
In Silico Identification and in Vitro Activity of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde-3-phosphate-dehydrogenase †


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. In Silico Prediction of Potential TbGAPDH Inhibitors
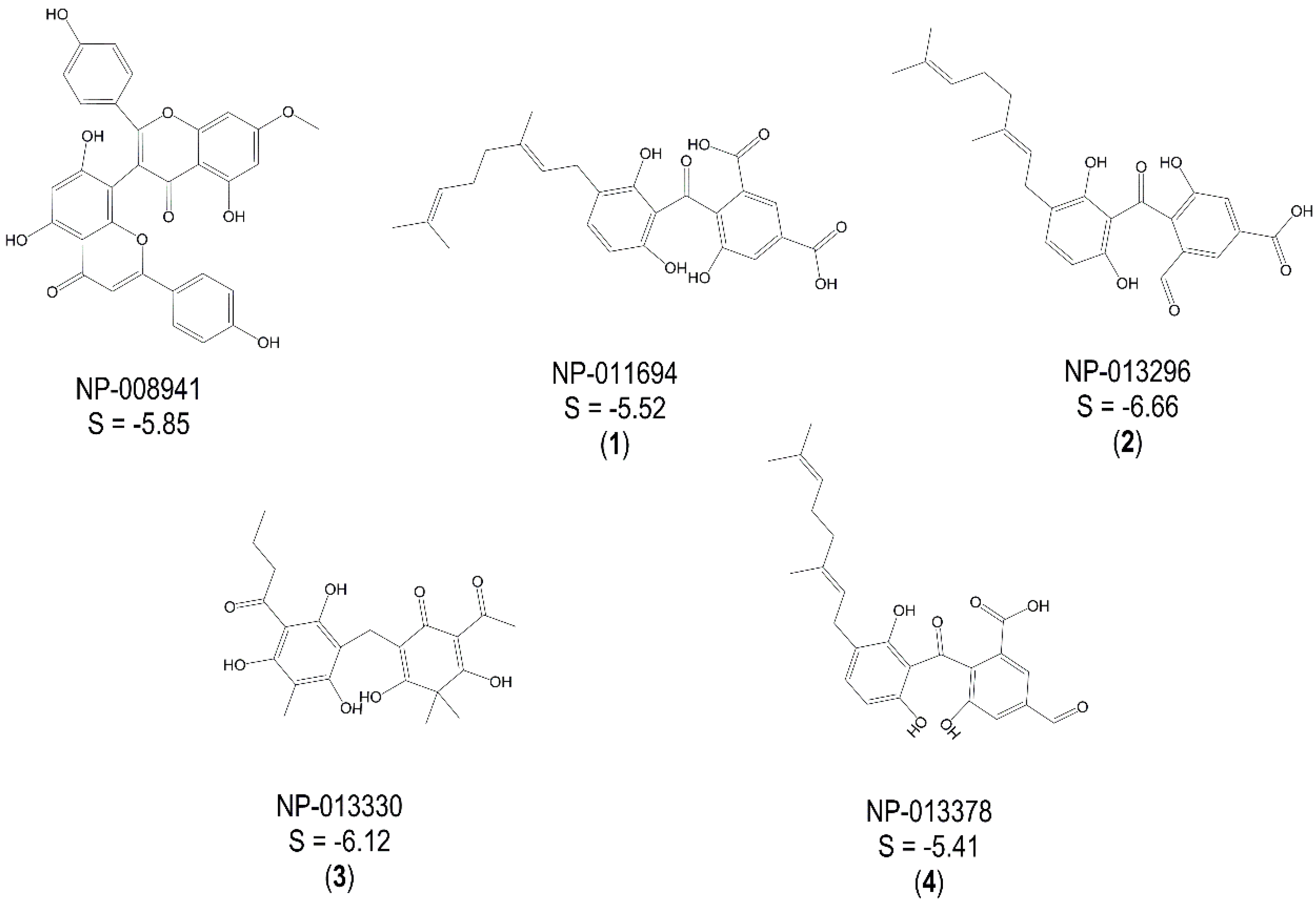
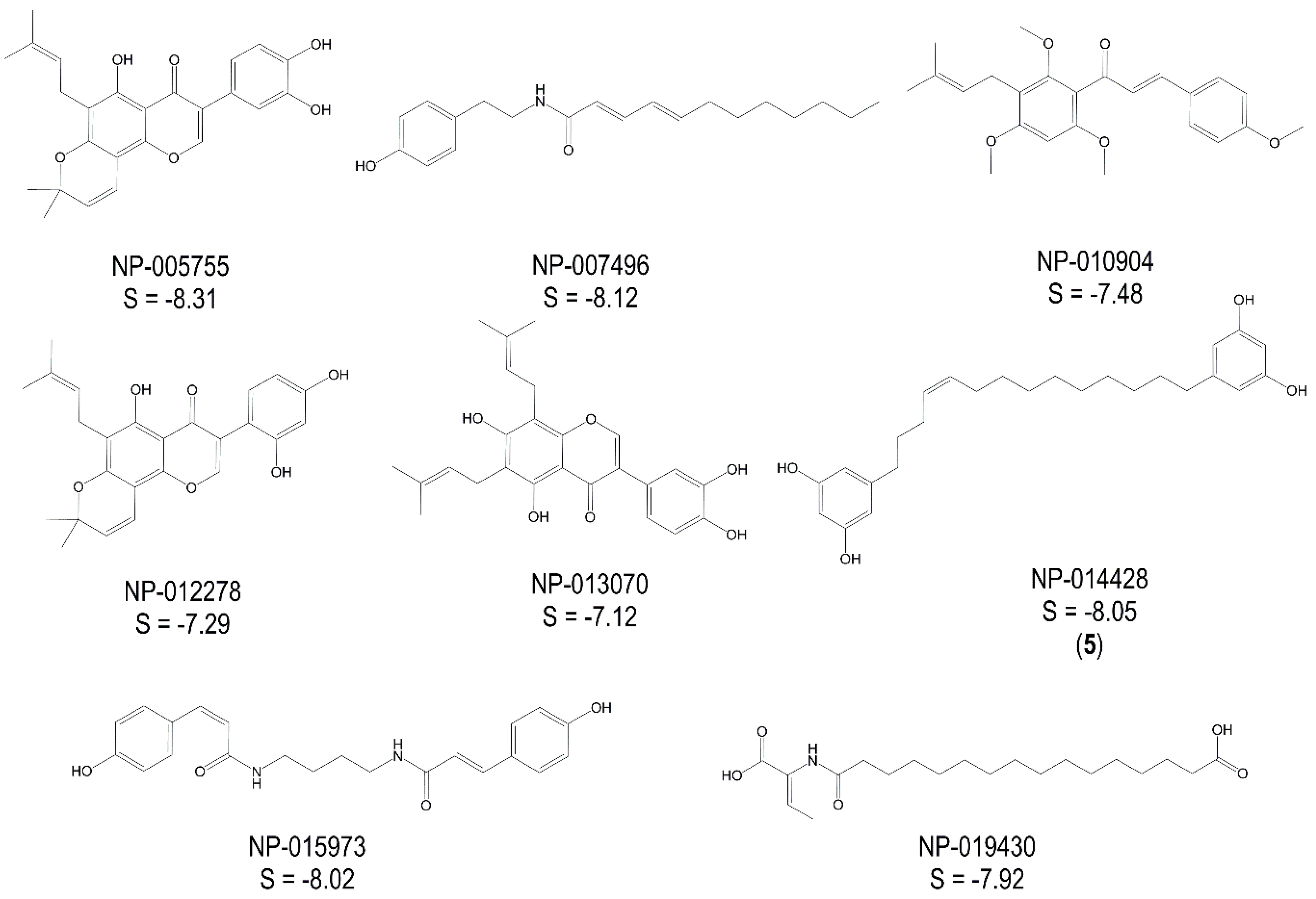
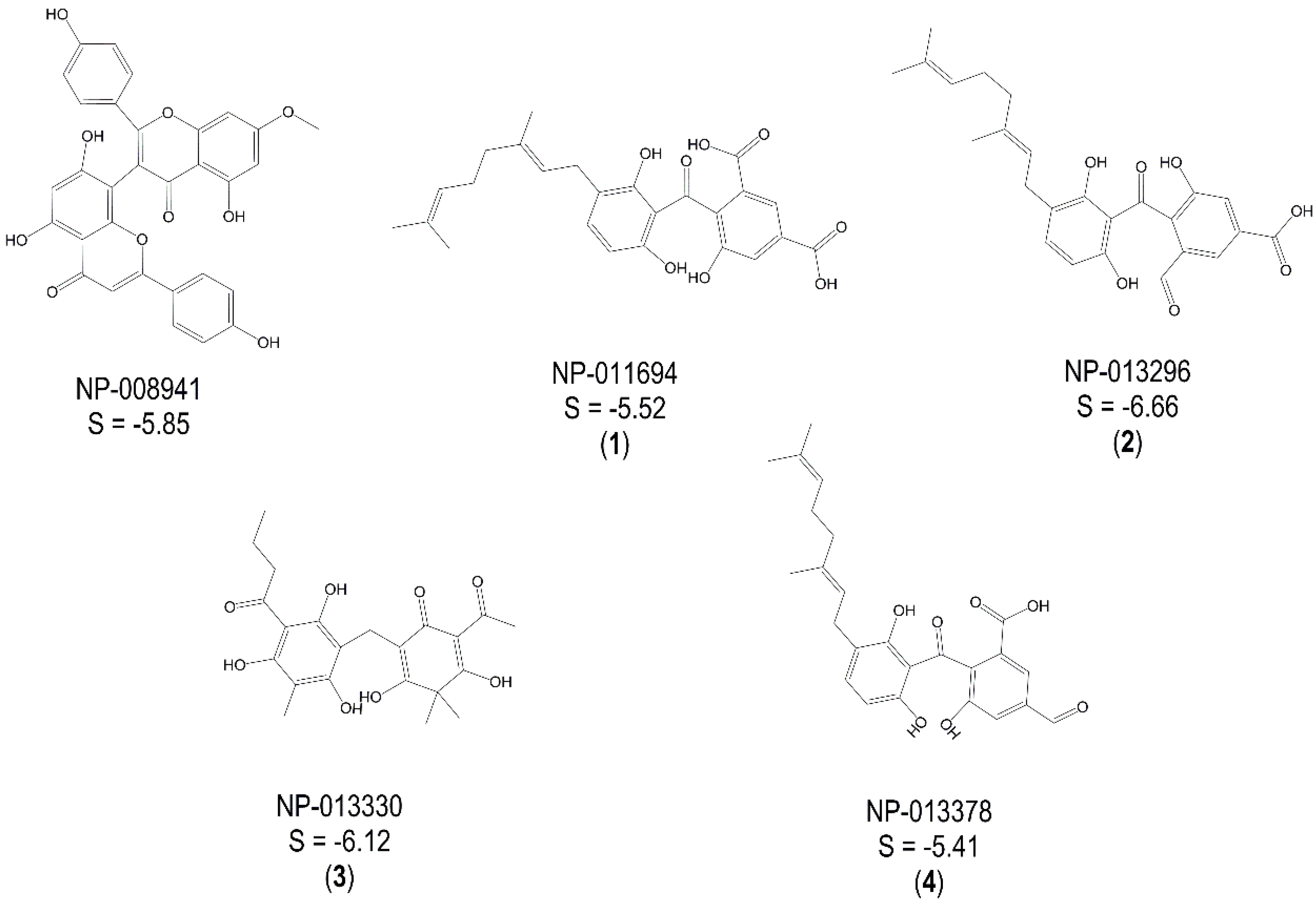
2.2. Experimental Validation of TbGAPDH Inhibition

{kind=link}
{kind=link}
{kind=link}
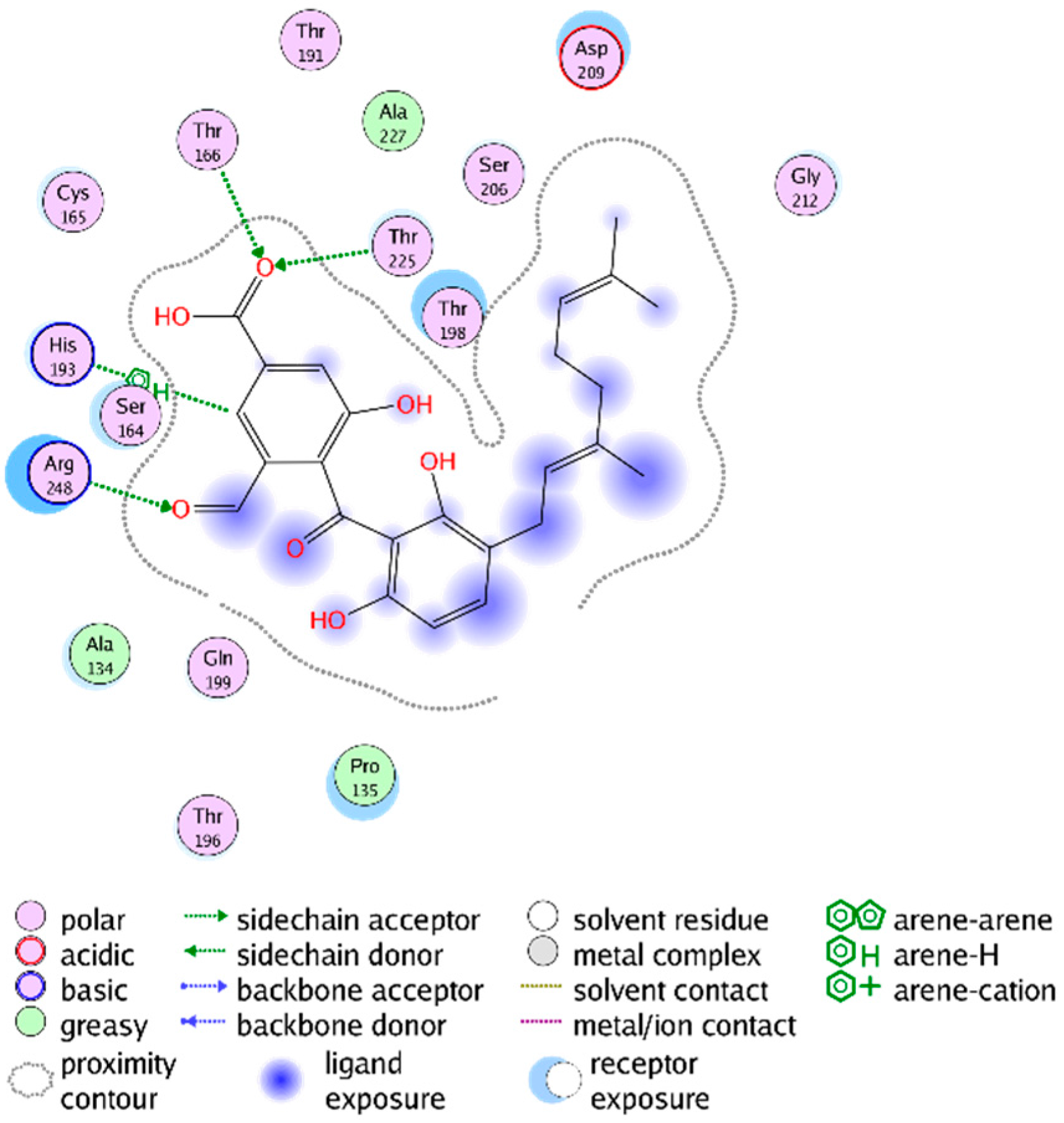{kind=link}
{kind=link}
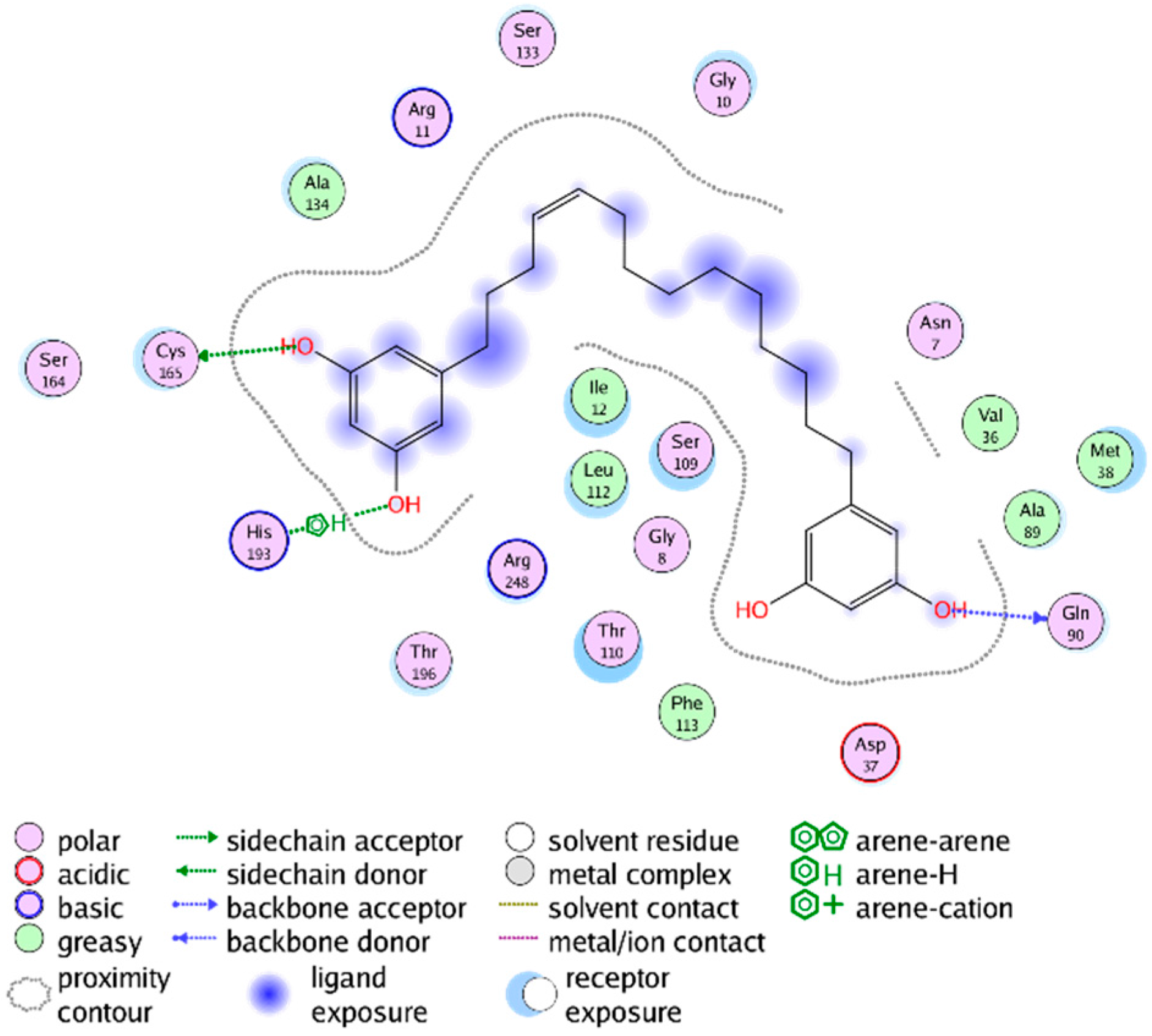{kind=link}
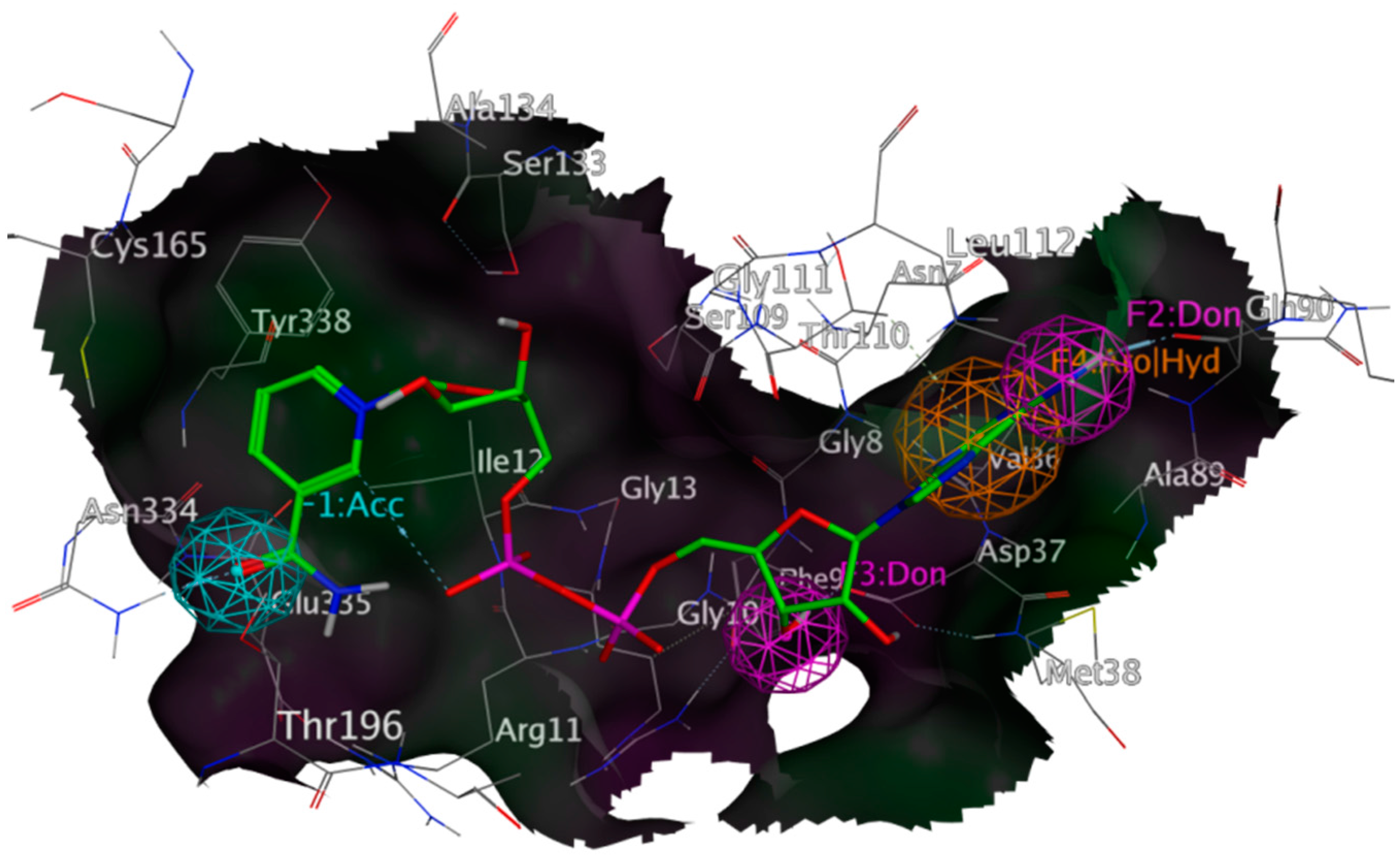{kind=link}
| Compound | % Inhibition at c = 50 µM | IC50 (µM) |
|---|---|---|
| NP-005755 | 44 | |
| NP-007496 | n.i. | |
| NP-008941 | n.i. | |
| NP-010904 | n.i. | |
| NP-011694 (1) | 66 | 24.56 ± 1.03 |
| NP-012278 | 24 | |
| NP-013070 | 33 | |
| NP-013296 (2) | 98 | 4.73 ± 1.04 |
| NP-013330 (3) | 88 | 21.97 ± 1.03 |
| NP-013378 (4) | >90 | 6.68 ± 1.04 |
| NP-014428 (5) | >90 | 22.79 ± 1.01 |
| NP-015973 | n.i. | |
| NP-019430 | 45 |
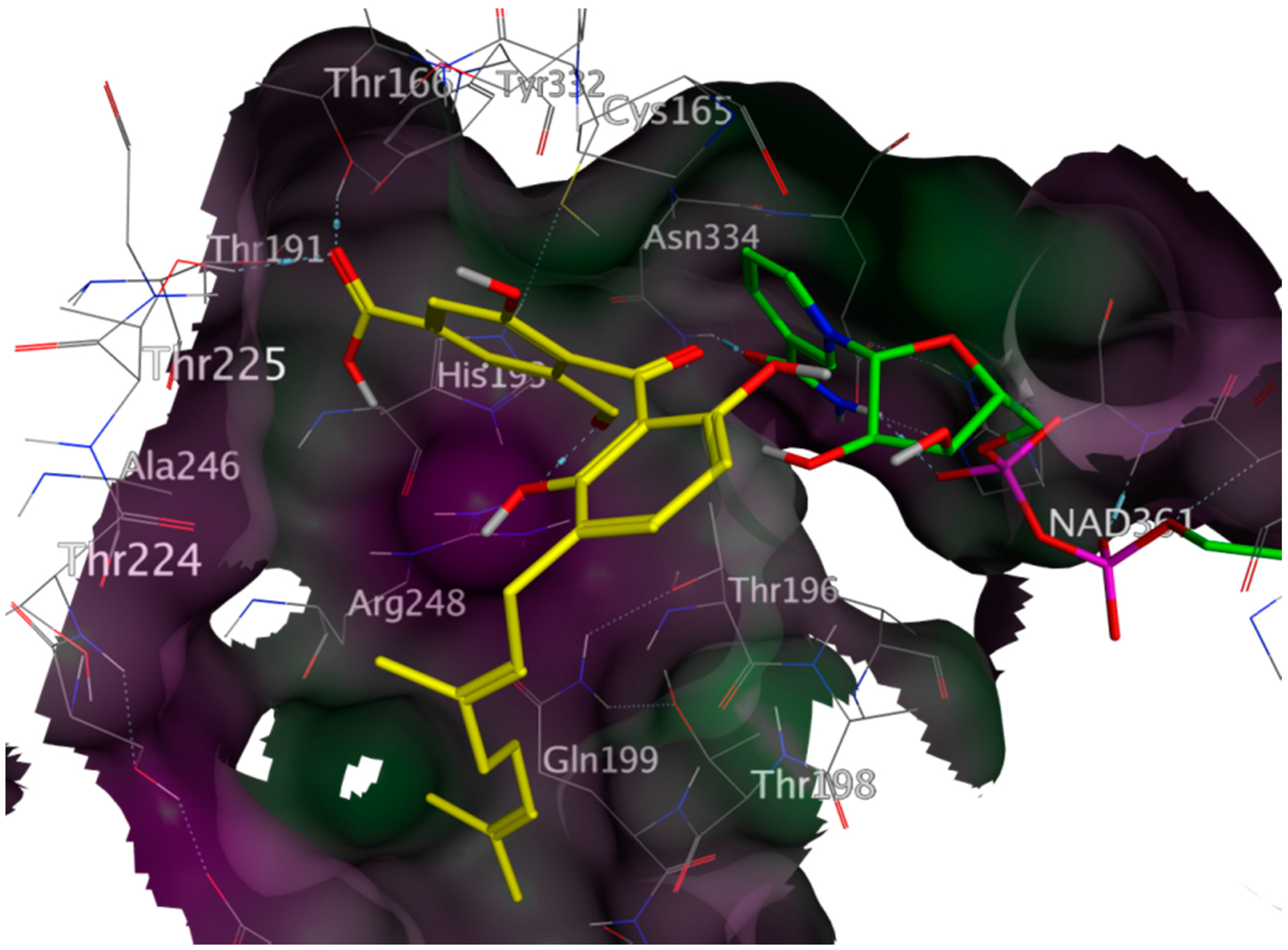
2.3. Mechanistic Considerations
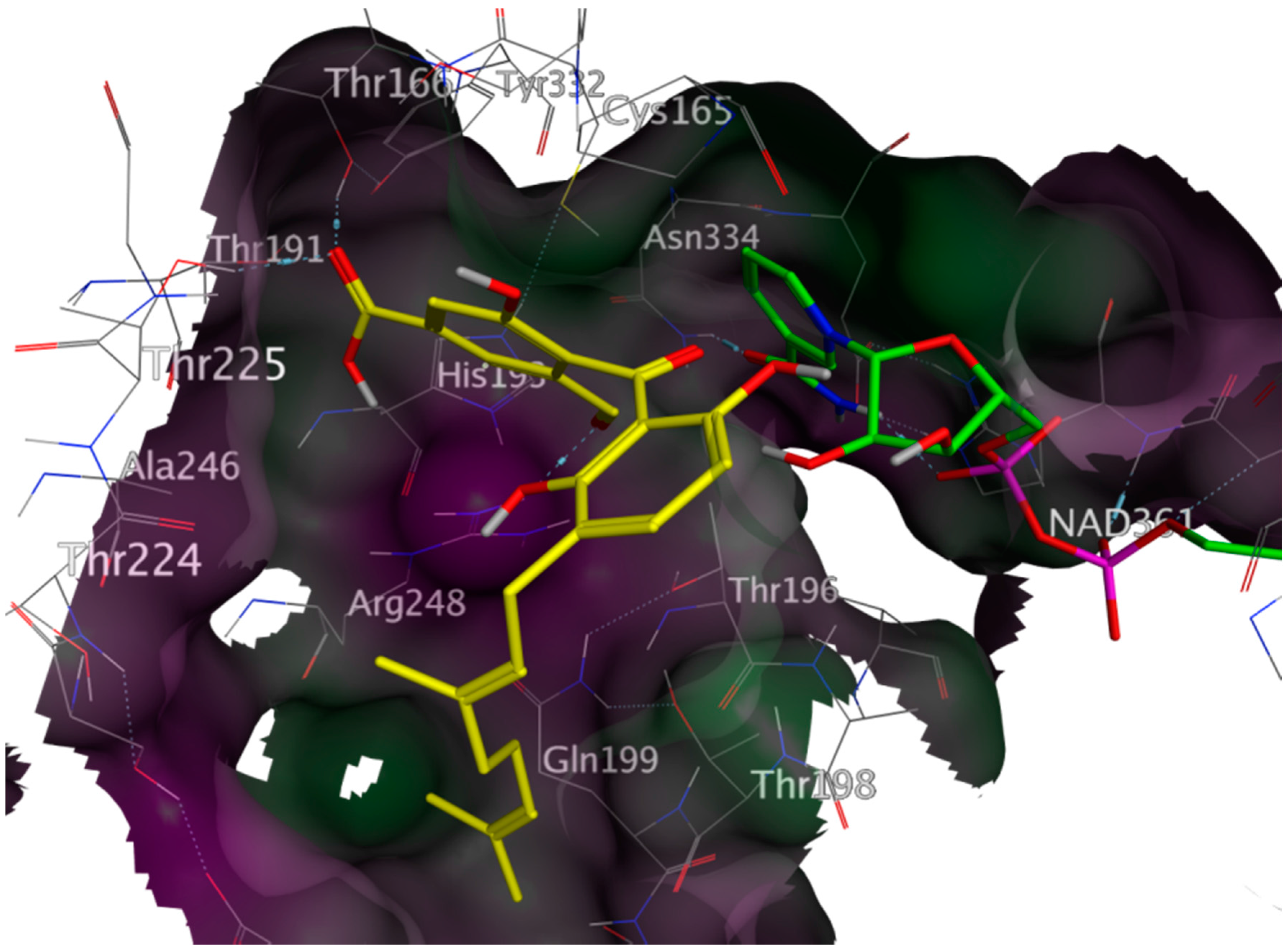
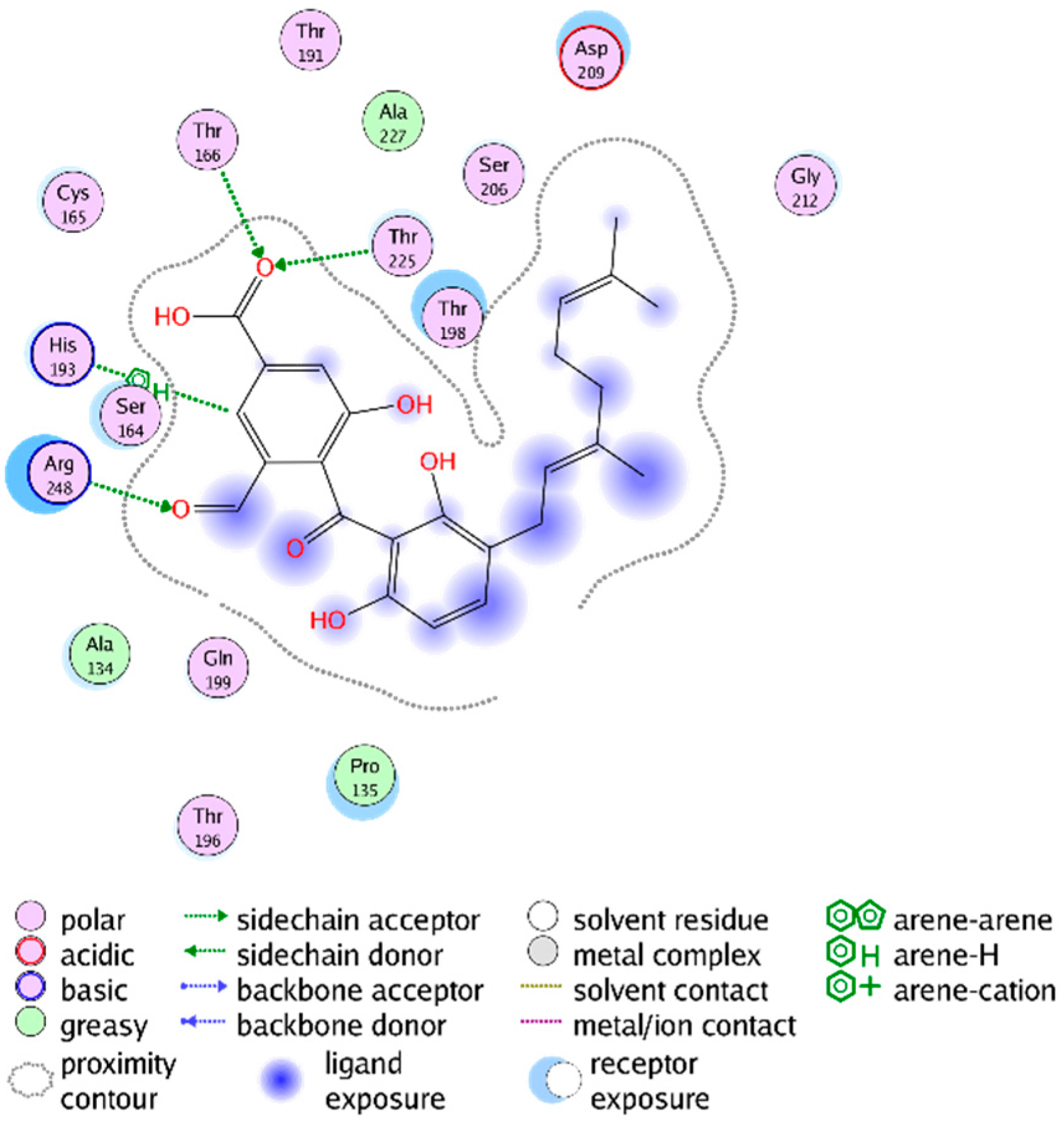
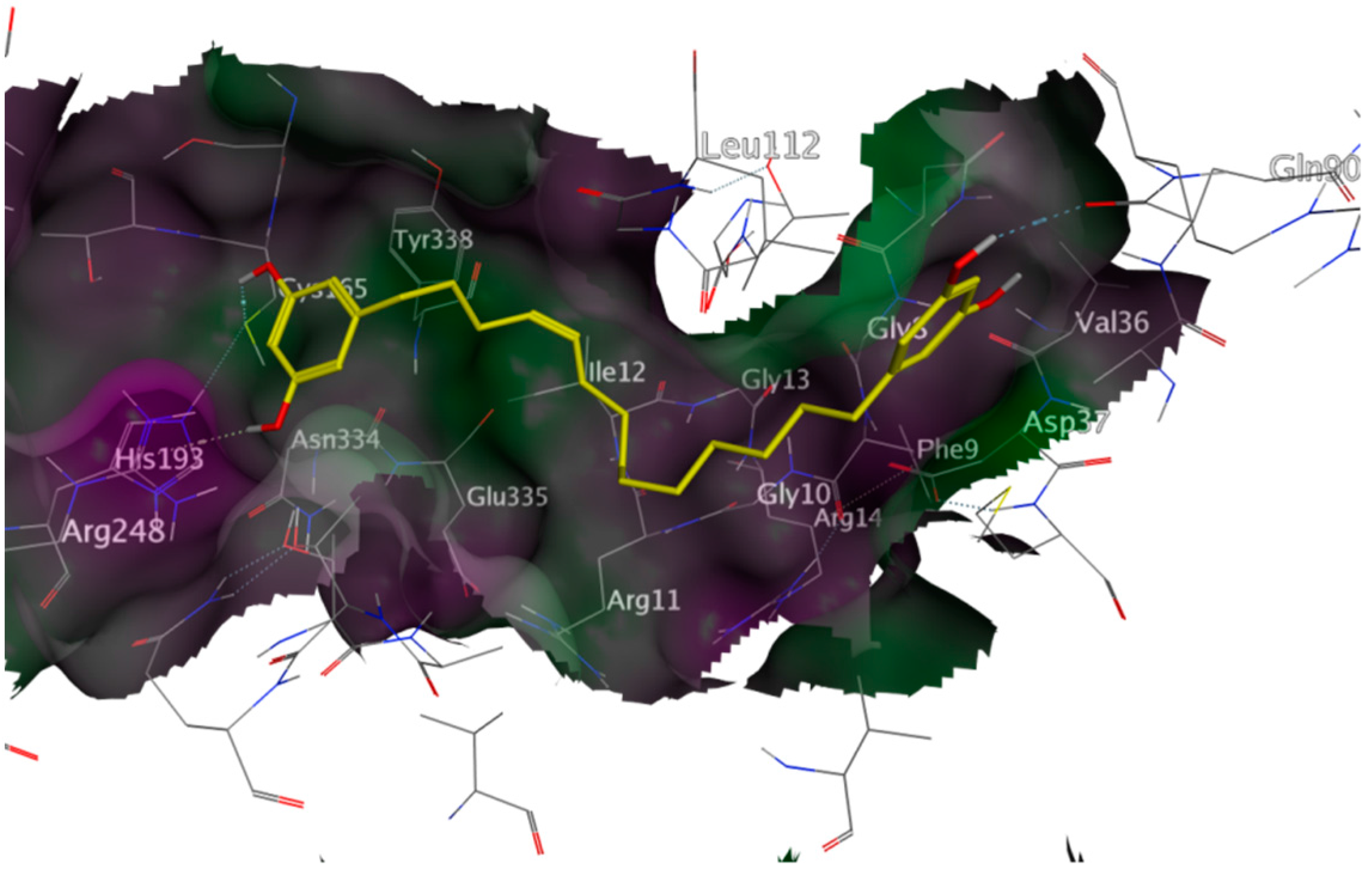
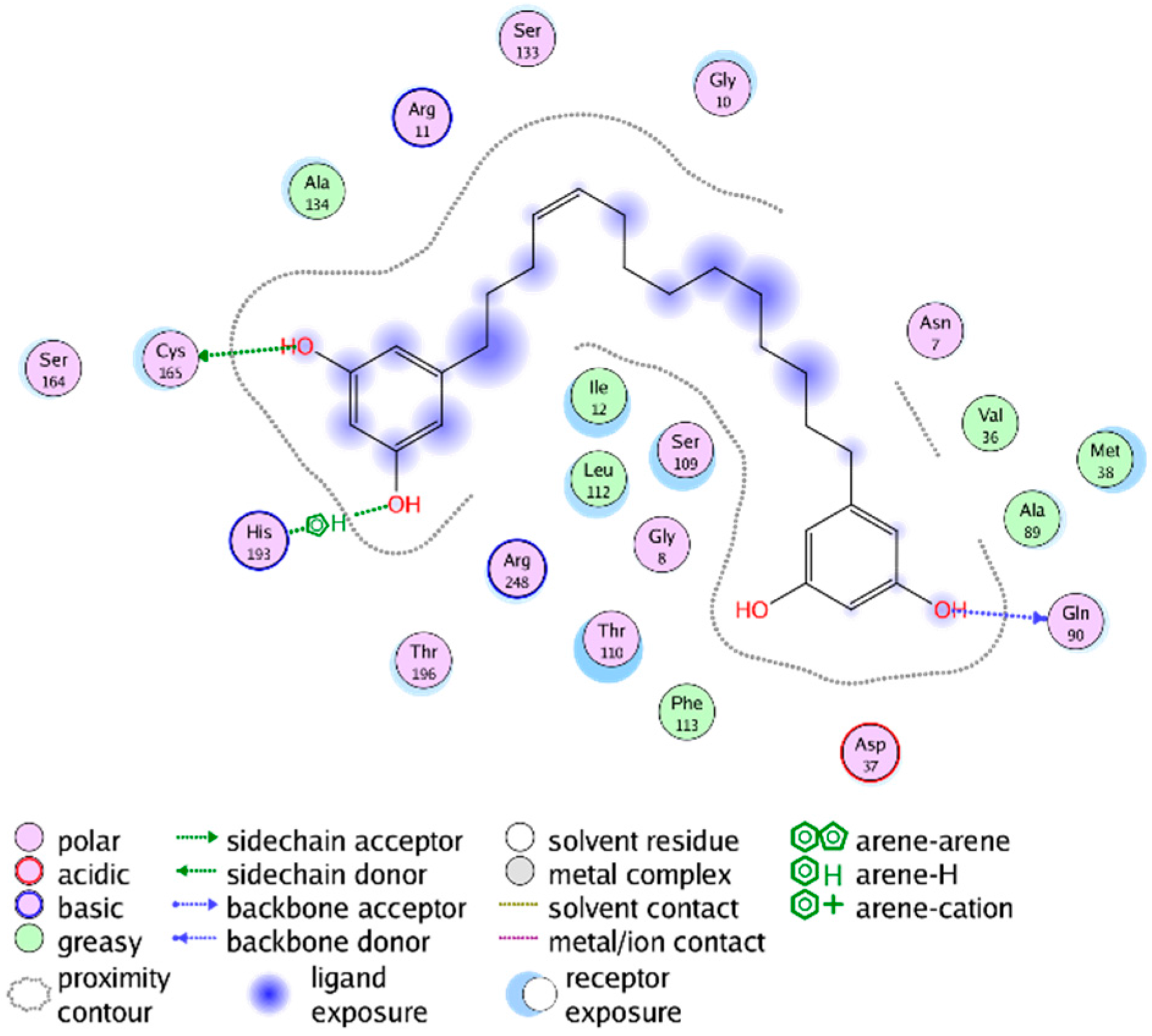
2.4. In Vitro Antitrypanosomal Activity of Compounds 1–5
| Compound | T. brucei rhodesiense (STIB 900) IC50 (µM) | Cytotoxicity (L6) IC50 (µM) | SI |
|---|---|---|---|
| NP-011694 (1) | 39.5 ± 3.1 | 50.6 ± 3.8 | 1.3 |
| NP-013296 (2) | 41.0 ± 0.5 | 48.7 ± 3.8 | 1.2 |
| NP-013330 (3) | 17.9 ± 1.6 | 54.2 ± 0.5 | 3.0 |
| NP-013378 (4) | 6.7 ± 2.2 | 48.9 ± 4.1 | 7.3 |
| NP-014428 (5) | 29.3 ± 6.3 | 15.5 ± 1.8 | 0.5 |
| Melarsoprol | 0.005 ± 0.001 | n.t. | n.d. |
| Podophyllotoxin | n.t. | 0.019 ± 0.002 | n.d. |
3. Experimental Section
3.1. Database Design
3.2. Acquisition and Preparation of the Proteins’ 3D-Structure
3.3. Pharmacophore Design and Virtual Screening
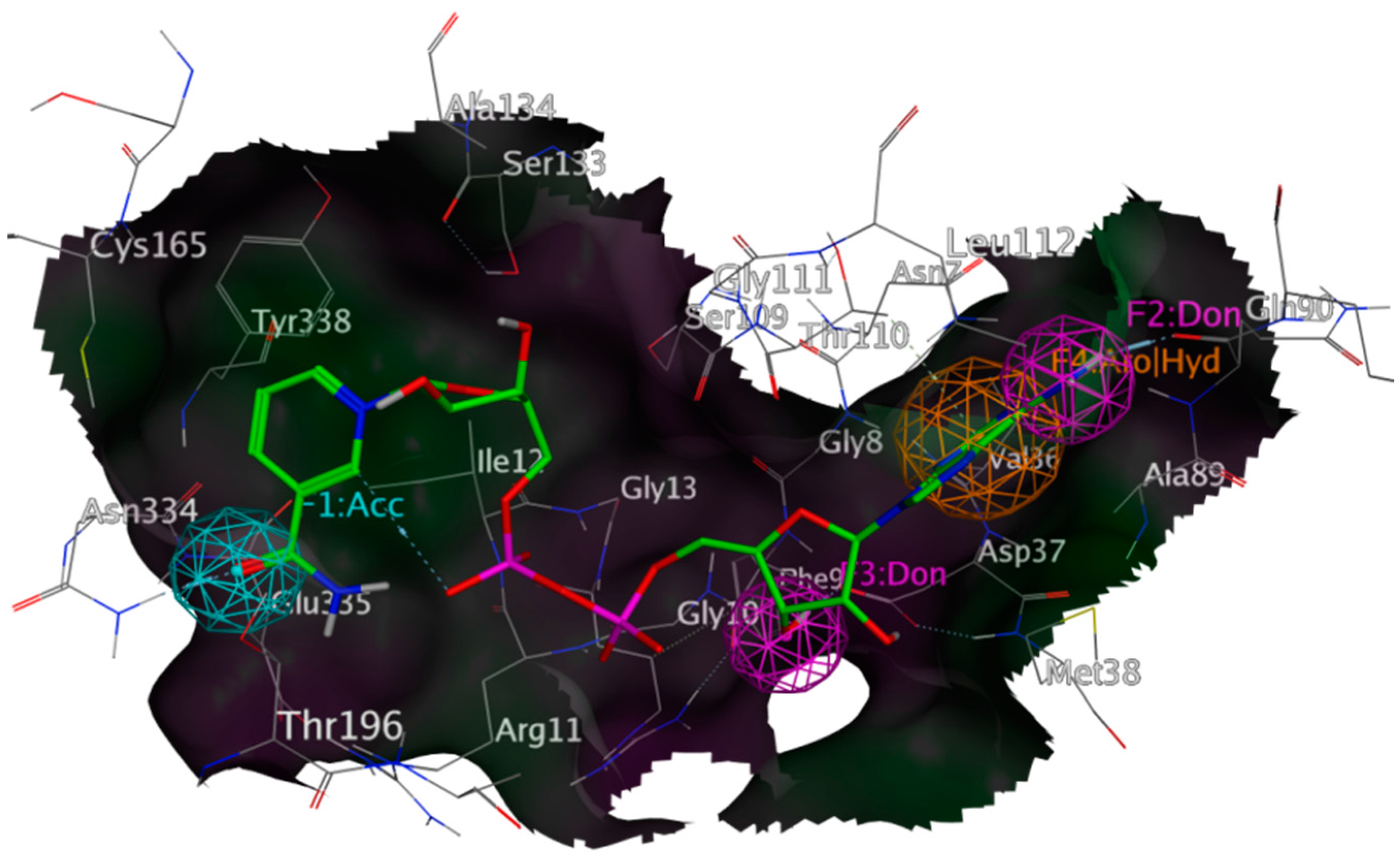
3.4. Docking Procedures
3.5. Tested Compounds
3.6. Recombinant Expression and Purification of Trypanosoma brucei GAPDH in E. coli
3.7. T. brucei Glyceraldehyde-3-phosphate Dehydrogenase Inhibition Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tissot, P. Third WHO Report on Neglected Tropical Diseases. Available online: http://apps.who.int/iris/bitstream/10665/152781/1/9789241564861_eng.pdf?ua=1 (accessed on 20 June 2015).
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.A.; Biavatti, M.W.; Brun, R.; Da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part I. Curr. Med. Chem. 2012, 19, 2128–2175. [Google Scholar] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.A.; Biavatti, M.W.; Brun, R.; Da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part II. Curr. Med. Chem. 2012, 19, 2176–2228. [Google Scholar] [CrossRef] [PubMed]
- Nour, A.M.M.; Khalid, S.A.; Brun, R.; Kaiser, M.; Schmidt, T.J. The antiprotozoal activity of sixteen Asteraceae species native to Sudan and bioactivity-guided isolation of xanthanolides from Xanthium brasilicum. Planta Med. 2009, 75, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Gökbulut, A.; Kaiser, M.; Brun, R.; Sarer, E.; Schmidt, T.J. 9β-Hydroxyparthenolide esters from Inula montbretiana DC. and their antiprotozoal activity. Planta Med. 2012, 78, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.B.; Kaiser, M.; Brun, R.; Schmidt, T.J. Antiprotozoal activity of Achillea ptarmica (Asteraceae) and its main alkamide constituents. Molecules 2014, 19, 6428–6438. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.B.; Jerz, G.; Winterhalter, P.; Kaiser, M.; Brun, R.; Schmidt, T.J. Antiprotozoal activity of Buxus sempervirens and activity-guided isolation of O-tigloyl-cyclovirobuxeine-B as main constituent active against Plasmodium falciparum. Molecules 2014, 19, 6184–6201. [Google Scholar] [CrossRef] [PubMed]
- Thao, N.P.; Luyen, B.T.T.; Brun, R.; Kaiser, M.; Kiem, P.V.; Minh, C.V.; Schmidt, T.J.; Kang, J.S.; Kim, Y.H. Anti-protozoal Activities of Cembrane-Type Diterpenes from Vietnamese Soft Corals. Molecules 2015, 20, 12459–12468. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; da Costa, F.B.; Lopes, N.P.; Kaiser, M.; Brun, R. In silico prediction and experimental evaluation of furanoheliangolide sesquiterpene lactones as potent agents against Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 2014, 58, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ellendorff, T.; Brun, R.; Kaiser, M.; Schmidt, T.J.; Sendker, J. PLS-Prediction and Confirmation of Hydrojuglone Glucoside as the Antitrypanosomal Constituent of Juglans Spp. Molecules 2015, 20, 10082–10094. [Google Scholar] [CrossRef] [PubMed]
- Bakker, B.M.; Michels, P.A.; Opperdoes, F.R.; Westerhoff, H.V. What controls Glycolysis in Bloodstream Form Trypanosoma brucei? J. Biol. Chem. 1999, 274, 14551–14559. [Google Scholar] [CrossRef] [PubMed]
- Opperdoes, F.R. Compartmentation of carbohydrate metabolism in Trypanosomes. Ann. Rev. Microbiol. 1987, 41, 127–151. [Google Scholar] [CrossRef] [PubMed]
- Gualdrón-López, M.; Michels, P.A.; Quiñones, W.; Cáceres, A.J.; Avilán, L.; Concepción, J.-L. Function of Glycosomes in the Metabolism of Trypanosomatid Parasites and the Promise of Glycosomal Proteins as Drug Targets. In Drug Discovery in Infectious Diseases; Trypanosomatid Diseases; Selzer, P.M., Jäger, T., Koch, O., Flohé, L., Eds.; Wiley-VCH: Weinheim, Germany, 2013; Volume 4, pp. 121–151. [Google Scholar]
- Cáceres, A.J.; Michels, P.A.M.; Hannaert, V. Genetic validation of aldolase and glyceraldehyde-3-phosphate dehydrogenase as drug targets in Trypanosoma brucei. Mol. Biochem. Parasitol. 2010, 169, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Holzhütter, H.G. Use of mathematical models for predicting the metabolic effect of large-scale enzyme activity alterations. Application to enzyme deficiencies of red blood cells. Eur. J. Biochem. 1995, 229, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M.; Suresh, S.; Buckner, F.S.; van Voorhis, W.C.; Verlinde, C.L.; Opperdoes, F.R.; Hol, W.G.; Gelb, M.H. Structure-based design of submicromolar, biologically active inhibitors of trypanosomatid glyceraldehyde-3-phosphate dehydrogenase. Proc. Nat. Acad. Sci. USA 1999, 96, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Nyasse, B.; Ngantchou, I.; Tchana, E.M.; Sonké, B.; Denier, C.; Fontaine, C. Inhibition of both Trypanosoma brucei bloodstream form and related glycolytic enzymes by a new kolavic acid derivative isolated from Entada abyssinica. Pharmazie 2004, 59, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Bero, J.; Beaufay, C.; Hannaert, V.; Hérent, M.-F.; Michels, P.A.; Quetin-Leclercq, J. Antitrypanosomal compounds from the essential oil and extracts of Keetia leucantha leaves with inhibitor activity on Trypanosoma brucei glyceraldehyde-3-phosphate dehydrogenase. Phytomedicine 2013, 20, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Ngantchou, I.; Nyasse, B.; Denier, C.; Blonski, C.; Hannaert, V.; Schneider, B. Antitrypanosomal alkaloids from Polyalthia suaveolens (Annonaceae): Their effects on three selected glycolytic enzymes of Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2010, 20, 3495–3498. [Google Scholar] [CrossRef] [PubMed]
- Tomazela, D.M.; Pupo, M.T.; Passador, E.A.P.; da Silva, M.F.; Vieira, P.C.; Fernandes, J.B.; Rodrigues Fo, E.; Oliva, G.; Pirani, J.R. Pyrano chalcones and a flavone from Neoraputia magnifica and their Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase-inhibitory activities. Phytochemistry 2000, 55, 643–651. [Google Scholar] [CrossRef]
- Pavão, F.; Castilho, M.S.; Pupo, M.T.; Dias, R.L.A.; Correa, A.G.; Fernandes, J.B.; da Silva, M.F.G.F.; Mafezoli, J.; Vieira, P.C.; Oliva, G. Structure of Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase complexed with chalepin, a natural product inhibitor, at 1.95 Å resolution. FEBS Lett. 2002, 520, 13–17. [Google Scholar] [CrossRef]
- Herrmann, F.C.; Schmidt, T.J. In silico screening of natural product databases reveals new potential leads against neglected diseases. Planta Med. 2013, 79, 1132. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); 2011.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2015.
- RCSB Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 28 March 2012 (2X0N), 29 April 2013 (3IDS) and 11 June 2013 (1GYP)).
- Lesk, A.M. NAD-binding domains of dehydrogenases. Curr. Opt. Struct. Biol. 1995, 5, 775–783. [Google Scholar] [CrossRef]
- Lambeir, A-M.; Loiseau, A.M.; Kuntz, D.A.; Vellieux, F.M.; Michels, P.A.; Opperdoes, F.R. The cytosolic and glycosomal glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei. Eur. J. Biochem. 1991, 198, 429–435. [Google Scholar]
- Kolossváry, I.; Guida, W.C. Low Mode Search. An Efficient, Automated Computational Method for Conformational Analysis: Application to Cyclic and Acyclic Alkanes and Cyclic Peptides. J. Am. Chem. Soc. 1996, 118, 5011–5019. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem. 1996, 490–519. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, F.C.; Lenz, M.; Jose, J.; Kaiser, M.; Brun, R.; Schmidt, T.J. In Silico Identification and in Vitro Activity of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde-3-phosphate-dehydrogenase. Molecules 2015, 20, 16154-16169. https://doi.org/10.3390/molecules200916154
Herrmann FC, Lenz M, Jose J, Kaiser M, Brun R, Schmidt TJ. In Silico Identification and in Vitro Activity of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde-3-phosphate-dehydrogenase. Molecules. 2015; 20(9):16154-16169. https://doi.org/10.3390/molecules200916154
Chicago/Turabian StyleHerrmann, Fabian C., Mairin Lenz, Joachim Jose, Marcel Kaiser, Reto Brun, and Thomas J. Schmidt. 2015. "In Silico Identification and in Vitro Activity of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde-3-phosphate-dehydrogenase" Molecules 20, no. 9: 16154-16169. https://doi.org/10.3390/molecules200916154
APA StyleHerrmann, F. C., Lenz, M., Jose, J., Kaiser, M., Brun, R., & Schmidt, T. J. (2015). In Silico Identification and in Vitro Activity of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde-3-phosphate-dehydrogenase. Molecules, 20(9), 16154-16169. https://doi.org/10.3390/molecules200916154