
Graveoline Analogs Exhibiting Selective Acetylcholinesterase Inhibitory Activity as Potential Lead Compounds for the Treatment of Alzheimer’s Disease

Abstract
:1. Introduction
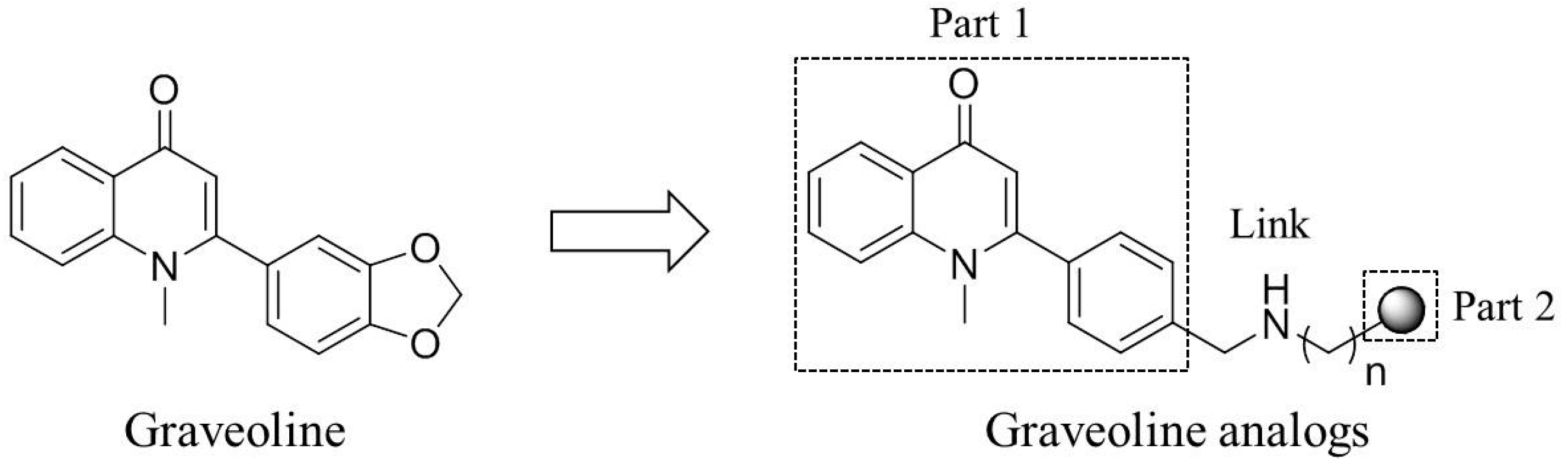
2. Results and Discussion
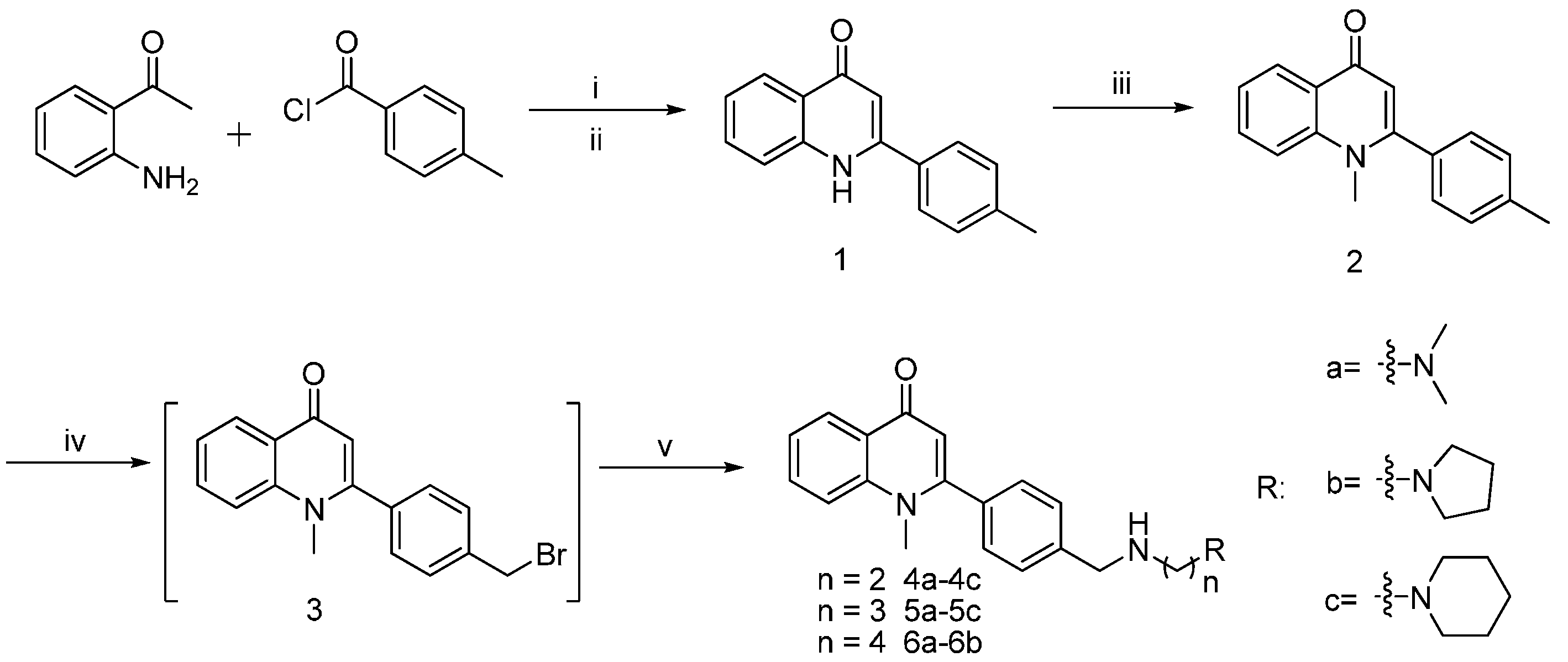
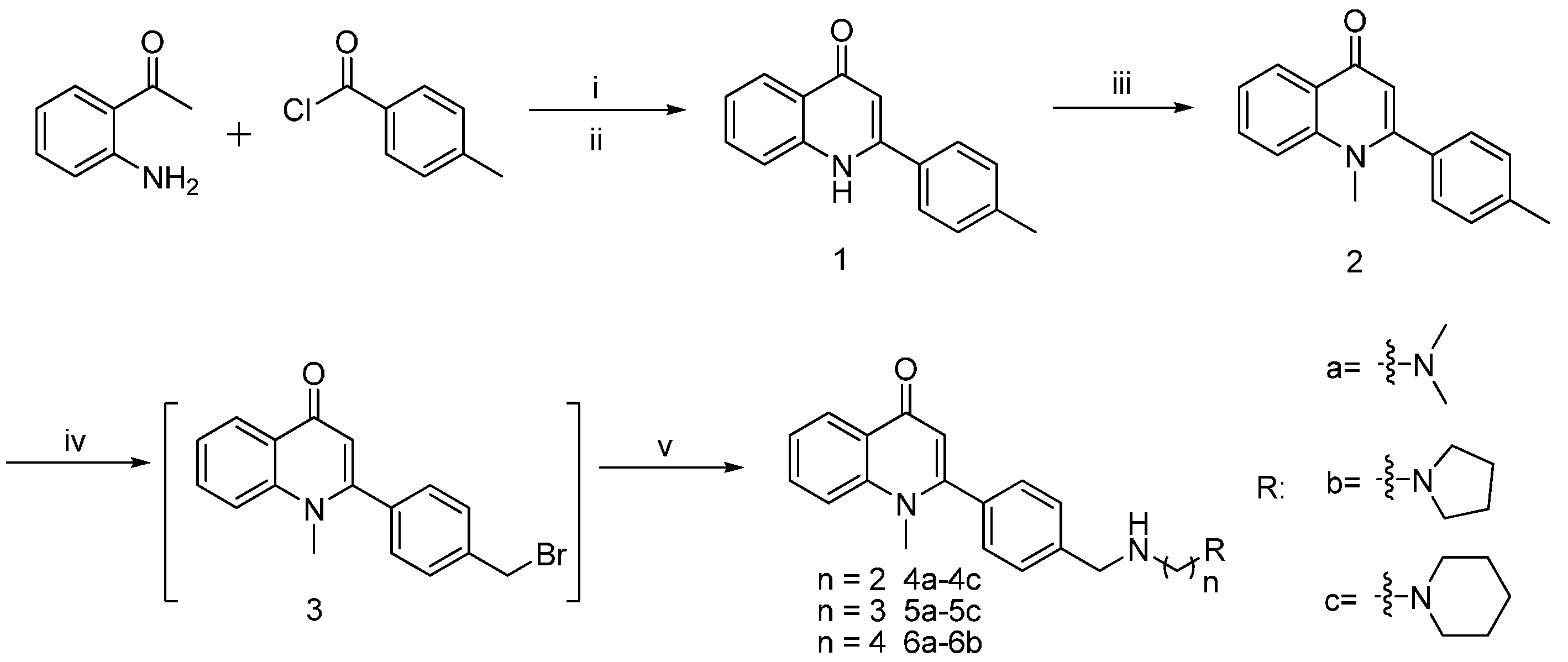
2.1. Chemistry
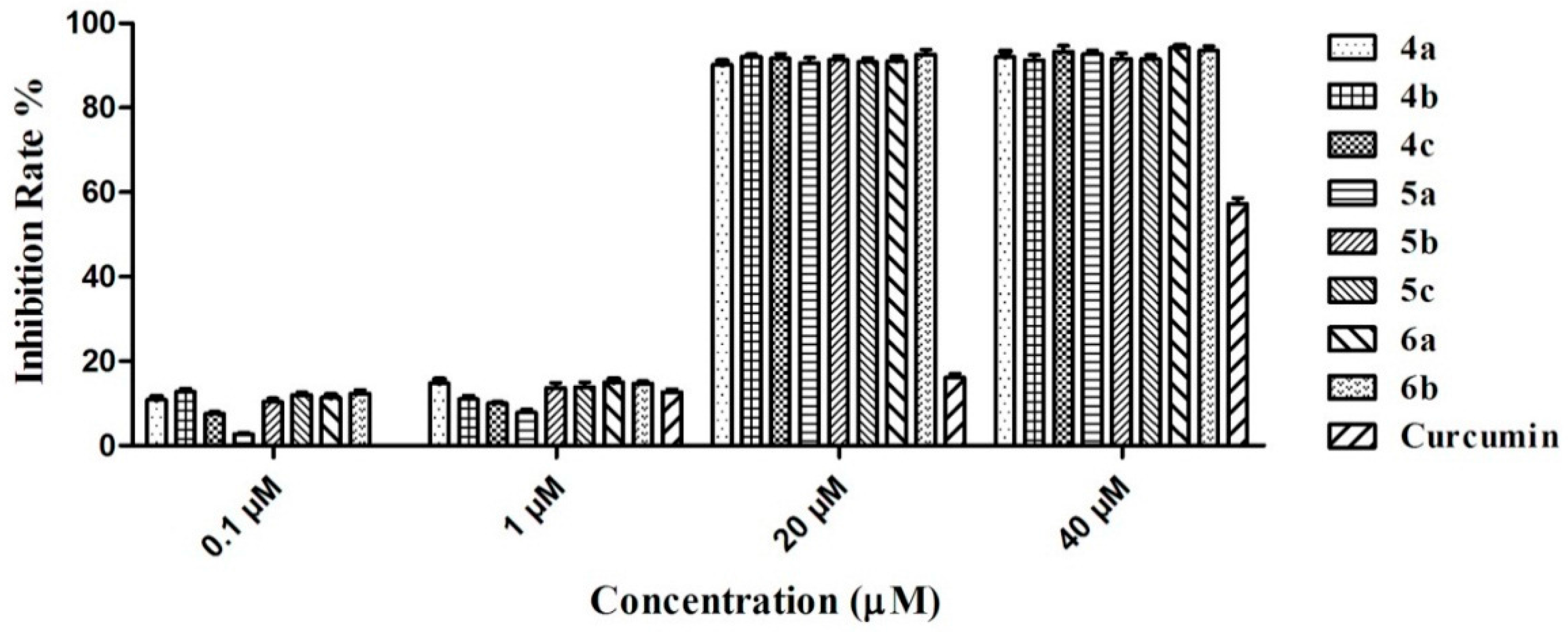
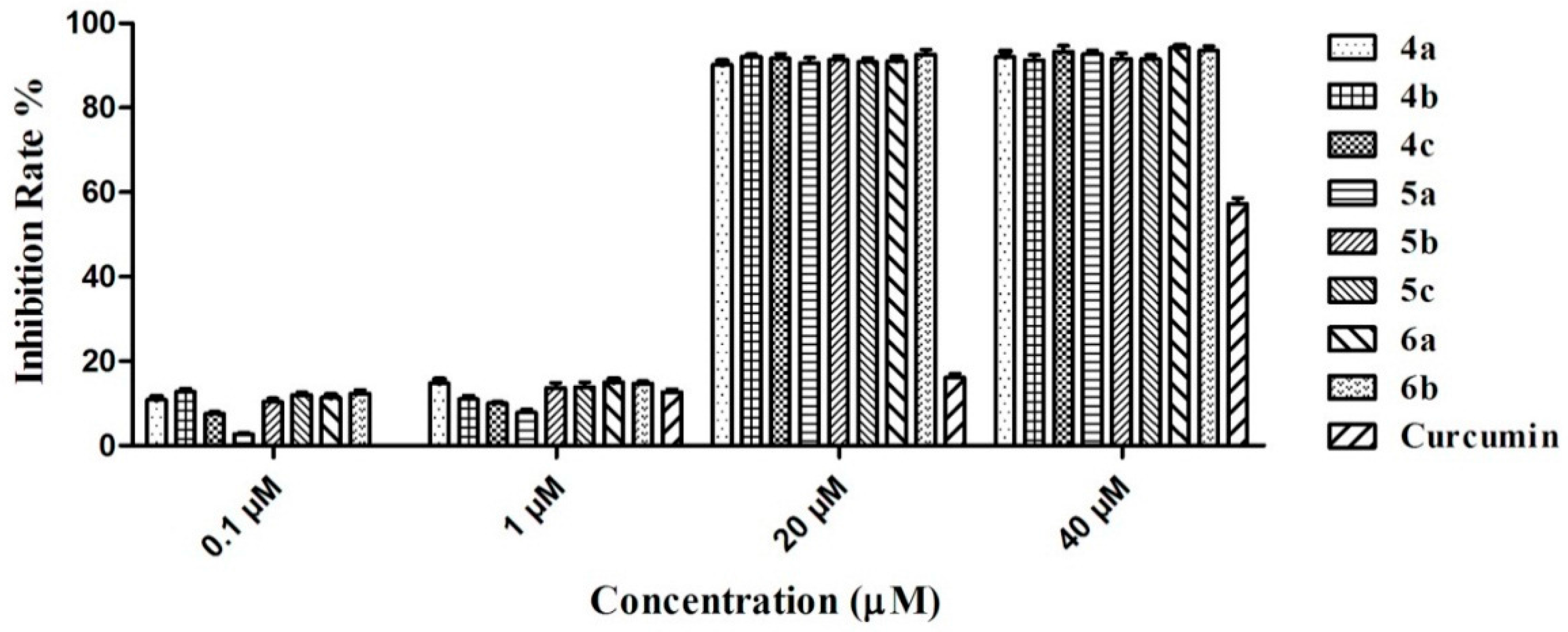
2.2. Inhibitory Activity of Graveoline Analogs on AChE and BuChE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | n | AChE Inhibition a, (IC50) nM | BuChE Inhibition b, (IC50) nM | Selectivity Index c |
|---|---|---|---|---|---|
| 4a | 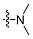 | 2 | 78.53 ± 1.76 | 3522 ± 46 | 45 |
| 5a | 3 | 33.36 ± 0.78 | 2680 ± 32 | 80 | |
| 6a | 4 | 35.51 ± 0.96 | 3674 ± 39 | 103 | |
| 4b | 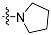 | 2 | 10.18 ± 1.33 | 2470 ±85 | 223 |
| 5b | 3 | 6.47 ± 0.15 | 1886 ± 54 | 291 | |
| 6b | 4 | 12.64 ± 0.57 | 2434 ± 49 | 193 | |
| 4c | 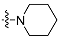 | 2 | 9.63 ± 0.76 | 1543 ± 29 | 160 |
| 5c | 3 | 2.36 ± 0.24 | 1147 ± 31 | 486 | |
| 2 | 3646 ± 38 | 9306 ± 76 | 3 | ||
| Tacrine | 224.7 ± 1.24 | 31.26 ± 0.34 | 0.1 |
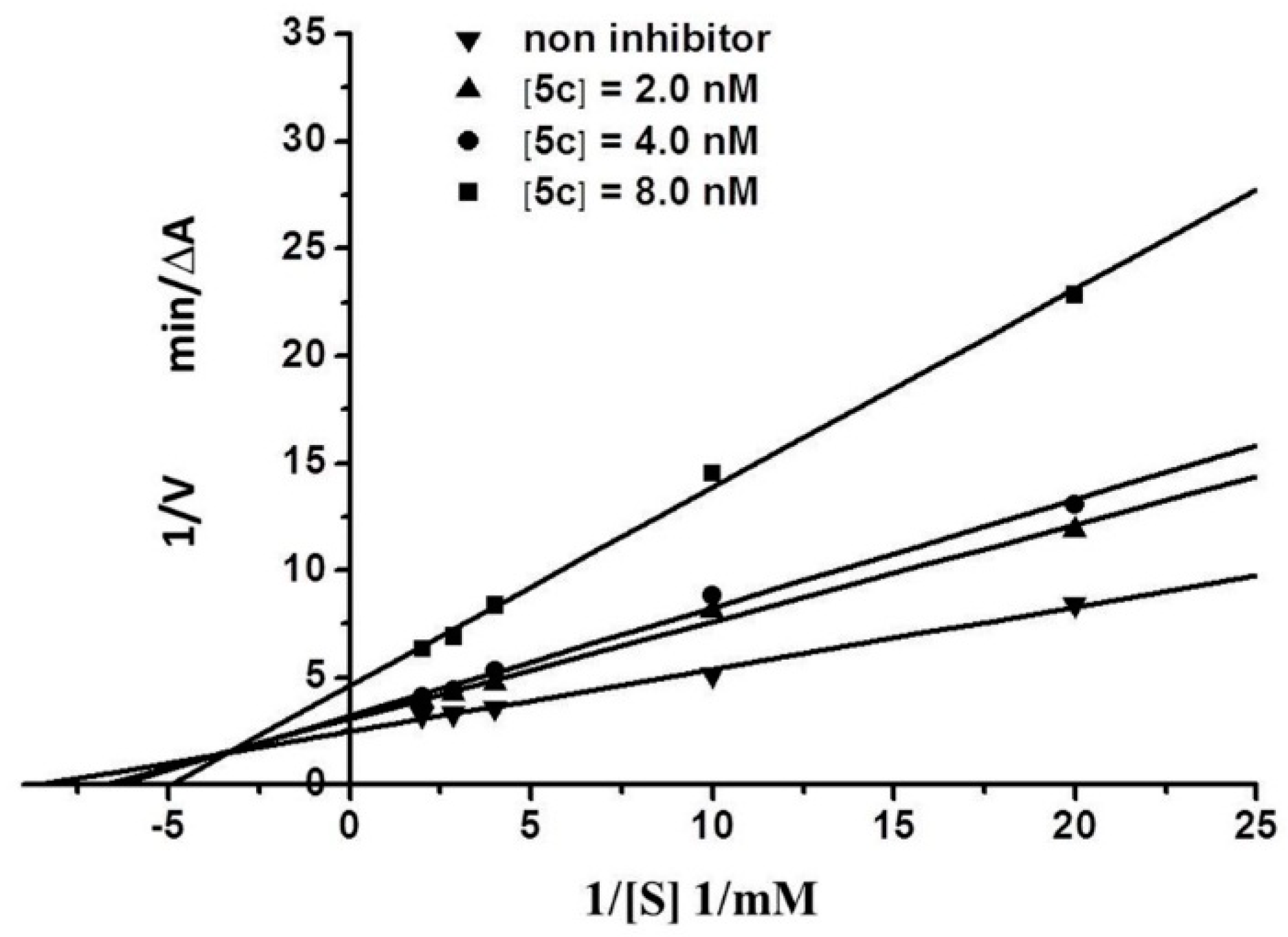
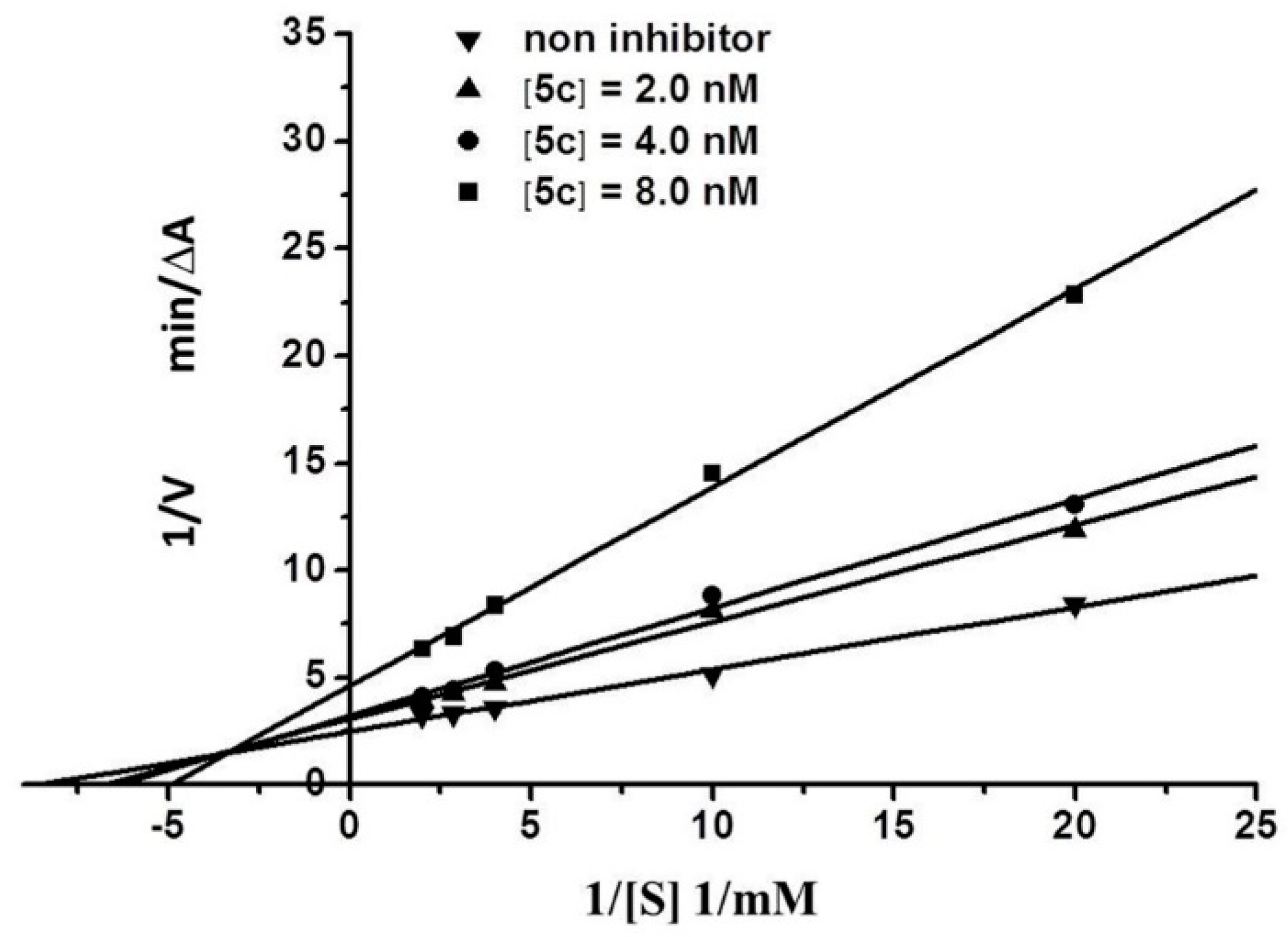
2.3. Kinetic Characterization of AChE Inhibition
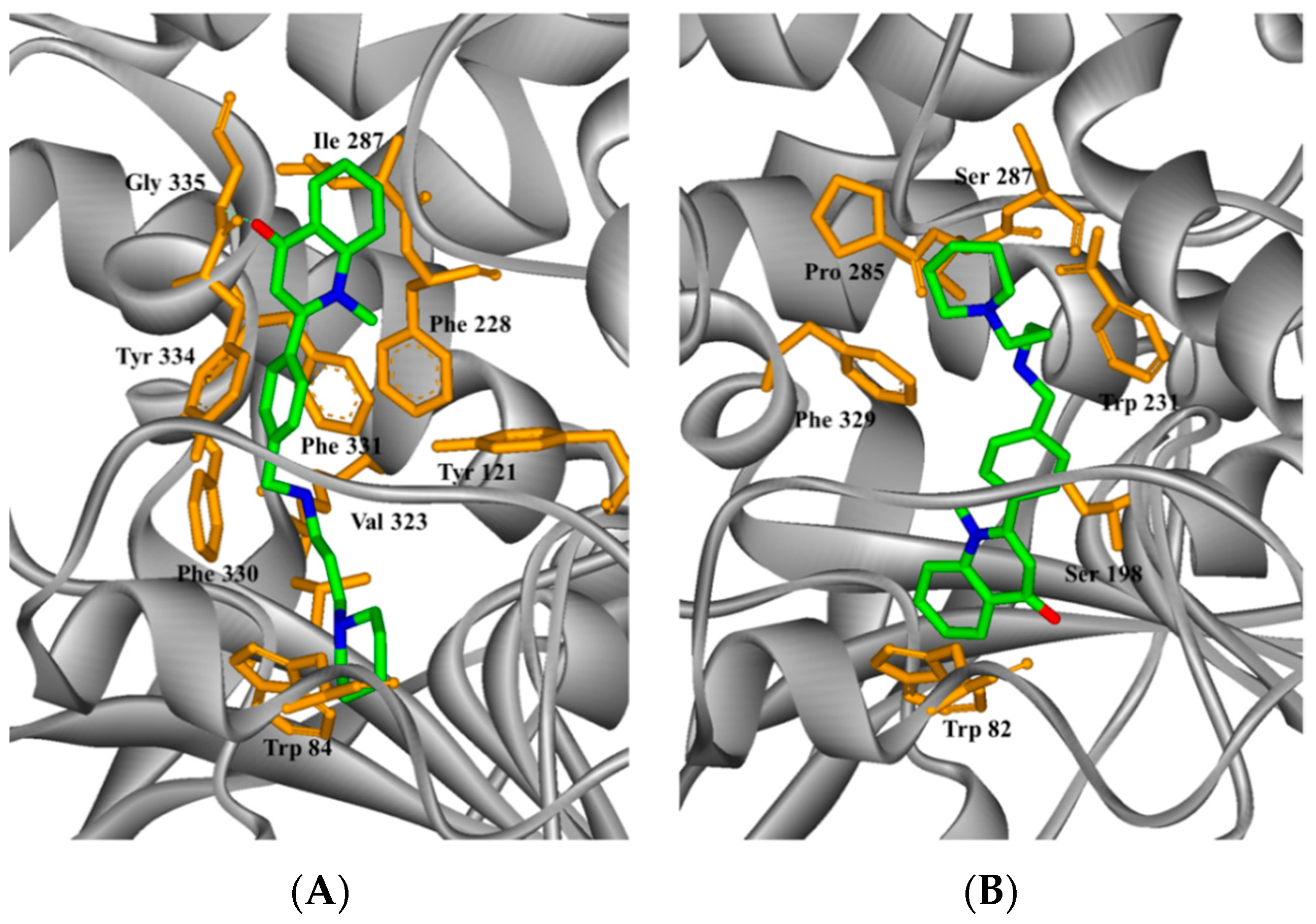
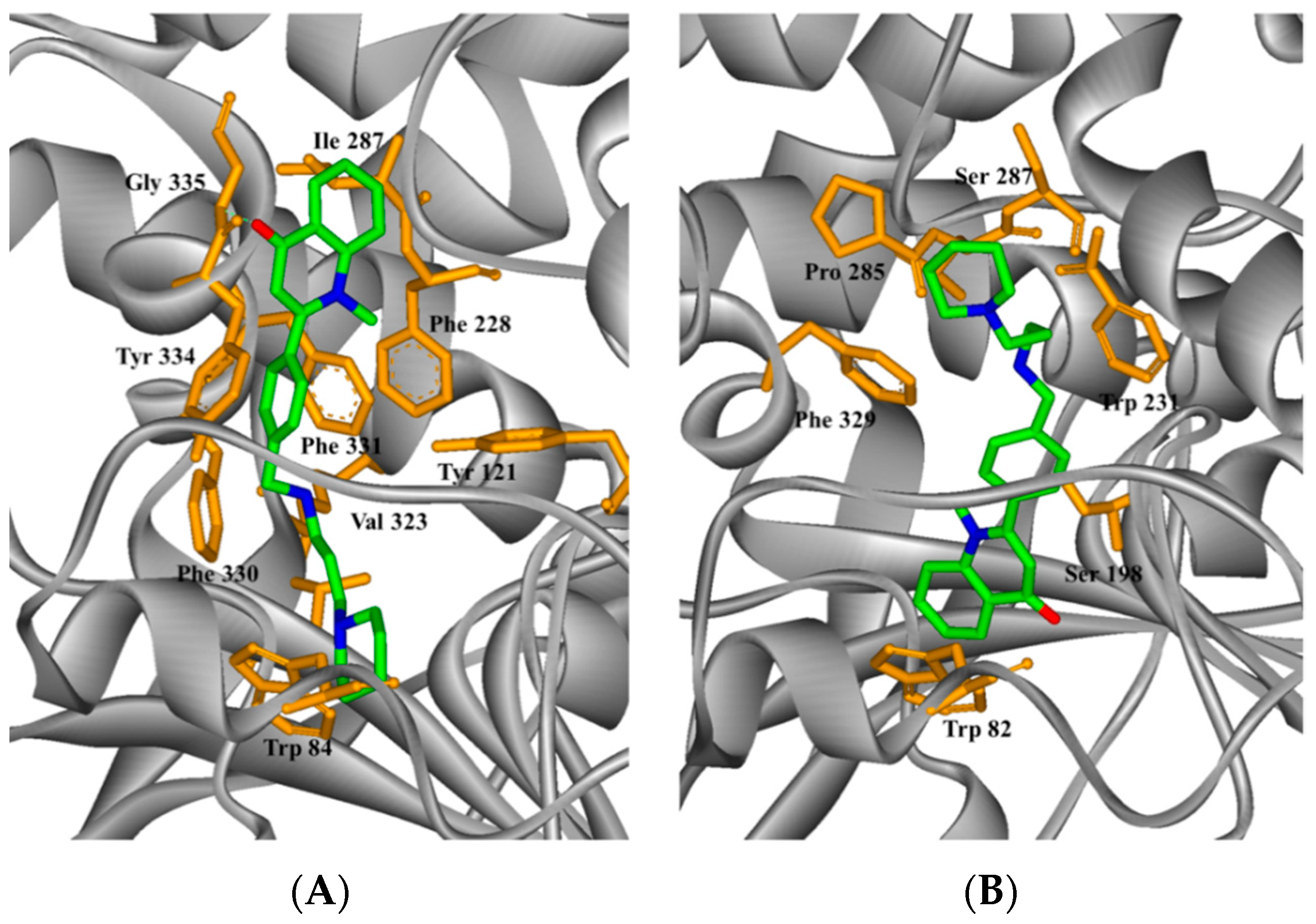
2.4. Study on the Interaction between 5c and Cholinesterase by Molecular Docking Method
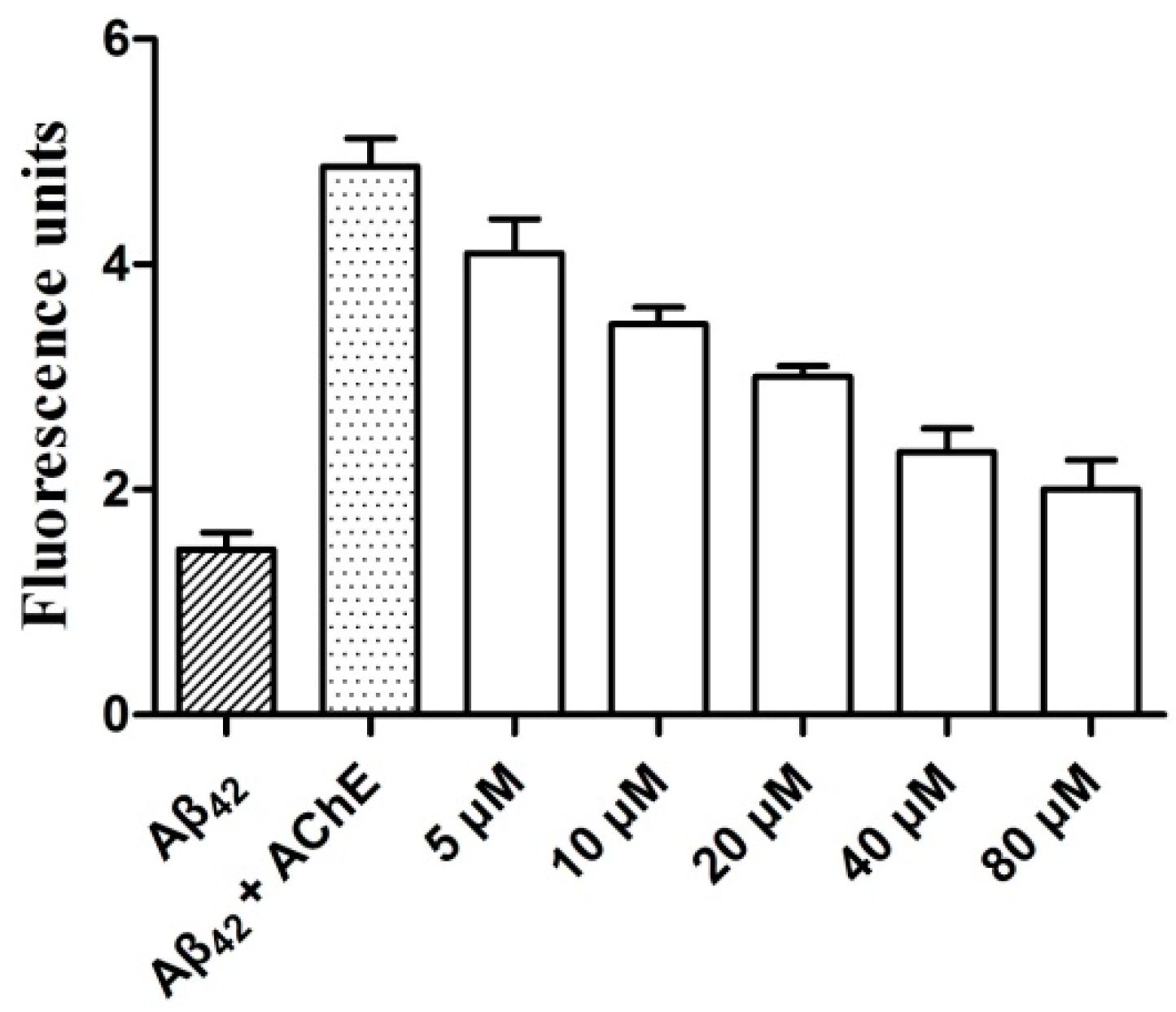
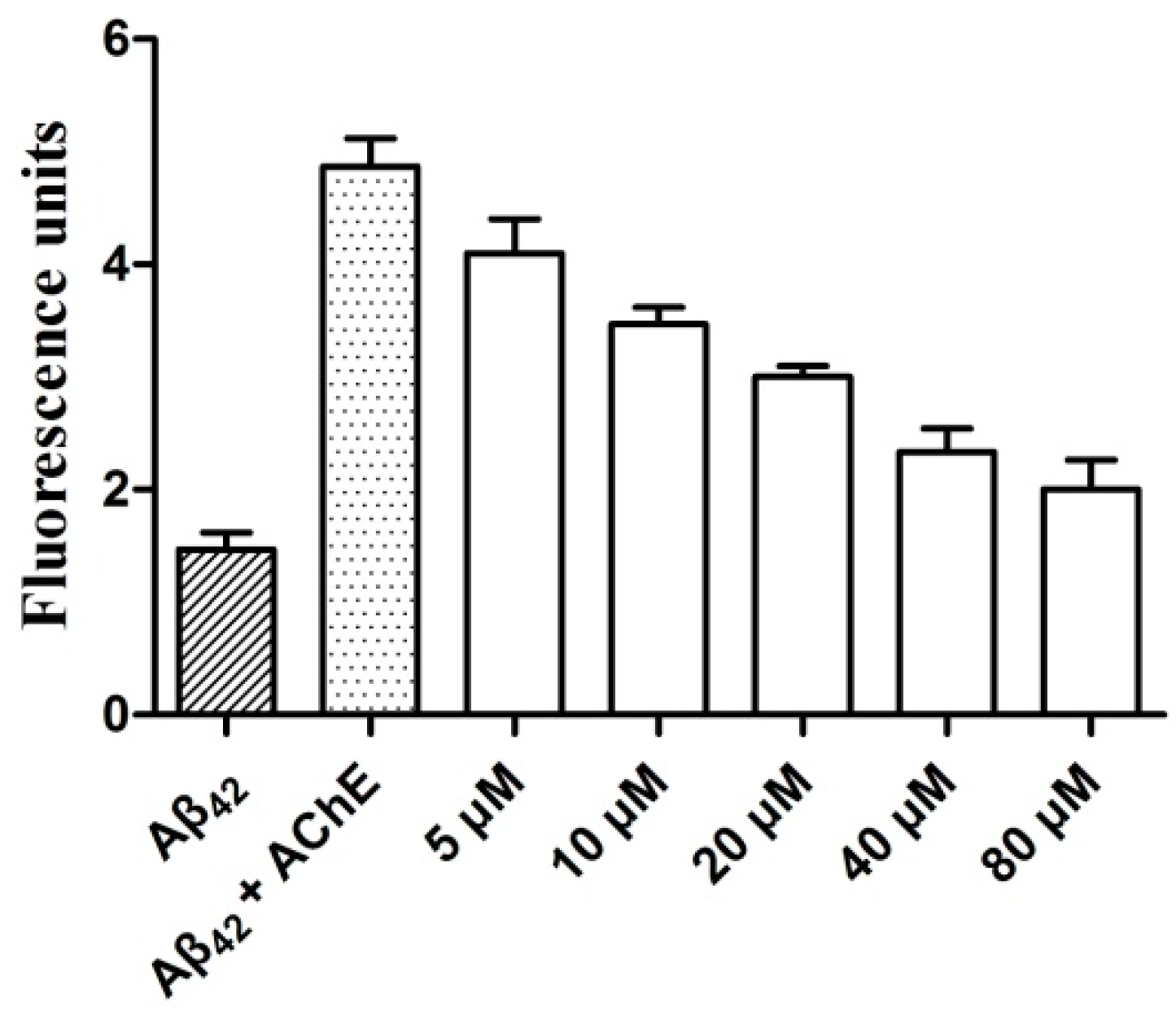
2.5. AChE-Induced Aβ Aggregation: Inhibition Studies
2.6. Effects of Graveoline Analogs on Neurocyte Viability
3. Materials and Methods
3.1. Chemistry
General Procedure for the Synthesis of Graveoline Analogs (4a–4c, 5a–5c, and 6a–6b)
3.2. Materials
3.3. In Vitro Inhibition of AChE and BuChE
3.4. Molecular Modeling
3.5. Thioflavin T-Based Fluorometric Assay
3.6. Cellular Toxicity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Amemori, T.; Jendelova, P.; Ruzicka, J.; Urdzikova, L.M.; Sykova, E. Alzheimer’s disease: Mechanism and approach to cell therapy. Int. J. Mol. Sci. 2015, 16, 26417–26451. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Sanchez, D.; Flores-Cuadrado, A.; Ubeda-Bañon, I.; de la Rosa-Prieto, C.; Martinez-Marcos, A. Interneurons in the human olfactory system in Alzheimer’s disease. Exp. Neurol. 2015, 276, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Klimova, B.; Maresova, P.; Valis, M.; Hort, J.; Kuca, K. Alzheimer’s disease and language impairments: social intervention and medical treatment. Clin. Interv. Aging 2015, 10, 1401–1407. [Google Scholar] [PubMed]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-β: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Henderson, Z. Responses of basal forebrain cholinergic neurons to damage in the adult brain. Prog. Neurobiol. 1996, 48, 219–254. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Kandemirli, F.; Saraçoglu, M.; Kovalishyn, V. Human acetylcholinesterase inhibitors: Electronic-topological and neural network approaches to the structure-activity relationships study. Mini Rev. Med. Chem. 2005, 5, 479–487. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid β-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Asuncion Morán, M. Cholinesterases within neurofibrillary tangles related to age and Alzheimer’s disease. Ann. Neurol. 1987, 22, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Doody, R.; Clark, C. Disease-modifying therapies for Alzheimer disease: Challenges to early intervention. Neurology 2007, 69, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, G.; Palacios-Esquivel, R.; Moss, D.E. Cholinesterase inhibitors proposed for treating dementia in Alzheimer’s disease: Selectivity toward human brain acetylcholinesterase compared with butyrylcholinesterase. J. Pharmacol. Exp. Ther. 1995, 274, 767–770. [Google Scholar] [PubMed]
- Liston, D.R.; Nielsen, J.A.; Villalobos, A.; Chapin, D.; Jones, S.B.; Hubbard, S.T.; Shalaby, I.A.; Ramirez, A.; Nason, D. Pharmacology of selective acetylcholinesterase inhibitors: Implications for use in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 486, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, D.E.; Koeppe, R.A.; Snyder, S.E.; Minoshima, S.; Frey, K.A.; Kilbourn, M.R. In vivobutyrylcholinesterase activity is not increased in Alzheimer’s disease synapses. Ann. Neurol. 2006, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, K.; Al-Said, M.S.; El-Feraly, F.S.; Ross, S.A. New quinoline alkaloids from Ruta chalepensis. J. Nat. Prod. 2000, 63, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.L.; Meepagala, K.M.; Oliva, A.; Aliotta, G.; Duke, S.O. Phytotoxins from the leaves of Ruta graveolens. J. Agric. Food Chem. 2004, 52, 3345–3349. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Bishayee, K.; Khuda-Bukhsh, A.R. Graveoline isolated from ethanolic extract of Ruta graveolens triggers apoptosis and autophagy in skin melanoma cells: A novel apoptosis-independent autophagic signaling pathway. Phytother. Res. 2014, 28, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhu, X.; Holloway, H.W.; Whittaker, N.F.; Brossi, A.; Greig, N.H. Anticholinesterase activity of compounds related to geneserine tautomers. N-oxides and 1,2-oxazines. J. Med. Chem. 2002, 45, 3684–3691. [Google Scholar] [PubMed]
- Takeuchi, Y.; Shibata, T.; Suzuki, E.; Iimura, Y.; Kosasa, T.; Yamanishi, Y.; Sugimoto, H. 1-Benzyl-4-[(5,6-dimethoxy-2-fluor-1-indanon)-2-y1]methyl Piperidine. European Patent WO 02/20482 A1, 14 March 2012. [Google Scholar]
- Martineza, A.; Fernandeza, E.; Castroa, A.; Condea, S.; Rodriguez-Francoa, L.; Banosb, J.E.; Badiab, A. N-Benzylpiperidine derivatives of 1,2,4-thiadiazolidinone as new acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2000, 35, 913–922. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.R., Jr.; Featherstone, M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Wang, B.; Mai, Y.C.; Li, Y.; Hou, J.Q.; Huang, S.L.; Ou, T.M.; Tan, J.H.; An, L.K.; Li, D.; Gu, L.Q.; et al. Synthesis and evaluation of novel rutaecarpine derivatives and related alkaloids derivatives as selective acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2010, 45, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Laura, P.; Carrie, W.; Meghan, L.; Jared, W. Comparison of motility, recovery, and methyl-thiazolyl-tetrazolium reduction assays for use in screening plant products for anthelmintic activity. Parasitol. Res. 2009, 105, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Mu, C.; Wang, B.; Jin, J. Graveoline Analogs Exhibiting Selective Acetylcholinesterase Inhibitory Activity as Potential Lead Compounds for the Treatment of Alzheimer’s Disease. Molecules 2016, 21, 132. https://doi.org/10.3390/molecules21020132
Li Z, Mu C, Wang B, Jin J. Graveoline Analogs Exhibiting Selective Acetylcholinesterase Inhibitory Activity as Potential Lead Compounds for the Treatment of Alzheimer’s Disease. Molecules. 2016; 21(2):132. https://doi.org/10.3390/molecules21020132
Chicago/Turabian StyleLi, Zeng, Chaoyu Mu, Bin Wang, and Juan Jin. 2016. "Graveoline Analogs Exhibiting Selective Acetylcholinesterase Inhibitory Activity as Potential Lead Compounds for the Treatment of Alzheimer’s Disease" Molecules 21, no. 2: 132. https://doi.org/10.3390/molecules21020132
APA StyleLi, Z., Mu, C., Wang, B., & Jin, J. (2016). Graveoline Analogs Exhibiting Selective Acetylcholinesterase Inhibitory Activity as Potential Lead Compounds for the Treatment of Alzheimer’s Disease. Molecules, 21(2), 132. https://doi.org/10.3390/molecules21020132