (δ
H/C 2.78/25.9 for CH
3-12) and two aromatic protons (δ
H 6.38 and 6.81 for H-1 and H-10, respectively). A UV maximum at 395 nm clearly evidenced that compound
1 has an extended aromatic system. Further two
1H-NMR singlet resonance signals arose from aromatic protons (δ
H 6.38 for H-1 and δ
H 6.81 for H-10). These aromatic protons (H-1 and H-10), each had a distinctive set of correlations in the
1H-
13C HMBC spectrum suggesting that each of these protons is attached to a different benzene ring. In the
1H-
13C HMBC spectrum, H-1 showed cross peak correlations with C-2, C-3, C-5, C-13, and C-14, whereas H-10 had correlations with C-7, C-8, C-9, C-12, and C-13. H
3-12 had heteronuclear couplings to C-10, C-11, and C-13. 2-OH showed cross peak correlations with C-1, C-2, and C-3; and 9-OH with C-8, C-9, and C-10. This pattern of heteronuclear correlations, together with the
1H- and
13C-NMR data indicated for a naphthalene-type compound of two connected penta-substituted benzene rings, substituted at C-2 and C-9 with phenolic groups and at C-11 with a methyl group. The presence of the 3-methyl-2-butenyl group in
1 was proven as follows: the
1H- and
13C-NMR spectrum contained two singlet resonances at δ
H/C 1.81/25.8 for CH
3-18 and δ
H/C 1.76/18.3 for CH
3-19 due to a geminal dimethyl group attached to an olefinic carbon. This was corroborated by the HMBC cross peak correlations between H
3-18 and H
3-19 and C-22. The downfield shifted doublet at δ
H/C 4.69/66.0 is assigned for the methylene protons CH
2-20 which is attached to oxygen and the methine triplet at δ
H/C 5.56/118.4 is assigned for CH-21. The
1H-
1H COSY spectrum showed cross peak correlations for a
1H-
1H-spin system ranging from both terminal methyl protons via H-21 to H
2-20. The prenylation occurred at the oxygenated carbon C-14 due to the HMBC correlation of H
2-20 to C-14 as depicted in
Figure 2. These chemical shifts and correlations are similar to those of the compound coniosclerodin [
4].
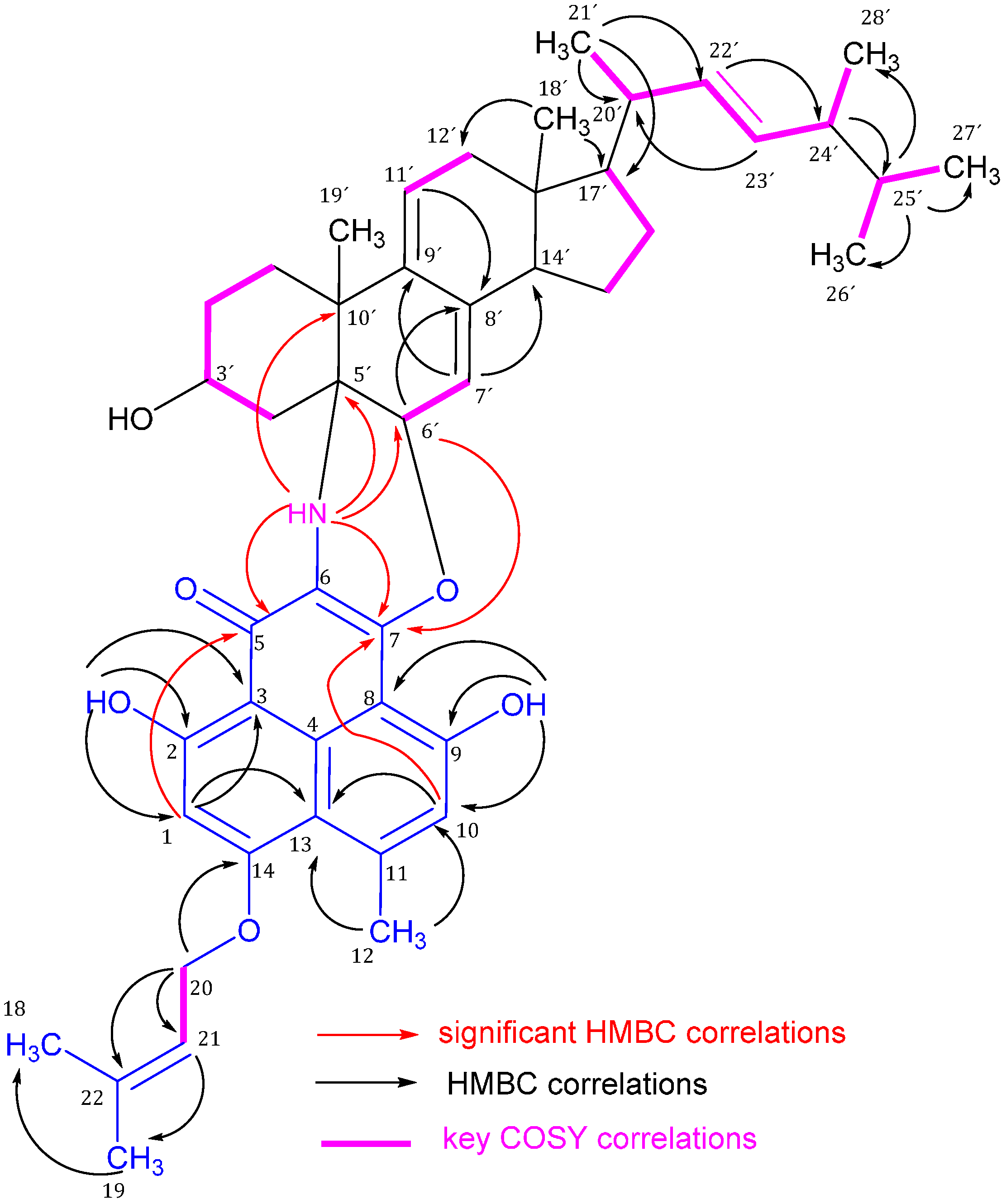
Figure 2.
Significant 1H-13C HMBC correlations (arrows, proton to carbon) and 1H-1H COSY (bold lines) of compound 1.
Figure 2.
Significant 1H-13C HMBC correlations (arrows, proton to carbon) and 1H-1H COSY (bold lines) of compound 1.
The sterol portion of
1 gave rise to
1H- and
13C-NMR signals (
Table 1) very similar to those of a sterol compound related to an ergosterol. Thus, the methyl groups of the sterol portion produced singlets at δ
H/C 0.50/11.6 and 1.23/23.6 for the angular tertiary CH
3-18′ and 19′, respectively, and four doublets at δ
H/C 0.96/20.7, 0.76/19.6, 0.78/19.9, and 0.85/17.6 for the secondary methyl groups CH
3-21′, 26′, 27′, and 28′, respectively, of the sterol side chain. The two alkenic CH groups of the side chain gave rise to double doublets at δ
H/C 5.07/135.2 and 5.17/132.2 for CH-22′ and 23′, respectively. The
1H-
1H COSY and
1H-
13C HMBC correlations (
Figures S9 and S10) resulted in a sterol side chain of nine carbons with one olefinic double bond to give an ergostene side chain (
Figure 2). Further two olefinic CH groups resonating at δ
H/C 5.00/116.9 and 5.68/124.9 are assigned to CH-7′ and 11′, respectively, to form an exocyclic diene system with the quaternary carbons C-8′ and C-9′ due to HMBC correlations as illustrated in
Figure 2. These structural features were confirmed by the HMBC correlations as illustrated in
Table 1 and
Figure 2. The methine multiplet at δ
H/C 4.10/66.7 has the expected complexity of steroidal 3-carbinol hydrogen which is characteristic for 3β-hydroxysterols. The steroidal nucleus is attached to the phenalenone part through two bonds. The first bond is from C-5′ to C-6 through a NH bridge and this is confirmed by the significant HMBC correlations of NH to C-5, 7, 4′, 5′, 6′, and 10′. The second bond is from CH-6′ to C-7 via oxygen due to the HMBC correlation of H-6′ to C-7 (
Figure 2 and
Table 2).
Table 1.
NMR (CDCl3) spectroscopic data for conio-azasterol (1).
Table 1.
NMR (CDCl3) spectroscopic data for conio-azasterol (1).
| No. | Mult. | δC (ppm) | δH (ppm), (mult, J in Hz) | No. | Mult. | δC (ppm) | δH (ppm), (mult, J in Hz) |
|---|
| 1 | CH | 97.4 | 6.38, s | 4′ | CH2 | 37.0 | a: 1.52, m
b: 2.12, m |
| 2 | C | 173.9 | | 5′ | C | 55.5 | |
| 3 | C | 104.4 | | 6′ | CH | 75.8 | 4.85, brs |
| 4 | C | 120.6 | | 7′ | CH | 116.9 | 5.00, brs |
| 5 | C | 174.4 | | 8′ | C | 139.0 | |
| 6 | C | 124.2 | | 9′ | C | 140.6 | |
| 7 | C | 141.5 | | 10′ | C | 40.2 | |
| 8 | C | 104.2 | | 11′ | CH | 124.9 | 5.68, brd (5.5) |
| 9 | C | 157.9 | | 12′ | CH2 | 41.9 | a: 2.12, m
b: 2.30, m |
| 10 | CH | 117.5 | 6.81, s | 13′ | C | 42.4 | |
| 11 | C | 143.0 | | 14′ | CH | 50.9 | 2.07, m |
| 12 | CH3 | 25.9 | 2.78, s | 15′ | CH2 | 22.9 | 1.53, m |
| 13 | C | 112.5 | | 16′ | CH2 | 28.6 | 1.60, m |
| 14 | C | 167.3 | | 17′ | CH | 55.7 | 1.20, m |
| 18 | CH3 | 18.3 | 1.76, s | 18′ | CH3 | 11.6 | 0.50, s |
| 19 | CH3 | 25.8 | 1.81, s | 19′ | CH3 | 23.6 | 1.23, s |
| 20 | CH2 | 66.0 | 4.69, d (6.2) | 20′ | CH | 40.3 | 1.95, m |
| 21 | CH | 118.4 | 5.56, brt (6.2) | 21′ | CH3 | 20.7 | 0.96, d (6.3) |
| 22 | C | 139.0 | | 22′ | CH | 135.2 | 5.07, dd (7.7, 15.0) |
| 2-OH | | | 16.91, s | 23′ | CH | 132.2 | 5.17, dd (7.3, 15.0) |
| 9-OH | | | 9.69, s | 24′ | CH | 42.8 | 1.78, m |
| NH | | | 3.93, s | 25′ | CH | 33.0 | 1.40, m |
| 1′ | CH2 | 29.7 | 1.98, m | 26′ | CH3 | 19.6 | 0.76, d (6.3) |
| 2′ | CH2 | 30.2 | 1.66, m | 27′ | CH3 | 19.9 | 0.78, d (6.3) |
| 3′ | CH | 66.7 | 4.10, m | 28′ | CH3 | 17.6 | 0.85, d (6.3) |
Table 2.
NMR (CDCl3) spectroscopic data for S-dehydroazasirosterol (2).
Table 2.
NMR (CDCl3) spectroscopic data for S-dehydroazasirosterol (2).
| No. | Mult. | δC (ppm) | δH (ppm), (mult, J in Hz) | No. | Mult. | δC (ppm) | δH (ppm), (mult, J in Hz) |
|---|
| 1 | C | 118.2 | | 5′ | C | 55.6 | |
| 2 | C | 169.9 | | 6′ | C | 75.6 | 4.85, brs |
| 3 | C | 105.4 | | 7′ | CH | 116.9 | 5.00, brs |
| 4 | C | 120.9 | | 8′ | C | 140.5 | |
| 5 | C | 174.8 | | 9′ | C | 139.0 | |
| 6 | C | 124.1 | | 10′ | C | 40.1 | |
| 7 | C | 140.6 | | 11′ | CH | 124.8 | 5.68 d (5.49) |
| 8 | C | 104.1 | | 12′ | CH2 | 41.9 | a: 2.13, m |
| 9 | C | 158.4 | | | | | b: 2.32, m |
| 10 | CH | 116.4 | 6.82, s | 13′ | C | 42.3 | |
| 11 | C | 142.3 | | 14′ | CH | 50.9 | 2.10, m |
| 12 | CH3 | 23.6 | 2.80, s | 15′ | CH2 | 22.7 | 1.53, m |
| 13 | C | 108.7 | | 16′ | CH2 | 28.5 | 1.64, m |
| 14 | C | 166.2 | | 17′ | CH | 55.7 | 1.23, m |
| 15 | CH | 91.2 | 4.67, q (6.6) | 18′ | CH3 | 11.6 | 0.51, s |
| 16 | CH3 | 14.6 | 1.46, d (6.6) | 19′ | CH3 | 23.5 | 1.25, s |
| 17 | C | 43.2 | | 20′ | CH | 40.3 | 1.98 |
| 18 | CH3 | 25.7 | 1.54, s | 21′ | CH3 | 20.6 | 0.98, d (6.6) |
| 19 | CH | 20.7 | 1.30, s | 22′ | CH | 135.3 | 5.10, dd (7.7, 15.0) |
| 2-OH | | | 16.84, s | 23′ | CH | 132.2 | 5.18, dd (7.0, 15.0) |
| 9-OH | | | 9.76, s | 24′ | CH | 42.8 | 1.82, m |
| NH | | | 3.98, s | 25′ | CH | 33.0 | 1.43, m |
| 1′ | CH2 | 29.4 | 2.01, m | 26′ | CH3 | 19.6 | 0.78, d (6.3) |
| 2′ | CH2 | 30.3 | 1.68, m | 27′ | CH3 | 19.9 | 0.80, d (6.3) |
| 3′ | CH | 66.8 | 4.11, m | 28′ | CH3 | 17.6 | 0.88, d (6.3) |
| 4′ | CH2 | 37.0 | a: 1.54, m | | | | |
| | | | b: 2.12, m | | | | |
Compound
2 is structurally similar to compound
1, except that the prenyl moiety is attached to the phenalenone part as a trimethyl dihydrofuran ring (
Figure 1) as follows. HMBC correlations (
Table 2) allowed connecting the C-15 to C-19 part of the molecule, which is an oxygenated hemiterpene unit. C-17 of this partial structure was attached to C-1 of the aromatic moiety due to heteronuclear long range couplings of H
3-18 and H
2-19 to C-1. Ring closure to a dihydrofuran ring occurred via the oxygen atom at C-15 to C-14 of the aromatic nucleus. The complete structural identification of
2 was done by extensive spectroscopic data (
Table 1 and
Table 2). Compound
2 was isolated in this work as an epimeric pure form for the first time (
Figure S7), and it was reported before as an epimeric mixture at C-15 under the name dehydroazasirosterol [
5]. Therefore, we gave the name conio-azasterol and
S-dehydroazasirosterol for compounds
1 and
2, respectively.
The LC/MS,
1H- and
13C-NMR spectra (
Figures S13 and S14) of compound
3 revealed an ergostane-type compound (ergosteroid) which has the same structure as (22
E,24
R)-ergosta-4,6,8(14)-22-tetraen-3-one [
6] (
Figure 1).
The structures of compounds
4 and
5 were established based on extensive spectroscopic measurements as in
Table 3 and
Table 4, respectively. Compound
4 could not be isolated in its genuine triketone form but instead as an acetone adduct (
4a) on C-6 (
Figure 1,
Figures S15 and S16, and
Table 3). The position of acetonyl moiety of the acetone adduct was confirmed by the HMBC correlation of H
2-23 to C-6. A pair of enantiomers (1:1 mixture) at C-6 was found which was confirmed by the absence of CD Cotton effects and zero optical rotation for
4a. The acetone adduct of compound
4 (
4a) is reported under the name rousselianone A′ [
7]. Also compound
5 could not be isolated in its genuine triketone form but instead as an acetone adduct (
5a) on C-6 (
Figure 1). The position of acetonyl moiety is confirmed by the HMBC correlation of H
2-23 to C-6. A pair of epimers (1:1 epimeric mixture) at C-6 was found which was confirmed by the presence of two sets of resonances for
5a in the
1H- and
13C-NMR spectra (
Table 4,
Figures S17 and S18). The complete spectroscopic data of
5a is listed in
Table 4 for the first time. Compound
5 (entatrovenetinone) was formerly separated from
Gremmeniella abietina [
8]. The genuine triketone compounds (
4 and
5) were recognized
via LC/MS (
m/z 340) and HRESIMS at 341.1037 [M + 1]
+, calc 341.1025 [M + 1]
+, using additionally prepared extract prepared under nitrogen atmosphere and without subjecting the extract to acetone during the extraction process (
Figure S20).
Table 3.
NMR (CDCl3) spectroscopic data for 4a.
Table 3.
NMR (CDCl3) spectroscopic data for 4a.
| No. | δC (ppm) | Multi. | δH (ppm), (mult, J in Hz) | 1H-1H COSY | 1H-13C HMBC | 1H-1H NOESY |
|---|
| 1 | 97.1 | CH | 6.37, s | | 2, 3, 13, 14 | 20 |
| 2 | 168.8 | C | | | | |
| 3 | 101.8 | C | | | | |
| 4 | 137.1 | C | | | | |
| 5 | 197.1 | C | | | | |
| 6 | 77.7 | C | | | | |
| 7 | 199.2 | C | | | | |
| 8 | 105.5 | C | | | | |
| 9 | 165.6 | C | | | | |
| 10 | 119.1 | CH | 6.77, s | 12 | 8, 9, 13 | 12 |
| 11 | 150.0 | C | | | | |
| 12 | 26.6 | CH3 | 2.78, s | 10 | 10, 11, 13 | 10 |
| 13 | 113.7 | C | | | | |
| 14 | 166.5 | C | | | | |
| 18 | 18.3 | CH3 | 1.78, s | 20, 21 | 19, 21, 22 | 20 |
| 19 | 25.8 | CH3 | 1.83, s | 20, 21 | 18, 21, 22 | 21 |
| 20 | 66.3 | CH2 | 4.68, d, 6.6 | 18, 19, 21 | 14, 21, 22 | 1, 18 |
| 21 | 117.7 | CH | 5.54, br t, 6.6 | 18, 19, 20 | | 19 |
| 22 | 139.9 | C | | | | |
| 23 | 52.0 | CH2 | 3.25, s | | 5, 6, 7, 24 | 25 |
| 24 | 205.6 | C | | | 23, 24 | |
| 25 | 31.1 | CH3 | 2.19, s | | | 23 |
| 2-OH | | | 13.24, s | | 1, 2, 3 | |
| 9-OH | | | 12.67, s | | 8, 9, 10 | |
Table 4.
NMR (CDCl3) spectroscopic data for 5a.
Table 4.
NMR (CDCl3) spectroscopic data for 5a.
| No. | δC (ppm) | Mult. | δH (ppm), (mult, J in Hz) | 1H-1H COSY | 1H-13C HMBC | 1H-1H NOESY |
|---|
| 1 | 118.4/118.5 | C | | | | |
| 2 | 165.5/165.4 | C | | | | |
| 3 | 102.5 | C | | | | |
| 4 | 137.4 | C | | | | |
| 5 | 196.9/196.8 | C | | | | |
| 6 | 77.1/77.3 | C | | | | |
| 7 | 199.2 | C | | | | |
| 8 | 105.4 | C | | | | |
| 9 | 166.2/166.1 | C | | | | |
| 10 | 117.9 | CH | 6.74, br s | 12 | 8, 9, 12, 13 | 12 |
| 11 | 149.3 | C | | | | |
| 12 | 24.3 | CH3 | 2.76, s | 10 | 10, 11, 13 | 10 |
| 13 | 109.7 | C | | | | |
| 14 | 166.1 | C | | | | |
| 15 | 91.7/91.6 | CH | 4.64, q (6.6) | 16 | 17, 18, 19 | 16, 18 |
| 16 | 14.7 | CH3 | 1.46, d (6.6) | 15 | 15, 17 | 15, 19 |
| 17 | 43.3 | C | | | | |
| 18 | 25.7 | CH3 | 1.51/1.52, s | | 1, 17, 19 | 15 |
| 19 | 20.6 | CH3 | 1.30/1.27, s | | 1, 15, 17, 18 | 16 |
| 23 | 51.8/52.1 | CH2 | 3.31/3.27, s | | 5, 6, 7, 24 | |
| 24 | 206.1/205.9 | C | | | | |
| 25 | 31.1/31.0 | CH3 | 2.20, s | | 24 | |
| 2-OH | | | 13.34/13.29, s | | 1, 2, 3, 5 | |
| 9-OH | | | 12.81/12.78, s | | 8, 9, 10 | |
Compound
6a was elucidated based on extensive spectroscopic measurements (
Figures S21–S25). Compound
6 has the same structural features as in compound
5, except that there is unique imine moiety at C-6 and lactone oxygen between C-8 and C-7. Compound
6 could not be isolated in the imine form, but in its hydrated one (
Figure 1). The latter was suspected due to the presence of epimeric mixture (1:1 ratio) at C-6 corroborated from the two sets of resonances in
1H- and
13C-NMR spectra (
Figures S21 and S22). The attachment of oxygen to C-8 was proven from its chemical shift at 130.2 ppm in the
13C-NMR spectrum (
Table 5). The latter is similar to the chemical shifts in the lactone and the ketolactone compounds reported by Elsebai
et al. [
4]. The presence of a nitrogen atom was suggested by the odd numbered molecular mass at 373 Da (HRESIMS
m/
z, found = 396.1057 [M + Na]
+). The position of the imine at C-6 is supported by the structure of compounds
1 and
2 where the nitrogen atom is at the same position which is the mid carbonyl group. This is also chemically the most plausible position because the mid carbonyl group of a triketone system is the most active one for a transamination reaction. In addition, the respective carbon chemical shifts were in agreement with the values calculated by the ACD NMR predictor software
® (ACD laboratories) for
6a. We report here the complete spectroscopic data (
Table 5) for the hydrated form of compound
6 (
6a).
Table 5.
NMR (CD3COCD3) spectroscopic data for 6a.
Table 5.
NMR (CD3COCD3) spectroscopic data for 6a.
| No. | δC (ppm) | Mult. | δH (ppm), (mult, J in Hz) | 1H-13C HMBC | 1H-1H NOESY |
|---|
| 1 | 117.9 | C | | | |
| 2 | 169.2 | C | | | |
| 3 | 94.9 | C | | | |
| 4 | 125.8 | C | | | |
| 5 | 189.1/189.2 | C | | | |
| 6 | 119.1 | C | | | |
| 7 | 157.1 | C | | | |
| 8 | 130.2 | C | | | |
| 9 | 146.7 | C | | | |
| 10 | 118.5 | CH | 6.81, s | 8, 9, 12, 13 | |
| 11 | 132.0 | C | | | |
| 12 | 22.1 | CH3 | 2.66, s | 10, 11, 13, 14 | |
| 13 | 109.8 | C | | | |
| 14 | 167.2 | C | | | |
| 15 | 92.06/92.13 | CH | 4.69, q (6.6) | 14, 17, 18, 19 | 16, 18 |
| 16 | 14.8/14.7 | CH3 | 1.51, d (6.6) | 15, 17 | 19 |
| 17 | 43.9/43.8 | C | | | |
| 18 | 25.8/26.0 | CH3 | 1.53, s | 1, 15, 17, 19 | 15 |
| 19 | 20.9/21.0 | CH3 | 1.30, s | 1, 15, 17, 18 | 16 |
| 2-OH | | | 12.99 | | |
| 9-OH | | | 11.98 | | |
For the stereochemistry of compounds 1–6, compound 2 has the all stereogenic centers as in compounds 1–6. Due to the similarity in NMR chemical shifts for both compounds 1 and 2 and their identical biogenetic origin, they should have the same stereochemistry of the sterol portion; and likewise for compounds 5 and 6, which have the phenalenone stereogenic centre at C-15 as in compound 2. Therefore, the stereochemistry of compound 2 is discussed in details as follows.
For the sterol portion, both the type and orientation of substituents in the steroid nucleus affect their
13C-NMR chemical shifts [
9]. Consequently, the orientation of 3′-OH is β due to the
13C chemical shift of C-3′ at δ
C 66.7 for compound
1 and δ
C 66.8 for compound
2 (the α-carbinol C-3′ has chemical shift around δ
C 71.0 [
10]. The NH bridge is α-oriented due to the NOESY correlation of NH to H-3′ (
Figures S11 and S12) which is further confirmed by enhancing the H-3′ resonance signal upon irradiation of NH resonance using 1D-NOE measurement. Also, the absence of a NOESY correlation between CH
3-19′ and NH further confirmed the α-orientation of the amine bond. A NOESY correlation between H
3-19′ and H-6′ indicated the oxygen bridge between C-7 and C-6′ to be α-oriented and in turn the whole phenalenone nucleus due to the planarity of the aromatic system. The phenalenone nucleus is therefore perpendicular to the flattish sterol part due to the aforementioned NOESY correlations and the
trans fusion of the steroid rings A/B and C/D in addition to the planar arrangements of the middle atoms of the sterol nucleus with the C-C double bonds. H
3-18′ has no NOESY correlation to H-14′ confirming the
trans fusion of the steroid ring C/D and this is matching with the
transoid nature of most natural steroids. H
3-19′ has a NOESY correlation to H
3-18′. The β-orientation of the CH
3-19′ group was further confirmed by its enhancement upon irradiation at the H
3-18′ using a 1D-NOE measurement.
The stereochemistry of the sterol side chain was determined to be as shown in
Figure 1. It was determined by comparison of the
13C-NMR data of compound
2 with those of the (22
E,24
R)-methyl-Δ
22-sterol side chain of known steroids [
11,
12]. Wright
et al.. [
12] studied the
13C-NMR spectra of diastereomeric C-24 alkyl sterols and they found that the differences in the
13C-NMR chemical shifts of side-chain carbons permitted the determination of the absolute configuration at C-24, and stated that the absolute configuration of the sterol side chain can be determined by the
13C chemical shifts of the respective carbons. Wright
et al.. [
12] found that specifically the resonance for the C-28′ methyl carbon appears at a characteristic value of 17.6 ± 0.1 ppm in the 24
R epimer. Interestingly, in compounds
1–
3, the
13C chemical shifts of C-28′ are the same which is 17.6 ppm, although the main sterol skeletons in compounds
1–
3 are not similar, indicating the
R configuration at C-24′ for all of them. This is possible since these chemical shifts are insensitive to structural changes remote from the C-24′ stereogenic centre [
12]. Further confirmation of the 24′
R configuration was described by Goad
et al.. [
13] who recognized that many sterols of plants and microorganisms contain a methyl or ethyl group at C-24′, and both C-24′ diastereoisomers have been found to occur naturally. Goad
et al.. [
13] established that there appears to be some phylogenetic significance to the configuration at C-24′, since, in general, algae and fungi produce sterols with the 24
′R configuration (24′
S if a saturated side chain, 24′
R in the Δ
22 derivative) whereas sterols in most vascular plants possess the 24′
S configuration. The presence of NOESY correlation between H
3-18′/H-20′ and between H-14′/H
3-21′ indicated that the sterol side chain is β-oriented to the main sterol nucleus and the
transoid nature of rings C/D (
Figure 1). We assume a
S configuration of the single stereogenic center of the phenalenone nucleus at C-15 for compounds
2,
5, and
6 based on the same biogenetic origin as the previously reported compounds from the same fungus [
4,
14].
All the compounds were evaluated for their antimicrobial and cytotoxic activity. All the compounds showed marginal antibacterial activities in agar diffusion assays against Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, and Candida albicans. The cytotoxicity of compounds 1, 2, 4a, 5a, and 6a was evaluated against SKM1, U266, and K562 cancer cell lines which are widely used for cytotoxicity assays, and the IC50 values in µM (XTT assay). Only compound 6a exhibited moderate cytotoxic activities with IC50 values between 75, 45, and 8.5 µM against SKM1, U266, and K562 cancer cell lines, respectively (n = 3).
Molecular Docking Study
In silico docking is a very famous method employed to investigate molecular association. The knowing of the binding capabilities of the active site residues to specific groups on the agonist or antagonist leads to various strategies for the synthesis of very specific agents with a high probability of biological action. In silico docking experiments were conducted for compounds
1 and
2 against the X-ray crystal structure of b-estradiol-bound ERα receptor ligand binding domain (ERα-LBD, PDB: 1A52) [
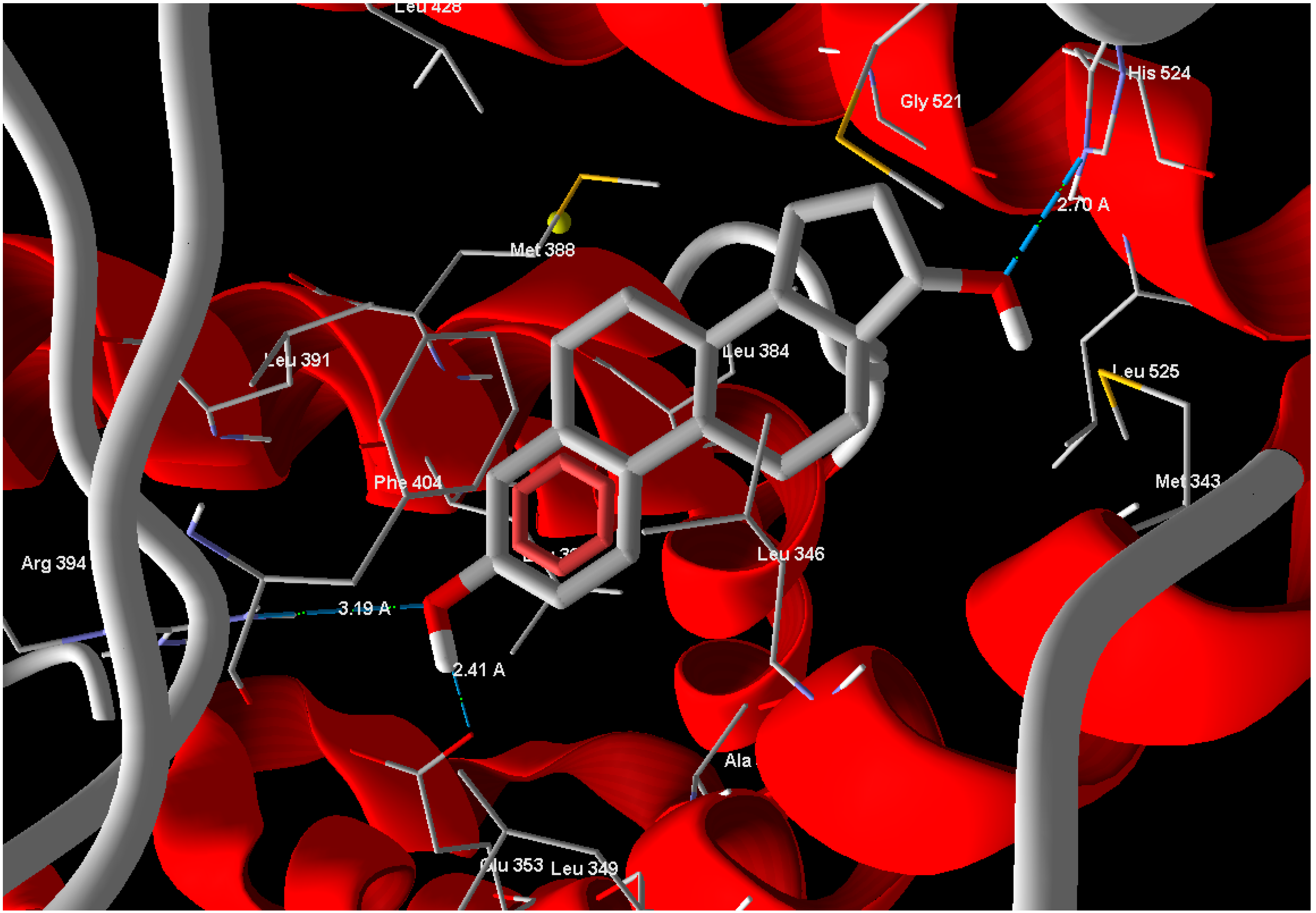
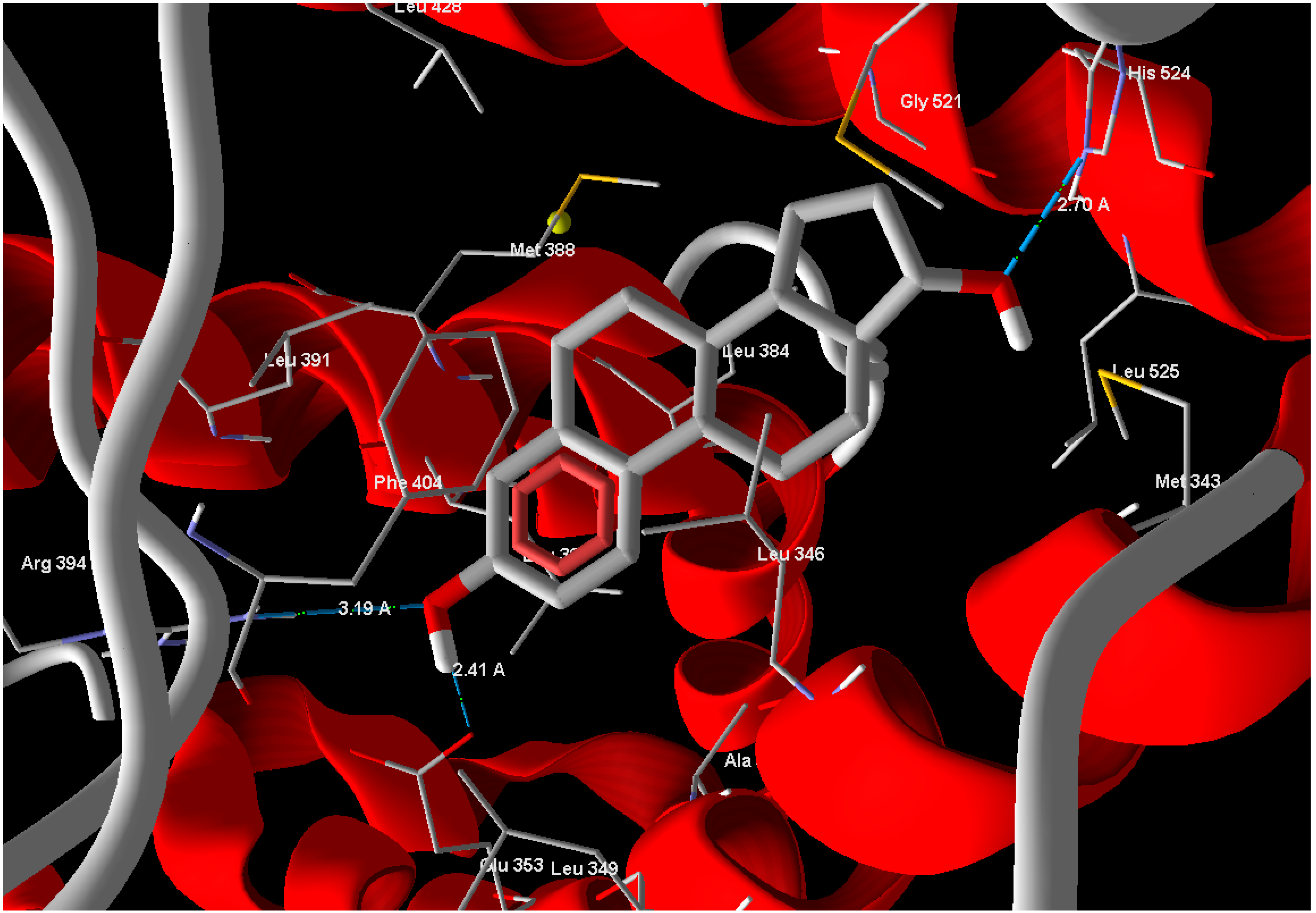
13], to compare the binding affinity of the tested compounds. Estrogen (standard drug) reveals Mol-Dock score of −105.9 and forms three hydrogen bonds between its phenolic OH moiety and Glu 353 with a bond distance of 2.41 Å and another bond with Arg 394 with bond distance of 3.19 Å and the alcoholic OH form one hydrogen bond with His 524 with bond distance of 2.70 Å (
Figure 3). Compounds
1 and
2 exhibited binding scores −77.61 and −18.54, respectively. Compound
1 has relatively good MolDock score and made two hydrogen bonds with same amino acids in the active site of ERα-LBD.
Figure 3.
Estrogen showed hydrogen bonds interactions with ERα-LBD active site.
Figure 3.
Estrogen showed hydrogen bonds interactions with ERα-LBD active site.
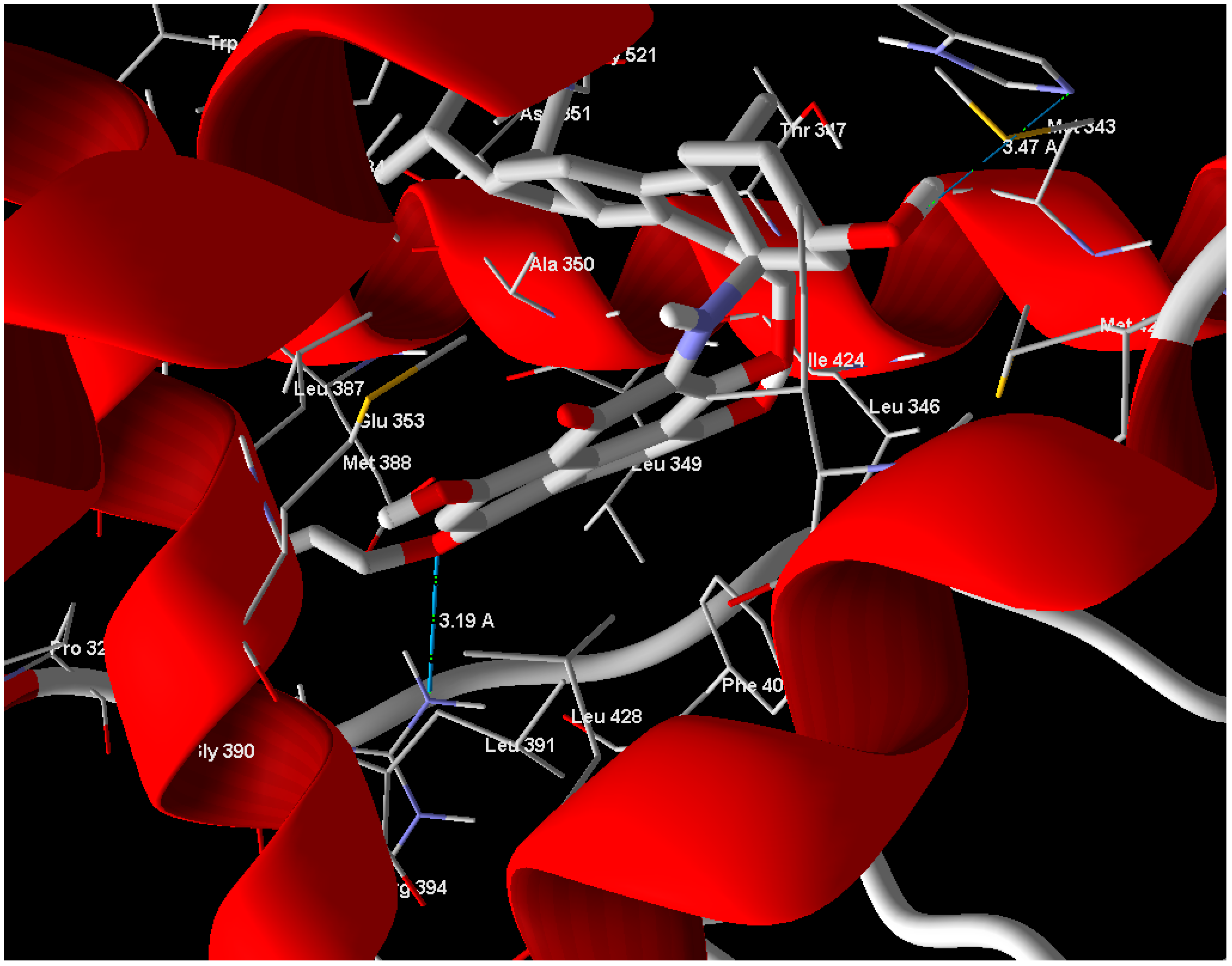
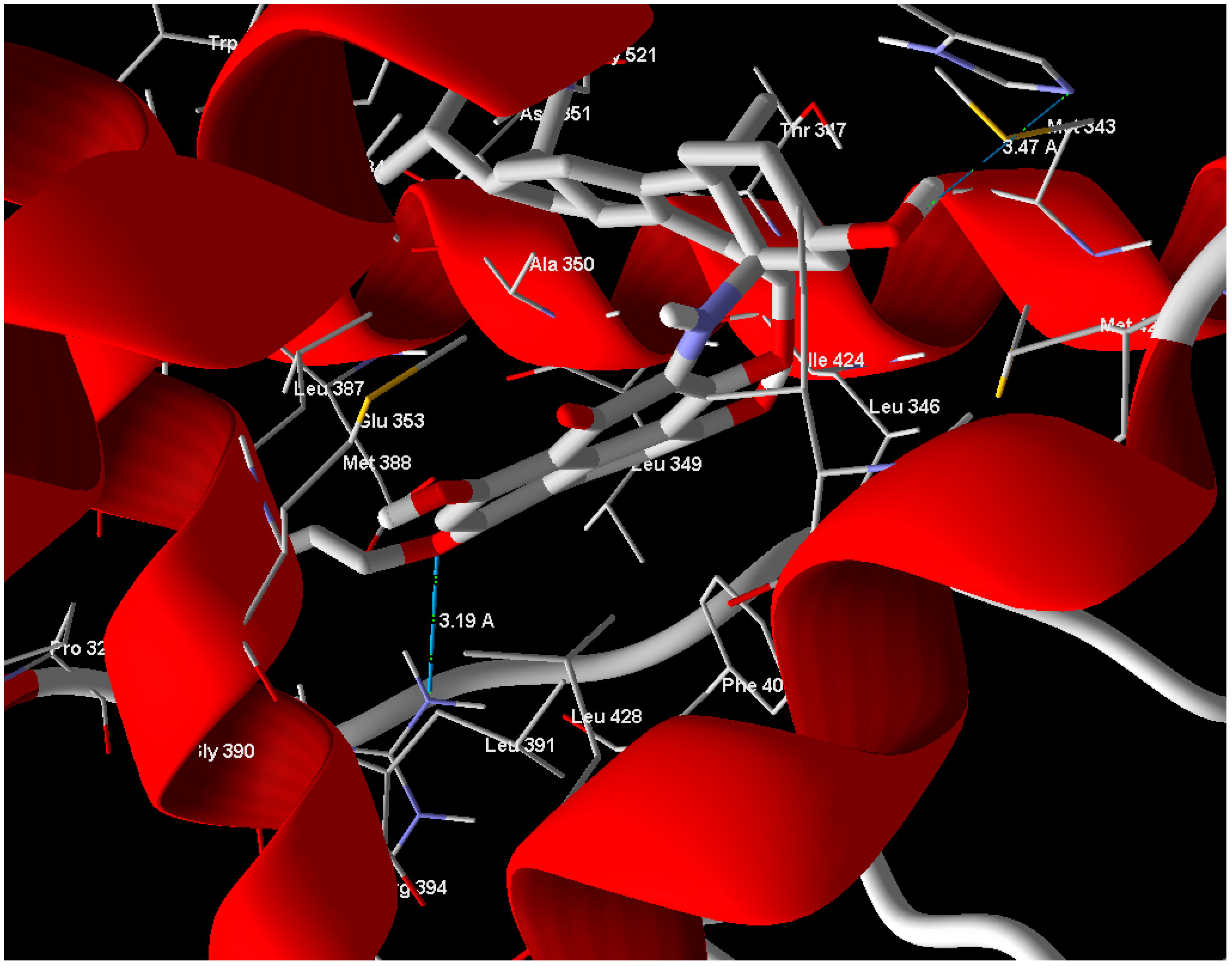
Arg 394 and His 524 with bond lengths 3.19 Å and 3.47 Å, respectively, in addition to hydrophobic interaction with Phe 404 and Trp 383 (
Figure 4). The position of compound
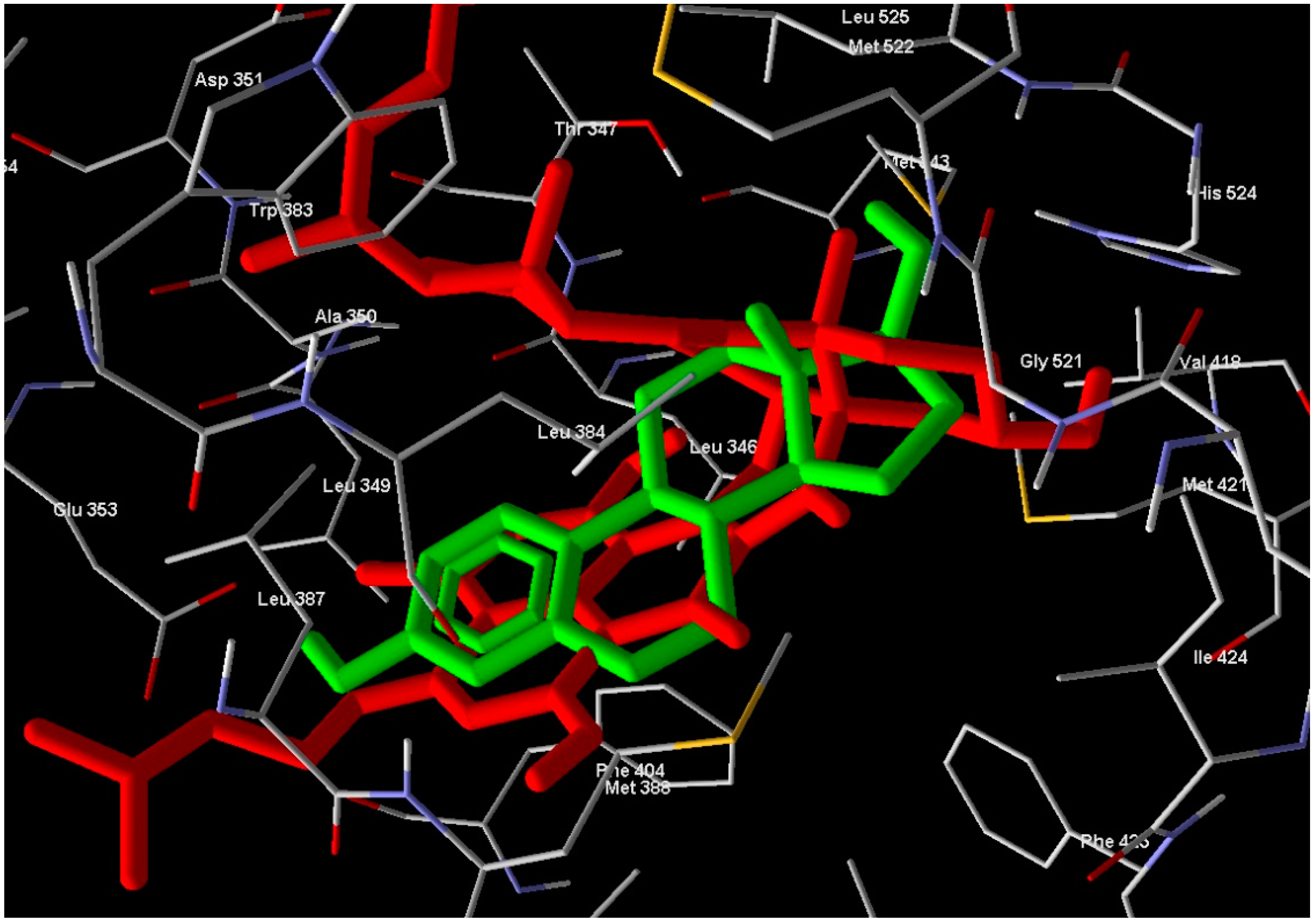
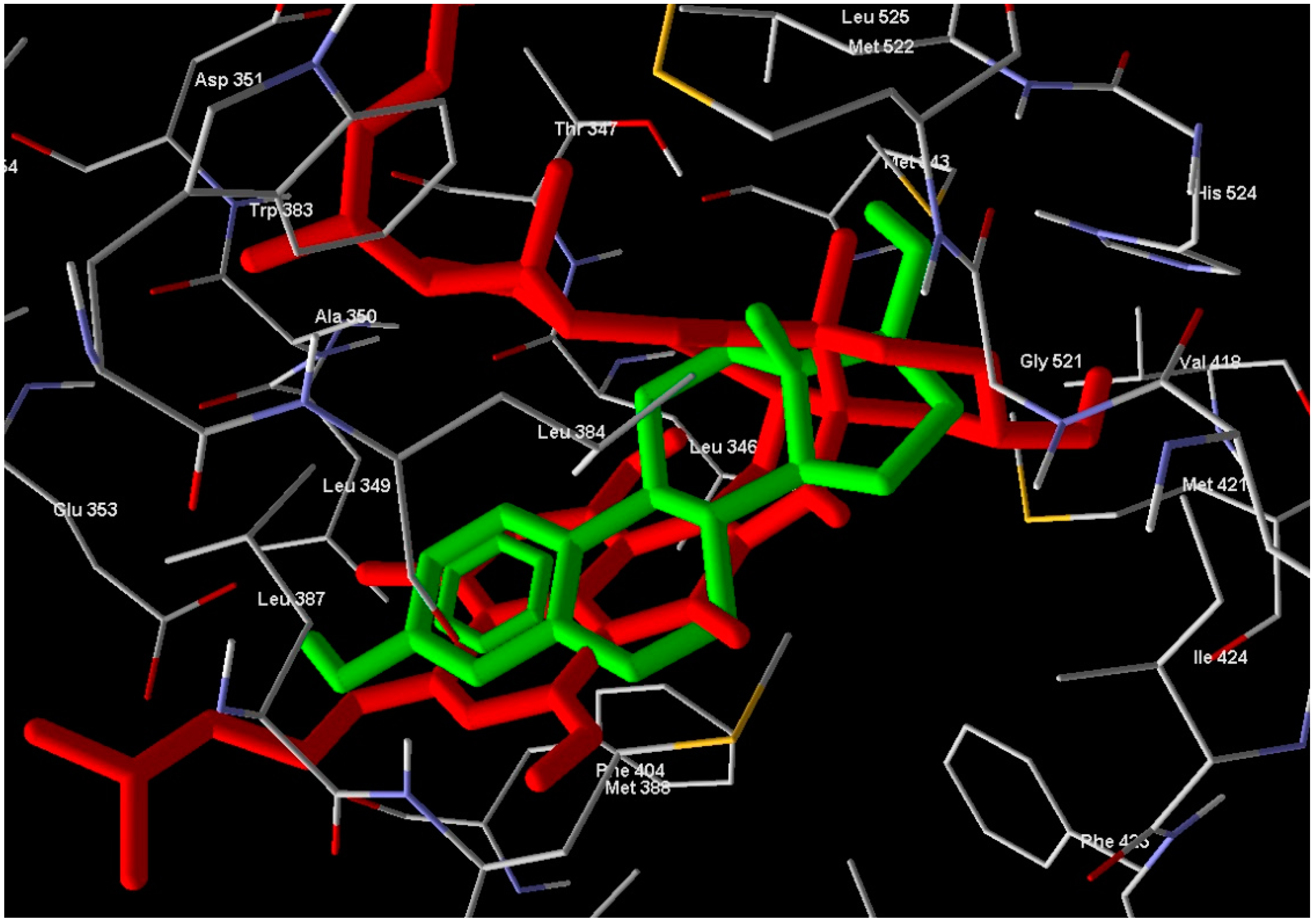
1 in the active site overlaps well with Estrogen (standard drug) (
Figure 5) and this means a good quality of the docking process. Compound
2 has relatively very low MolDock score and weak interaction with receptor.
Figure 4.
Compound 1 showed hydrogen bonds interaction with ERα-LBD active site.
Figure 4.
Compound 1 showed hydrogen bonds interaction with ERα-LBD active site.
Figure 5.
Compound 1 (red molecule) superimposed with standard drug (green molecule) in ERα-LBD active site (Color figure online).
Figure 5.
Compound 1 (red molecule) superimposed with standard drug (green molecule) in ERα-LBD active site (Color figure online).
3. Materials and Methods
3.1. General Experimental Procedures
UV and IR spectra were obtained employing Perkin-Elmer Lambda 40 and Perkin-Elmer Spectrum BX instruments (PerkinElmer, Inc., Waltham, USA), respectively. VLC grade (Macherey-Nagel, Polygoprep 60-50C18) was used for vacuum liquid chromatography. All organic solvents were distilled prior to use. HPLC was carried out using a Waters system, controlled by Waters Millenium software, consisting of a 600E pump, a 996 PDA, and a 717 plus autosampler (Waters Corporation, Milford, MA, USA). All NMR spectra were recorded on a Bruker Avance 300 and 500 DRX spectrometers (Bruker Corporation, Rheinstetten, Germany). Spectra were referenced to the residual solvent signals with resonances at δH/C 2.04/29.8 (acetone-d6) and δH/C 7.26/77.0 (CDCl3). ESI mass spectra were obtained on an Applied Biosystems/MDS Sciex API 2000 MS spectrometer (Applera Corporation and MDS Inc., Foster, CA, USA). HRESIMS were recorded on a Bruker Daltonik micrOTOF-Q Time-of-Flight mass spectrometer with ESI source, and UPLC-Synapt G2 HDMS mass spectrometer (Bruker CorporationCompany, Rheinstetten, Germany).
3.2. Origin of the Algal Sample and Isolation of the Fungus
An algal sample Enteromorpha sp. was collected from Fehmarn, Baltic Sea. The isolation of the fungus from the host tissues was carried out through single colony isolation method as follows. Algal samples were rinsed three times with sterile H2O. After surface sterilization with 70% EtOH for 15 s the algae was rinsed in sterile artificial seawater (ASW). Subsequently, the algae were aseptically cut into small pieces and placed on agar plates containing isolation medium: agar 15 g/L, ASW 800 mL/L, glucose 1 g/L, peptone from soymeal 0.5 g/L, yeast extract 0.1 g/L, benzyl penicillin 250 mg/L, and streptomycin sulfate 250 mg/L. The fungus growing out of the algal tissue was separated on biomalt medium (biomalt 20 g/L, agar 10 g/L, ASW 800 mL/L) until the culture was pure.
3.3. Cultivation, Extraction and Isolation of Compounds 1–6
The fungal strain Coniothyrium cereale was cultivated for 40 days on 10 L solid BMS medium (Gesundheitsprodukte GmbH, Kirn, Germany) with agar (15 g/L) at room temperature in 40 Fernbach flasks. Afterwords, the fungal biomass and media were homogenized using an Ultra-Turrax apparatus and extracted with 8 L EtOAc to yield 10.0 g of crude extract. This material was fractionated by RP VLC using a stepwise gradient solvent system of increasing polarity starting from 50% methanol and 50% water to 100% methanol which yielded 12 fractions. RP-HPLC separation of the subfraction 11 (column: Waters Atlantis C18, 250 × 4.6 mm, 5 µm; methanol/H2O (95:5), flow rate 2 mL/min) afforded compounds 1–3. RP-HPLC separation of the subfraction 6 (column: Waters Atlantis C18, 250 × 4.6 mm, 5 µm; acetonitrile/H2O (70:30), flow rate 1.5 mL/min) afforded compounds 4–6.
Conio-azasterol (
1): Yellowish brown powder (7.5 mg; 0.7 mg·L
−1); UV λ
max MeOH (log ε): 395 (4.90), 245 (4.11) nm;
= +90 (C 0.3, CHCl
3); IR νmax (ATR): 3354, 2919, 2850, 1741, 1711, 1609, 1461, 1371, 1193, 1064, 1032, 977 cm
−1;
1H- and
13C-NMR see supp. info.
Table 1; (+)-HRESIMS:
m/
z found = 756.4235 [M + Na]
+ and
m/
z calcld = 756.4240 [M + Na]
+.
S-Dehydroazasirosterol (
2): Yellowish brown powder (9.3 mg; 0.9 mg·L
−1); UV λ
max MeOH (log ε): 395 (4.81), 240 (4.50) nm;
= +35 (C 0.6, CHCl
3); IR ν
max (ATR): 3382, 2919, 2850, 2358, 1737, 1711, 1608, 1461, 1371, 1295, 1193, 1064, 1033, 977, 861 cm
−1;
1H- and
13C-NMR see supp. info.
Table 2; (+)-HRESIMS:
m/
z found = 756.4235 [M + Na]
+ and
m/
z calcld = 756.4240 [M + Na]
+.
(22E,24R)-Ergosta-4,6,8(14)22-tetraen-3-one (3): Yellowish brown powder (8.5 mg; 0.8 mg·L−1); 13C-NMR (75 MHz; CDCl3, 25 °C): δ 34.0 (CH2-1), 18.9 (CH2-2), 200.5 (C-3), 122.6 (CH-4), 165.6 (C-5), 124.3 (CH-6), 134.6 (CH-7), 124.3 (C-8), 44.2 (CH-9), 36.8 (C-10), 25.4 (CH2-11), 34.0 (CH2-12), 44.0 (C-13), 156.7 (C-14), 35.5 (CH2-15), 27.7 (CH2-16), 55.6 (CH-17), 18.9 (CH3-18), 16.6 (CH3-19), 39.3 (CH-20), 21.2 (CH3-21), 134.9 (CH-22), 132.2 (CH-23), 42.8 (CH-24), 33.1 (CH-25), 19.6 (CH3-26), 19.9 (CH3-27), 17.6 (CH3-28); (−)ESIMS m/z = 391.6 [M − H]−.
The acetone adduct of compound 4 (
4a): Yellowish brown powder (10 mg; 1.0 mg·L
−1); UV λ
max MeOH (log ε): 331 (4.01), 255 (3.91), 216 (3.81) nm; IR ν
max (ATR): 2919, 2850, 2359, 1708, 1608, 1460, 1379, 1206, 836, 720, 668, 534 cm
−1;
1H- and
13C-NMR see supp. info.
Table 3; (+)-HRESIMS:
m/
z found = 421.1258 [M + Na]
+ and
m/
z calcld = 421.1264 [M + Na]
+.
The acetone adduct of compound 5 (
5a): Yellowish brown powder (12 mg; 1.2 mg·L
−1);
1H- and
13C-NMR see supp. info.
Table S4; (+)-HRESIMS:
m/
z found = 421.1260 [M + Na]
+ and
m/
z calcld= 421.1264 [M + Na]
+.
The hydrated form of compound 6 (
6a): Yellowish green powder (6.5 mg; 0.6 mgL
−1); UV λ
max MeOH (log ε): 385 (4.91), 242 (4.21) nm; IR ν
max (ATR): 3380, 2920, 2853, 2458, 1740, 1717, 1610, 1465, 1373, 1300, 1195, 1065, 1030, 970, 862 cm
−1;
1H- and
13C-NMR see supp. info.
Table 5; (+)-HRESIMS:
m/
z found = 396.1057 [M + Na]
+ and
m/
z calcld = 396.1059 [M + Na]
+.
3.4. Biological Activity
Cell Lines. The human cancer cell lines K562 (chronic myelogenous leukemia), U266 (myeloma), SKM1 (myelodysplastic symdrom), and Kasumi (acute myeloid leukemia) were provided by ATCC and were grown at 37 °C under 10% CO2 in RPMI 1640 medium (Gibco BRL, Paisley, UK) supplemented with 10% FCS (Gibco BRL, Paisley, UK) completed with 50 units/mL penicillin, 50 mg/mL streptomycin, and 1 mM sodium pyruvate.
Measurement of Cell Metabolism (XTT). Cells (10 × 104/mL) were incubated with 1 or 2 for the times indicated. 50 µL of XTT kit (sodium 39-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzene sulfonic acid hydrate) was added to each well, which contain 100 µL of medium. Absorbance of the formazan dye produced by metabolically active cells was measured at 490 nm. Each assay was performed in quadruplicate.
3.5. Molecular Modeling
All the modeling studies were carried out on a laptop PC, Intel
© Core™ i7-3630QM CPU @ 2.40 GHz, RAM 8 GB operating under Windows 7 professional. It consists of several steps. First, the 3D crystal structures of ERα-LBD with PDB code 1A52 [
15] were downloaded from Brookhaven Protein Data loaded to Molegro Virtual Docker (MVD 2013.6.0.0 [Win32], CLC Bio Company, Aarhus, Denmark) program fully functional free trial version with time limiting license (Molegro Virtual Docker (MVD 2013.6.0). ChemBio3D Ultra 10 [
16] was used to draw the 3D structures of different ligands. Ligands were further pre-optimized using a free version of Marvinsketch 4.1.13 from Chemaxon Ltd (Marvinsketch, version 6.1.0, ChemAxon company cheminformatics technology products services, Budapest, Hungary, 2013,
http://www.chemaxon.com) with MM force field and saved in Tripos mol2 file format. MolDock score functions were used with a 0.3 Å grid resolution. Prior to the calculations of the subject compounds, the MVD software was benchmarked docking the Estrogen.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}