
The Guareschi Pyridine Scaffold as a Valuable Platform for the Identification of Selective PI3K Inhibitors

 and
and 
Abstract
: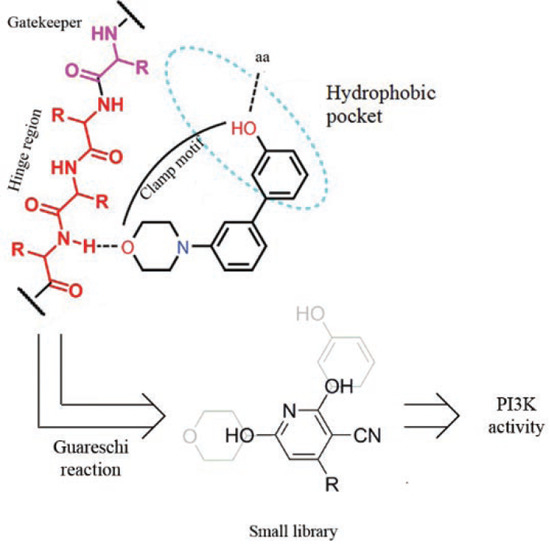
1. Introduction
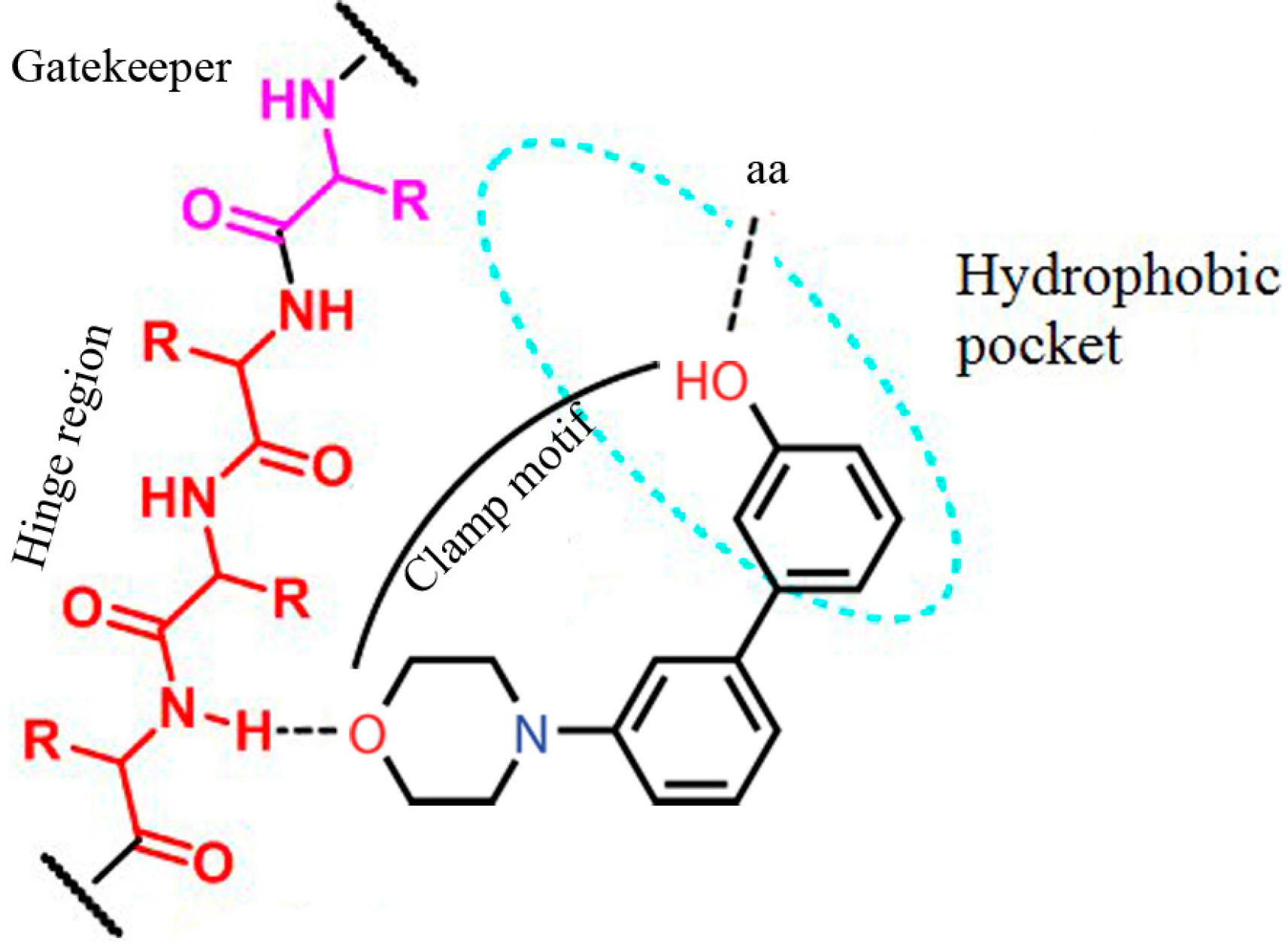
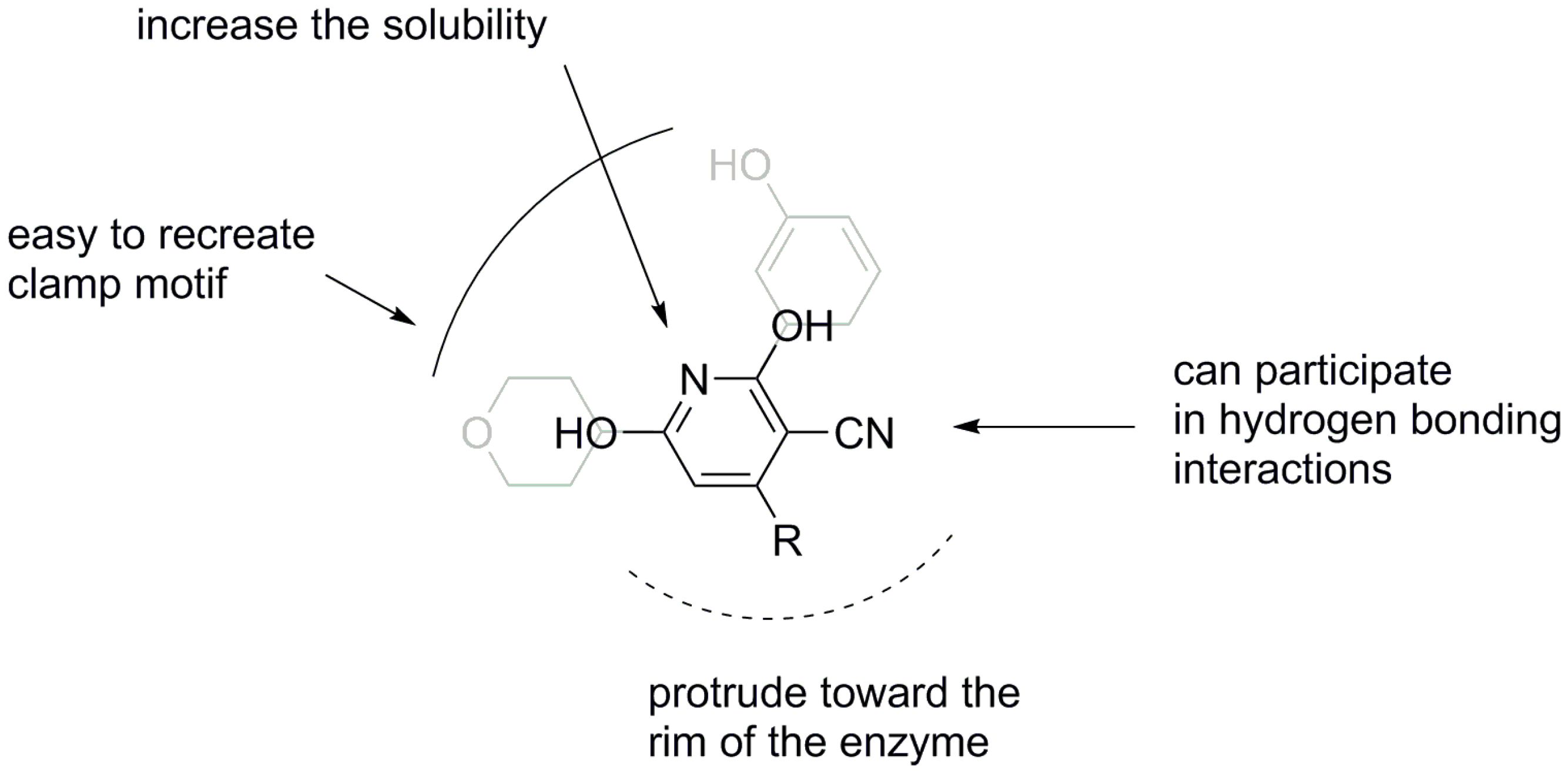
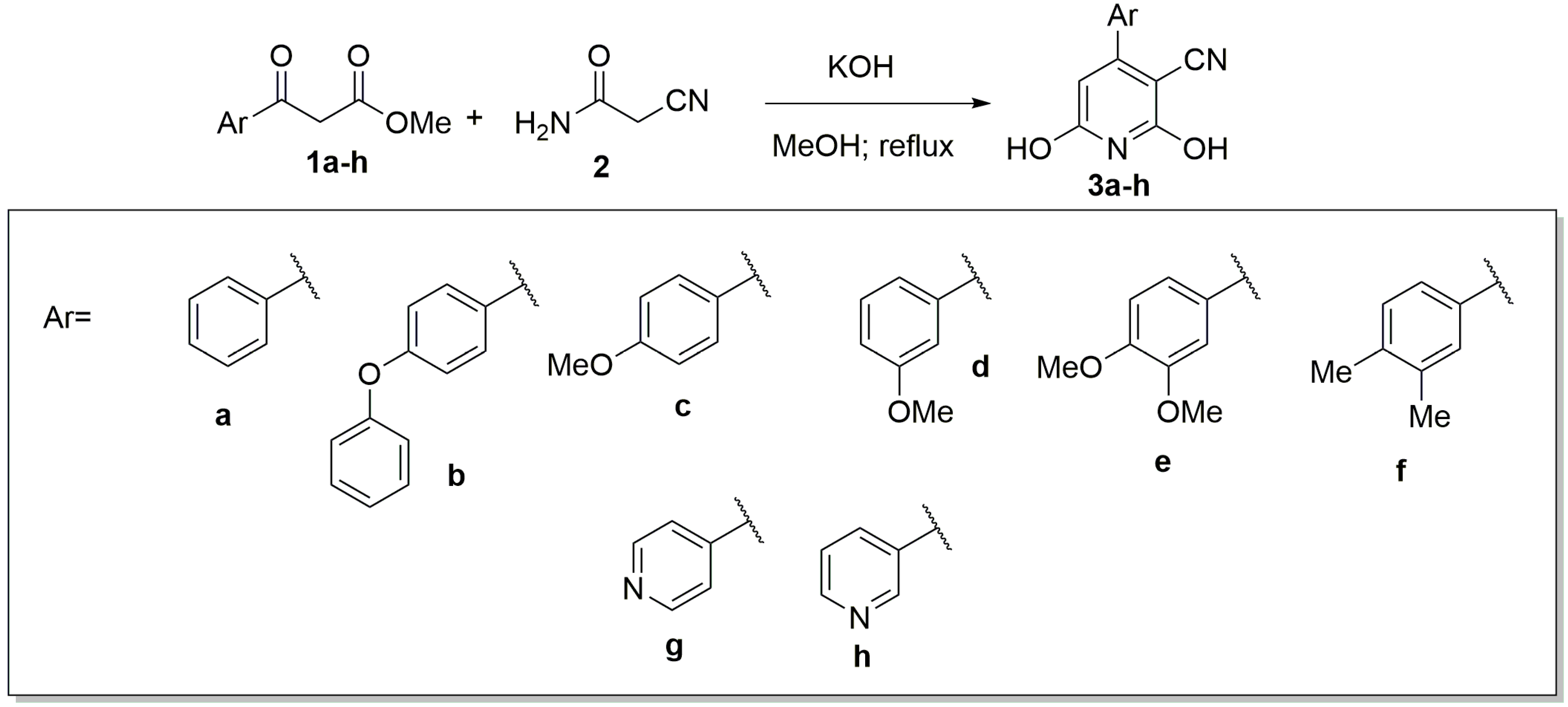
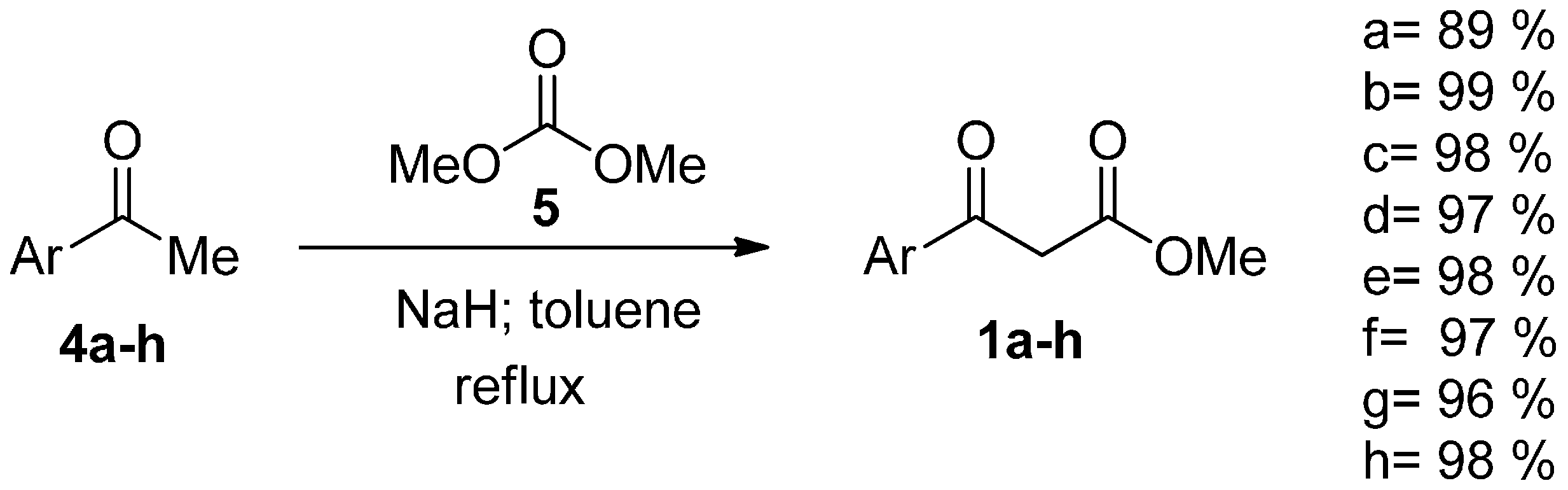
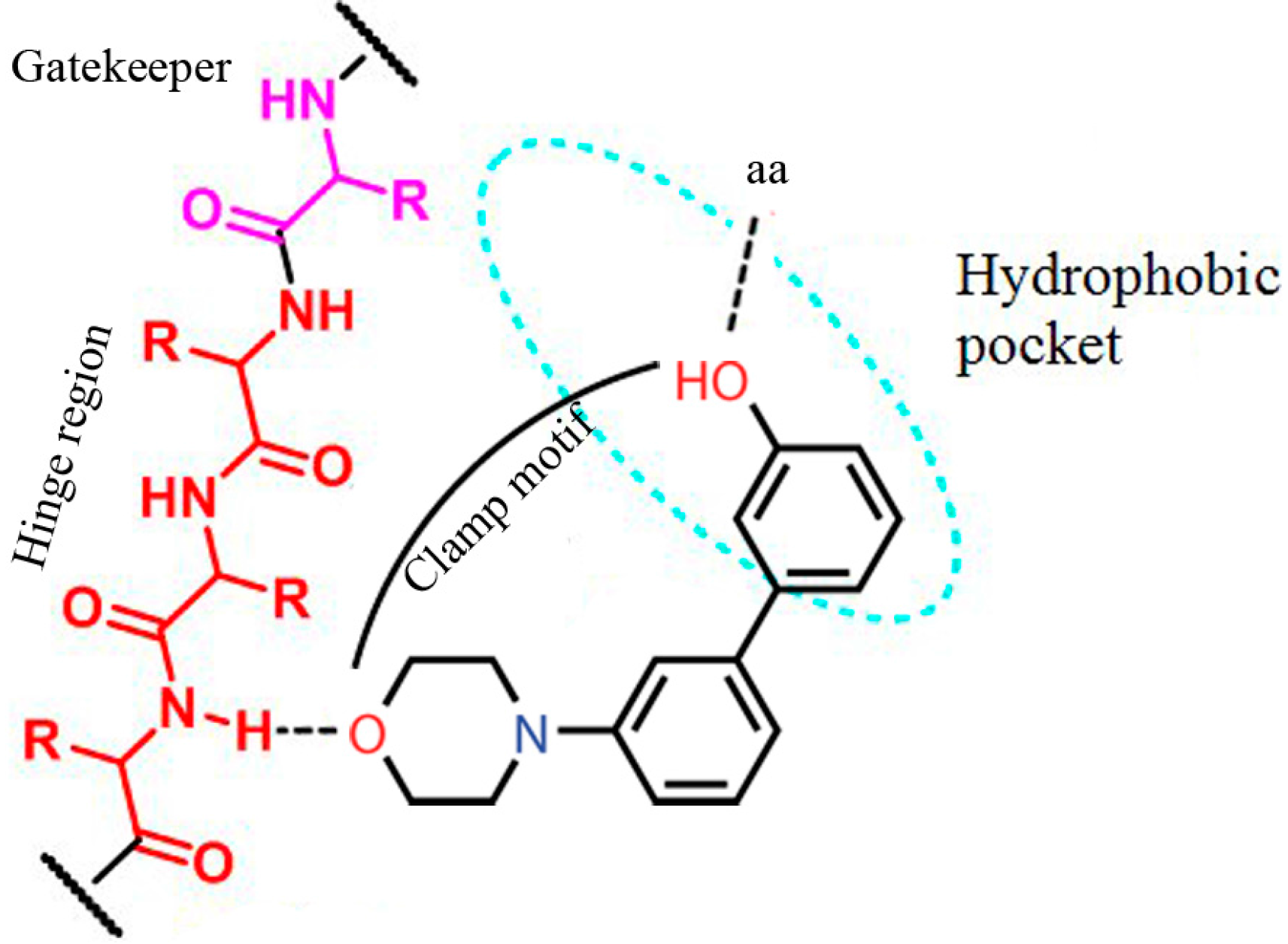
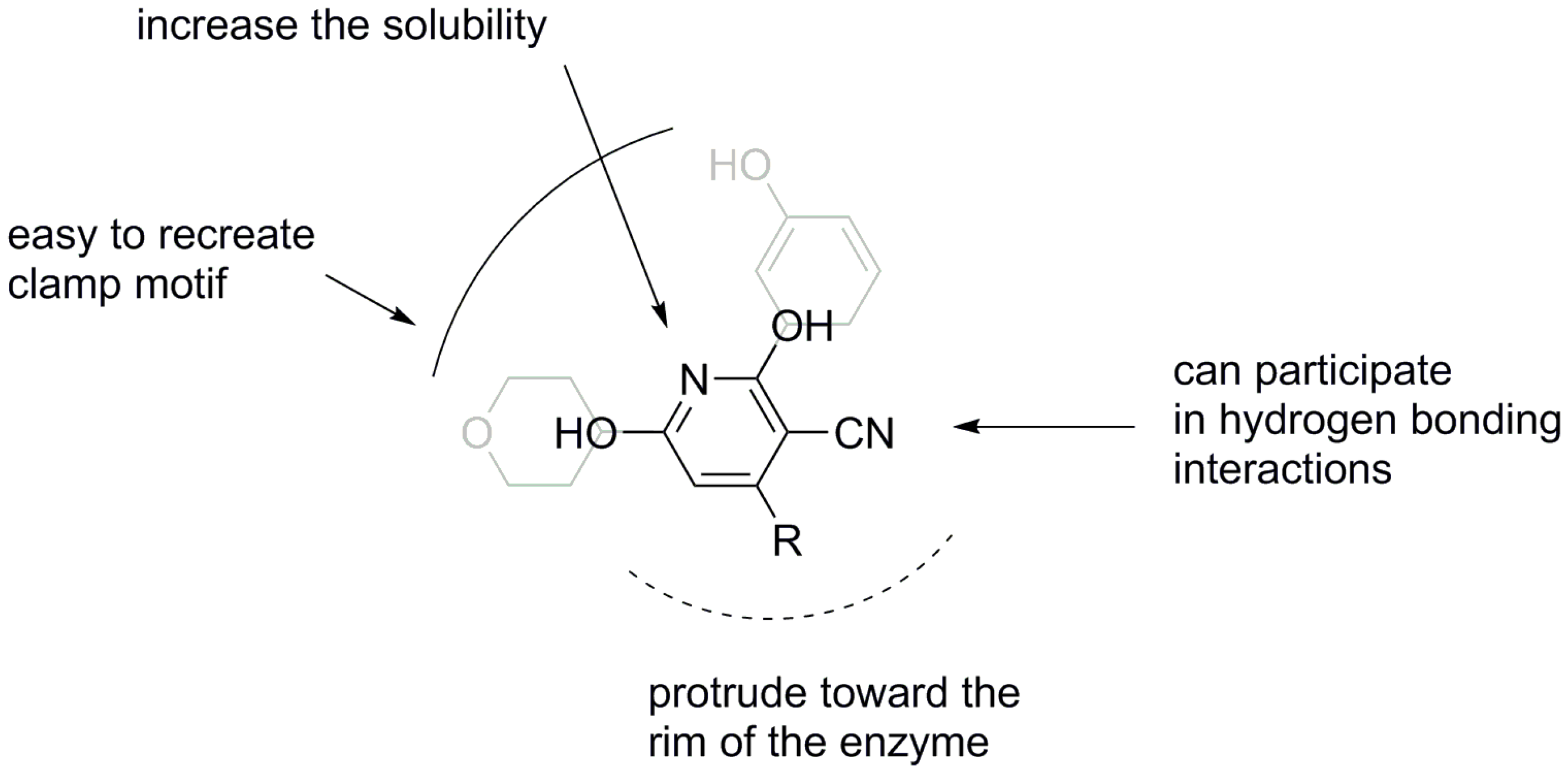
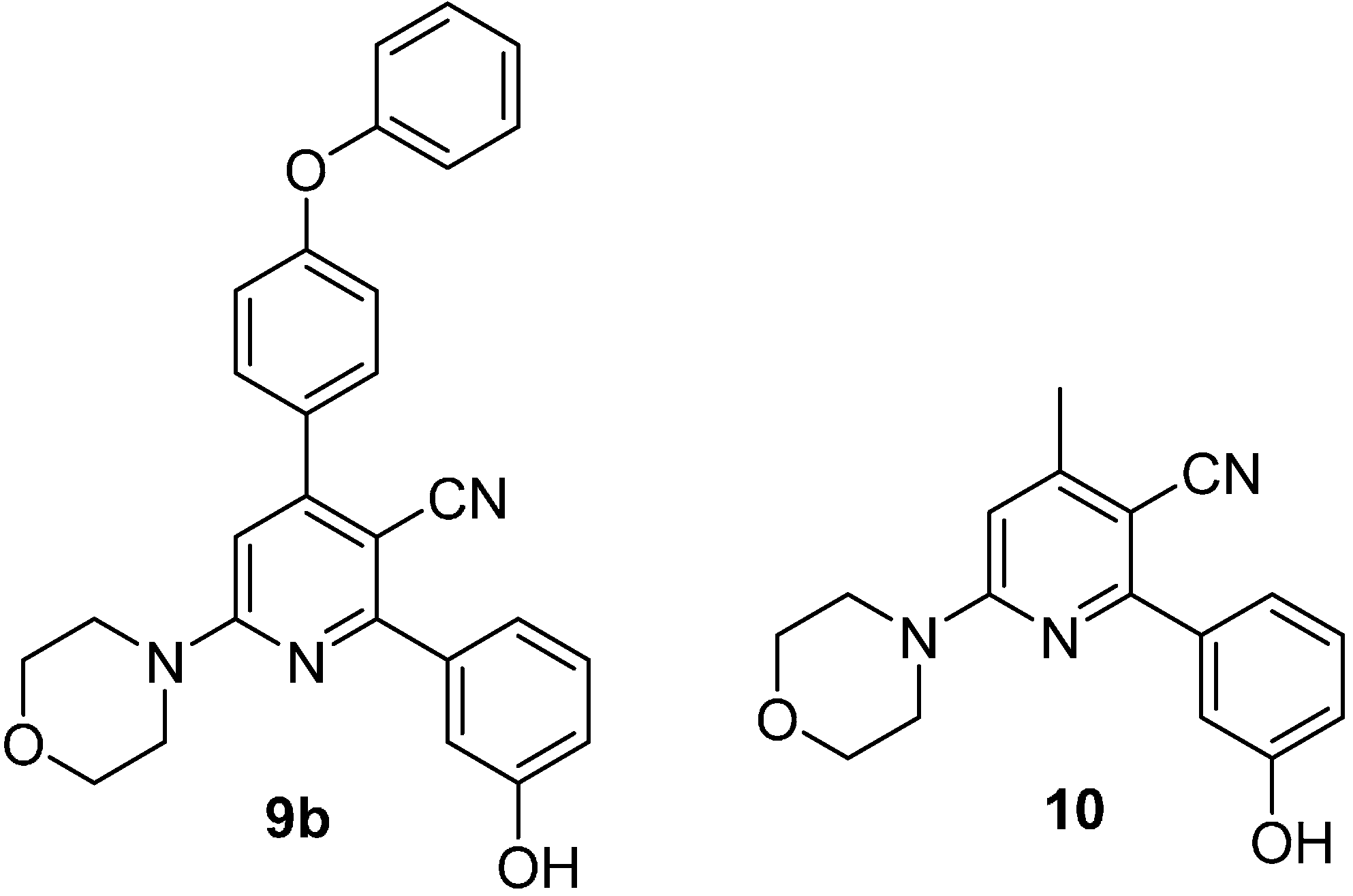
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
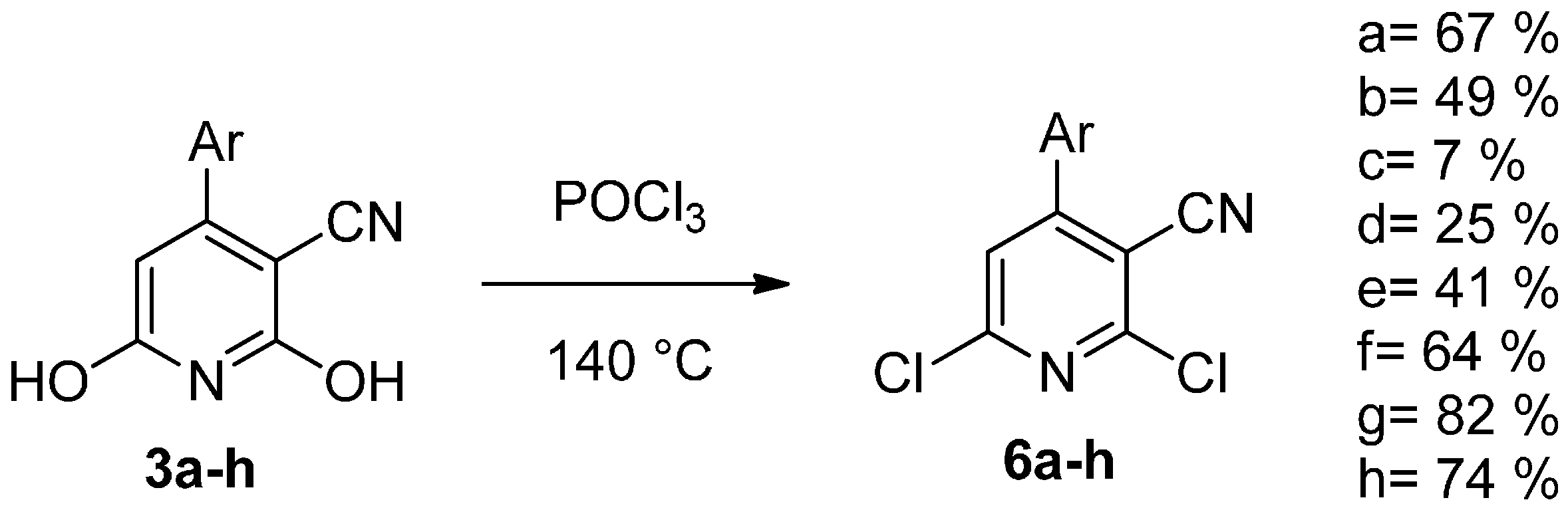
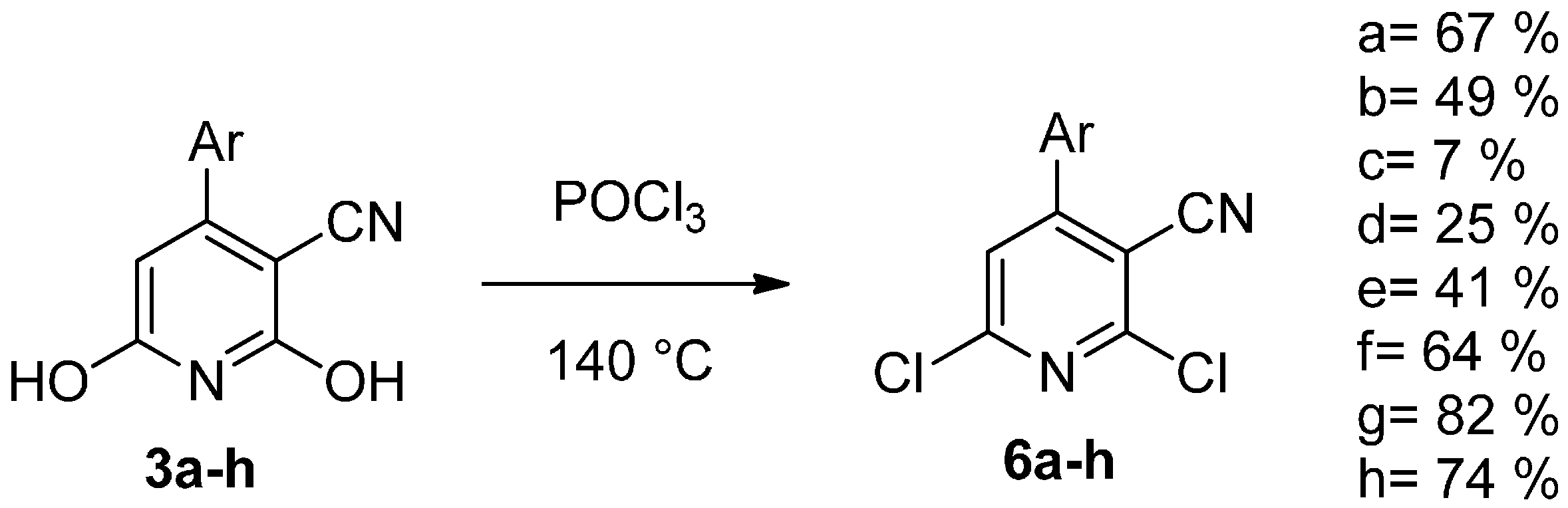{kind=link}
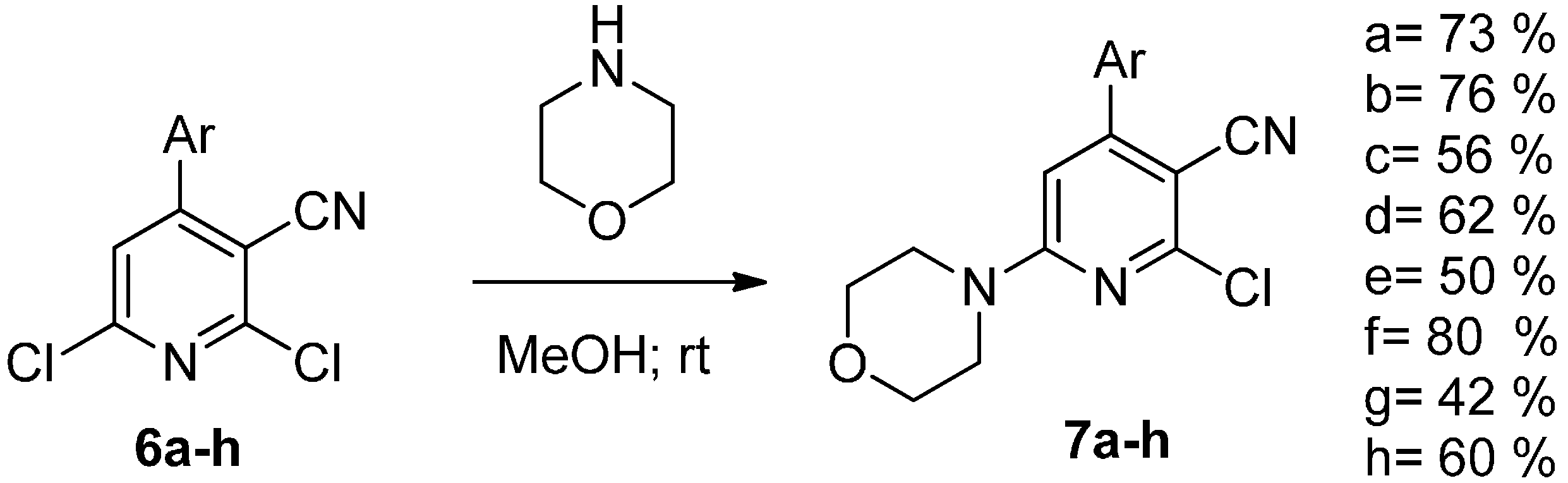{kind=link}
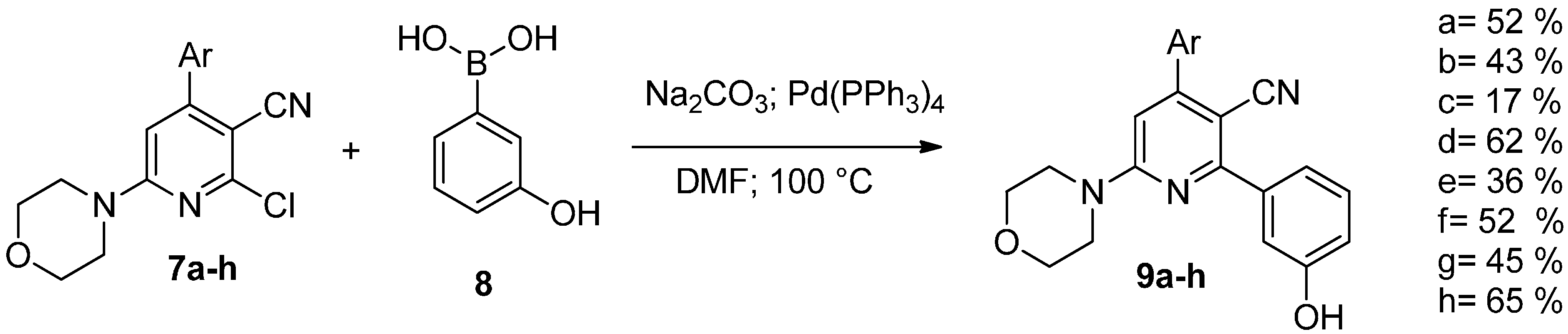{kind=link}
| Compound | Yield (%) |
|---|---|
| 3a | 41 |
| 3b | 15 |
| 3c | 14 |
| 3d | 28 |
| 3e | 9 |
| 3f | 27 |
| 3g | 26 |
| 3h | 10 |
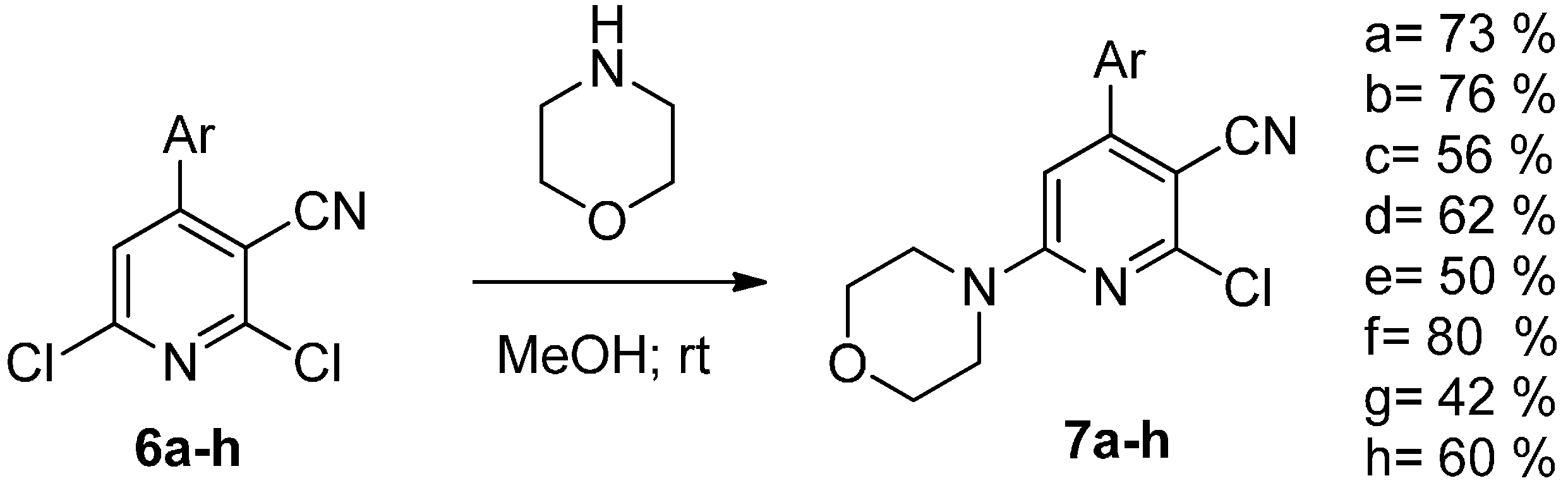
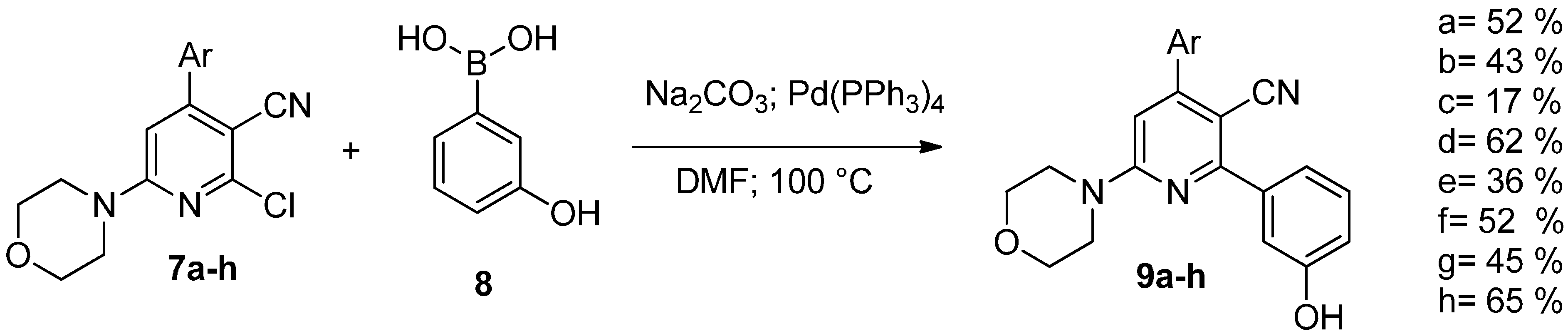
| Compound | IC50 Values (µM) | |||
|---|---|---|---|---|
| PI3Kα | PI3Kβ | PI3Kγ | PI3Kδ | |
| 9a | 0.13 (SE ± 0.03) | 1.99 (SE ± 0.79) | >10 | 1.22 (SE ± 0.47) |
| 9b | 0.63 (SE ± 0.13) | >10 | >10 | >10 |
| 9c | 1.79 (SE ± 0.47) | >10 | 2.18 (SE ± 0.51) | 1.37 (SE ± 0.37) |
| 9d | 0.63 (SE ± 0.15) | 2.34 (SE ± 0.54) | 1.64 (SE ± 0.29) | 1.09 (SE ± 0.19) |
| 9e | 0.83 (SE ± 0.15) | 6.48 (SE ± 1.85) | 2.91 (SE ± 0.56) | 0.90 (SE ± 0.11) |
| 9f | >10 | >10 | 4.43 (SE ± 0.69) | 1.33 (SE ± 0.19) |
| 9g | 1.23 (SE ± 0.14) | >10 | 1.56 (SE ± 0.27) | 0.85 (SE ± 0.14) |
| 9h | 0.98 (SE ± 0.12) | >10 | 2.43 (SE ± 0.41) | 0.71 (SE ± 0.13) |
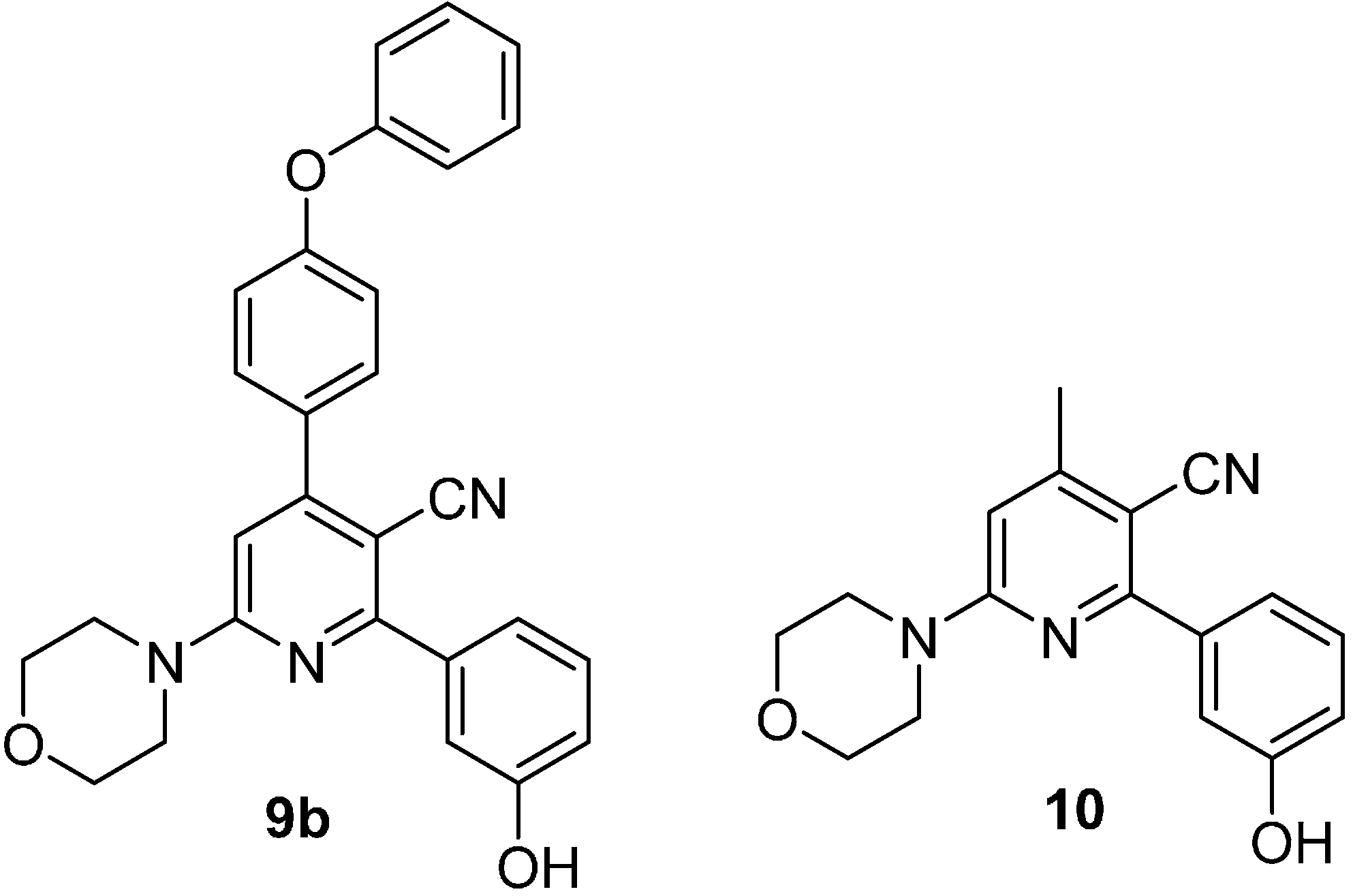
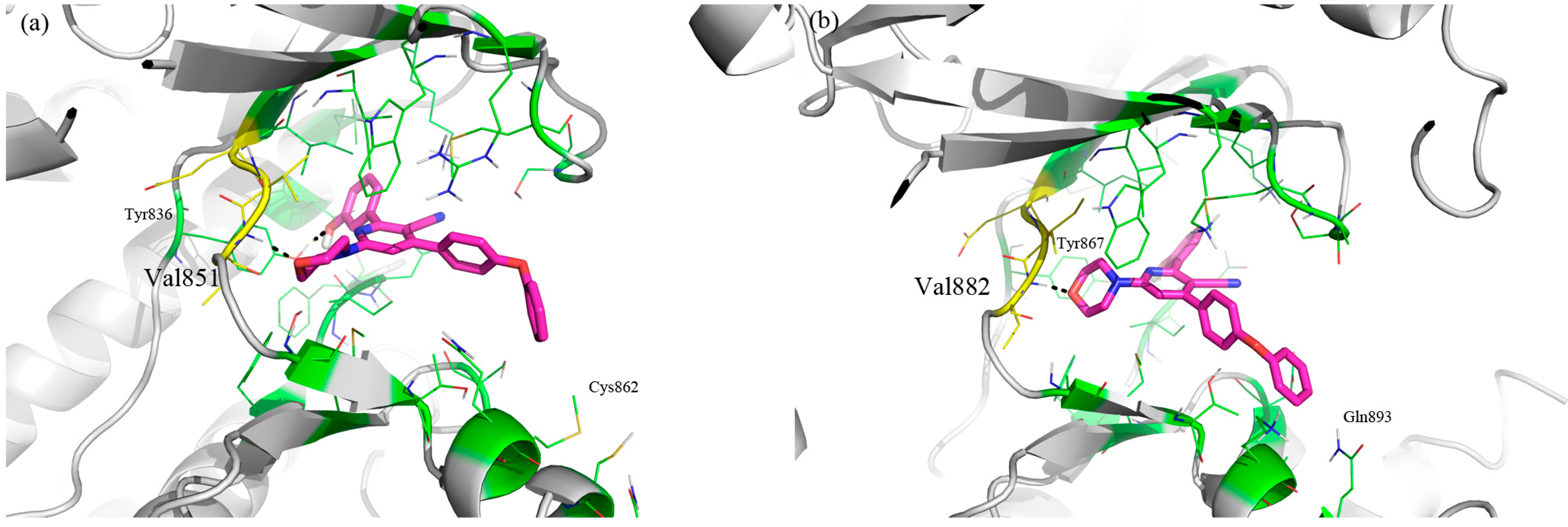
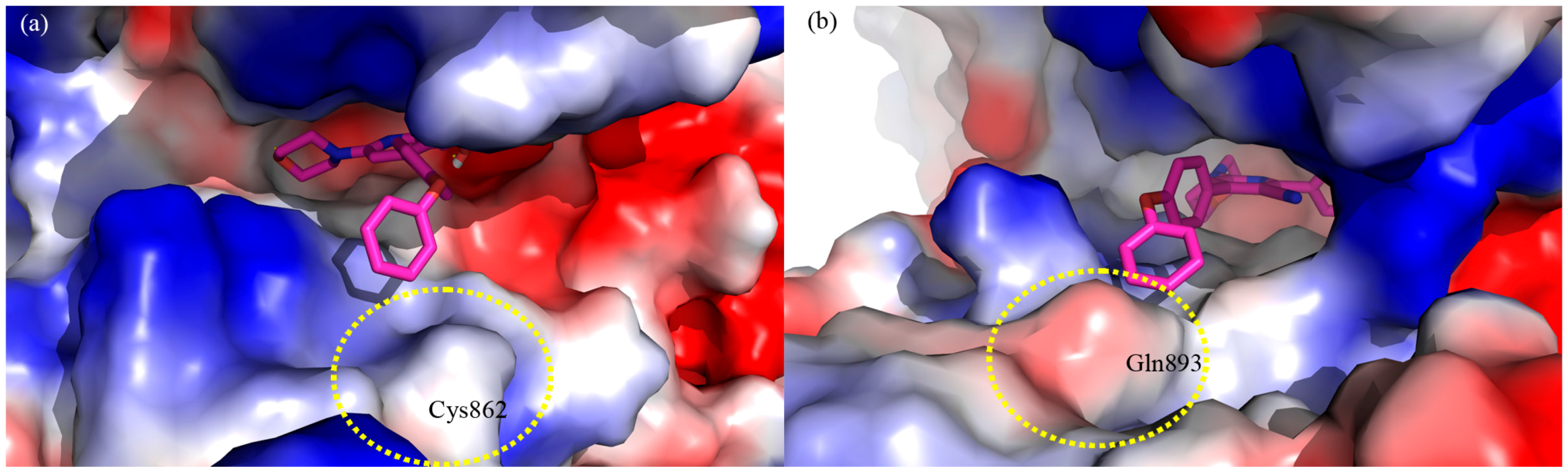
3. Experimental Section
3.1. Molecular Modeling
3.2. Chemistry
3.3. In Vitro Enzyme Inhibition
3.4. Cell-Based Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Blajecka, K.; Borgström, A.; Arcaro, A. Phosphatidylinositol 3-kinase isoforms as novel drug targets. Curr. Drug Targets 2011, 12, 1056–1081. [Google Scholar] [CrossRef] [PubMed]
- Ciraolo, E.; Morello, F.; Hirsch, E. Present and future of PI3K pathway inhibition in cancer: Perspectives and limitations. Curr. Med. Chem. 2011, 18, 2674–2685. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in cancer: Any good news? Front. Oncol. 2013, 3, 108. [Google Scholar] [CrossRef] [PubMed]
- Ihle, N.T.; Powis, G. The biological effects of isoform-specific PI3-kinase inhibition. Curr. Opin. Drug Discov. Dev. 2010, 13, 41–49. [Google Scholar]
- Brana, I.; Siu, L.L. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Ciraolo, E.; Iezzi, M.; Marone, R.; Marengo, S.; Curcio, C.; Costa, C.; Azzolino, O.; Gonella, C.; Rubinetto, C.; Wu, H.; et al. Phosphoinositide 3-kinase p110beta activity: Key role in metabolism and mammary gland cancer but not development. Sci. Signal. 2008, 1, ra3. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.M.; Lengauer, C. Pten-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [PubMed]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor cal-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Perrone, G.; Miura, N.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Santo, L.; Vallet, S.; et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood 2010, 116, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Meadows, S.A.; Vega, F.; Kashishian, A.; Johnson, D.; Diehl, V.; Miller, L.L.; Younes, A.; Lannutti, B.J. PI3Kdelta inhibitor, gs-1101 (cal-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of hodgkin lymphoma. Blood 2012, 119, 1897–1900. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, P.S.; Whye, D.W.; Efimenko, E.; Chen, J.; Tosello, V.; de Keersmaecker, K.; Kashishian, A.; Thompson, M.A.; Castillo, M.; Cordon-Cardo, C.; et al. Targeting nonclassical oncogenes for therapy in t-all. Cancer Cell 2012, 21, 459–472. [Google Scholar] [PubMed]
- Verheijen, J.C.; Zask, A. Phosphatidylinositol 3-kinase (PI3K) inhibitors as anticancer drugs. Drugs Future 2007, 32. [Google Scholar] [CrossRef]
- Carnero, A. Novel inhibitors of the PI3K family. Expert Opin. Investig. Drugs 2009, 18, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.R.; Degterev, A. Small-molecule inhibitors of the pi3k signaling network. Future Med. Chem. 2011, 3, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Hu, Y. Targeting PI3K/AKT/MTOR cascade: The medicinal potential, updated research highlights and challenges ahead. Curr. Med. Chem. 2013, 20, 2991–3010. [Google Scholar] [CrossRef] [PubMed]
- Ruckle, T.; Schwarz, M.K.; Rommel, C. PI3Kgamma inhibition: Towards an “aspirin of the 21st century”? Nat. Rev. Drug Discov. 2006, 5, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Rewcastle, G.W.; Gamage, S.A.; Flanagan, J.U.; Frederick, R.; Denny, W.A.; Baguley, B.C.; Kestell, P.; Singh, R.; Kendall, J.D.; Marshall, E.S.; et al. Synthesis and biological evaluation of novel analogues of the pan class I phosphatidylinositol 3-kinase (pi3k) inhibitor 2-(difluoromethyl)-1-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]-1h-benzimidazole (zstk474). J. Med. Chem. 2011, 54, 7105–7126. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, I. Sintesi di composti piridinici dagli eteri chetonici coll’etere cinacetico in presenza dell’ammoniaca e delle ammine. Acc. R. Sci. Torino Ser. II 1896, 24, 1–29. [Google Scholar]
- Mao, Y.; Zhu, W.; Kong, X.; Wang, Z.; Xie, H.; Ding, J.; Terrett, N.K.; Shen, J.; Shen, J. Design, synthesis and biological evaluation of novel pyrimidine, 3-cyanopyridine and m-amino-n-phenylbenzamide based monocyclic egfr tyrosine kinase inhibitors. Bioorg. Med. Chem. 2013, 21, 3090–3104. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Chiang, G.G.; Alaimo, P.J.; Kenski, D.M.; Ho, C.B.; Coan, K.; Abraham, R.T.; Shokat, K.M. Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg. Med. Chem. 2004, 12, 4749–4759. [Google Scholar] [CrossRef] [PubMed]
- Bobbitt, J.M.; Scola, D.A. Synthesis of isoquinoline alkaloids. II. The synthesis and reactions of 4-methyl-3-pyridinecarboxaldehyde and other 4-methyl-3-substituted pyridines1,2. J. Org. Chem. 1960, 25, 560–564. [Google Scholar] [CrossRef]
- Zheng, H.J.; Chen, W.B.; Wu, Z.J.; Deng, J.G.; Lin, W.Q.; Yuan, W.C.; Zhang, X.M. Highly enantioselective synthesis of β-amino acid derivatives by the lewis base catalyzed hydrosilylation of β-enamino esters. Chem. Eur. J. 2008, 14, 9864–9867. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Tahershamsi, L.; Oskooie, H.A.; Baghernejad, B. 1,4-Diaza-bicyclo[2,2,2]octane as a novel and efficient catalyst for the synthesis of 3,4,6-trisubstituted 2-pyridone derivatives. Chin. J.Chem. 2010, 28, 670–672. [Google Scholar] [CrossRef]
- Harrington, P.J.; Johnston, D.; Moorlag, H.; Wong, J.W.; Hodges, L.M.; Harris, L.; McEwen, G.K.; Smallwood, B. Research and development of an efficient process for the construction of the 2,4,5-substituted pyridines of NK-1 receptor antagonists. Org. Process Res. Dev. 2006, 10, 1157–1166. [Google Scholar] [CrossRef]
- Hirsch, E.; Costa, C.; Ciraolo, E. Phosphoinositide 3-kinases as a common platform for multi-hormone signaling. J. Endocrinol. 2007, 194, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Omega, version 2.4.6; OpenEye Scientific Software: Santa Fe, NM, USA.
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with omega: Algorithm and validation using high quality structures from the protein databank and cambridge structural database. J. Chem. Inf. Model 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with omega: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Fred, version 3.0.0; OpenEye Scientific Software: Santa Fe, NM, USA.
- McGann, M. Fred pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- The Pymol Molecular Graphics System, version 1.3; Schrödinger LLC: New York, NY, USA, 2010.
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Nacht, M.; Qiao, L.; Sheets, M.P.; St Martin, T.; Labenski, M.; Mazdiyasni, H.; Karp, R.; Zhu, Z.; Chaturvedi, P.; Bhavsar, D.; et al. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase pi3kalpha. J. Med. Chem. 2013, 56, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The identification of 2-(1h-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (gdc-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S.; et al. Molprobity: All-atom contacts and structure validation for proteins and nucleic acids. Nucl. Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Ciraolo, E.; Perino, A.; Hirsch, E. Measuring PI3K lipid kinase activity. Methods Mol. Biol. 2012, 795, 55–67. [Google Scholar] [PubMed]
- Ciraolo, E.; Gulluni, F.; Hirsch, E. Methods to measure the enzymatic activity of PI3Ks. Methods Enzymol. 2014, 543, 115–140. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds 9a–h and 10 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, U.; Ciraolo, E.; Massarotti, A.; Margaria, J.P.; Sorba, G.; Hirsch, E.; Tron, G.C. The Guareschi Pyridine Scaffold as a Valuable Platform for the Identification of Selective PI3K Inhibitors. Molecules 2015, 20, 17275-17287. https://doi.org/10.3390/molecules200917275
Galli U, Ciraolo E, Massarotti A, Margaria JP, Sorba G, Hirsch E, Tron GC. The Guareschi Pyridine Scaffold as a Valuable Platform for the Identification of Selective PI3K Inhibitors. Molecules. 2015; 20(9):17275-17287. https://doi.org/10.3390/molecules200917275
Chicago/Turabian StyleGalli, Ubaldina, Elisa Ciraolo, Alberto Massarotti, Jean Piero Margaria, Giovanni Sorba, Emilio Hirsch, and Gian Cesare Tron. 2015. "The Guareschi Pyridine Scaffold as a Valuable Platform for the Identification of Selective PI3K Inhibitors" Molecules 20, no. 9: 17275-17287. https://doi.org/10.3390/molecules200917275
APA StyleGalli, U., Ciraolo, E., Massarotti, A., Margaria, J. P., Sorba, G., Hirsch, E., & Tron, G. C. (2015). The Guareschi Pyridine Scaffold as a Valuable Platform for the Identification of Selective PI3K Inhibitors. Molecules, 20(9), 17275-17287. https://doi.org/10.3390/molecules200917275