
Preparation and Reaction Chemistry of Novel Silicon-Substituted 1,3-Dienes

Abstract
: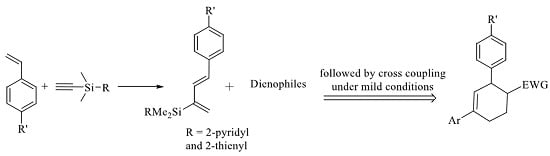
1. Introduction
2. Results and Discussion
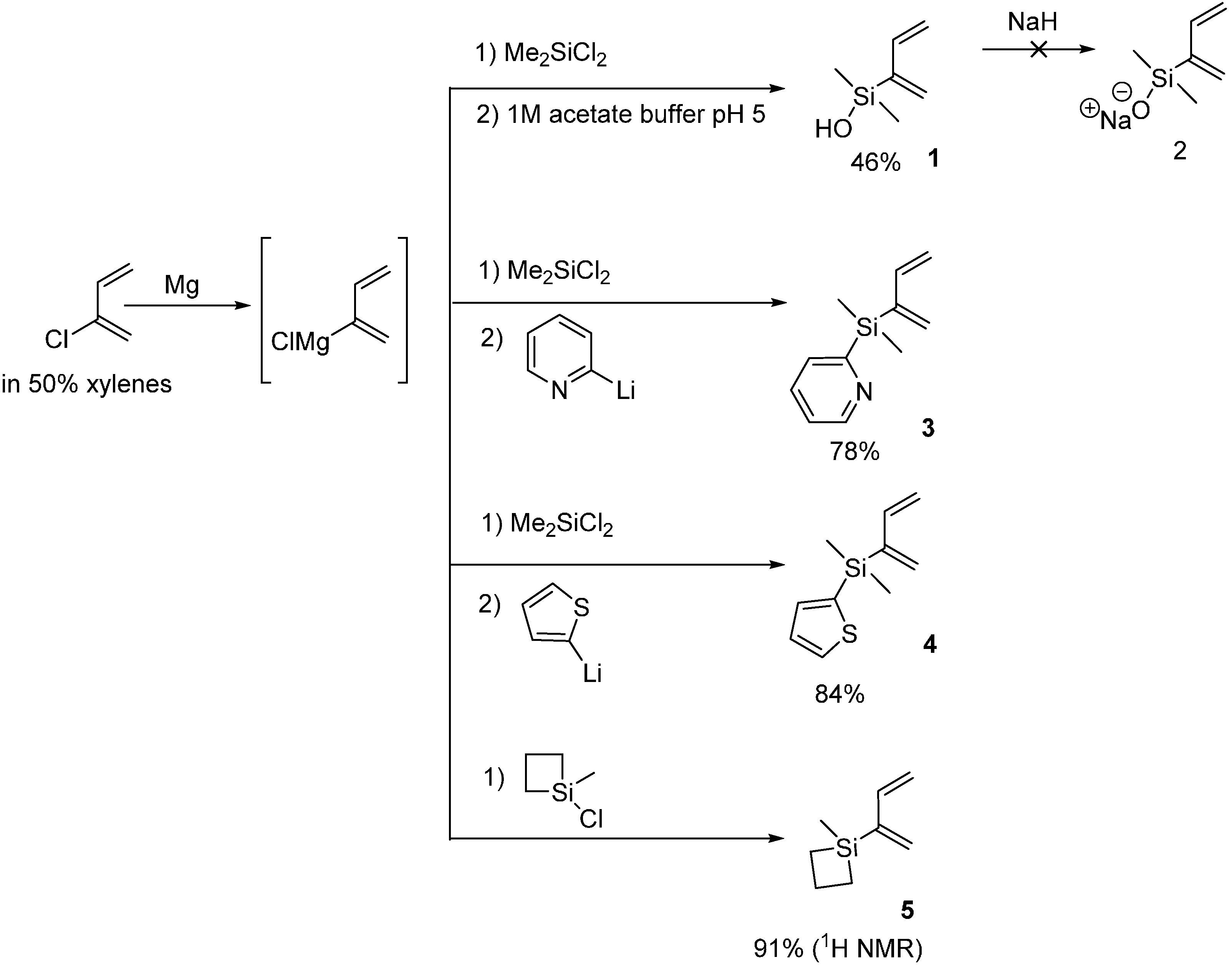
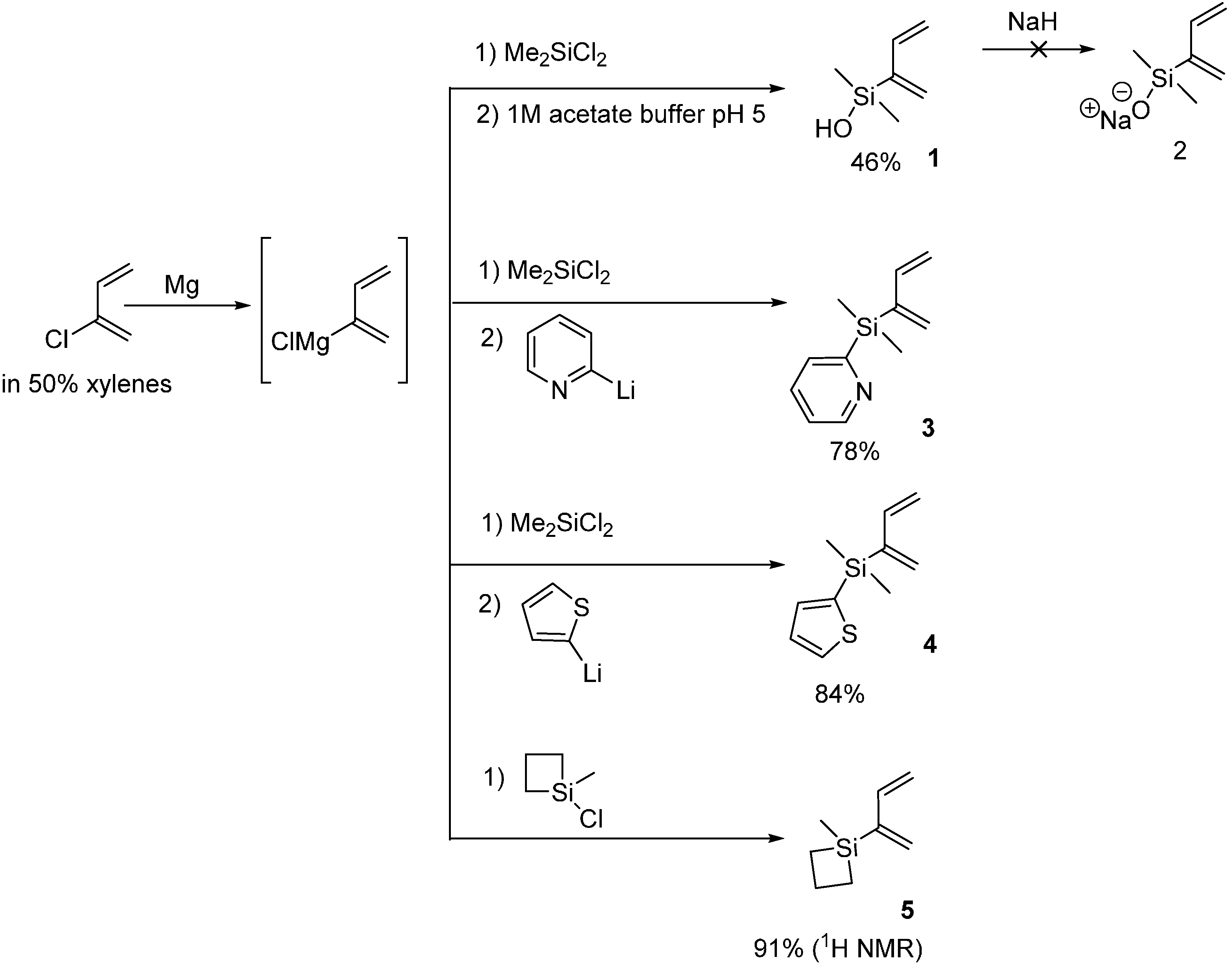
2.1. 2-Silicon-Substituted Diene Synthesis via Grignard Chemistry
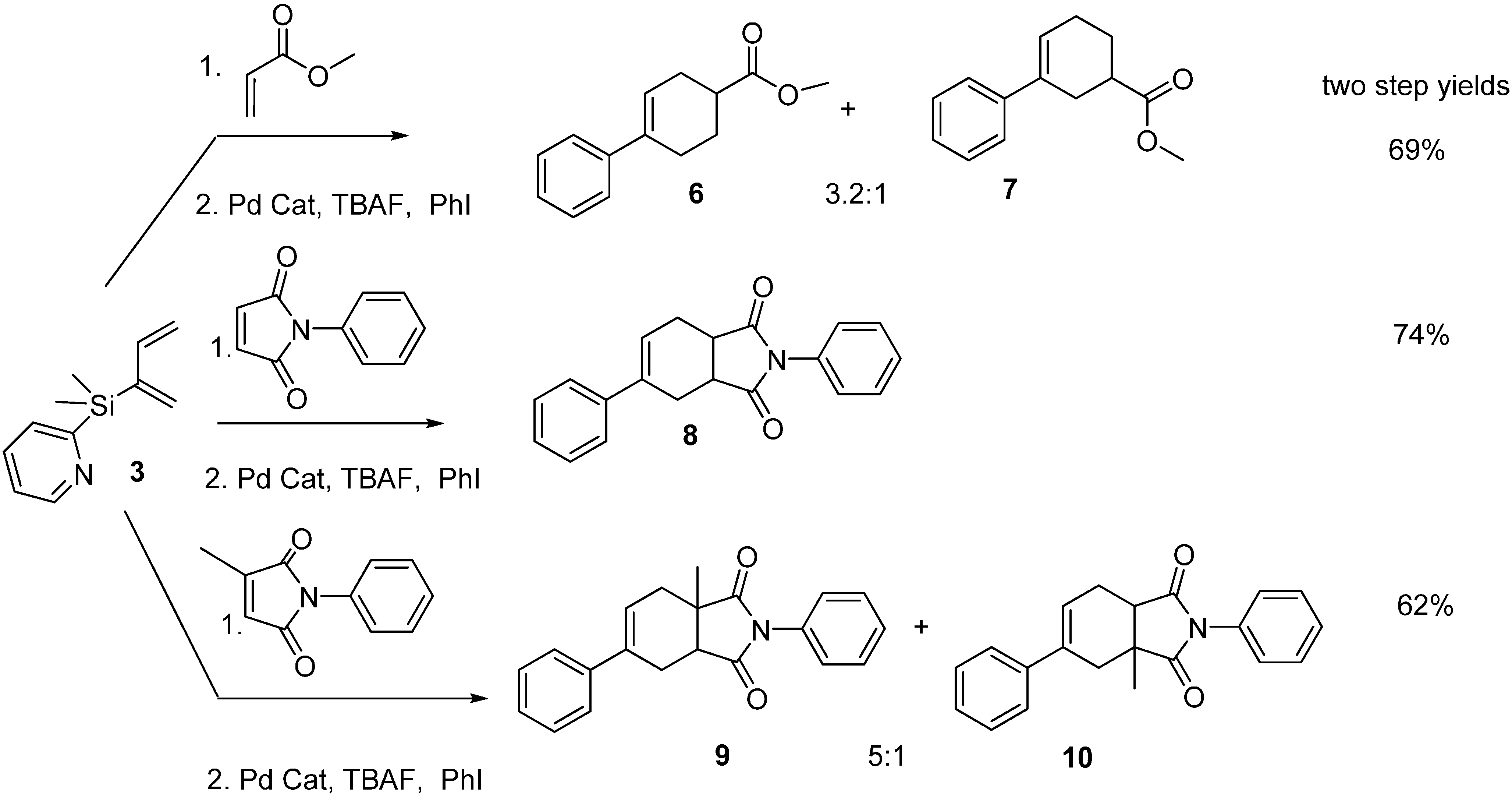
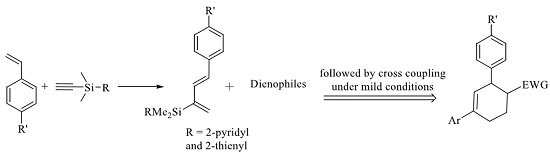
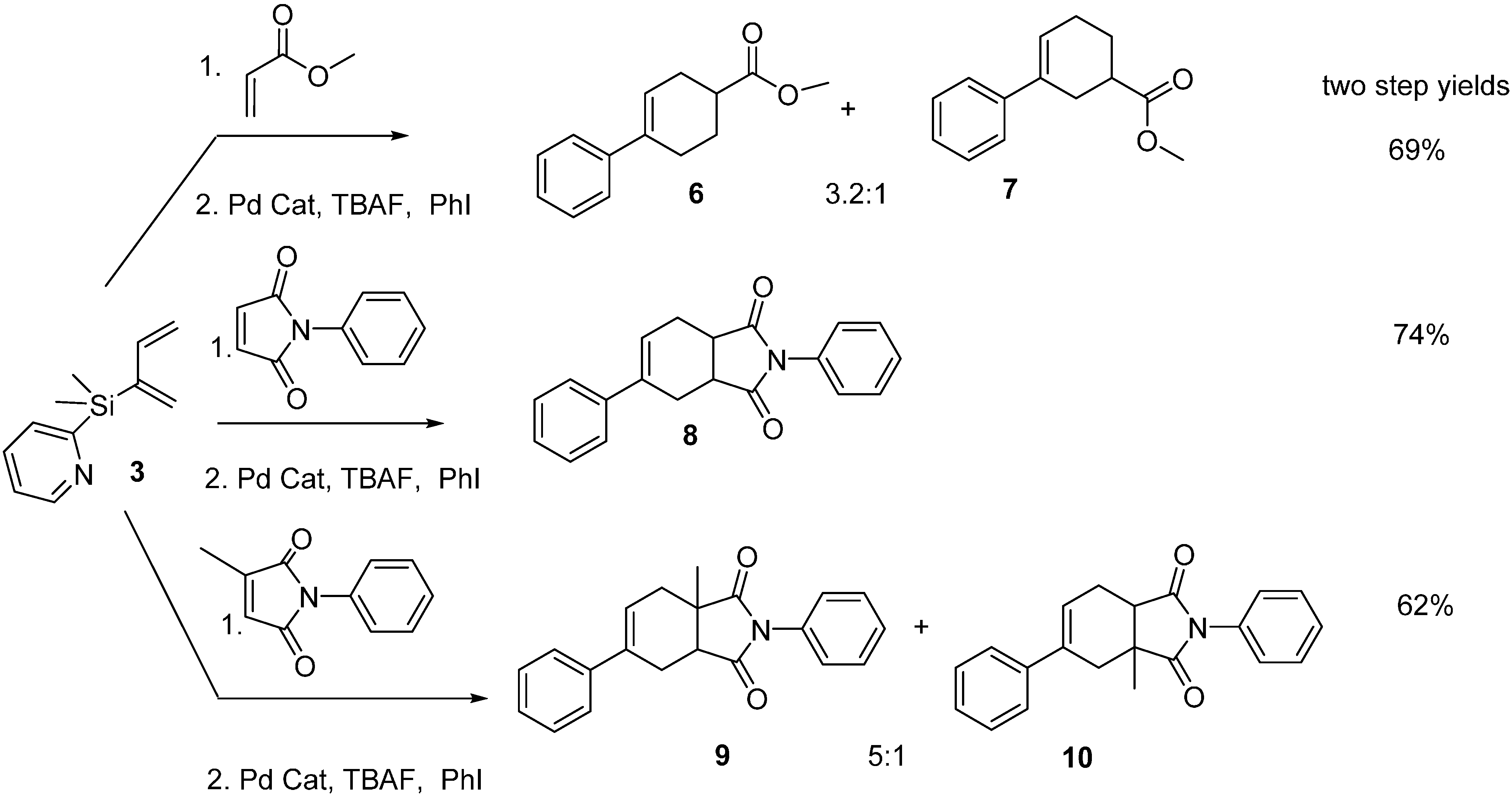
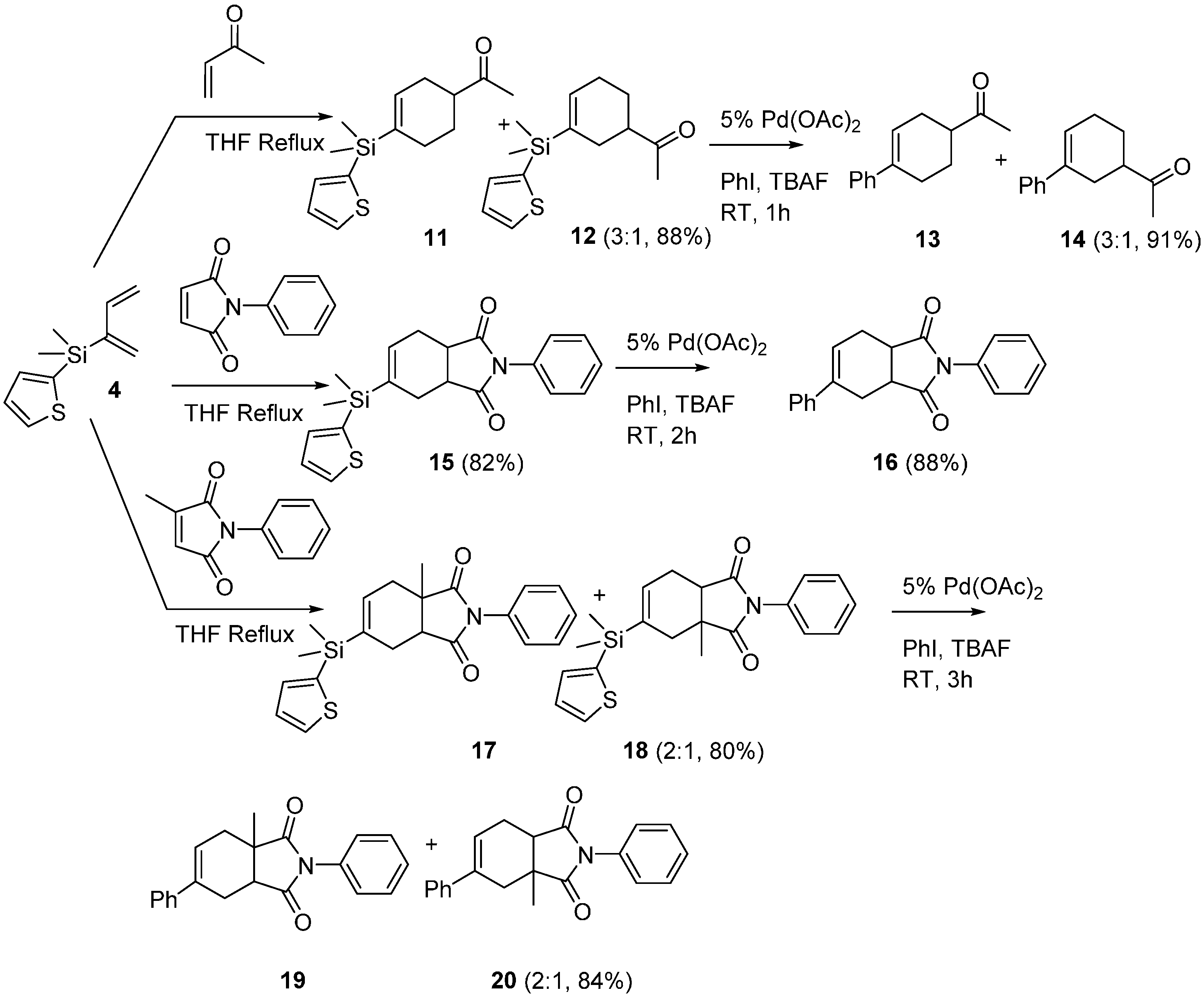
2.2. Diels-Alder/Cross-Coupling Reactions

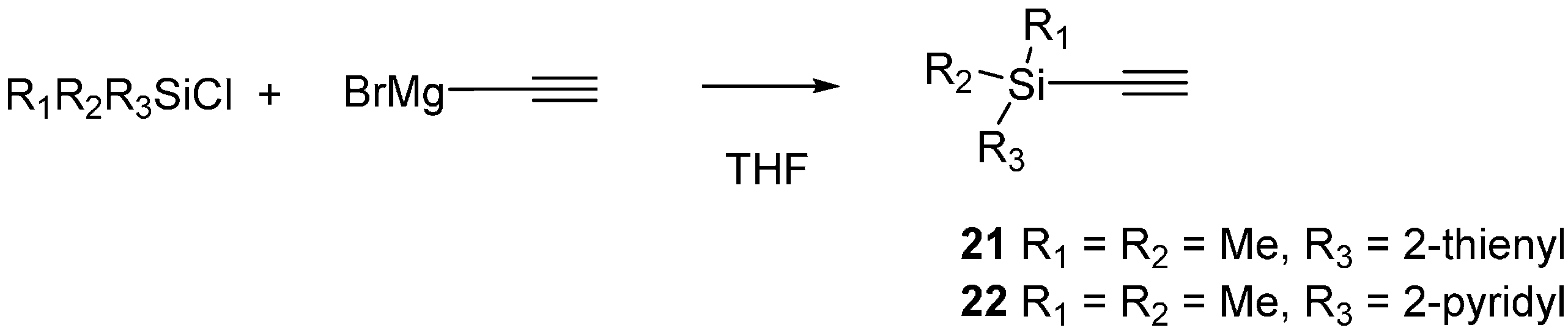
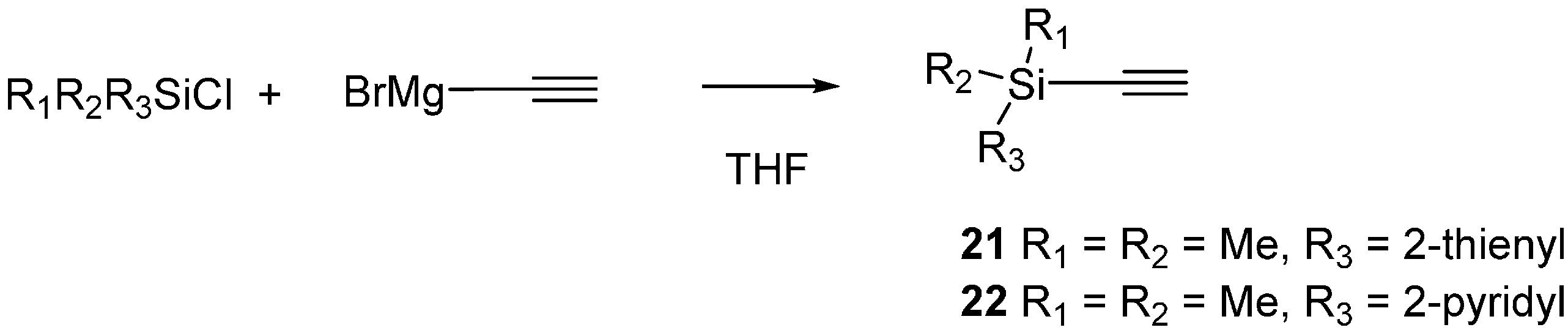
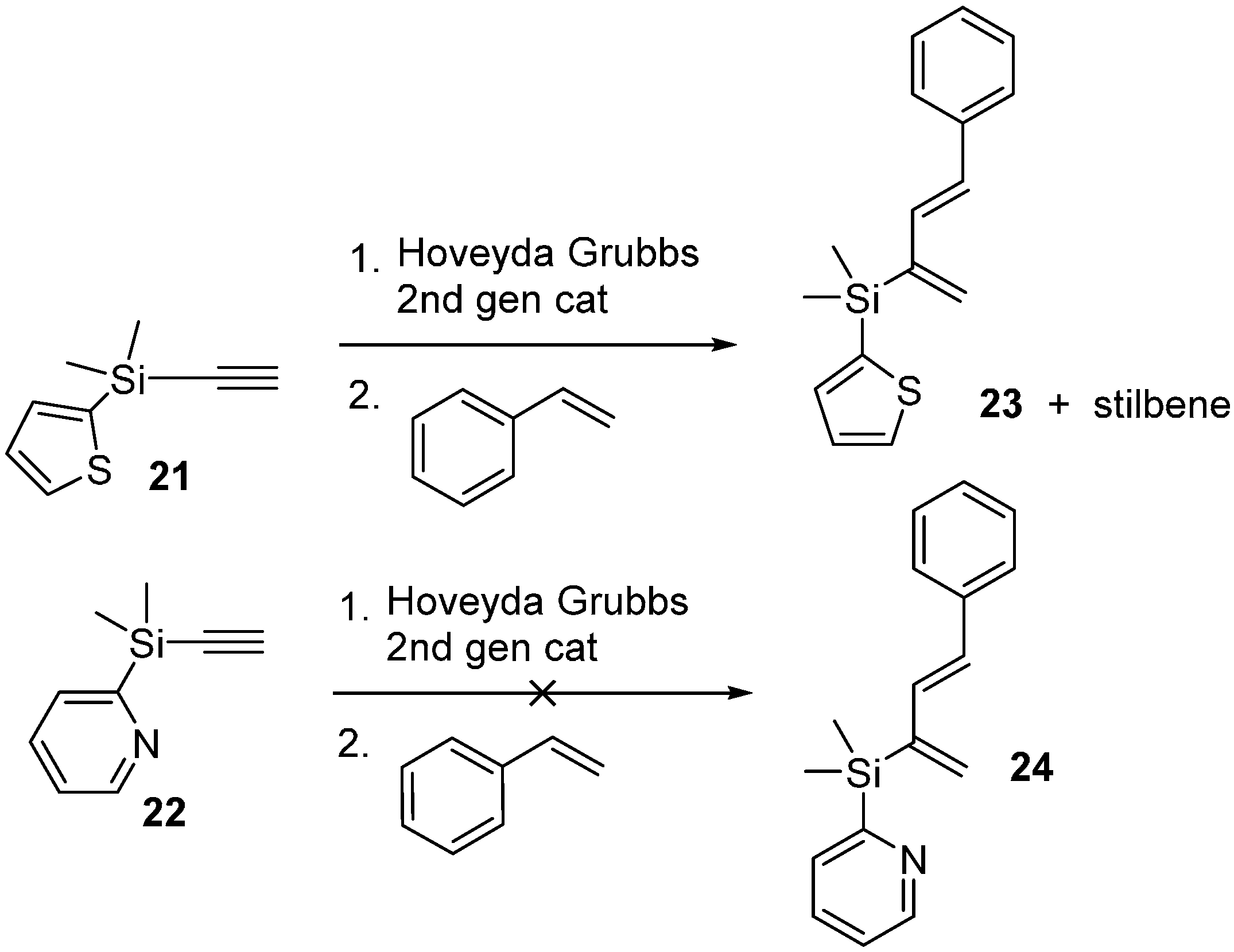
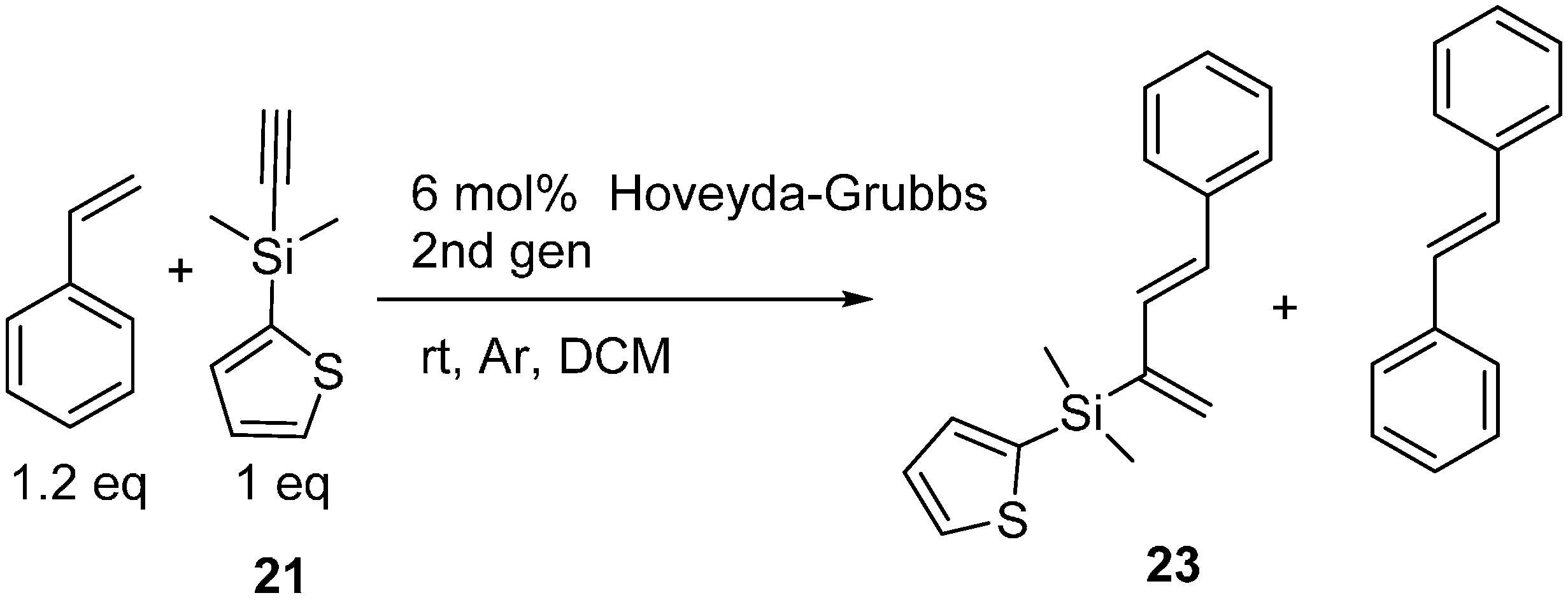
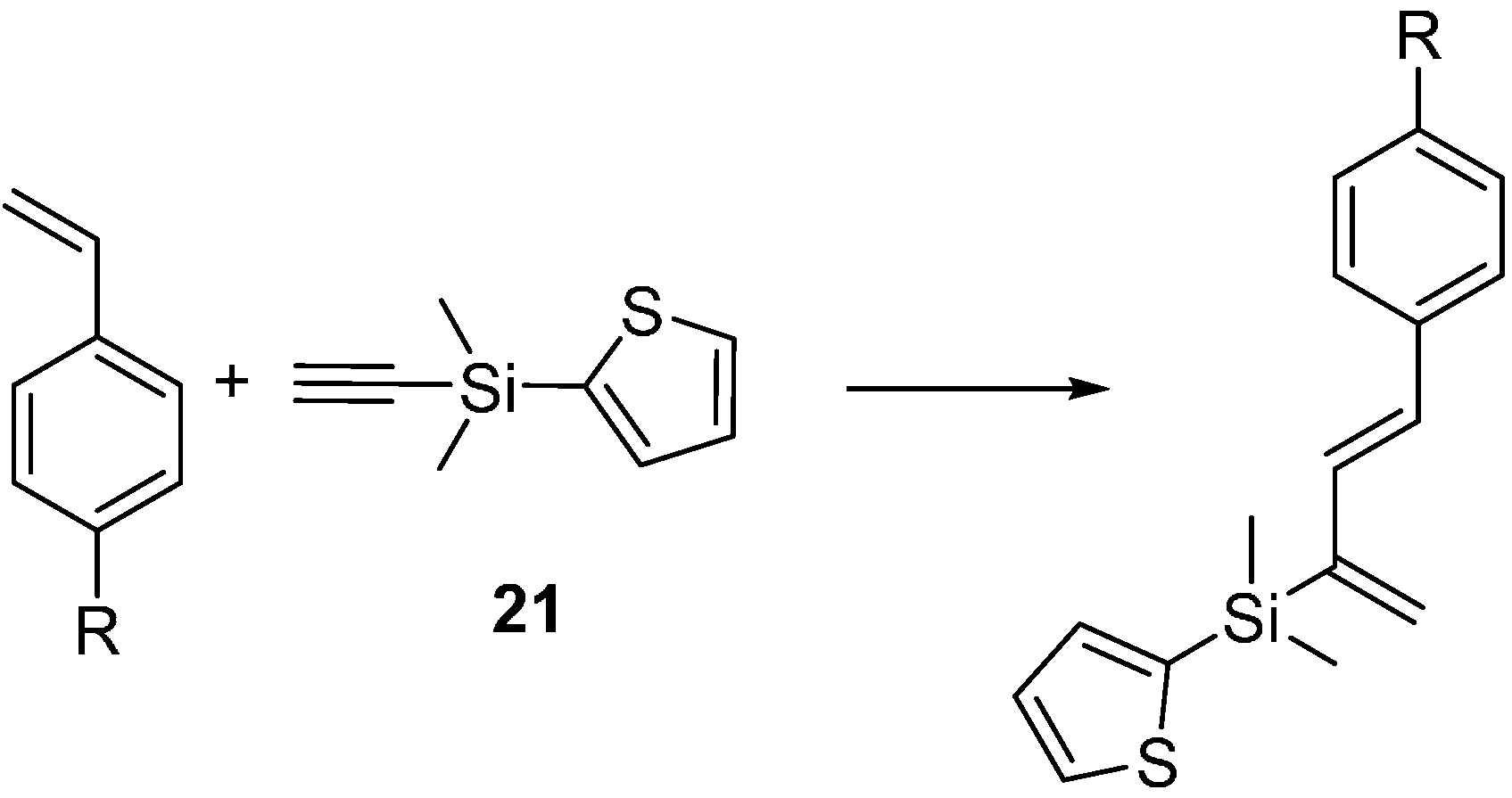
2.3. 2-Silicon-Substituted Diene Synthesis via Enyne Metathesis
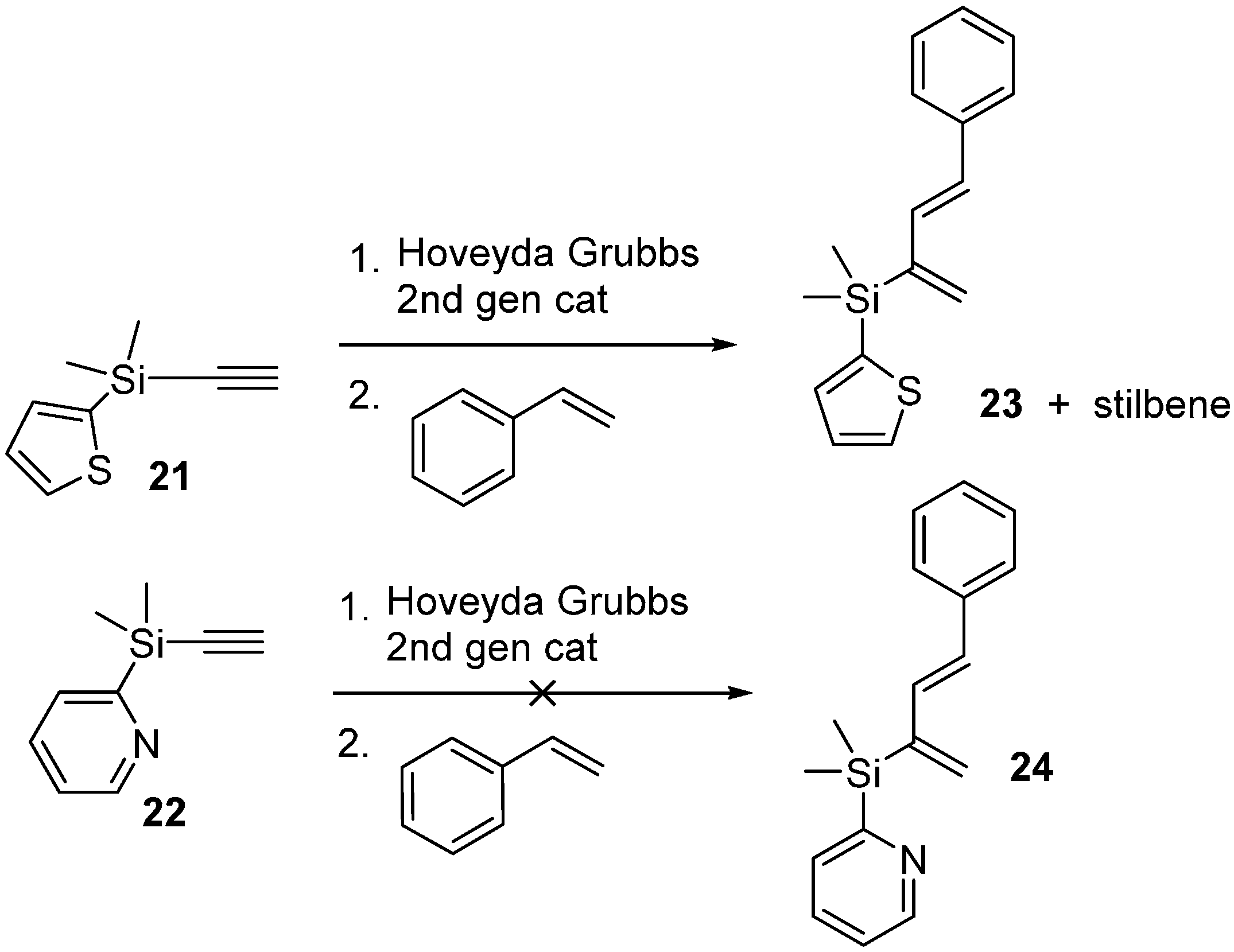
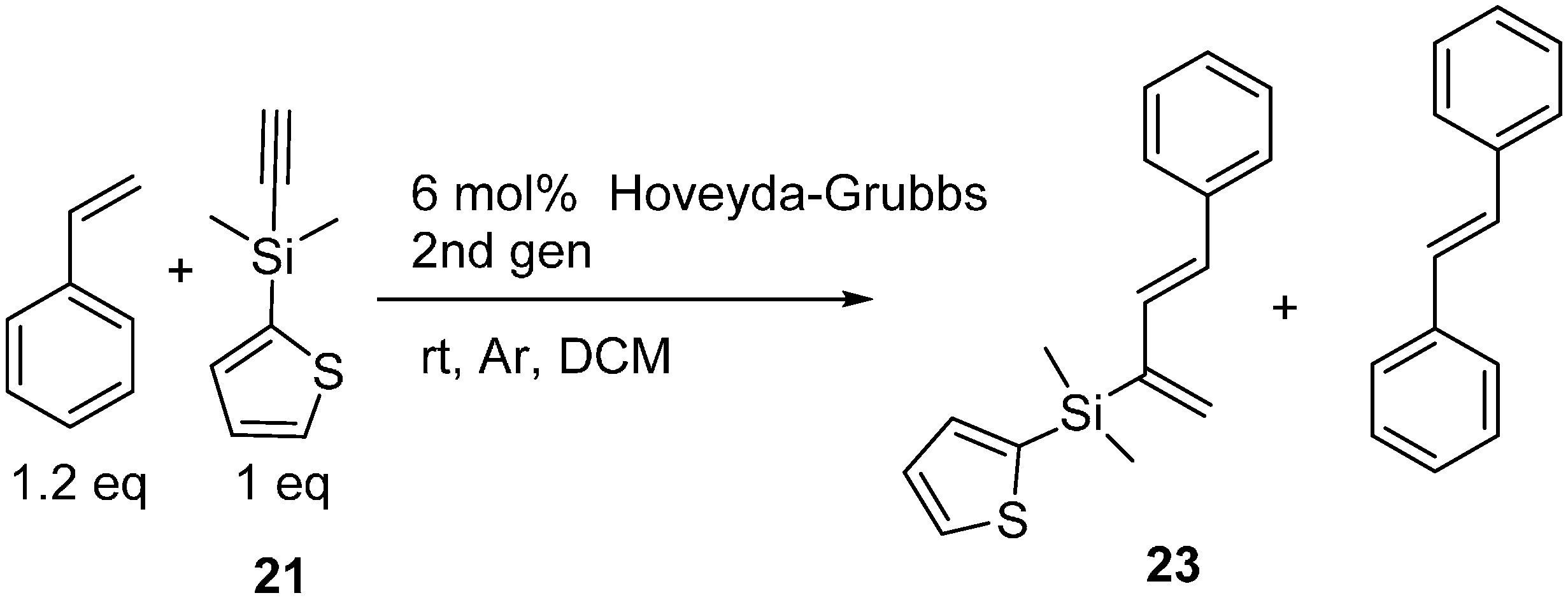
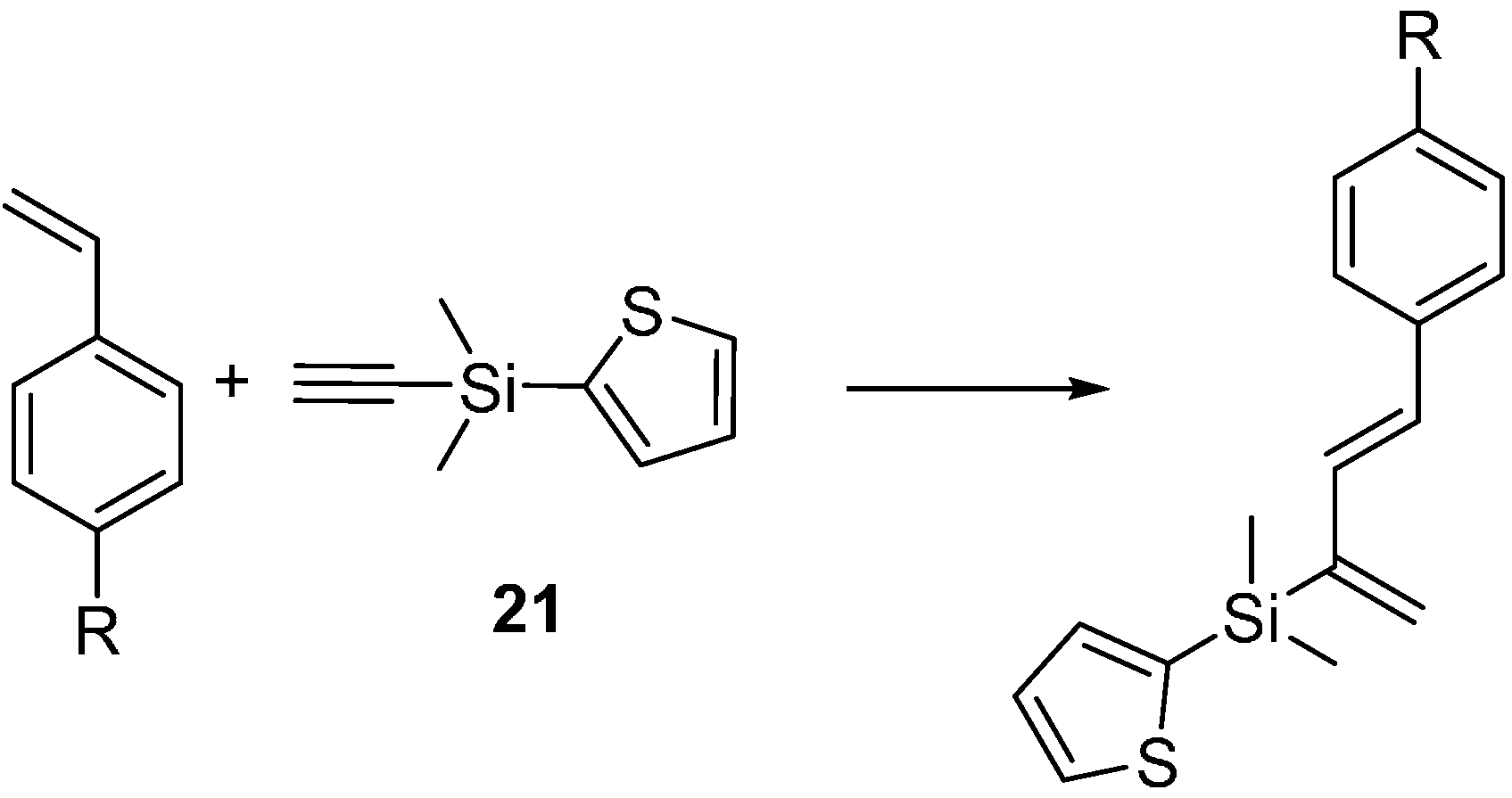

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
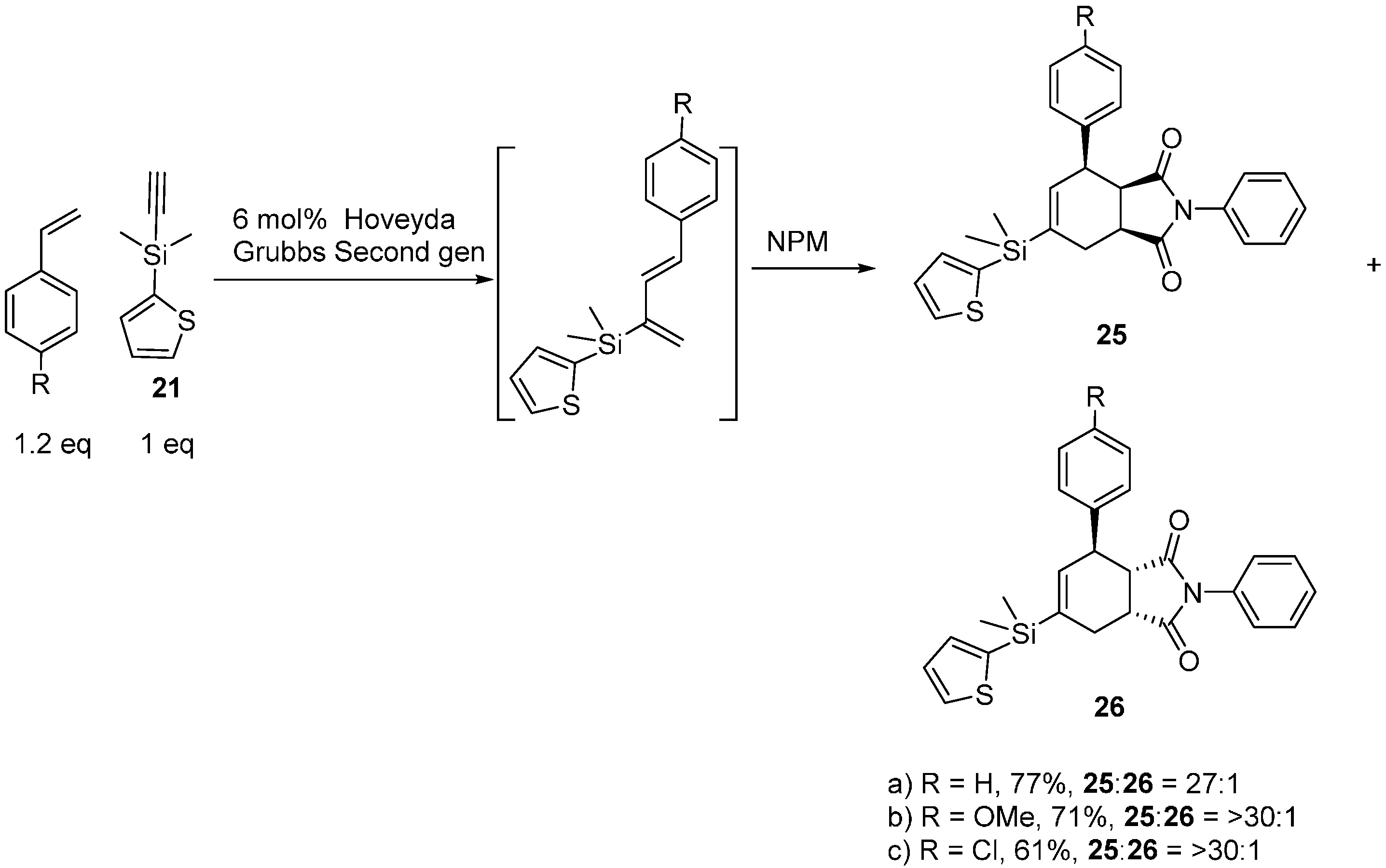
| R | Catalyst Loading (mol %) | Temperature | Time (Disappearance of the Alkyne Peak in 1H-NMR) |
|---|---|---|---|
| -OCH3 | 7 | RT | 3 h |
| -OCH3 | 3 | reflux | 2 h |
| -H | 6 | reflux | 12 h |
| -Cl | 6 | reflux | 30 h |
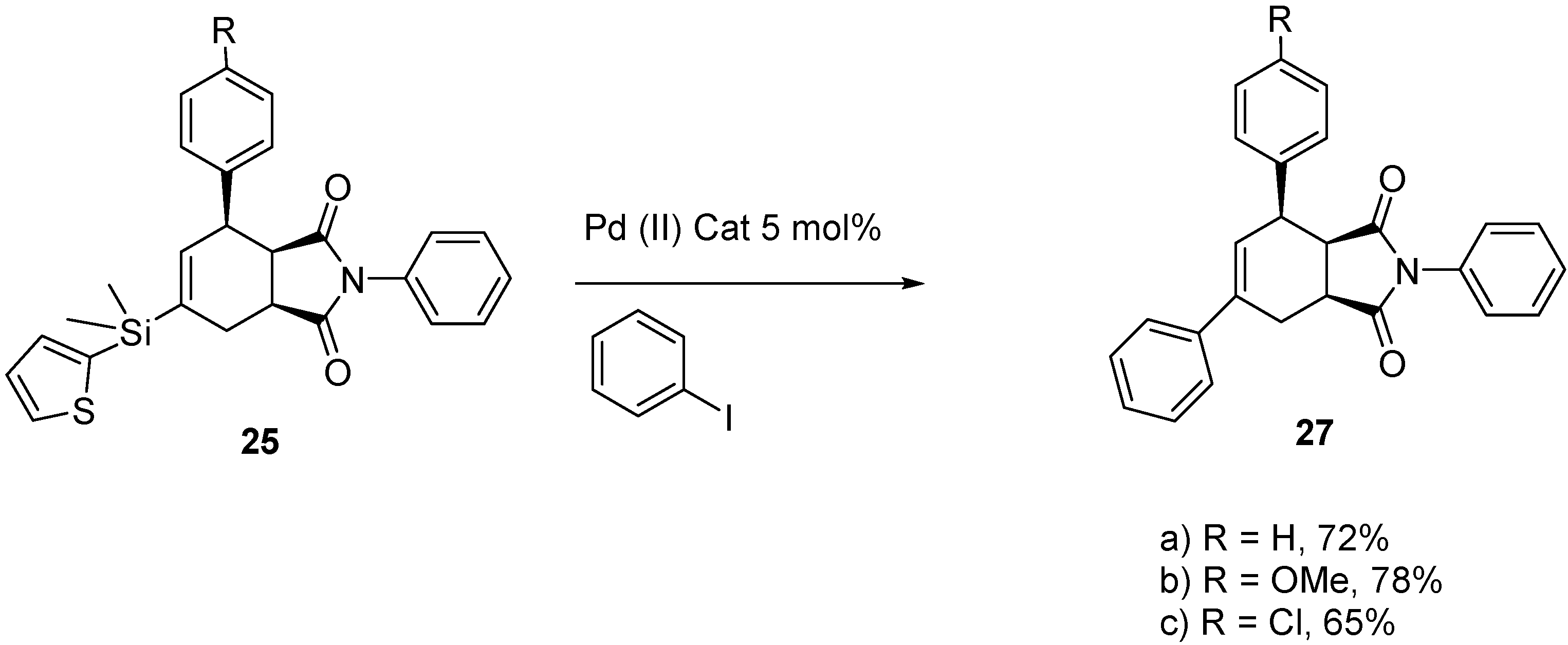
2.4. Tandem Methathesis/Diels-Alder Reactions and Subsequent Cross-Coupling
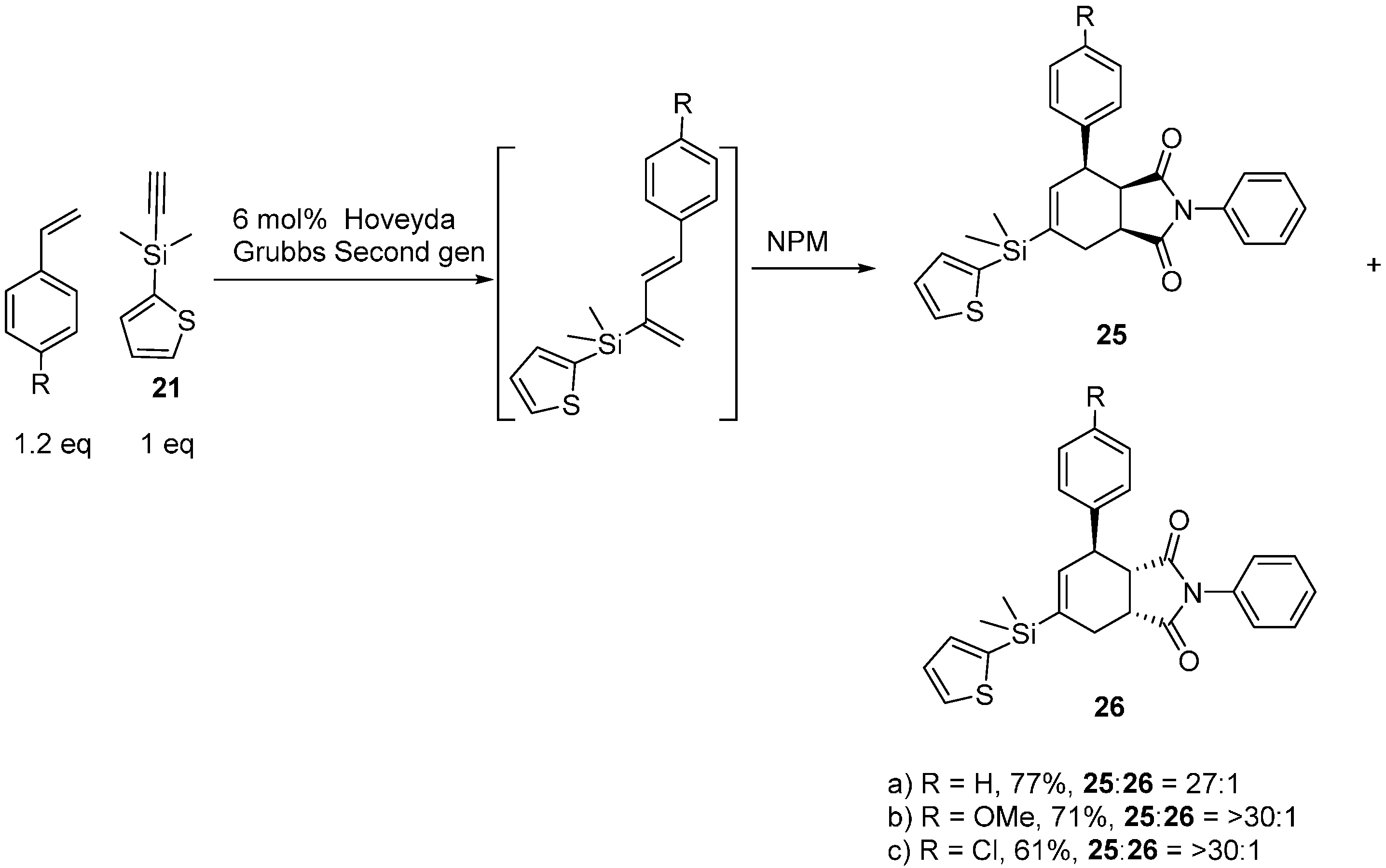
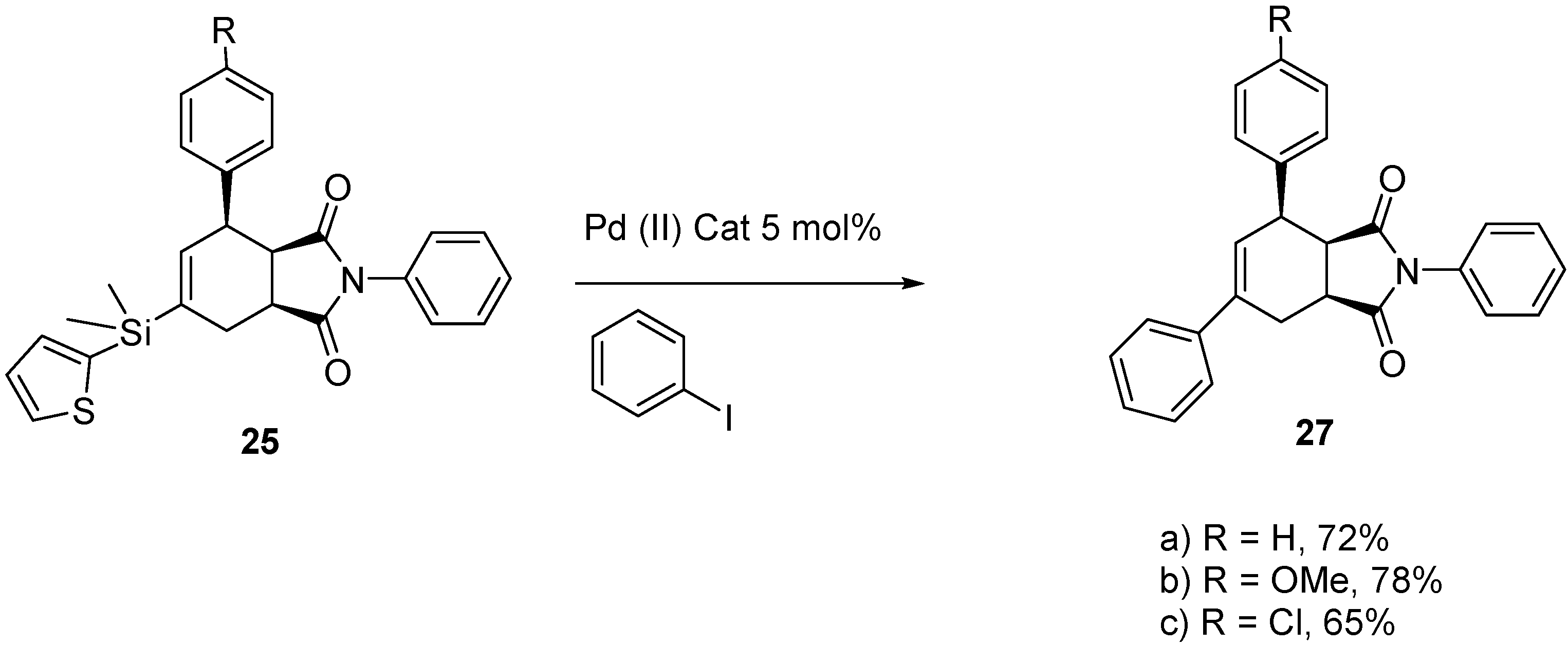
3. Experimental Section
3.1. General Information
3.2. Silyl Diene Synthesis via Grignard Chemistry
3.2.1. 2-(Buta-1,3-dien-2-yldimethylsilyl)pyridine 3
3.2.2. 2-(Buta-1,3-dien-2-yldimethylsilyl)thiophene 4
3.2.3. 1-(Buta-1,3-dien-2-yl)-1-methylsiletane 5
3.3. General Procedure of One-Pot Diels-Alder and Cross-Coupling Reactions of 3
3.3.1. Methyl 2,3,4,5-Tetrahydro-[1,1′-biphenyl]-4-carboxylate 6 and Methyl 2,3,4,5-Tetrahydro-[1,1′-biphenyl]-3-carboxylate 7
3.3.2. 2,5-Diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 8
3.3.3. 3a-Methyl-2,6-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 9 and 3a-Methyl-2,5-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 10
3.4. General Procedure of Diels-Alder Reactions of Diene 4
3.4.1. 1-(4-(Dimethyl(thiophen-2-yl)silyl)cyclohex-3-en-1-yl)ethan-1-one 11 and 1-(3-(Dimethyl(thiophen-2-yl)silyl)cyclohex-3-en-1-yl)ethan-1-one 12
3.4.2. 5-(Dimethyl(thiophen-2-yl)silyl)-2-phenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 15
3.4.3. 6-(Dimethyl(thiophen-2-yl)silyl)-3a-methyl-2-phenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 17 and 5-(Dimethyl(thiophen-2-yl)silyl)-3a-methyl-2-phenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 18
3.5. General Procedure for Cross-Coupling Diels-Alder Adducts to Produce Phenyl-Substituted Cyclohexenes (13, 14, 16, 19, 20)
3.5.1. 1-(2,3,4,5-Tetrahydro-[1,1′-biphenyl]-4-yl)ethan-1-one (13) and 1-(2,3,4,5-Tetrahydro-[1,1′-biphenyl]-3-yl)ethan-1-one (14)
3.5.2. 2,5-Diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (16)
3.5.3. 3a-Methyl-2,6-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (19) and 3a-Methyl-2,5-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (20)
3.6. Silyl Alkyne Preparation
Ethynyldimethyl(pyridin-2-yl)silane (22)
3.7. General Procedure for Tandem Ene-Yne Cross-Metathesis and Diels-Alder Reactions
3.8. General Procedure for Cross-Coupling of 25a–c
3.8.1. (3aR,4R,7aS)-2,4,6-Triphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (27a)
3.8.2. (3aR,4R,7aS)-4-(4-Methoxyphenyl)-2,6-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (27b)
3.8.3. (3aR,4R,7aS)-4-(4-Chlorophenyl)-2,6-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (27c)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Welker, M.E. Preparation and exo-selective [4 + 2] cycloaddition reactions of cobaloxime-substituted 1,3-dienes. Adv. Cycloaddit. 1997, 4, 149–206. [Google Scholar]
- Welker, M.E. Organocobalt complexes in organic synthesis. Curr. Org. Chem. 2001, 5, 785–807. [Google Scholar] [CrossRef]
- Welker, M.E. Recent advances in syntheses and reaction chemistry of boron and silicon substituted 1,3-dienes. Tetrahedron 2008, 64, 11529–11539. [Google Scholar] [CrossRef]
- Lim, D.S.W.; Anderson, E.A. Synthesis of vinylsilanes. Synth. Stuttg. 2012, 44, 983–1010. [Google Scholar]
- Herndon, J.W. The chemistry of the carbon-transition metal double and triple bond: Annual survey covering the year 2010. Coord. Chem. Rev. 2012, 256, 1281–1376. [Google Scholar] [CrossRef]
- Sore, H.F.; Galloway, W.; Spring, D.R. Palladium-catalysed cross-coupling of organosilicon reagents. Chem. Soc. Rev. 2012, 41, 1845–1866. [Google Scholar] [CrossRef] [PubMed]
- Puri, J.K.; Singh, R.; Chahal, V.K. Silatranes: A review on their synthesis, structure, reactivity and applications. Chem. Soc. Rev. 2011, 40, 1791–1840. [Google Scholar] [CrossRef] [PubMed]
- Pidaparthi, R.R.; Welker, M.E. Preparation of siloxacyclopentene containing 1,3-dienes and their Diels-Alder reactions. Tetrahedron Lett. 2007, 48, 7853–7856. [Google Scholar] [CrossRef]
- Pidaparthi, R.R.; Welker, M.E.; Day, C.S.; Wright, M.W. Preparation of 2-trialkylsiloxy-substituted 1,3-dienes and their Diels-Alder/cross-coupling reactions. Org. Lett. 2007, 9, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Pidaparthi, R.R.; Junker, C.S.; Welker, M.E.; Day, C.S.; Wright, M.W. Preparation of 2-silicon-substituted 1,3-dienes and their Diels-Alder/cross-coupling reactions. J. Org. Chem. 2009, 74, 8290–8297. [Google Scholar] [CrossRef] [PubMed]
- Junker, C.S.; Welker, M.E.; Day, C.S. Synthesis of 4-aryl- and 4-alkyl-2-silyl-1,3-butadienes and their Diels-Alder/cross-coupling reactions. J. Org. Chem. 2010, 75, 8155–8165. [Google Scholar] [CrossRef] [PubMed]
- Junker, C.S.; Welker, M.E. Ruthenium carbenes as catalysts in stereoselective ene-yne metathesis/Diels-Alder and ene-yne metathesis/Diels-Alder/cross coupling multicomponent reactions. Tetrahedron 2012, 68, 5341–5345. [Google Scholar] [CrossRef]
- Choudhury, P.P.; Junker, C.S.; Pidaparthi, R.R.; Welker, M.E. Syntheses of 2-silicon-substituted 1,3-dienes. J. Organomet. Chem. 2014, 754, 88–93. [Google Scholar] [CrossRef]
- Hanson, J.R. Diterpenoids. Nat. Prod. Rep. 2003, 20, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Choi, J.Y. Highly stereospecific, cross-coupling reactions of alkenylsilacyclobutanes. J. Am. Chem. Soc. 1999, 121, 5821–5822. [Google Scholar] [CrossRef]
- Itami, K.; Mitsudo, K.; Kamei, T.; Koike, T.; Nokami, T.; Yoshida, J. Highly efficient carbopalladation across vinylsilane: Dual role of the 2-pyme2si group as a directing group and as a phase tag. J. Am. Chem. Soc. 2000, 122, 12013–12014. [Google Scholar] [CrossRef]
- Hosoi, K.; Nozaki, K.; Hiyama, T. Alkenyldimethyl(2-thienyl)silanes, excellent coupling partner for the palladium-catalyzed cross-coupling reaction. Chem. Lett. 2002, 31, 138–139. [Google Scholar] [CrossRef]
- Ostrowska, S.; Powała, B.; Jankowska-Wajda, M.; Żak, P.; Rogalski, S.; Wyrzykiewicz, B.; Pietraszuk, C. Regio- and steroselective cross-metathesis of silylacetylenes with terminal olefins and alpha, omega-dienes. J. Organomet. Chem. 2015, 783, 135–140. [Google Scholar] [CrossRef]
- Denmark, S.E.; Tymonko, S.A. Sequential cross-coupling of 1,4-bissilylbutadienes: Synthesis of unsymmetrical 1,4-disubstituted 1,3-butadienes. J. Am. Chem. Soc. 2005, 127, 8004–8005. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.R.; Diver, S.T. Atom economy in the metathesis cross-coupling of alkenes and alkynes. Org. Lett. 2011, 13, 2896–2899. [Google Scholar] [CrossRef] [PubMed]
- Giessert, A.J.; Diver, S.T. Cross enyne metathesis of para-substituted styrenes: A kinetic study of enyne metathesis. Org. Lett. 2005, 7, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Sakakibara, N.; Kinoshita, A. Remarkable effect of ethylene gas in the intramolecular enyne metathesis of terminal alkynes. J. Org. Chem. 1998, 63, 6082–6083. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Sakakibara, N.; Mori, M. Novel 1,3-Diene Synthesis from alkyne and ethylene by ruthenium-catalyzed enyne metathesis. J. Am. Chem. Soc. 1997, 119, 12388–12389. [Google Scholar] [CrossRef]
- Gregg, T.M.; Keister, J.B.; Diver, S.T. Inhibitory effect of ethylene in ene-yne metathesis: The case for ruthenacyclobutane resting states. J. Am. Chem. Soc. 2013, 135, 16777–16780. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Welker, M.E. Preparation of 2-BF3-substituted 1,3-dienes and their Diels−Alder/cross-coupling reactions. Org. Lett. 2005, 7, 2481–2484. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choudhury, P.P.; Welker, M.E. Preparation and Reaction Chemistry of Novel Silicon-Substituted 1,3-Dienes. Molecules 2015, 20, 16892-16907. https://doi.org/10.3390/molecules200916892
Choudhury PP, Welker ME. Preparation and Reaction Chemistry of Novel Silicon-Substituted 1,3-Dienes. Molecules. 2015; 20(9):16892-16907. https://doi.org/10.3390/molecules200916892
Chicago/Turabian StyleChoudhury, Partha P., and Mark E. Welker. 2015. "Preparation and Reaction Chemistry of Novel Silicon-Substituted 1,3-Dienes" Molecules 20, no. 9: 16892-16907. https://doi.org/10.3390/molecules200916892
APA StyleChoudhury, P. P., & Welker, M. E. (2015). Preparation and Reaction Chemistry of Novel Silicon-Substituted 1,3-Dienes. Molecules, 20(9), 16892-16907. https://doi.org/10.3390/molecules200916892