1. Introduction
Technologies based on the exponential amplification of nucleic acids are widely used in molecular diagnostics and biosensing. For example, the quantitative polymerase chain reaction (qPCR) is a sensitive and precise method for detecting target nucleic acids [
1,
2]. Due to its capacity for exponential amplification, qPCR can detect minute amounts of a target. The starting concentration of the target can be determined based on the number of thermal cycles required to reach an amplification threshold. Other methods can be used to detect non-nucleic-acid targets based on the binding of the target to an antibody, aptamer, or other capture reagent. In such cases the binding event must usually be coupled to a means for signal amplification, such as enzyme turnover or nucleic acid amplification. A different approach is to employ a capture reagent that itself undergoes exponential amplification upon binding the target. Such a system has been demonstrated based on aptamer-containing, self-replicating RNA enzymes [
3,
4]. A major shortcoming of that system is that, unlike qPCR, it does not provide quantitation throughout the course of amplification. That shortcoming has now been addressed, enabling the real-time, ligand-dependent, exponential amplification of RNA.
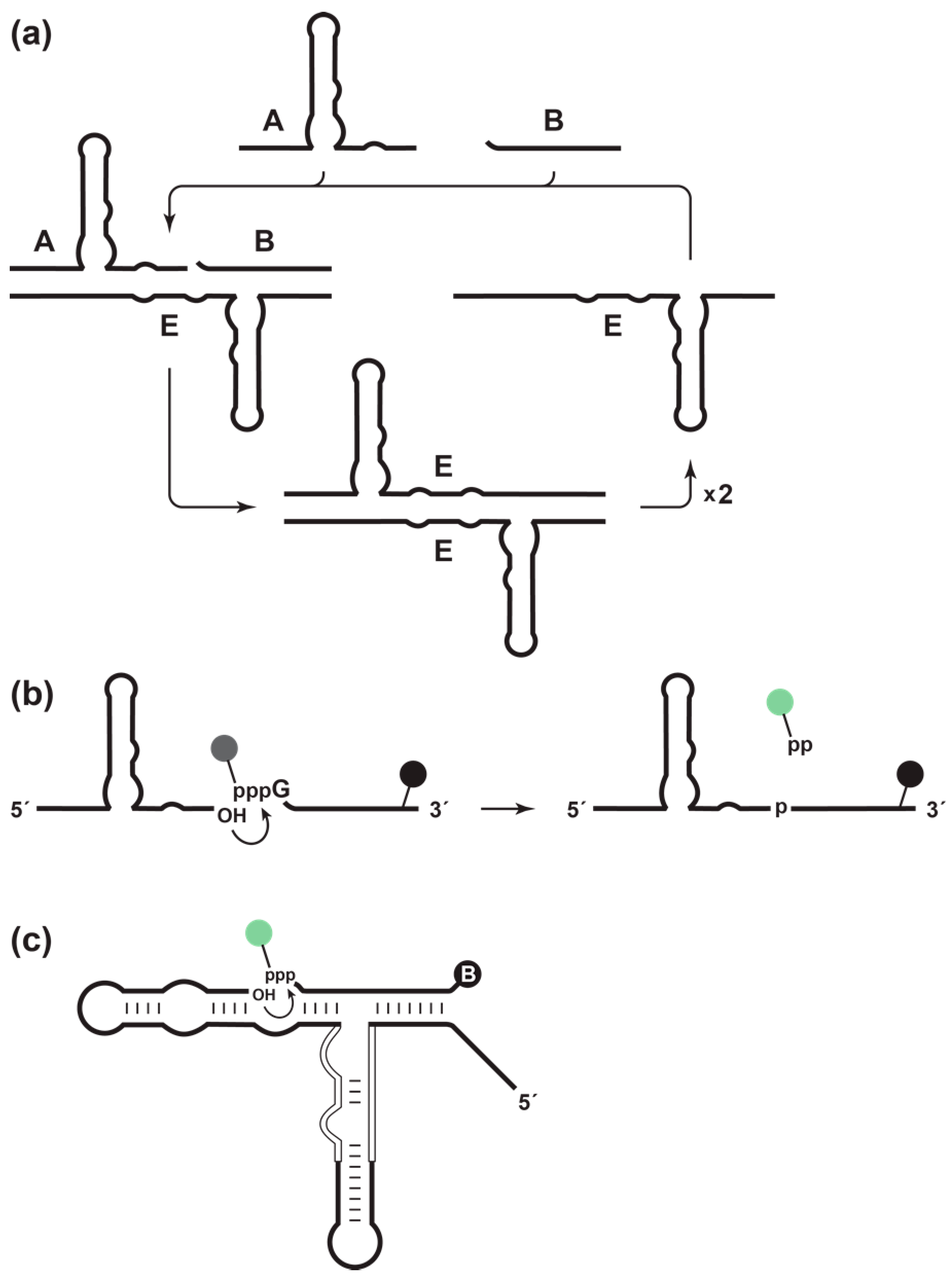
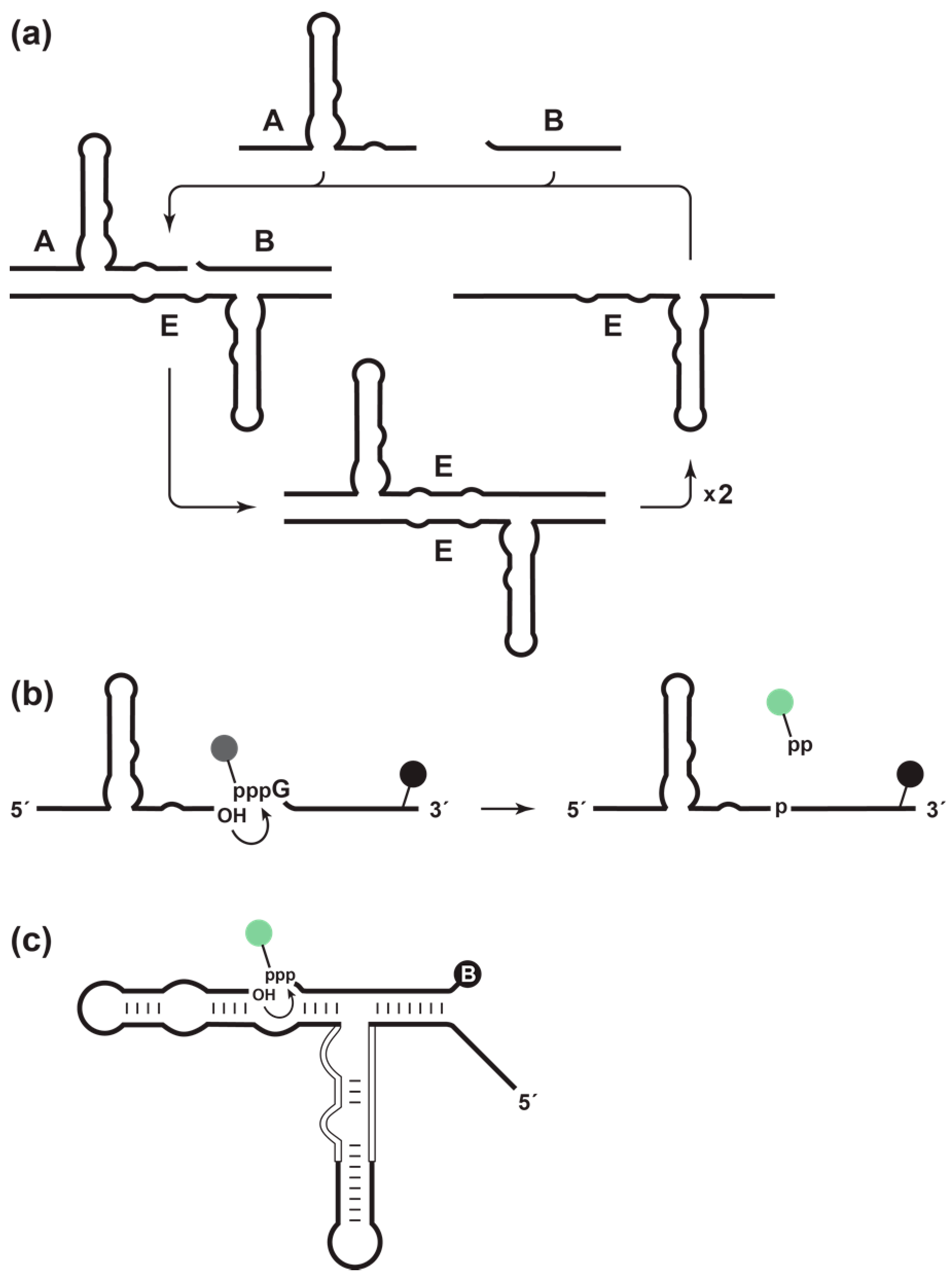
The self-replicating RNA enzyme (E) is an RNA ligase that catalyzes the joining of two RNA substrates (A and B) to form another copy of itself [
5,
6]. The ligation reaction involves an attack of the 3′-OH of substrate A on the 5′-triphosphate of substrate B, forming a 3′,5′-phosphodiester linkage and releasing inorganic pyrophosphate. The resulting E•E complex dissociates in a non-rate-limiting manner [
7] to yield two copies of E, each of which can enter another replication cycle (
Figure 1a). Replication continues at a constant temperature until the supply of substrates is exhausted. The rate of exponential amplification depends on the inherent catalytic properties of the enzyme. For the original self-replicating enzyme, this rate is 0.03 min
–1, corresponding to a doubling time of 23 min [
6]. For an improved form of the enzyme, the exponential amplification rate is 0.14 min
–1, corresponding to a doubling time of 5 min [
8].
The catalytic activity of the RNA enzyme can be made dependent on a target ligand by linking a ligand-binding aptamer domain to the catalytic center of the enzyme [
3]. The resulting “aptazyme” [
9] binds the ligand in a manner that stabilizes the catalytic center, thereby triggering enzymatic activity and causing exponential amplification to be dependent on the presence of the ligand. The concentration of the ligand relative to the
Kd of the aptamer domain determines the degree of saturation of the aptazyme, which, as previously shown [
3,
4], determines the rate of exponential amplification relative to the rate at saturation. The faster the rate of amplification, the more quickly the system reaches a defined amplification threshold. Thus, like qPCR, the system can be used to measure precisely the concentration of the target ligand; however, unlike qPCR, it can be generalized to any target for which a suitable aptamer is available.
A previous limitation of the RNA replication system was that it could not be applied to biological samples without first removing ribonucleases that would otherwise degrade the RNA. This limitation was overcome by preparing the enzyme and its substrates in the form of enantiomeric
l-RNA [
10], which is impervious to ribonucleases. The
d- and
l-RNA enzymes have identical biochemical properties, but only the latter have been shown to operate in the presence of biological samples such as human serum [
10]. This is important, for example, if one wants to measure the concentration of serum proteins.
For the ligand-dependent exponential amplification of RNA to be a practical method, it would be important to track the course of amplification in real time, based on the development of a fluorescent signal or other simple readout. Previous analyses were conducted after the reaction had been completed, using either polyacrylamide gel electrophoresis (PAGE) to determine the amount of ligated products or a luciferase assay that is driven by the release of inorganic pyrophosphate [
3].
In this study, a fluorimetric method was developed to monitor the amplification of the self-replicating RNA enzyme in real time. The B substrate was labeled with a fluorophore linked to the γ-phosphate of the 5′-triphosphate, and a quencher was placed at the 3′-end (
Figure 1b). Prior to ligation, fluorescence is quenched; however, upon ligation, the pyrophosphate is released together with the fluorophore, separating the fluorophore from the quencher and giving rise to a fluorescent signal. This is analogous to “pyrosequencing” methods for DNA sequencing, which employ modified dNTPs that contains both a fluorophore linked to the γ-phosphate and a quencher linked to the base [
11]. Just as pyrosequencing required engineering the DNA polymerase to accept the modified dNTPs, it was necessary to engineer the self-replicating RNA enzyme to accept the modified B substrate. This was achieved using in vitro selection, which resulted in a variant form of the enzyme that can undergo exponential amplification with the modified substrate, can function as either
d- or
l-RNA, and can be made to operate in a ligand-dependent manner.
3. Discussion
The self-replicating RNA enzyme is the only known macromolecule that can produce additional copies of itself and undergo exponential amplification in a self-sustained manner. This enzyme has been used to study processes of molecular evolution relevant to the origins of life [
6,
8,
16]. It also has been investigated for potential applications in molecular diagnostics and biosensing [
3,
4]. The latter make use of the ability of the enzyme to operate in a ligand-dependent manner, where the rate of exponential amplification is dependent on the concentration of the target ligand.
Following initial reports of ligand-dependent exponential amplification of RNA, three key shortcomings of the system were identified. First, because the enzyme and its substrate are composed of RNA, they are susceptible to degradation by ribonucleases that are present in biological samples. While it is possible to remove or inactivate these ribonucleases, such processing would affect other proteins in the sample that may be the target for detection. This shortcoming was addressed by preparing the enzyme and its substrates in the form of
l-RNA, which is resistant to ribonuclease degradation [
10]. A second shortcoming is that the original form of the self-replicating RNA enzyme has a doubling time of 23 min, requiring assay times of a few hours for the quantitative detection of a target ligand. Accordingly, an improved form of the enzyme was developed that has a doubling time of only 5 min and an exponential growth rate of 0.14 min
−1 [
8]. This rate is close to the limit of 0.21 min
–1 imposed by the rate of product release of the E•E complex [
7].
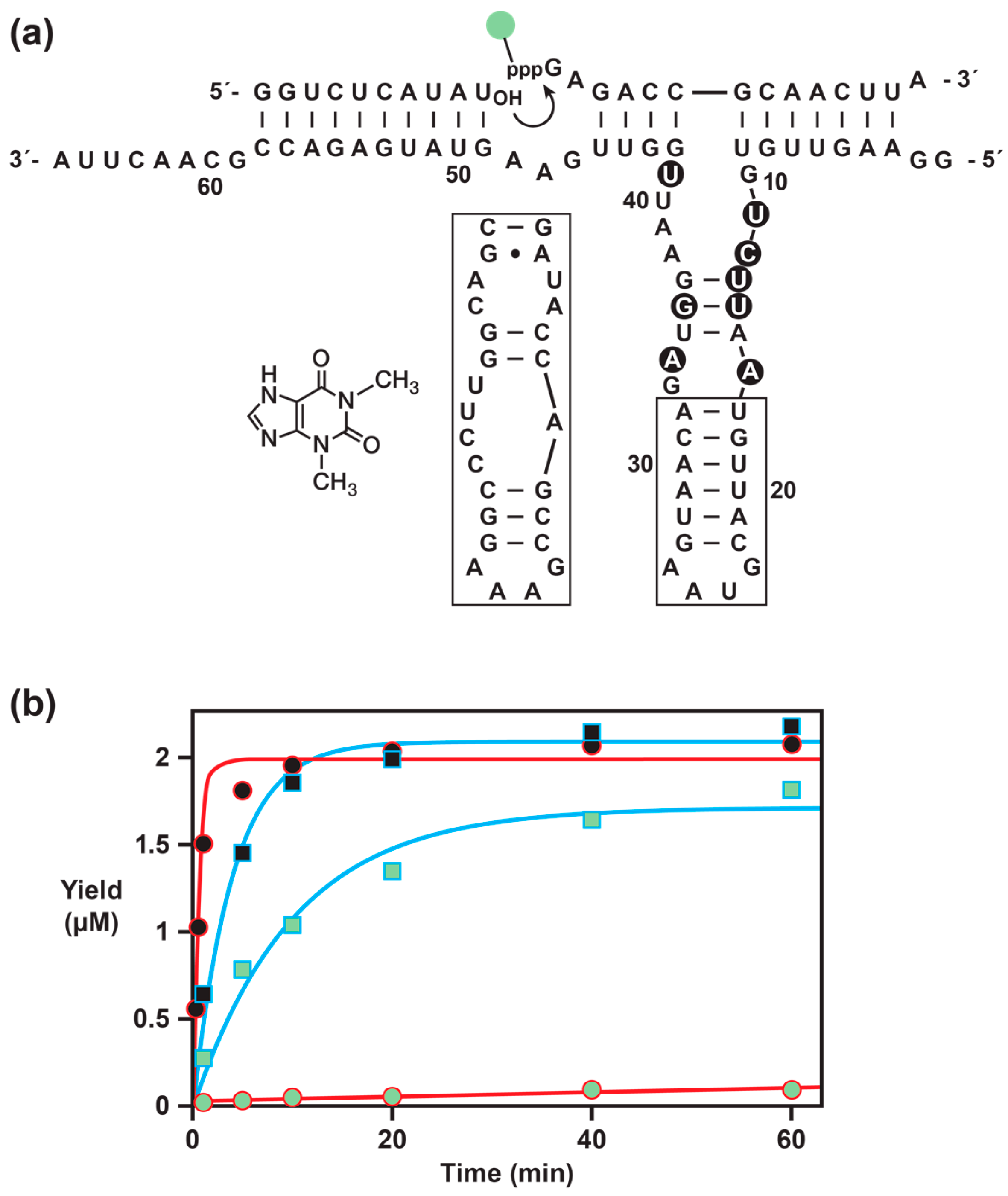
The present study sought to address the third shortcoming of the ligand-dependent exponential amplification system, which is the need to provide a convenient real-time assay, preferably one based on a simple fluorimetric readout. Methods based on FRET signaling and molecular beacons were explored, but suffered from high background and potential false positive signals. Instead, by linking the fluorescent reporter directly to the pyrophosphate leaving group, one obtains a direct measurement of the RNA ligation event that underlies exponential amplification. By also placing a fluorescence quencher at a suitable location within the substrate, the level of background fluorescence is very low. The key challenge was that, because the 5′-triphosphate is directly involved in the ligation reaction, it is generally intolerant of substitution. This limitation was overcome by using in vitro selection to obtain a variant enzyme that can operate efficiently when a fluorescent label is linked to the 5′-γ-phosphate of the substrate.
Exponential amplification of the selected enzyme with the labeled substrate is only three-fold slower than for the original self-replicating enzyme with the unmodified substrate. However, the rate-enhanced form of the enzyme described above is another five-fold faster when reacting with the unmodified substrate [
8]. The constellation of eight mutations present in the fluorophore-accommodating substrate are not compatible with the six mutations present in the rate-enhanced enzyme. Thus, further efforts will be needed to combine the advantageous properties of both enzymes.
Regardless of the sequence of the enzyme, it can be readily prepared in the form of enantiomeric
l-RNA having the same catalytic properties. This is why it was essential to prepare the fluorophore-modified substrate by chemical synthesis, making it available for use as either
d- or
l-RNA. Even for the
d-RNA substrate, the synthetic approach is preferred because it does not require competition between γ-(6-aminohexyl)-GTP and GTP during in vitro transcription to install a derivatizable group on the 5′-γ-phosphate. Chemical synthesis also avoids the 5′-terminal sequence heterogeneity that commonly occurs during in vitro transcription [
17,
18].
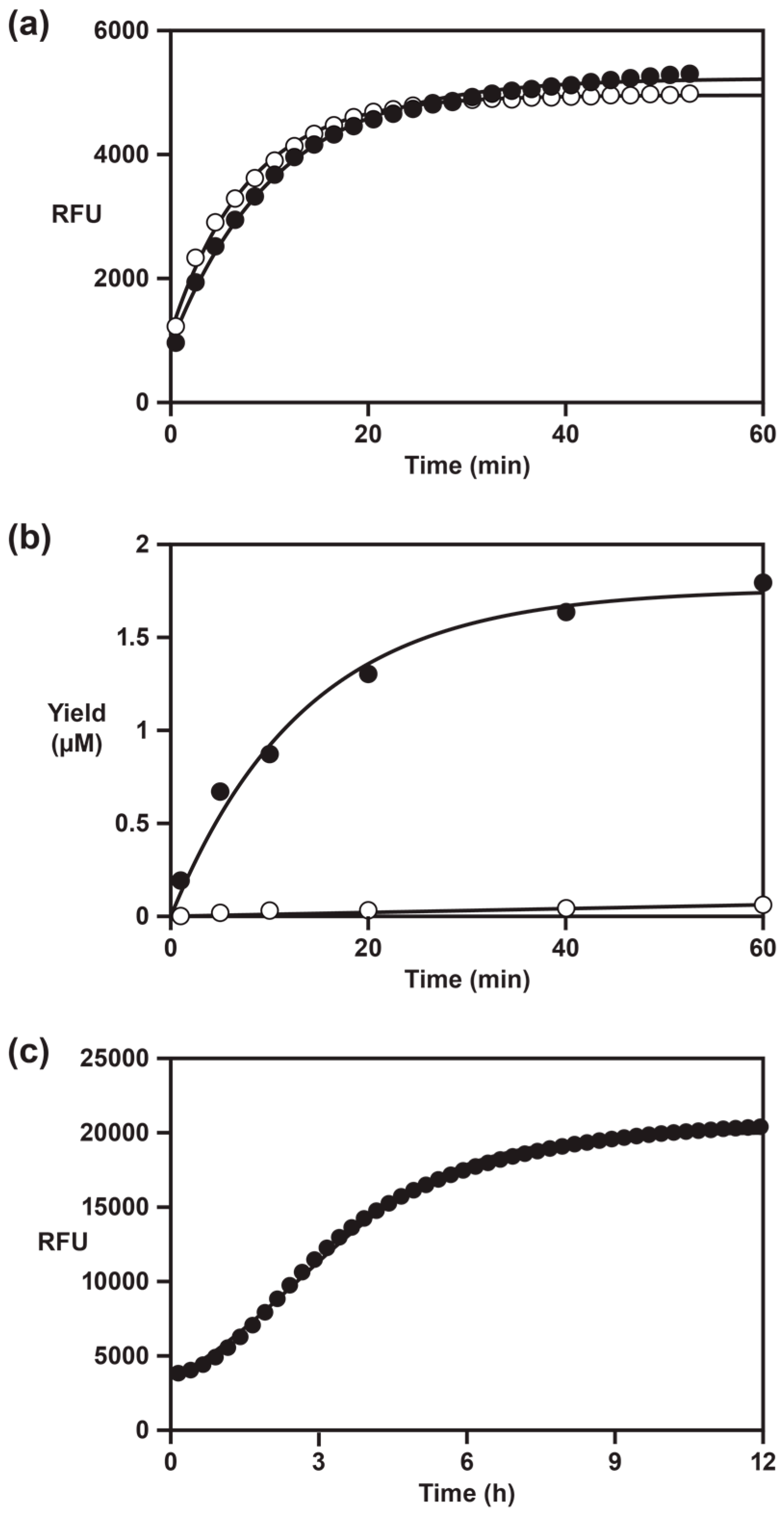
A potential application of the fluorescent-signaling, self-replicating RNA enzyme is to monitor ligand-dependent exponential amplification in real time. The instrument used in this study was a standard qPCR device with 96-well format, although maintaining a constant reaction temperature rather than thermal cycling. The time required to reach an amplification threshold that is distinguishable from background is about 2–3 h (
Figure 3c), which is longer than typically required for qPCR. The rate-enhanced self-replicating enzyme has an exponential growth rate comparable to PCR, but is not capable of direct fluorescent signaling. Despite the slower rate of the present system compared with PCR, it has the advantage of potentially being used to detect a wide variety of ligands, including small molecules, peptides, and proteins. As the inventory of known aptamers, including
l-RNA aptamers, continues to expand, the opportunities to combine those reagents with the real-time exponential amplification system will expand accordingly.
4. Materials and Methods
4.1. Materials
Synthetic oligonucleotides were prepared by solid-phase synthesis using an Expedite 8909 DNA/RNA synthesizer. All nucleoside phosphoramidites and 2′-
O-propargyl uridine 3′-Lcaa CPG were purchased from ChemGenes (Wilmington, MA, USA). Both T7 RNA polymerase and RNaseP M1 RNA were prepared as described previously [
6]. Bovine pancreatic DNase I was purchased from Roche Life Science (Indianapolis, IN, USA), Superscript II RNase H
– reverse transcriptase and Alexa Fluor 488 NHS ester were from Thermo Fisher (Carlsbad, CA, USA), and yeast poly(A) polymerase was from Affymetrix (Santa Clara, CA, USA). Tributylammonium pyrophosphate, 6-(Boc-amino)-1-hexanol, TMR-NHS ester, biotin-PEG4-alkyne, NTPs, dNTPs, and theophylline were purchased from Sigma-Aldrich (St. Louis, MO, USA), γ-(6-aminohexyl)-GTP was from Jena Bioscience (Jena, Germany), 2′-azido,2′-deoxyadenosine 5′-triphosphate was from TriLink Biotechnologies (San Diego, CA, USA), and Tide Quencher™ 3 (TQ3) azide was from AAT Bioquest (Sunnyvale, CA, USA).
The TMR-B-biotin substrate was prepared by in vitro transcription of an extra-length RNA, which was cleaved using
E. coli RNase P M1 RNA and an external guide RNA to yield RNA with a homogeneous end and free 2′- and 3′-hydroxyls [
3,
8,
19]. The transcription mixture contained 2 mM of GTP and 5 mM each of γ-(6-aminohexyl)-GTP, ATP, and CTP. The external guide RNA had the sequence 5′-GGAG
UAAGUUGCGGUCUCACCA-3′ (region of hybridization underlined). Following RNA cleavage, the RNA was extended by a single 2′-azido,2′-deoxyadenosine residue using yeast poly(A) polymerase, then coupled to biotin-PEG4-alkyne, as described previously [
8].
In vitro transcribed RNA enzymes were prepared by run-off transcription, as described previously [
4,
6]. Double-stranded DNA templates encoding the enzyme were prepared by cross-extension of two fully-complementary synthetic oligodeoxynucleotides. For the selected enzyme the sense strand had the sequence 5′-GGC
TAATACGACTCACTATAGGAAGTTGTGTCTTAATGTTACGTAAGTAACAGATGGAATTGGTTGAAGTATGAGACCGCAACTTA-3′; for E
theo, the sense strand had the sequence 5′-GGC
TAATACGACTCACTATAGGAAGTTGTGTCTTAA
GATACCAGCCGAAAGGCCCTTGGCAGCGATGGAATTGGTTGAAGTATGAGACCGCAACTTA-3′ (T7 RNA polymerase promoter sequence underlined; aptamer domain in bold).
In vitro transcribed substrate A containing the eight mutations present in the selected enzyme was prepared by first generating the enzyme, as described above. Then, the B-substrate portion was removed using M1 RNA and the same external guide RNA described above.
Synthetic d- and l-RNA enzymes and both the full-length and simplified forms of substrate A were prepared by automated solid-phase synthesis using 2′-TBDMS phosphoramidites. The RNAs were deprotected using a 1:1 mixture of aqueous ammonia and methylamine at 65 °C for 10 min, then lyophilized. Then, the 2′-TBDMS group was removed by incubation with tetrabutylammonium fluoride for 16 h. The resulting materials were mixed with an equal volume of 1 M Tris (pH 7.5) and desalted, and the RNAs were purified by PAGE.
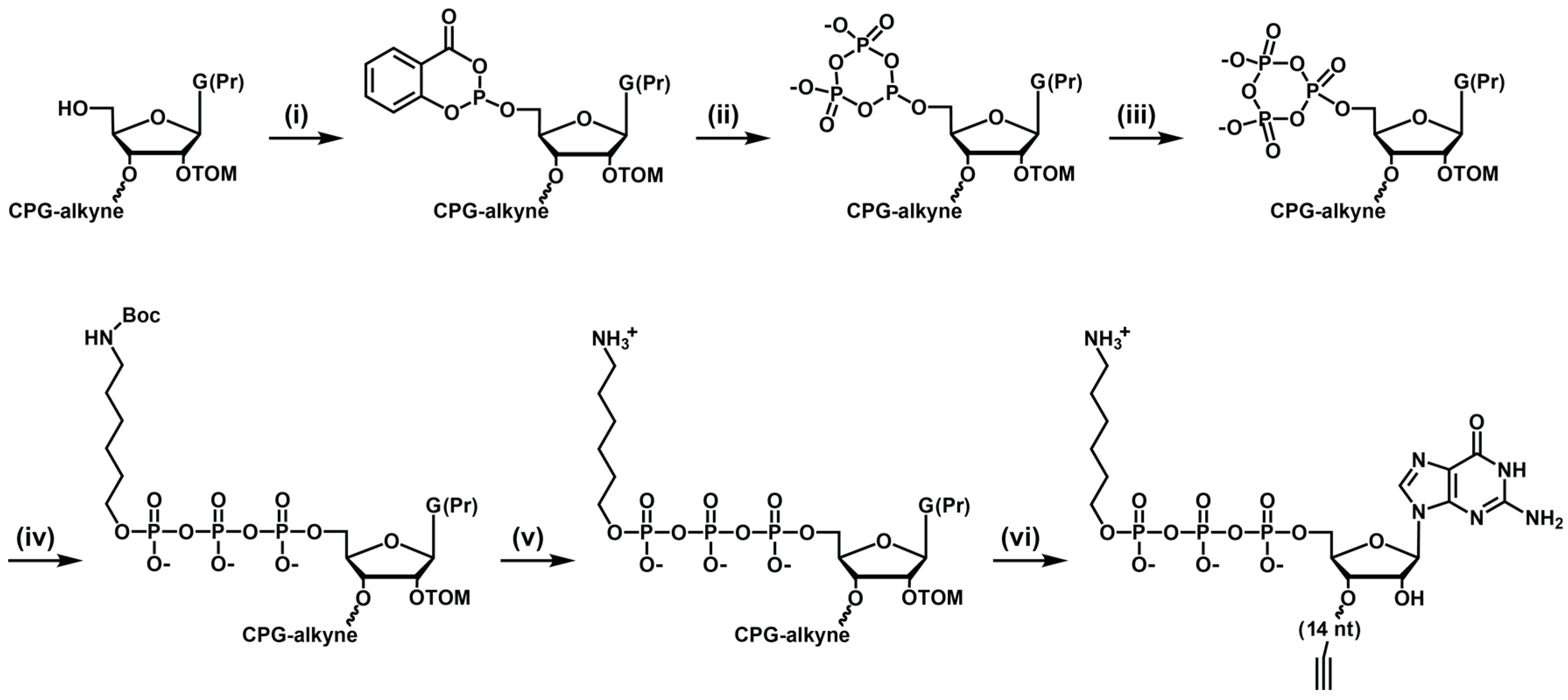
4.2. Synthesis of TMR-B and TMR-B-Q Substrates
Both the
d- and
l-RNA forms of substrate B were prepared by automated solid-phase synthesis using 2′-TOM phosphoramidites. For substrates containing the TQ3 quencher, synthesis was carried out on a 2′-
O-propargyl uridine CPG column. The substrates were 5′-triphosphorylated on the column, based on a modification of previous methods [
10,
14] (
Scheme 1). Following removal of the 5′-dimethoxytrityl protecting group, the column was removed from the DNA/RNA synthesizer and connected at each end to a syringe, one fitted with a 3-way stopcock linked to both an argon source and an injection port, and the other fitted with a plunger to allow mixing and withdrawal of waste material. The 5′-hydroxyl was phosphitylated with salicyl phosphorochloridite in 3:1 dioxane/pyridine for 15 min, this solution having been prepared just prior to use by dissolving 10 mg of dry salicyl phosphorochloridite in 100 µL of dioxane/pyridine. The resulting material is extremely sensitive to water and must be maintained under dry argon. It was converted to the cyclic trimetaphosphite using 250 µL of dry tributylammonium pyrophosphate in DMF for 20 min, then washed twice with 500 µL of dry ACN/hexanes. Then, the cyclic trimetaphosphite was oxidized to the cyclic trimetaphosphate using 250 µL of dry 1 M
t-butyl peroxide in hexanes for 15 min. The resulting material was washed with 500 µL of dry ACN and allowed to react with dry 3.5 M of 6-(Boc-amino)-1-hexanol in ACN for 72 h [
15], and then again washed with dry ACN. The 5′-terminal amine was deprotected using 9:1 TFA/hexanes, and the column was then washed with 100 mM of Tris (pH 7.5), washed with ACN, and dried under argon.
The oligonucleotide was deprotected as described above, and the 5′-amine was then labeled with TMR-N-hydroxysuccinimidyl ester according to the manufacturer’s directions, followed by ethanol precipitation. For the TMR-B-Q substrate, the 3′-terminal alkyne was coupled with TQ3 azide in a click reaction, employing 25 µM of RNA, 150 µM of TQ3 azide, 0.4 mM of Cu(II)SO4, 2 mM of Tris(3-hydroxypropyltriazolylmethyl)amine, 10 mM of sodium ascorbate, 50 mM of Na2HPO4 (pH 7.4), and 10% dimethyl sulfoxide, which was incubated at 23 °C for 2 h. The resulting materials were purified by PAGE and ethanol precipitation.
4.3. In Vitro Selection
The in vitro selection procedure was based on a previously reported method [
8]. The starting DNA library had the sequence 5′-GGC
TAATACGACTCACTATAGGAGCGAGAACGTTGT-N
7-TGTTACGTAAGTAACA-N
9-GTTGAAGTATGAGACCGGAAGGTTGAAGTATGAGACCGCAACGTA (T7 RNA polymerase promoter sequence underlined; N indicates nucleotides that were fully randomized). The DNA was made double-stranded by extending a primer having the sequence 5′-TACGTTGCGGTCTCATACTTCAACCTTCCGGTCTCATACTTCAACC-3′; then, 1.7 × 10
13 DNA molecules (282 pmol) were used to transcribe the corresponding pool of RNAs. These were cleaved using M1 RNA and an external guide RNA having the same sequence as described above for the processing of substrate B. Following purification by PAGE, 6.0 × 10
12 RNA molecules (100 pmol) were mixed with TMR-B-biotin substrate and 50 mM of EPPS (pH 8.5), heated to 70 °C for 2 min, and then cooled to 4 °C over 3 min. The mixture was then heated to 42 °C, and the ligation reaction was initiated by adding MgCl
2 and then quenched by adding 50 mM of EDTA. During the first round of selection the reaction mixture contained 2 µM of TMR-B-biotin, 1 µM of E-A, and 25 mM of MgCl
2, and the reaction time was 1 h. During rounds 2 and 3, the concentration of reactants was the same, and reaction time was reduced to 7.5 min in round 2 and to 30 s in round 3. During rounds 4 and 5, the reaction mixture contained 50 nM of TMR-B-biotin and 50 nM of E-A, the concentration of MgCl
2 was 25 mM in round 4 and 5 mM in round 5, and the reaction time was 30 s in round 4 and 15 s in round 5.
The ligated products were captured on a streptavidin-agarose resin and washed successively with the following: 10 mL of a solution containing 100 mM of NaCl, 1 mM of EDTA, and 50 mM of Tris (pH 7.5); 20 mL of a solution containing 1 M of NaCl, 1 mM of EDTA, 0.1% Triton X-100, and 50 mM of Tris (pH 7.5); 10 mL of a solution containing 100 mM of NaCl, 1 mM of EDTA, and 50 mM of Tris (pH 7.5); and 20 mL of a solution containing 1 mM of EDTA and 10 mM of Tris (pH 7.5). The ligated RNA was reverse transcribed on the resin by extending a primer having the sequence 5′-TAAGTTGCGGTCTC-3′. The DNA (as an agarose slurry) was then PCR amplified using the same reverse primer and a forward primer having the sequence 5′-GGCTAATACGACTCACTATAGGAGCGAGAACGTTGT-3′ (T7 RNA polymerase promoter sequence underlined). The amplified DNA was in vitro transcribed, and the resulting RNA was purified by PAGE and then used to initiate the next round of selection.
4.4. Ligation Assay
Ligation was carried out by first mixing 5 µM of substrate A, 2 µM of substrate B (unlabeled B, TMR-B, or TMR-B-Q), 6 µM of enzyme (E or Etheo), and 50 mM of EPPS (pH 8.5), which were heated at 70 °C for 2 min and then cooled to 4 °C over 3 min. The mixture was then heated to 42 °C, and the reaction was initiated by adding MgCl2 to a final concentration of 25 mM. Aliquots were taken from the mixture at various times. The products were analyzed by PAGE, and imaging fluorescence with a BioRad Pharos FXPlus Molecular Imager. For the real-time assays, the reaction mixture was monitored continuously using a BioRad CFX-96 Real-Time PCR Detection System.
4.5. Exponential Amplification Assay
Amplification was carried out by first mixing 20 µM of substrate A, 20 µM of TMR-B-Q, 50 nM of E, and 50 mM of EPPS (pH 8.5), which were heated to 70 °C for 2 min and then cooled to 4 °C over 3 min. The mixture was then heated to 42 °C, and the reaction was initiated by adding MgCl2 to a final concentration of 100 mM. The course of the reaction was monitored using a BioRad CFX-96 Real-Time PCR Detection System.


{kind=link}
{kind=link}
{kind=link}
{kind=link}