2. Results and Discussion
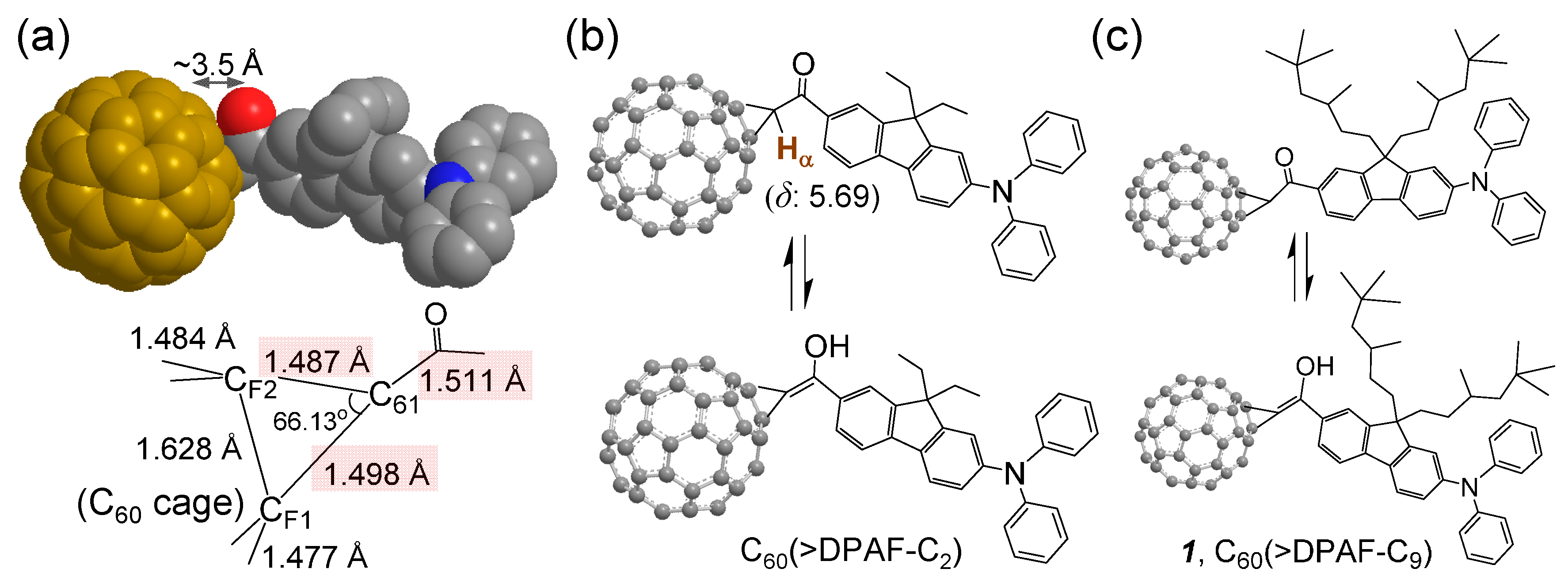
The C
60 cage is highly electronegative, a feature associated with its strongly electron-accepting ability. In the presence of a covalently bound electron-donating chromophore, such as 9,9-di(3,5,5-trimethylhexyl)-2-diphenylaminofluorene (DPAF-C
9), high molecular e
‒-polarizability characteristics can be achieved in the conjugate upon photoexcitation [
16]. The phenomena arise from intramolecular charge-transfer event among e
‒-donating DPAF-C
9 moieties and the e
‒-accepting C
60, leading to the formation of a negatively charged (C
60>)
−· cage and positively charged (DPAF)
+·-C
9. In our recent studies of femtosecond transient absorption measurements, we have synthesized and presented an approach of C
60/or C
70-(antenna)
x type fullerene-chromophore dyads [
14,
15], triads, and pentaads [
17,
18] using similar C
60/or C
70(A)–
keto–DPAF-C
9(D) assembly structures. This A‒D design as that of C
60(>DPAF-C
2) (
Figure 1) takes the advantage of both C
60> and DPAF-C
9 being active in linear and nonlinear photonic processes [
14]. Its photoresponsive activity was verified by intramolecular energy/or electron-transfer processes and found to occur in an ultrafast rate of <130 fs [
15]. In PhCN, electron-transfer process was found to dominate photophysical events giving the charge-separated state C
60−·(>DPAF
+·-C
n)
1or2 in a long life-time [
17]. A closely linked molecular nanostructure with periconjugation between C
60 and the carbonyl moiety of DPAF-C
n in a spacing distance of only ~3.5 Å [
15], as shown in
Figure 1a, and the concurrence of keto-enol tautomerism at the conjugation bridge in
Figure 1b were reasoned for the observed ultrafast photoresponsive rate. The latter leads to the creation of partial conjugation between C
60 and covalently linked DPAF-C
n separated by only one carbon atom (C
61). This makes the photoinduced intramolecular electron-transfer very efficient and ultrafast.
Figure 1.
Bridging structure of a C
60-DPAF-C
2 conjugate with (
a) carbon-carbon bond lengths at the C
60 cage (C
F1 and C
F2) bridging unit indicated showing a shorter length of C
61‒C(=O) than a normal single C‒C bond, as determined by X-ray single crystal structural analysis reported previously [
15]; (
b,
c) Keto-enol tautomerism of the bridging unit structure showing a large downfielded chemical shift of
α-proton H
α from the normal α-proton chemical shift of δ 2.1‒2.3. This downfield shift is also caused partly by the fullerenyl ring current.
Figure 1.
Bridging structure of a C
60-DPAF-C
2 conjugate with (
a) carbon-carbon bond lengths at the C
60 cage (C
F1 and C
F2) bridging unit indicated showing a shorter length of C
61‒C(=O) than a normal single C‒C bond, as determined by X-ray single crystal structural analysis reported previously [
15]; (
b,
c) Keto-enol tautomerism of the bridging unit structure showing a large downfielded chemical shift of
α-proton H
α from the normal α-proton chemical shift of δ 2.1‒2.3. This downfield shift is also caused partly by the fullerenyl ring current.
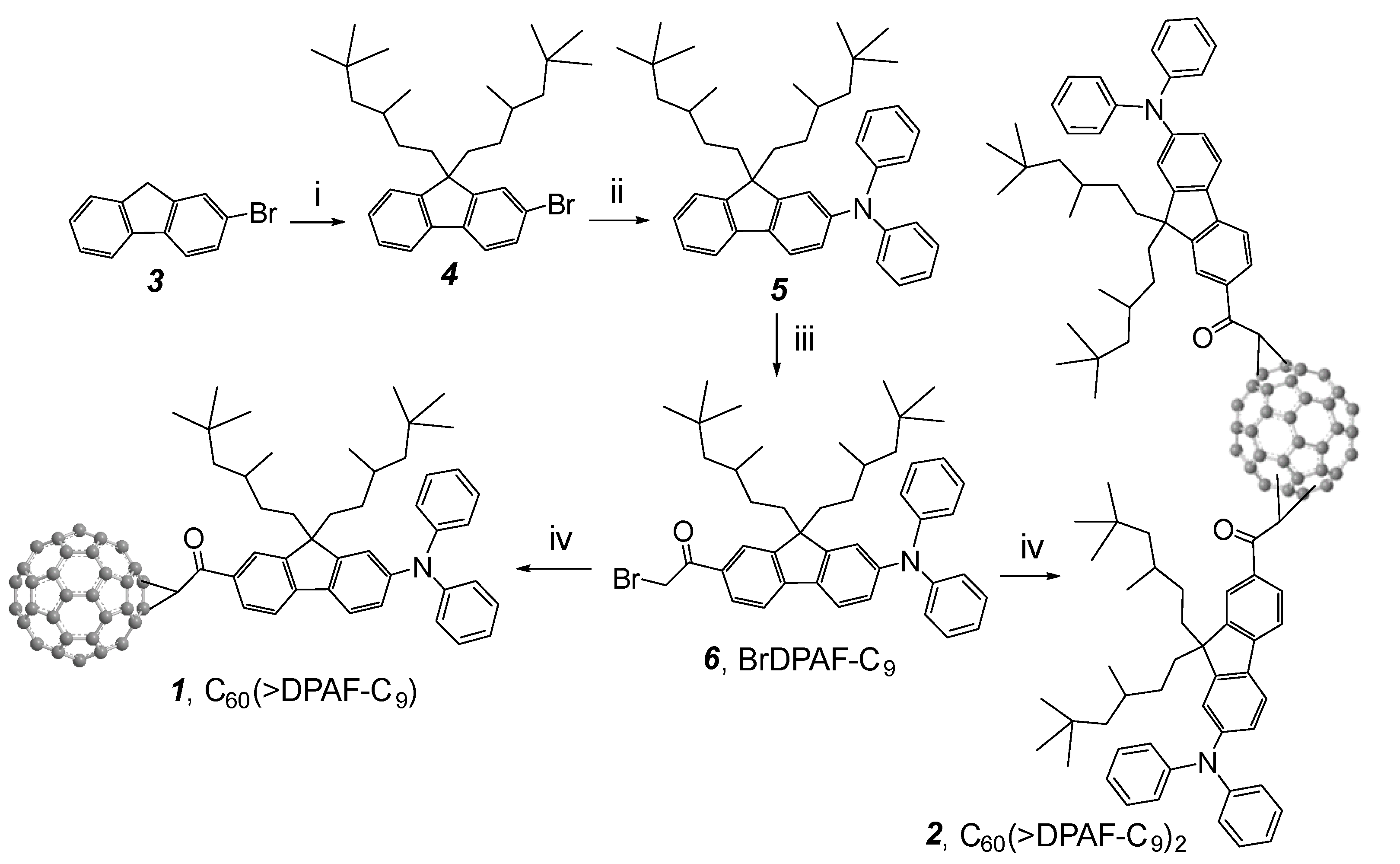
Synthesis of electronically polarizable push-pull conjugative structures as methano[60]ullerene dyad C
60(>DPAF-C
9)
1 and triads C
60(>DPAF-C
9)
2 2 was performed by using modified synthetic procedures reported recently [
15], as outlined in
Scheme 1 with the reagent and conditions indicated. The key precursor intermediate BrDPAF-C
9 6 was synthesized from 9,9-di(3,5,5-trimethylhexyl)-2-diphenylaminofluorene (
5) with α
-bromoacetyl bromide in the presence of Lewis acid for a period of 4.0 h. Subsequent fullerene condensation reaction was carried out by the treatment of
6 with C
60 using DBU as a base to yield both
1 and
2 in a yield of 70 and 14%, respectively, after chromatographic purification on SiO
2.
Scheme 1.
Synthesis of C
60-(antenna)
x nanostructures
1 (x = 1) and
2 (x = 2) [
14]. Reagents and reaction conditions: (i) 1-C
9H
37-OMs,
t-BuOK in THF, 0 °C‒r.t., 4 h; (ii) diphenylamine, tris(dibenzylideneacetone)dipalladium(0) (cat.),
rac-BINAP (cat.),
t-BuONa, toluene, 110 °C, 8.0 h; (iii) α
-bromoacetyl bromide, AlCl
3, ClCH
2CH
2Cl, 0 °C, 4.0 h; (iv) C
60, DBU, toluene, r.t., 5.0 h.
Scheme 1.
Synthesis of C
60-(antenna)
x nanostructures
1 (x = 1) and
2 (x = 2) [
14]. Reagents and reaction conditions: (i) 1-C
9H
37-OMs,
t-BuOK in THF, 0 °C‒r.t., 4 h; (ii) diphenylamine, tris(dibenzylideneacetone)dipalladium(0) (cat.),
rac-BINAP (cat.),
t-BuONa, toluene, 110 °C, 8.0 h; (iii) α
-bromoacetyl bromide, AlCl
3, ClCH
2CH
2Cl, 0 °C, 4.0 h; (iv) C
60, DBU, toluene, r.t., 5.0 h.
Spectroscopic characterization of monoadduct 1 and bisadduct 2 was made by their positive ion fast atom bombardment mass spectrum (FAB+‒MS) showing clearly the molecular ion mass at m/z 1345/1346 (M+/MH+) and 1971 (M+) for 1 and 2, respectively. Both spectrum displayed a fragmentation peak at m/z 720/721 corresponding to the ion mass of C60 cage with no major detectable intermediate fragments indicating the main bond breakage occurring at the cyclopropanyl carbon conjunction to C60 cage.
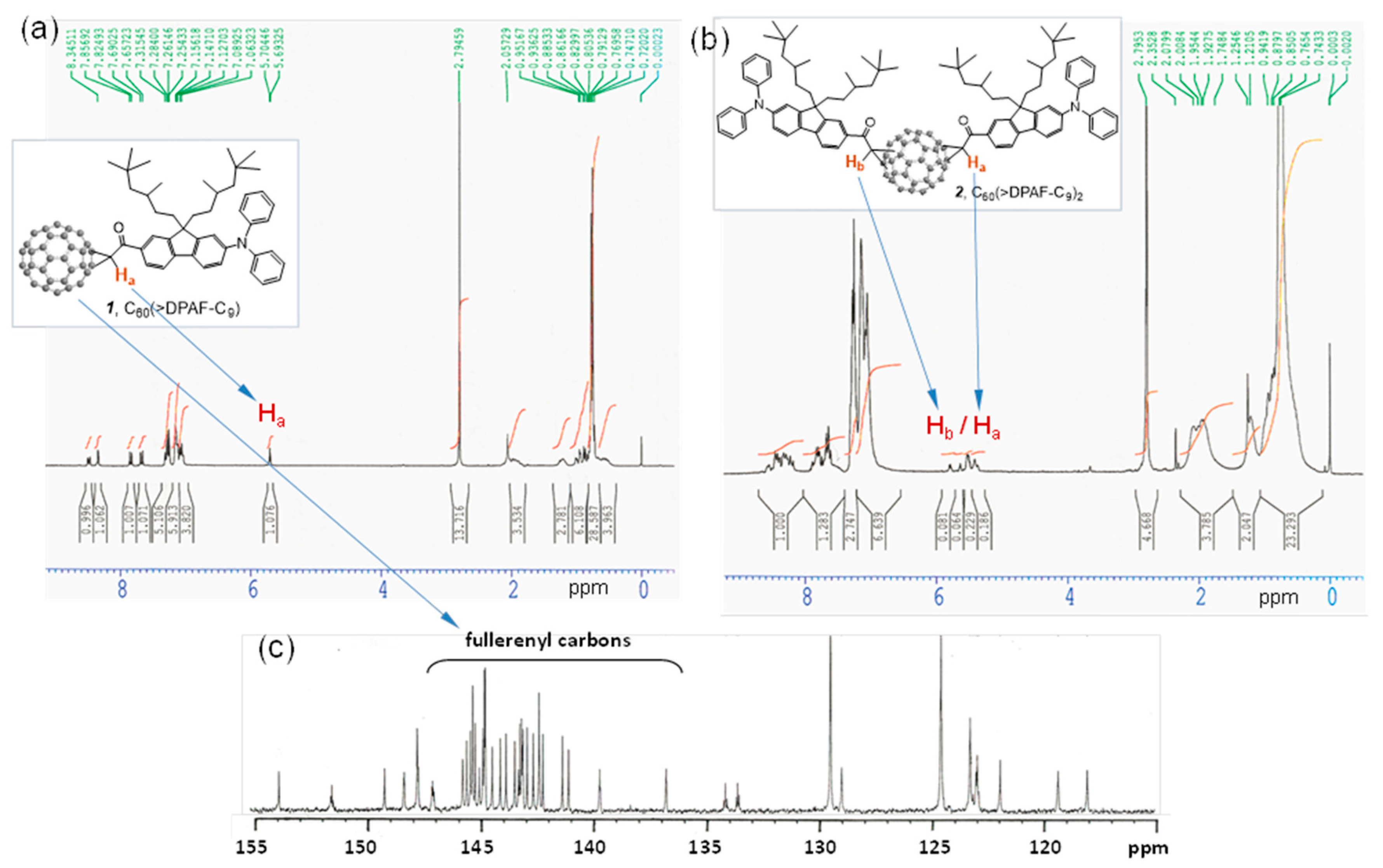
We assigned all major proton peaks in
1H-NMR spectrum of
1 and
2. Since the compound
1 is a single component, its spectrum can be used as a reference to assist chemical shift assignments of
2. The α-proton (H
a next to the carbonyl group) peak of
1 appeared as a triplet (via through space coupling with two fluorenyl protons in vicinity) at δ 5.69 (
Figure 2a) with a large downfield shift of roughly 1.2 ppm from that of
6. It was also accompanied with downfield shift of all fluorenyl phenyl protons to 7.83 (d,
J = 8 Hz), 8.48 (d,
J = 8 Hz), and 8.34 (s). In
Figure 2b, we assigned the multiplet peaks at δ 7.7‒7.9 (2H), 8.2‒8.35 (2H), and 8.35‒8.6 (2H) to the chemical shift of three types of fluorenyl protons. There are two groups of proton peaks at δ 5.35‒5.6 and 5.6‒5.85 in a different integration intensity accounted for a total of two types of α-protons each with H
a and H
b in a slightly different chemical shift value. These revealed only two regioisomers possible for the bisadduct
2 in a molecular ratio of roughly 1:2.8 in a chromatographically non-separable fraction due to close similarity of the molecular polarity between these two regioisomers. Highly steric hindrance of DPAF-C
9 antenna was reasoned for the isolation of a very limit number of regioisomers.
Figure 2.
1H-NMR spectra of (a) the monoadduct C60(>DPAF-C9) 1 and (b) the bisadduct C60(>DPAF-C9)2 2. Chemical shift of the Ha proton of 1 was used to assign Ha and Hb protons of 2 that revealed only one major and one minor regioisomer existing in the chromatographically purified sample; (c) 13C-NMR spectrum of 1 indicating a C2 molecular symmetry with a total of 29 fullerenyl sp2 carbons.
Figure 2.
1H-NMR spectra of (a) the monoadduct C60(>DPAF-C9) 1 and (b) the bisadduct C60(>DPAF-C9)2 2. Chemical shift of the Ha proton of 1 was used to assign Ha and Hb protons of 2 that revealed only one major and one minor regioisomer existing in the chromatographically purified sample; (c) 13C-NMR spectrum of 1 indicating a C2 molecular symmetry with a total of 29 fullerenyl sp2 carbons.
Preparation of
1 or
2-encapsulated γ-FeO
x@AuNP trilayered core-shell nanoparticles, (γ-FeO
x@AuNP)@[C
60(>DPAF-C
9)
x]
n 3–
1 (x = 1) or
3–
2 (x = 2) was made first by the synthesis of magnetic γ-FeO
x nanoparticles using either the method A1 or A2 described in the experimental section. The latter method carried out at 320 °C gave monodisperse nanocrystals. The former method performed at 150 °C resulted in smaller NPs, as shown in
Figure 3a. Two methods B1 and B2, as described in the experimental section, were applied for deposition of the gold layer. The latter method involves dissolution of iron oxide NPs by (CH
3)
4N
+OH
‒ in H
2O, followed by the attachment of sodium citrate upon stirring for the reduction of hydrogen tetrachloroaurate(III) trihydrate (HAuCl
4∙3H
2O) along with hydroxylamine hydrochloride. Since a magnetic core NP was used as the substrate for providing a magnetic field to interact with electromagnetic waves, all coated core-shell γ-FeO
x@AuNPs can be removed from solution easily by an external magnet and purified. Surface of the resulting gold layer was stabilized by the addition of 1-octanethiol in toluene to furnish the products of core-shell Au-nanoparticles-coated γ-FeO
x NPs. Sufficient binding of 1-octanethiol as a capping agent rendered moderate solubility of NPs in organic solvents ready for encapsulation by
1 and
2 under ultrasonication. The resulting relatively pure trilayered core-shell nanoparticles can be retrieved easily from the washing solution (ethanol and ether) by an external permanent magnet. The procedure provided separation of core-shell nanoparticles from the residual capping agent and an excessive amount of C
60(>DPAF-C
9) in solution. The binding force of
1 and
2 to the nanoparticle surface was controlled by the strong hydrophobic‒hydrophobic interaction forces of C
60‒C
60 cages that resulted in molecular self-assembly of C
60(>DPAF-C
9) to form a partial bilayer or bilayer configuration at the surface, resembling that found on the formation of bilayered
fullerosome nanovesicles of water-soluble C
60(>DPAF-EG
x) reported recently [
19]. Accordingly, initial interactions of
1 at the nanoparticle surface was designated to occur on two 3,5,5-trimethylhexyl moieties owing to low solubility of C
60> in alkanes that should push fullerene cages outward at the solvent interfacial area. The sonication treatment provides sonochemical energy to invert
1 with the favor of C
60 cage in contact with Au NPs that gives the monolayer capping of the surface by C
60(>DPAF-C
9) molecules [
20]. They become accessible for the binding of the second layer of
1 in the solution based on strong C
60‒C
60 cage and C
60‒DPAF aromatic-ring attraction forces. This is proposed to be the formation mechanism of a partial bilayered or bilayered fullerosome membrane covering γ-FeO
x@AuNP nanoparticles as the nanostructure configuration of (γ-FeO
x@AuNP)@[C
60(>DPAF-C
9)
x]
n 3.
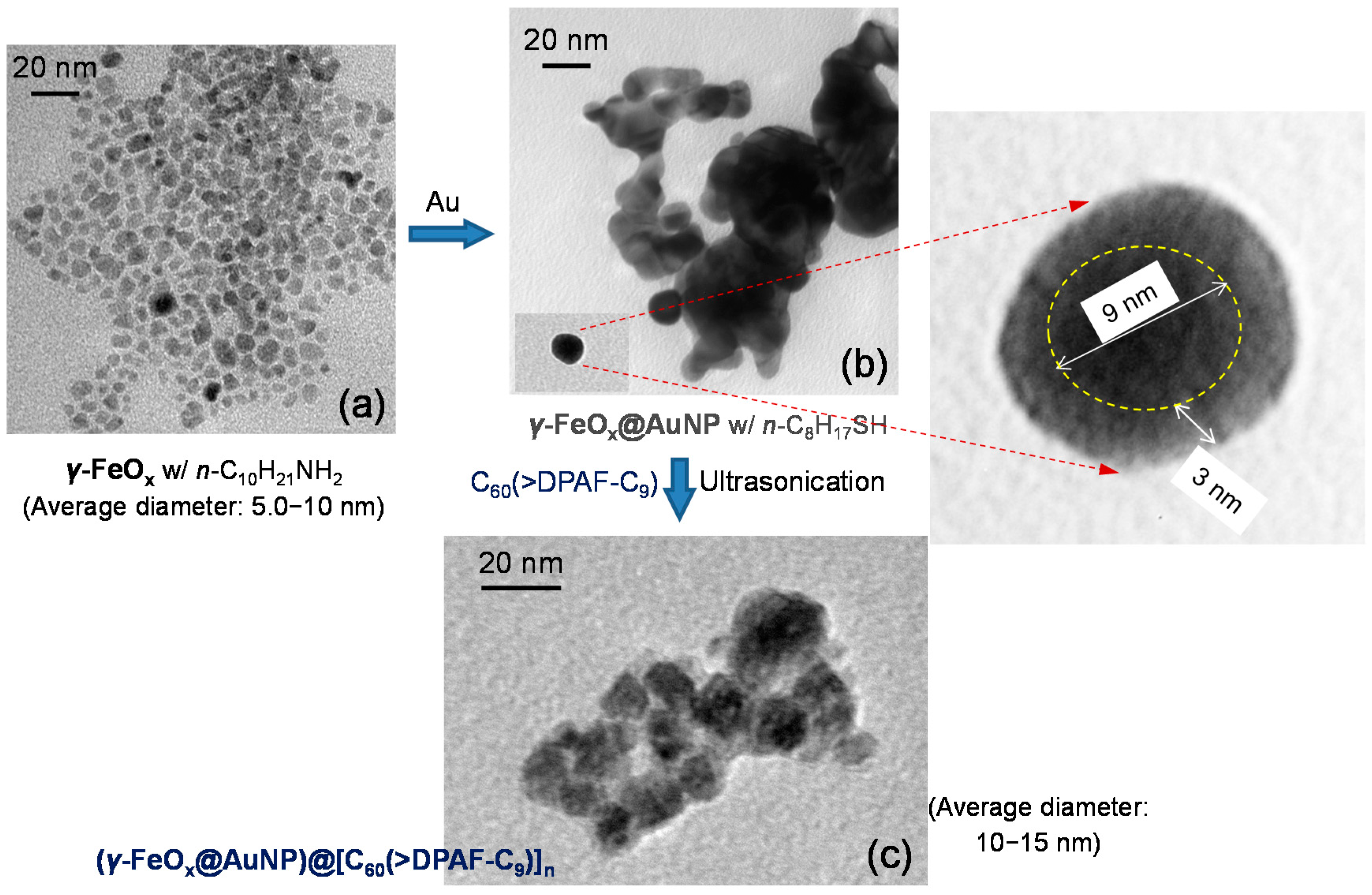
Figure 3.
TEM micrographs of (a) γ-FeOx (prepared by the method A1); (b) γ-FeOx@AuNP, and (c) C60(>DPAF-C9)-encapsulated γ-FeOx@AuNP nanoparticles 3–1 showing evolution of particle morphology changes.
Figure 3.
TEM micrographs of (a) γ-FeOx (prepared by the method A1); (b) γ-FeOx@AuNP, and (c) C60(>DPAF-C9)-encapsulated γ-FeOx@AuNP nanoparticles 3–1 showing evolution of particle morphology changes.
Morphology and topography of all NPs was investigated by tunneling electron microscopic (TEM) micrographs. As a result, the average size of the parent γ-FeO
x nanoparticles was measured to be small as 5–10 nm in diameter (
Figure 3a) in a narrow distribution. Upon deposition of a gold layer, only a slight layer contrast difference between Au and FeO
x was observed in
Figure 3b that is still possible to differentiate a darker core iron oxide particle diameter of ~9 nm with a slightly lighter gold shell thickness of ~3.0 nm (inset of
Figure 3b). Further coating of
1 at the outer shell led to encapsulation of many nanoparticles by a layer of organic substance in light soft amorphous image (
Figure 3c) that was measured in a shell thickness of few nm. This thickness corresponds well to a proposed partial bilayered or bilayered packing configuration of molecules
1 on the surface of γ-FeO
x@AuNP. It is plausible to assume the strong attraction force between the C
60> cage and AuNP in directing the initial orientation of molecules
1 having the fullerene cage directly associated with interfacial binding on Au surface that resulted in the dialkylfluorene ring moiety facing outward to the solution media. Subsequent packing of more molecules of
1 on the layer of DPAF-C
9 is expectable to form a layer of fullerosome membrane owing to the stronger hydrophobic–hydrophobic interaction forces of (C
60>)–(C
60>) than (C
60>)–(DPAF-C
9). By using the MM2 energy minimization technique to simulate the 3D molecular configuration with the assumption of a C
60 cage diameter width of 1.0 nm, we estimated the molecular long-axis length of
1 in roughly 3.5 nm. This fits well with the sum of an observed bilayer packing thickness of few nm, consisting of a head-to-head orientation of
1 in the fullerosome membrane region.
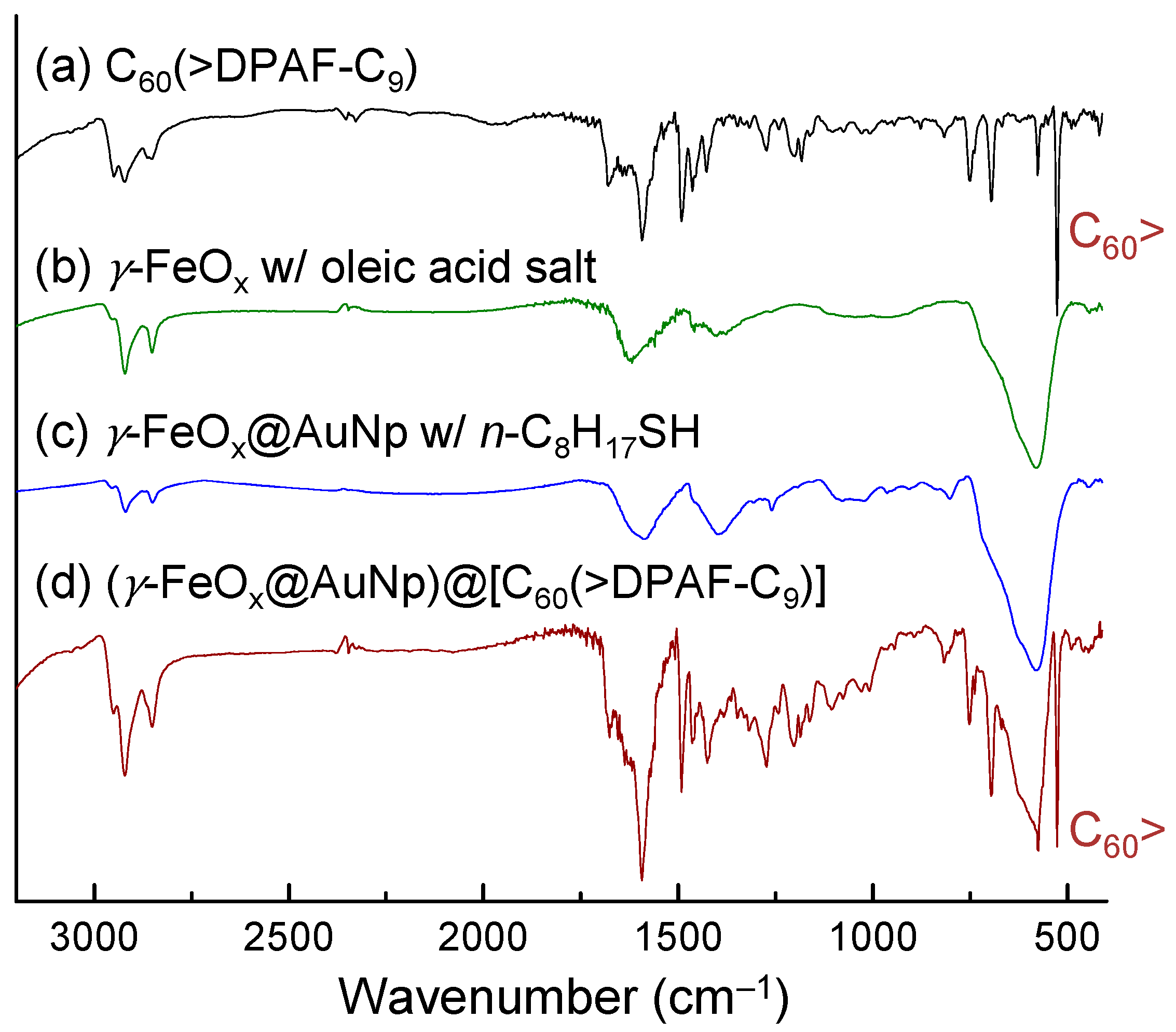
Successful construction of core-shell nanomaterials was verified by their infrared spectra of purified samples. Oleic acid sodium salt stabilized γ-FeO
x NPs (
Figure 4b) displayed three medium to weak bands at 1618 (s), 1459 (w), and 1407 (m) cm
‒1 along with the main Fe-O absorption band centered at 578 (vs) cm
‒1. Incorporation of an Au layer on top does not change much on the main peak of spectrum in
Figure 4c. Deposition of the fullerosome layer of
1 on the outer layer obviously incurred major absorption band changes in
Figure 4d to show carbonyl (C=O) bands at 1741 (m), 1675 (m), and 1637 (m) cm
‒1 that revealed various bonding interactions of at least two moieties, one from oleic acid and the other carbonyl group in solid state of
1. Olefin (C=C) absorption band of
1 centered at 1591 cm
‒1 can be detected very clearly. The most indicative strong sharp band at 526 cm
‒1 of
Figure 4d can be assigned to the half-cage absorption of C
60> that is consistent with that of
1 in
Figure 4a.
Figure 4.
Infrared absorption spectra of four samples indicated, showing a consecutive sum of absorption bands on final product 3–1 in (d).
Figure 4.
Infrared absorption spectra of four samples indicated, showing a consecutive sum of absorption bands on final product 3–1 in (d).
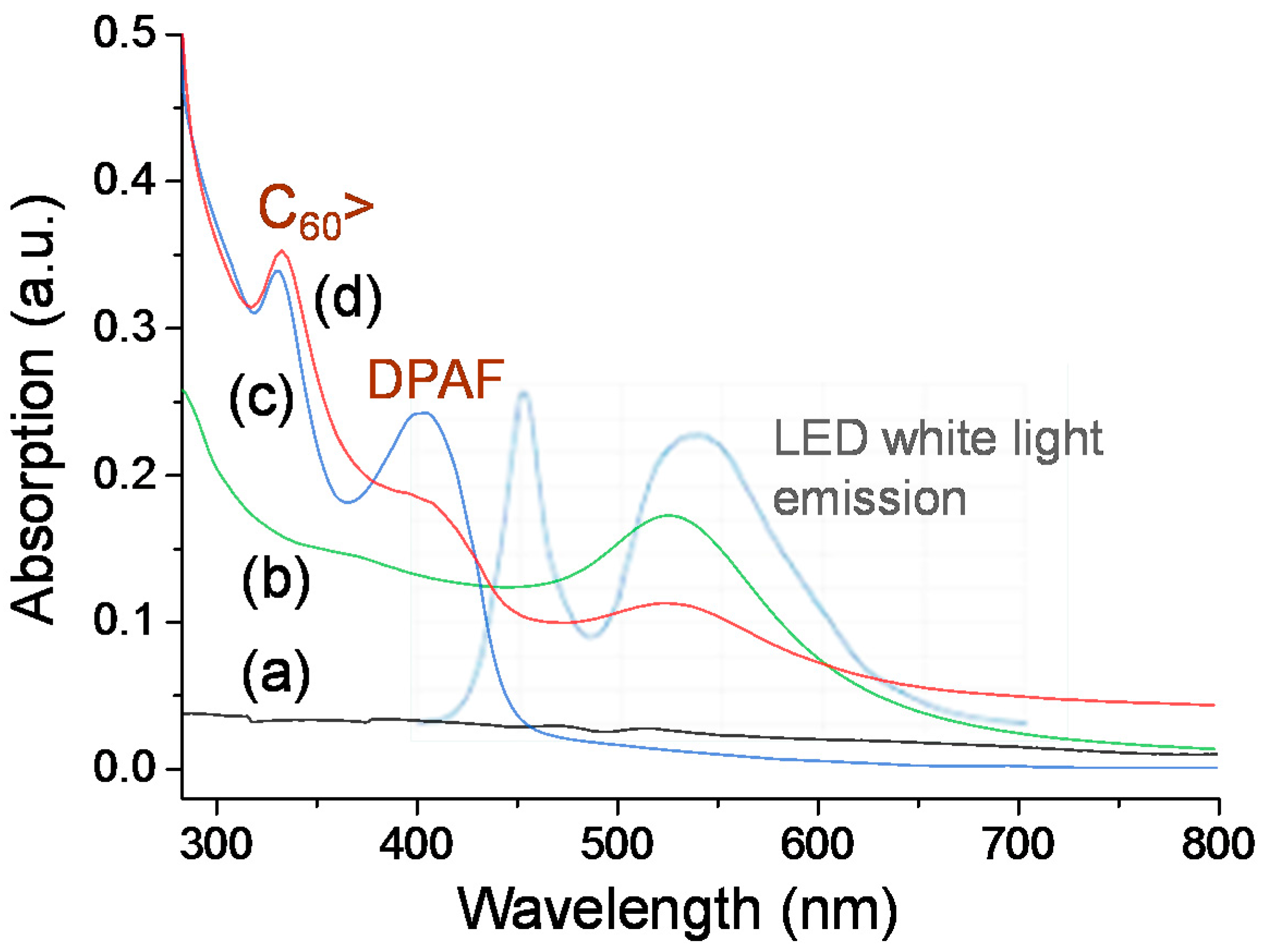
Optical absorption of the Au nanoparticle subshell layer is crucial for this study to allow us executing and introducing photoinduced surface plasmon resonance energy generation at UV to visible light wavelengths (300‒550 nm), followed by subsequent near-field SPR energy-transfer from the inner layer to the outer C
60(>DPAF-C
9)
x-derived fullerosome layer. This is in addition to the ability of
1 or
2 to perform photoinduced intramolecular charge-separation themselves to the corresponding transient C
60−·(>DPAF
+·-C
9) state for
1 and C
60−·(>DPAF
+·-C
9)(>DPAF-C
9) or C
60−2(>DPAF
+·-C
9)
2 states for
2 at the same wavelength range covering roughly 250‒380 nm and 350‒430 nm corresponding to absorptions of C
60> and DPAF-C
9 moieties, respectively. These absorption bands remain unchanged during the fabrication procedure of
3–
1, as shown in comparison between
Figure 5c,d). The broad absorption band (
Figure 5b) of the Au layer covers the most of the spectrum wavelength range at 280–630 nm with the extinction coefficient higher than that of γ-FeO
x (
Figure 5a).
Figure 5.
UV-vis absorption of (a) γ-FeOx NPs stabilized by n-C10H21NH2; (b) γ-FeOx@AuNP stabilized by n-C8H17SH; (c) C60(>DPAF-C9) 1 in CHCl3; and (d) 3–1 in CHCl3‒THF (1:5) with their absorption correlation to emission bands (light blue) of white LED light used.
Figure 5.
UV-vis absorption of (a) γ-FeOx NPs stabilized by n-C10H21NH2; (b) γ-FeOx@AuNP stabilized by n-C8H17SH; (c) C60(>DPAF-C9) 1 in CHCl3; and (d) 3–1 in CHCl3‒THF (1:5) with their absorption correlation to emission bands (light blue) of white LED light used.
Frequency-dependent dielectric and permittivity measurements of
3–
1 and
3–
2 were carried out using an open-ended coaxial probe and a network analyzer in the range of 0.5‒4.5 GHz to study properties including the relative dielectric constant (ε
r′, the real part of complex permittivity) and the relative dielectric loss factor (ε
r′′, the imaginary part of complex permittivity). It was conducted inside a custom-built chamber to create a circumferentially uniform illumination environment. Inner chamber temperature was maintained and controlled using several fans. Illumination periods were chosen to be 10, 30, and 60 min. The objective of these measurements was to investigate the dielectric amplification of nanoparticles in various core-shell layered configurations in response to external photoinduced generation of SPR effects within the gold-shell layer. We selected a LED white light with the output power of 2.0 W and a UVB-enhanced visible lamp with the output power density of 20 mW/cm
2 for the study. With the use of a white light, the optical absorption should occur mainly on the gold layer since the absorption peak maximum (λ
max) of C
60> cage and DPAF-C
9 moieties are centered at 326 and 410 nm, respectively.
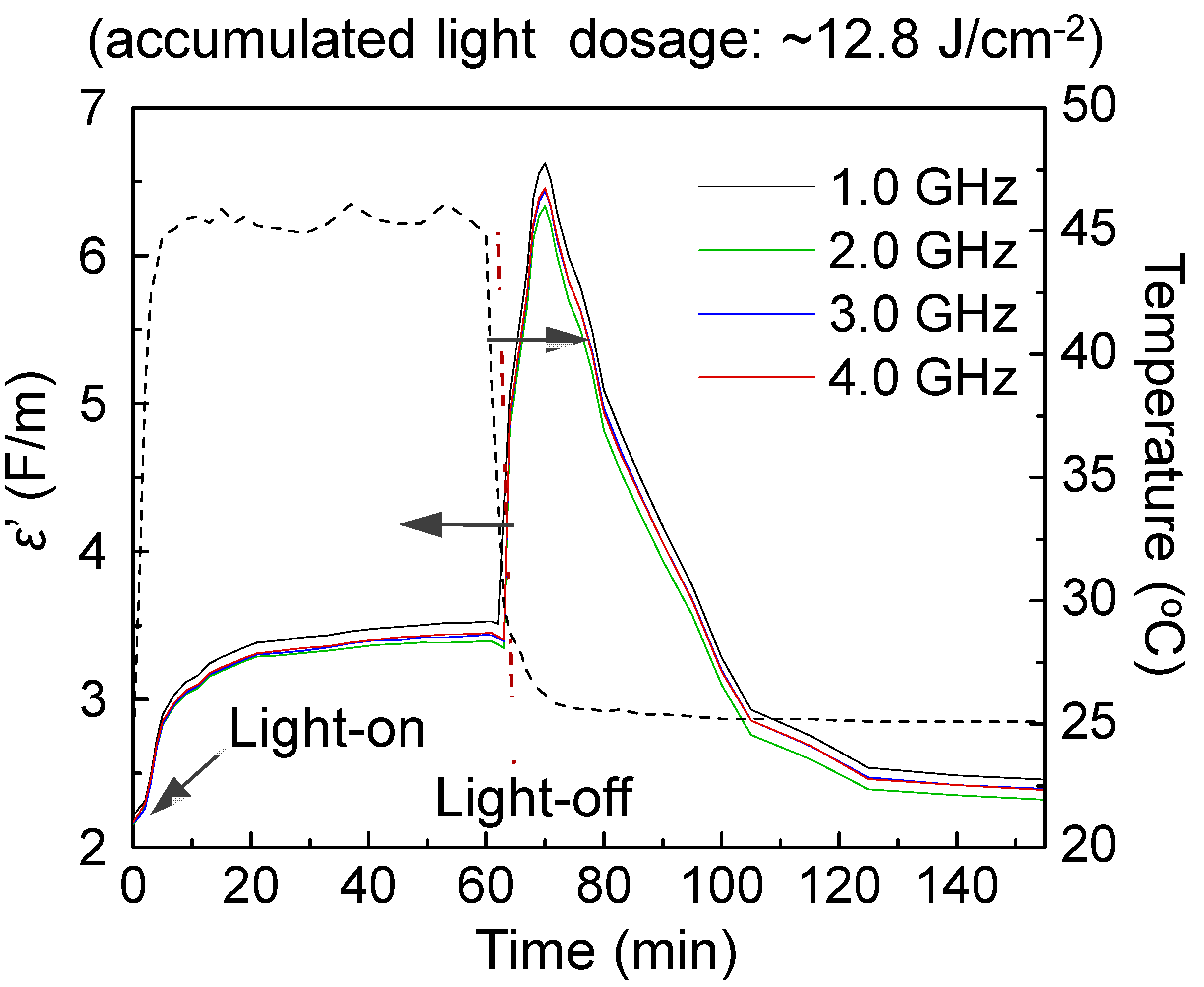
Figure 6 shows relative dielectric constant (ε
r′) curves of
3–
1, as an example, with the data taken at the frequency of 1.0‒4.0 GHz during and after 60 min illumination. The inner chamber temperature was monitored constantly during the irradiation period at the sample tube wall. In all experiments, white light illumination led to a slight rise in temperature to 39‒52 °C in the first 10 min. Increase of chamber temperatures from 25 °C resulted in corresponding small increases of the ε
r′ value of samples in a different extent, as shown in
Figure 6. The value remains relatively constant during further irradiation from 10 to 60 min. In separate similar experiments using three reference samples of
1 alone, γ-FeO
x NPs, and core-shell γ-FeO
x@[C
60(>DPAF-C
9)]
n NPs without the Au layer, no detectable large change of ε
r′ values was observed during or after the irradiation period. Therefore, observed large ε
r′ changes with the core-shell sample
3–
1 can be correlated to the ε
r′ value increase of C
60(>DPAF-C
9)-derived fullerosome membrane, as a result of photoinduced plasmon-energy generation in the gold layer.
The sharp increase of permittivity can be accounted by a ratio of εmax′/εo′ (εo′ = 2.25 at 1.0 GHz and time zero) in a roughly 2.89-fold or 1.91-fold (εmax′/εr′, based on εr′ = 3.4 at 60 min) at the epak maximum reached at roughly at the time of 70 min (10 min after the start of light-off state). This is considered to be a novel switch-on permittivity phenomenon that we have observed to date. We hypothesized that the release of SPR energy from the intermediate gold layer sandwiched between the layers of fullerosome membrane and core γ-FeOx Np caused activation of the molecular polarization of C60(>DPAF-C9)x molecules to its transient charge-separate (CS) state. This CS state consists of a positively charged electron-donor moiety (DPAF)+∙-C9 and a negatively charged electron-acceptor moiety (C60>)‒ -. This is in a large contrast to no clear change of εr′ values of core-shell nanoparticles without incorporation of an Au-layer at the light switch-off stage. Subsequently, the amplified εr′ underwent a relaxation process back to the initial values within the next 20 min, revealing good recyclability and photoswitchability during light-on and light-off cycles.
Figure 6.
Time-dependent relative dielectric constant (εr′) profiles of trilayered sample 3–1 at the frequency range of 1.0‒4.0 GHz under illumination using a LED white light, showing large photoswitchable amplification of εr′ at the light-off state.
Figure 6.
Time-dependent relative dielectric constant (εr′) profiles of trilayered sample 3–1 at the frequency range of 1.0‒4.0 GHz under illumination using a LED white light, showing large photoswitchable amplification of εr′ at the light-off state.
3. Experimental Section
3.1. Materials
Reagents and solvents: aluminum chloride, 1,2-dichloroethane, α-bromoacetyl bromide, sodium sulfate, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), FeCl3·6H2O, FeCl2·4H2O, sodium acetate, n-octylamine, 1,2-hexadecanediol, 1-oleylamine, 1,2-propanediol, sodium oleate, 1-oleic acid, 1-octadecene, Au(ac)3, hydrogen tetrachloroaurate(III) trihydrate (HAuCl4∙3H2O), phenyl ether, tetramethylammonium hydroxide, 1-octanethiol, and hydroxylamine hydrochloride were purchased from Aldrich Chemicals (St. Louis, MO, USA) and used without further purification. All other chemicals were purchased from Acros Chemicals Ltd. (New Brunswick, NJ, USA). The anhydrous grade solvent of THF was refluxed over sodium and benzophenone overnight and distilled under reduced pressure (10−1 mmHg).
3.2. Spectroscopic Measurements
1H-NMR and 13C-NMR spectra were recorded on either an Avance Spectrospin–200 or AC-300 spectrometer (Bruker, Billerica, MA, USA). UV-vis spectra were recorded on a U-3410 UV spectrometer (Hitachi, Chiyoda, Tokyo, Japan). Infrared spectra were recorded as KBr pellets on a 750 series FT-IR spectrometer (Thermo Scientific Nicolet, Waltham, MA, USA). Photoluminescence (PL) spectra were measured using PTI Fluorescence Master Systems (Photon Technology International, Edison, NJ, USA) connected with a photomultiplier (914 Photomultiplier Detection System) with a xenon short arc lamp as the excitation source. Mass spectroscopic measurements were performed by the use of positive ion matrix-assisted laser desorption ionization (MALDI–TOF) technique on a M@LDI-LR mass spectrometer (Micromass, Cary, NC, USA). The sample blended or dissolved in the matrix material was irradiated by nitrogen UV laser at 337 nm with 10 Hz pulses under high vacuum. Mass ion peaks were identified for the spectrum using the MassLynx v4.0 software. In a typical experiment, the samples of 5, 6, and 7, were dissolved in CHCl3 in a concentration of 1.0 mg/mL. The matrix of 3,5-dimethoxy-4-hydroxycinnamic acid (sinapic acid) was dissolved in THF in a concentration of 10 mg/mL. The solution of matrix (1.0 mL) was taken and mixed with the sample solution (0.1 mL) prior to the deposition on a stainless-steel MALDI target probe. It was subsequently dried at ambient temperature.
3.3. Synthetic Procedures
Synthesis of 7-α-Bromoacetyl-9,9-di(3,5,5-trimethylhexyl)-2-diphenylaminofluorene BrDPAF-C
9 (
6). A similar method as that of a recently reported procedure was used [
14].
Synthesis of 7-(1,2-Dihydro-1,2-methanofullerene[60]-61-carbonyl)-9,9-di(3,5,5-trimethylhexyl)-2-diphenylaminofluorene, Monoadduct C
60(>DPAF-C
9) (
1) and Bisadduct, C
60[methanocarbonyl-7-(9,9-di(3,5,5-trimethylhexyl)-2-diphenylaminofluorene)]
2, C
60(>DPAF-C
9)
2 (
2). A similar method as that of a recently reported procedure was used [
14]. Spectroscopic data of
2: FAB
+‒MS calcd for
12C
1501H
11014N
216O
2 m/
z 1970.8; found,
m/
z 1971; MALDI−MS (TOF) found,
m/
z 1974, 1973, 1972, 1971 (M
+), 1347, 1346, 870, 861, 710, 705, 672, 666, 630, 629, 612, and 583; UV-vis (CHCl
3, 2.0 × 10
‒5 M) λ
max (ε) 255 (1.9 × 10
5), 310 (1.1 × 10
5), and 406 nm (8.5 × 10
4 L/mol-cm); FT-IR (KBr)
νmax 3429 (br), 3163, 2951 (s), 2922, 2863, 1680, 1595 (vs), 1492 (vs), 1465, 1420, 1400, 1316(w), 1277, 1200, 1156 (w), 1093, 1030 (w), 816 (w), 753, 670 (s), and 527 (s) cm
‒1;
1H-NMR (500 MHz, CDCl
3, ppm) δ 8.5−8.1 (m, 4H), 7.9−7.4 (m, 4H), 7.24 (m, 12H), 7.12 (m, 8H), 7.05 (m, 4H), 5.8−5.3 (m, 2H), 2.1−1.8 (m, 8H), 1.35−1.15 (m, 8H), and 1.1−0.5 (m, 60H).
13C-NMR (500 MHz, CDCl
3, ppm) δ 189.8, 154.1, 151.8, 151.7, 149.4, 148.1, 148.0, 147.7, 147.5, 146.9, 146.8, 146.6, 146.2, 146.0, 145.9, 145.7, 145.5, 145.0, 145.0, 144.9, 144.8, 144.7, 144.5, 144.3, 144.0, 143.9, 143.7, 143.6, 143.3, 143.3, 143.1, 143.0, 143.0, 142.9, 142.6, 142.5, 142.3, 142.3, 142.2, 142.1, 140.9, 140.5, 140.0, 139.8, 139.7, 139.5, 134.5, 134.0, 129.8, 129.5, 129.3, 128.65, 124.9, 123.6, 122.3, 119.6, 118.5, 73.4, 73.2, 55.6, 51.3, 51.2, 44.4, 44.4, 42.9, 38.2, 38.2, 38.1, 33.6, 33.5, 33.5, 31.4, 30.4, 29.9, 27.7, 23.1, and 22.9.
3.3.1. Synthesis of γ-FeOx (1.0 < x < 1.5) Nanoparticles
Method A1. In a typical reaction, a mixture of FeCl3·6H2O (2.70 g, 0.010 mol), FeCl2·4H2O (1.98 g, 0.010 mol), sodium acetate (4.92 g, 0.060 mol, as a hydrolyzing agent), H2O (4.0 mL), and n-octylamine (7.3 mL, as a capping agent) in 1,2-propanediol (66 mL) were prepared and stirred under reflux at 150 °C for a period of 5.0 h. It was followed by precipitation upon the addition of 2-propanol to afford the product in a nearly quantitative yield. Spectroscopic data: FT-IR (KBr) νmax 2970 (w), 2924 (s), 2852 (m), 1743 (w), 1630 (s), 1458 (m), 1383 (m), 1134 (m), 1041 (m), 922 (w), 879 (w), 839 (w), 627 (vs), and 584 (vs) cm‒1.
Method A2. Iron oleate was firstly prepared by heating iron chloride, FeCl3·6H2O (5.4 g, 20 mmol) and FeCl2·4H2O (2.0 g, 10 mmol), and sodium oleate (36.5 g, 120 mmol) at 70 °C in a mixture solvent, composed of ethanol (80 mL), distilled water (60 mL), and hexane (140 mL), for a period of 4.0 h. At the end of reaction, the upper organic layer containing the iron oleate complex was washed three times with distilled water (30 mL each), dried over sodium sulfate, and solvent evaporated off to yield iron oleate complex in a waxy solid form. To this resulting organometallic complex, 1-oleic acid (4.3 g, 15 mmol) and 1-octadecene (200 g, act as the solvent) were added at room temperature and refluxed at 320 °C for 1.5 h. Upon the temperature reaching 320 °C, a severe exothermic reaction occurred with the initial transparent solution turning to turbid and brownish black. The product mixture was cooled to room temperature, followed by precipitation with ethanol (500 mL). Resulting nanoparticles were collected using a permanent magnet, washed twice with both 2-propanol (100 mL) and ethanol (100 mL). They were re-dispersed in nonpolar solvent, such as hexane and toluene, via ultrasonication. In general, dark solid particles of γ-FeOx (Fe3O4) obtained by this method were found to be highly magnetic in response to the external magnet and soluble in many nonpolar organic solvents. Distribution of the particle size is fairly homogenous and was measured via TEM as ~20 nm in diameter on average. Spectroscopic data: FT-IR (KBr) νmax 2960 (w), 2921 (s), 2852 (m), 1618 (s), 1459 (w), 1407 (m), and 578 (vs) cm‒1.
3.3.2. Synthesis of Au-Nanoparticles Coated γ-FeOx Nanoparticles, γ-FeOx@AuNP
Method B1. Our first trial was involving the direct reduction and deposition of gold acetate, Au(ac)3, on the surface of iron oxide NPs (10‒20 nm, prepared by either methods A1 or A2) in the presence of 1,2-hexadecanediol and 1-oleylamine. Briefly, to the suspension of γ-FeOx NPs (0.1 g) in phenyl ether (20 mL) was added Au(ac)3 (0.45 g, 1.2 mmol), 1,2-hexadecanediol (3.0 g, 25 mmol), and oleylamine (4 mL) under N2 atmosphere. The mixture was heated to 180‒190 °C with vigorously stirred for 2.0 h while maintain the temperature constant. After cooling to room temperature, the NPs were collected by a permanent magnet, washed twice with both 2-propanol (100 mL) and ethanol (100 mL). Subsequently, the NPs were collected with a permanent magnet to yield a dark brown solid powder.
Method B2. To the γ-FeOx nanoparticles (prepared by the method A1 stated above, 100 mg) was added D.I. water (100 mL) and tetramethylammonium hydroxide (1.0 M, 10 mL, 10 mmol). The mixture was ultrasonicated for 1.0 h to dissolve particles and yield a transparent brown solution. The solution was filtered through 0.4-μm PTFE syringe filter to remove insoluble large particles. The solution was then diluted to 2.0 L by D.I water, added by sodium citrate (0.5 M, 100 mL, 50 mmol), and stirred for 30 min at ambient temperature. While kept stirring, the solution of hydroxylamine hydrochloride (1.0 M, 2.0 mL, 2.0 mmol) and hydrogen tetrachloroaurate(III) trihydrate (HAuCl4∙3H2O, 0.025 M, 2.0 mL, 0.05 mmol) were added portionwise in 1/10 quantity each by an interval period of 15 min. After finishing addition, a magnet was used to separate and remove the particles containing γ-FeOx from the solution. They were washed with D.I water (×2) and ethanol (×2) and, finally, washed with methanol and decanted the supernatant. The suspension solution was then added acetone (10 mL), methanol (10 mL), and 1-octanethiol (1.0 mL) and ultrasonicated for 1.0 h to replace hydrophilic stabilizer (tertiary amine or sodium citrate) on the surface by hydrophobic thiol hydrocarbons. The resulting solids were filtered and washed by water and ethanol to remove remaining residual hydrophilic stabilizers and excessive 1-octanethiol to yield core-shell Au-NPs coated γ-FeOx nanoparticles. Spectroscopic data: FT-IR (KBr) νmax 2958 (m), 2928 (s), 2852 (m), 1724 (w), 1626 (s), 1496 (m), 1463 (s), 1380 (m), 1330 (w), 1263 (w), 1168 (m), 1130 (s), 1078 (s), 877 (w), 804 (m), 730 (s), 696 (m), 609 (m), 584 (s), and 557(m) cm‒1.
3.3.3. Preparation of C60(>DPAF-C9)-Encapsulated γ-FeOx@Au Trilayered Core-Shell Nanoparticles, (γ-FeOx@AuNP)@[C60(>DPAF-C9)]n
We carried out an encapsulation procedure by dissolving γ-FeOx@AuNP nanoparticles (100 mg) in toluene with the aid of a small quantity of 1-octanethiol, followed by the addition of C60(>DPAF-C9) (100 mg) and subsequent sonication for a period of 30 min. After filtration of the solution through celite, an excessive amount of C60(>DPAF-C9) (est. in <5 mg) remaining in the solution was removed. All encapsulated magnetic nanoparticles filtered off at the top celite surface as brown solids were recovered by toluene extraction and physically removed from the solution by an internal permanent magnet. Solids were washed repeatedly by ethanol and ether, followed by drying in vacuo.
Alternatively, a modified method was used to prevent the possible material loss during celite filtration. In this procedure, both γ-FeOx@AuNP nanoparticles (100 mg) and C60(>DPAF-C9) 1 or C60(>DPAF-C9)2 2 in a predefined weight ratio amount were dissolved in toluene (30 mL) with stirring for 30 min and then ultrasonicated for an additional more than 30 min until a clear solution being obtained to homogenize nanoparticles dispersion and increase their interactions with 1 or 2. The solution was concentrated via rotary evaporation to less than 3.0 mL in volume to increase the molecular contact of 1 or 2 to γ-FeOx@AuNPs. It was then diluted by toluene again to a volume of 30 mL with stirring and subsequent sonication for 10 min to dissolve excessive 1 or 2. All encapsulated magnetic nanoparticles were physically removed from the container solution with the assistance of an internal permanent magnet. The nanoparticles were washed repeatedly by ethanol and ether, followed by drying in vacuo to afford dark brown solids of (γ-FeOx@AuNP)@[C60(>DPAF-C9)x]n (x = 1 or 2) 3. Spectroscopic data of 3–1 prepared based on the nanoparticle methods A1 and B2: FT-IR (KBr) νmax 2952 (m), 2920 (s), 2848 (m), 1741 (m), 1675 (m), 1637 (m), 1618 (m), 1591 (s), 1490 (m), 1458 (s), 1429 (m), 1382 (m), 1271 (m), 1203 (s), 1168 (m), 1110 (m), 1076 (m), 1029 (m),.875 (w), 811(w), 748 (m), 694 (m), 617 (m), 582 (s), and 526 (s) cm‒1.
3.4. Tunneling Electron Microscopic (TEM) Measurements
Both formvar and carbon-copper film grids in a 200-mesh size were used for the topography investigation of molecularly assembled structures, derived from 1 and 2, by TEM images. Samples were prepared by direct coating the grid with a sample solution of 1.0 × 10−6 M diluted from the master nanoparticle solution (5.0 × 10−4 M in H2O). Different concentrations (10−4–10−6 M) may be used and evaluated to ensure full separation of particles from each other without stacking prior to the collection of microimages. It was followed by the freeze-dry technique via placing the sample-coated grid on a metal dish that was in thermal equilibrium with liquid nitrogen. This apparatus was then placed inside a vacuum chamber for overnight to remove all solvents completely prior to the measurement that will retain the nanoparticle shape on the grid. To prevent particle aggregations during the solvent removal procedure, a low concentration of 10−5–10−6 M in either H2O or benzene is essential.
3.5. Dielectric and Permittivity Measurements
Dielectric property measurements were carried out by an Agilent Network Analyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) equipped with an open-ended Agilent 85070E dielectric probe kit (200 MHz to 50 GHz). Calibration was conducted by using open-ended, close-ended, and attenuated calibrators prior to each measurement to remove the cable-related instability and system drift errors. In the experiment, a complex scattering parameter, defined as S11, was measured and converted to relative complex electric dielectric constant values using Agilent 85071E Materials Measurement Software. This complex form is compdose of a real and an imaginary part. The former represents the value of dielectric constant and the latter is defined as the loss factor.
Two light sources were used in the measurement including a LED white light with the output power of 2.0 W and a UVB-enhanced visible lamp (Uvitron 400 W with the output power density of 20 mW/cm2). Poly(dimethylsiloxane) (PDMS, 1.0 g) semi-solid was applied as a polymer matrix host that is capable of forming a paste-like sample with 2. A mixture of PDMS and either C60(>DPAF-C9), γ-FeOx NPs, γ-FeOx@AuNPs, or (γ-FeOx@AuNP)@[C60(>DPAF-C9)]n (100 mg) was prepared by dissolving both components in ethyl acetate (20 mL) in the testing tube under sonication until a clear solution was obtained. Ethyl acetate was then completely evaporated under vacuum to yield brown paste-like semi-solid materials.
A custom-built chamber was applied for permittivity measurements to be conducted under a circumferentially uniform illumination environment. It was achieved by the installation of a reflective half-circular aluminum plate at the back-wall side surrounding the testing tube which is located at the center of the chamber. A light was allowed to pass through a small window at the front side of the chamber to irradiate the sample tube. Reflected light beams were design to refocus from the back-side aluminum mirror plate back to the tube. Four fans were installed at the top and side-walls of the chamber to control and prevent temperature building-up inside the chamber. Illumination periods were chosen to be 10, 30, and 60 min.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}