
Non-Nucleosidic Analogues of Polyaminonucleosides and Their Influence on Thermodynamic Properties of Derived Oligonucleotides


Abstract
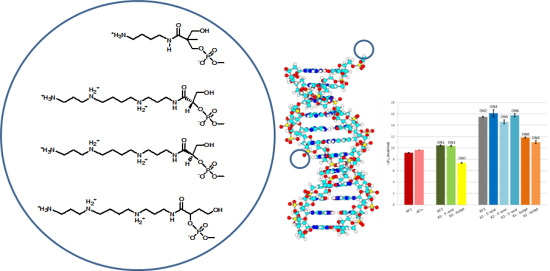
:
1. Introduction
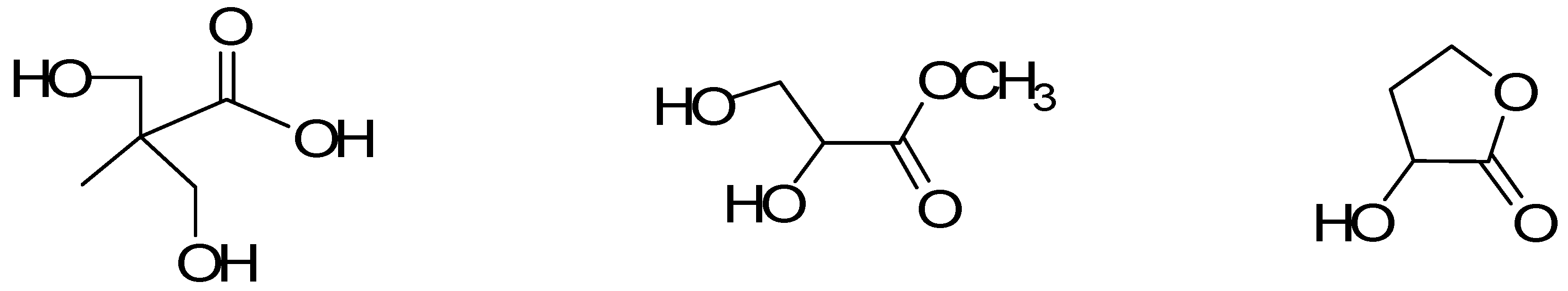
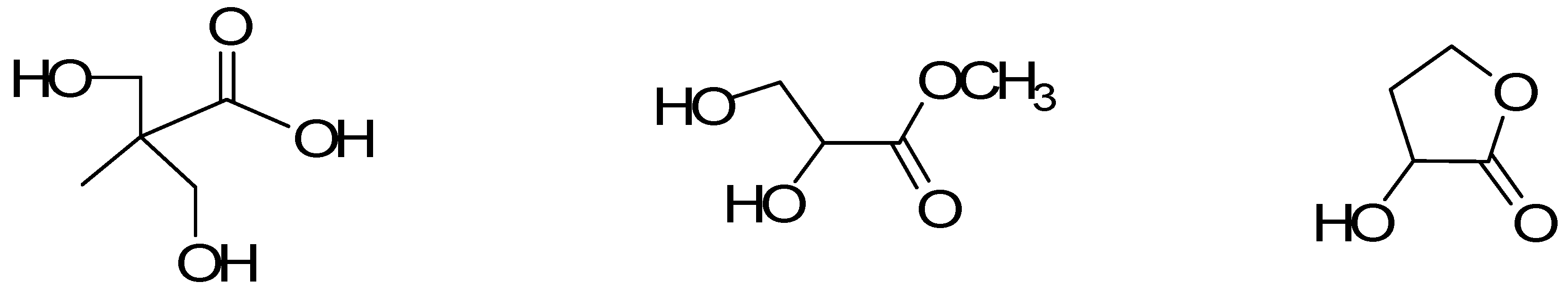
2. Results and Discussion
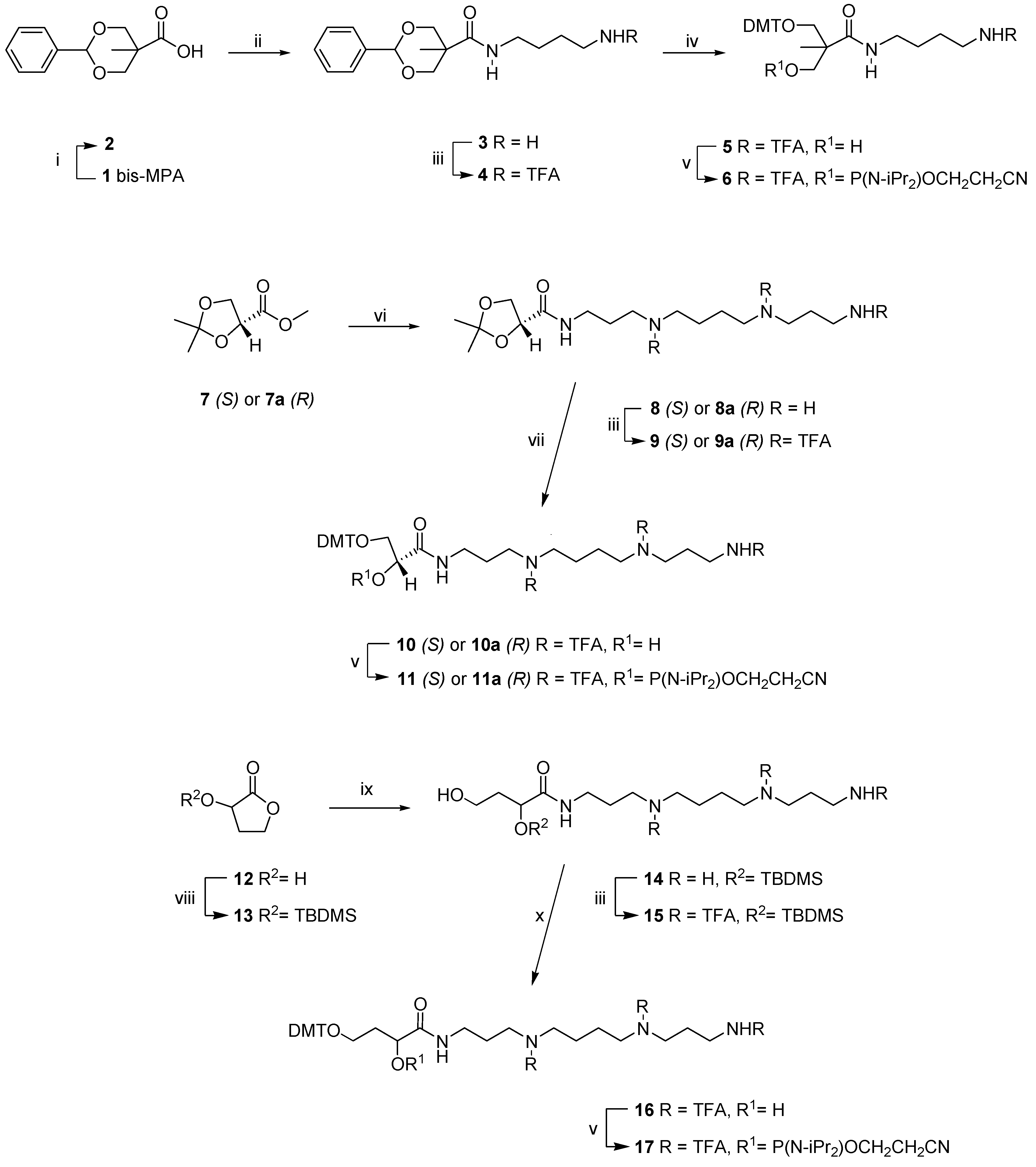
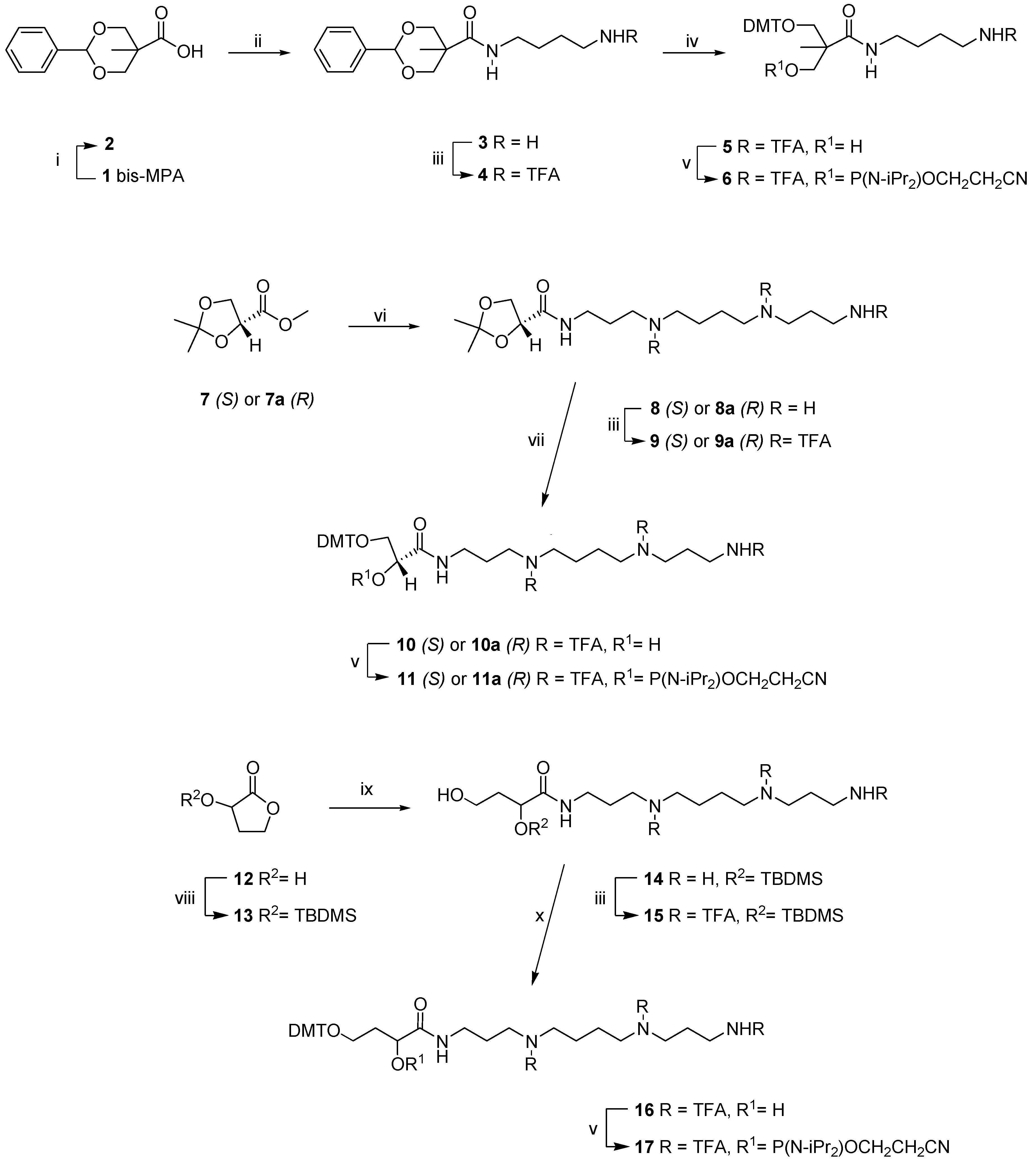
2.1. Chemistry
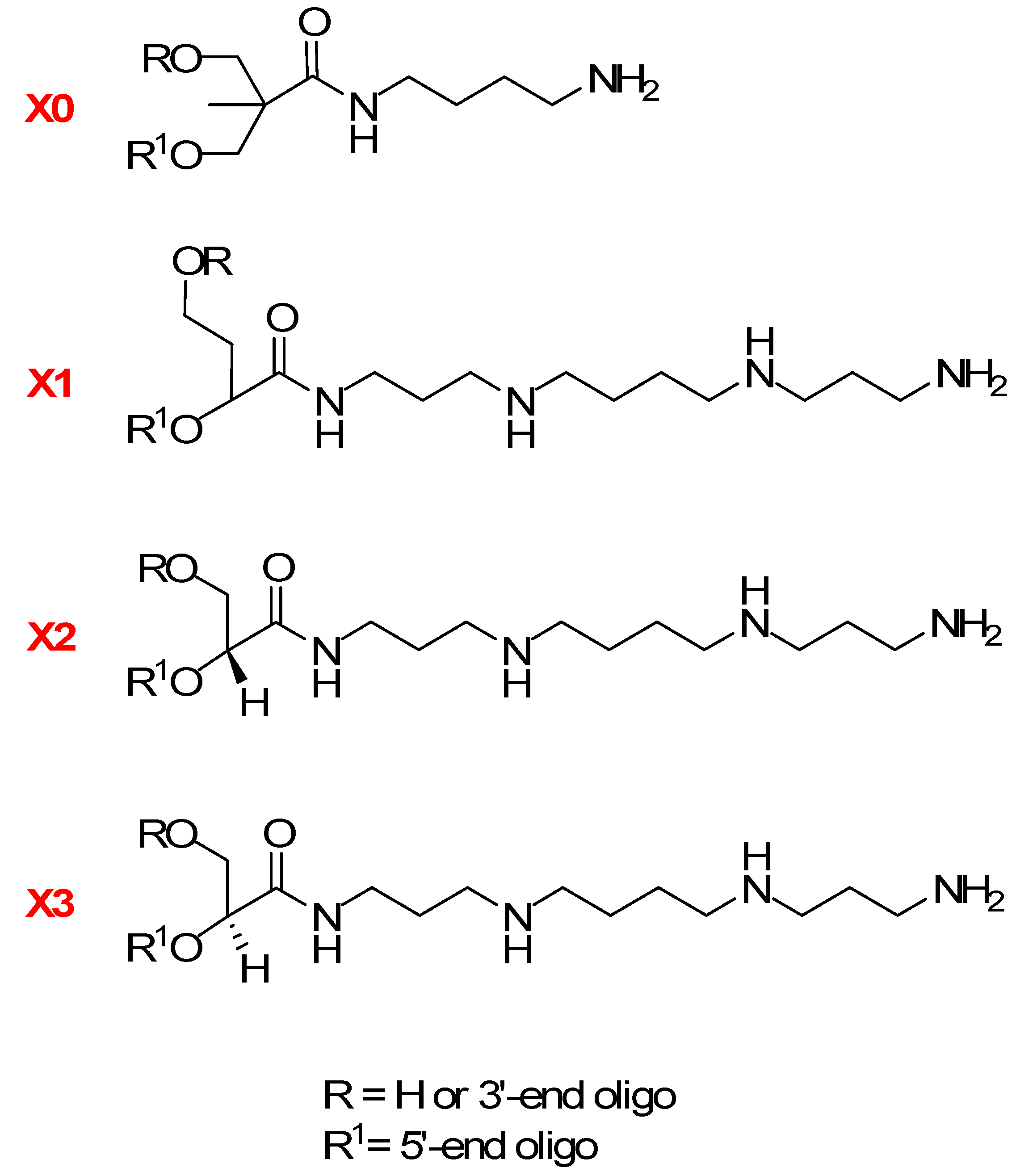
2.2. Oligonucleotides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence 5′ → 3′ a | m/z [M − H]− | |
|---|---|---|---|
| Calcd | Found | ||
| ON1 | CTC AAG CAA GCT | 3614.42 | 3613.08 |
| ON2 | CTC ACA TGC GCG | 3606.40 | 3605.54 |
| ON3 | CTC AAG CAA GCT | 3994.42 | 3993.19 |
| ON4 | CTC ACA TGC GCG | 3973.12 | 3972.23 |
| ON5 | CTC ACA TGC GCG | 3959.54 | 3959.24 |
| ON6 | CTC ACA TGC GCG | 3959.54 | 3959.57 |
| ON7 | CTC AAG CAA GCT | 3994.42 | 3993.25 |
| ON8 | CTC ACA TGC GCG | 3973.12 | 3973.12 |
| ON9 | CTC ACA TGC GCG | 3959.54 | 3957.45 |
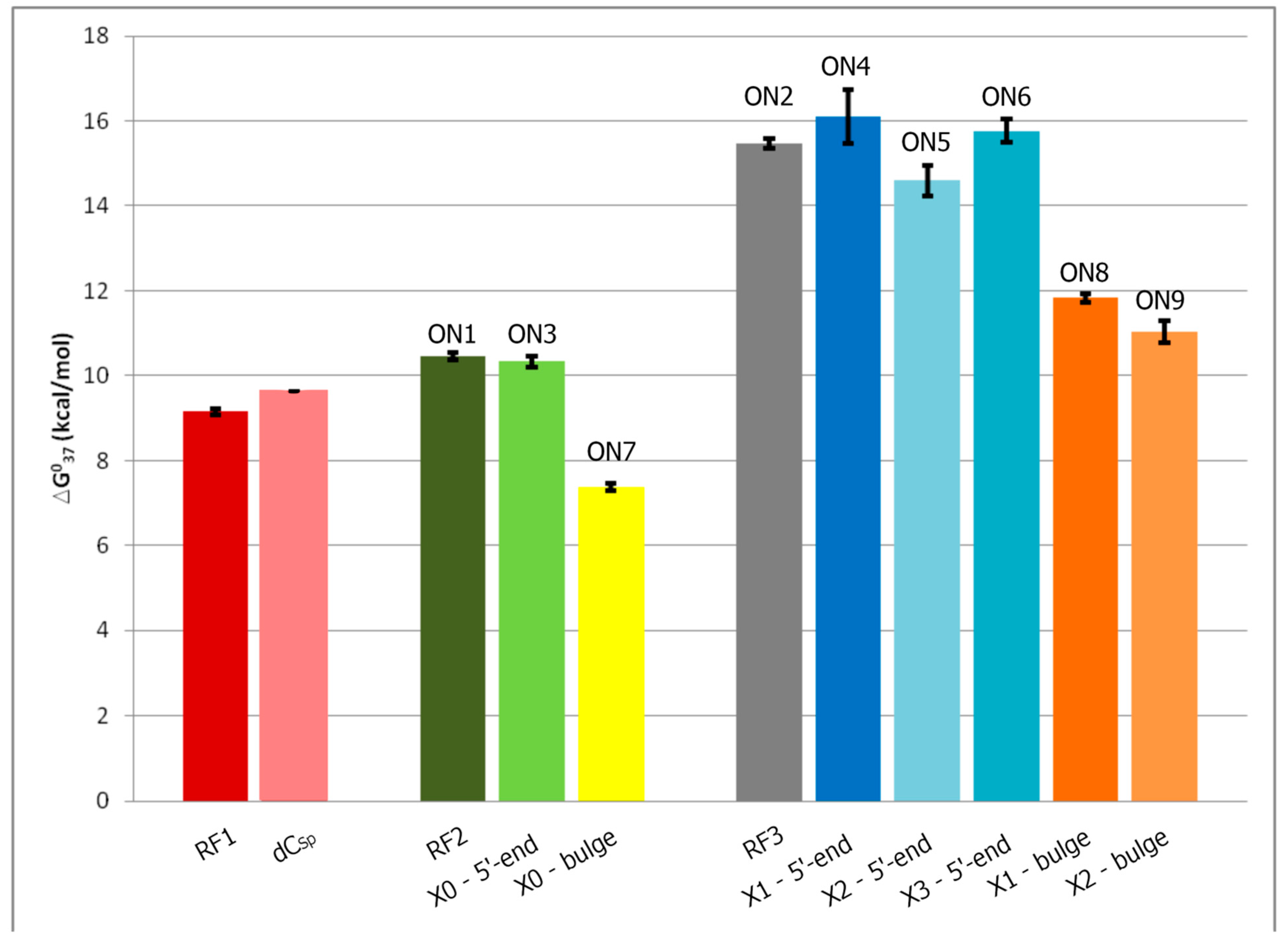
2.3. Stability of Duplexes with Modified Oligodeoxyribonucleotides
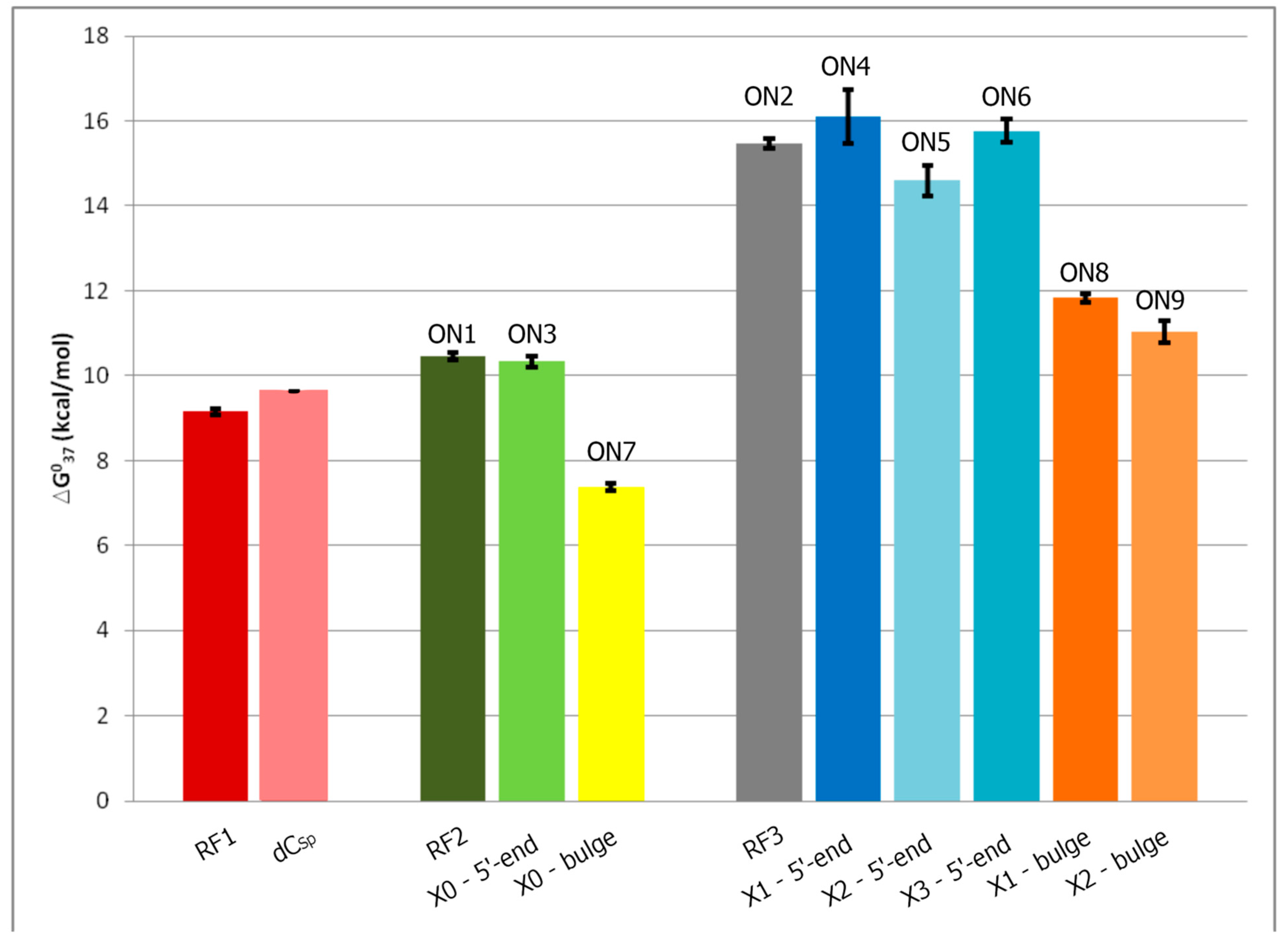
| Oligo | Average of Curve Fits | TM−1 vs. log (CT/4) Plots | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| −ΔH° [kcal/mol] | −ΔS° [eu] | −ΔG°37 [kcal/mol] | TM [°C] | −ΔH° [kcal/mol] | −ΔS° [eu] | −ΔG°37 [kcal/mol] | TM [°C] | ΔΔG°37 [kcal/mol] | ΔTM b [°C] | |
| A. Thermodynamic parameters of references duplexes | ||||||||||
| ON1 c | 67.8 ± 9.0 | 185.6 ± 27.8 | 10.27 ± 0.44 | 55.1 | 72.6 ± 2.0 | 200.6 ± 6.2 | 10.45 ± 0.08 | 54.7 | | |
| ON2 d | 115.9 ± 4.8 | 322.1 ± 14.2 | 16.00 ± 041 | 64.6 | 108.5 ± 1.8 | 300.1 ± 5.4 | 15.46 ± 0.12 | 64.8 | | |
| B. Thermodynamic parameters of duplexes with the polyamine analog as a dangling end | ||||||||||
| ON3 c | 63.3 ± 6.8 | 171.5 ± 20.9 | 10.12 ± 0.37 | 55.7 | 69.09 ± 3.1 | 189.4 ± 9.7 | 10.33 ± 0.13 | 55.0 | −0.3 | |
| ON4 d | 131.8 ± 10.4 | 368.8 ± 31.0 | 17.47 ± 0.80 | 65.1 | 114.0 ± 8.3 | 315.9 ± 24.9 | 16.10 ± 0.64 | 65.4 | − | |
| ON5 d | 97.8 ± 9.4 | 268.4 ± 28.1 | 14.57 ± 0.68 | 64.8 | 98.9 ± 5.1 | 271.9 ± 15.4 | 14.59 ± 0.36 | 64.5 | −0.3 | |
| ON6 d | 106.1 ± 4.7 | 292.4 ± 14.0 | 15.43 ± .0.37 | 65.4 | 110.9 ± 3.9 | 307.0 ± 11.7 | 15.76 ± 0.27 | 65.1 | − | |
| C. Thermodynamic parameters of duplexes with the polyamine analog as a bulge | ||||||||||
| ON7 c | 62.7 ± 16.7 | 178.2 ± 53.8 | 7.50 ± 0.20 | 41.9 | 59.0 ± 4.2 | 166.5 ± 13.7 | 7.38 ± 0.08 | 41.5 | −13.2 | |
| ON8 d | 71.6 ± 6.0 | 194.4 ± 18.3 | 11.31 ± 0.35 | 59.2 | 82.0 ± 2..0 | 226.3 ± 6.2 | 11.83 ± 0.10 | 58.4 | −6.4 | |
| ON9 d | 81.2 ± 11.5 | 225.0 ± 35.6 | 11.45 ± 0.48 | 57.0 | 70 ± 6.4 | 192.1 ± 20.0 | 11.03 ± 0.25 | 58.1 | −6.7 | |
3. Experimental Section
3.1. General Methods
3.2. Synthesis of Monomers
3.3. Oligonucleotide Preparation
3.4. Thermodynamic Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Thomas, T.; Wada, M.; Sigal, L.H.; Shirahata, A.; Thomas, T.J. Facilitation of the cellular uptake of a triplex-forming oligonucleotide by novel polyamine analogues: Structure-activity relationships. Biochemistry 1999, 38, 13328–13337. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Barawkar, D.A.; Vaijayanti, K.; Ganesh, K.N. Synthesis of site-specific oligonucleotide-polyamine conjugates. Bioorg. Med. Chem. Lett. 1994, 4, 1733–1738. [Google Scholar] [CrossRef]
- Horn, T.; Chaturvedi, S.; Balasubramaniam, T.N.; Letsinger, R.L. Oligonucleotides with alternating anionic and cationic phosphoramidate linkages: Synthesis and hybridization of stereo-uniform isomers. Tetrahedron Lett. 1996, 37, 743–746. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Singman, C.N.; Histand, G.; Salunkhe, M. Cationic oligonucleotides. J. Am. Chem. Soc. 1988, 110, 4470–4471. [Google Scholar] [CrossRef]
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; Berg, A.V.D.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Delair, T.; Brua, C.; Veron, L.; Pavirani, A.; Kolbe, H.V. Synthesis of polyallylamine derivatives and their use as gene transfer vectors in vitro. Bioconjug. Chem. 1999, 10, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Bhuchar, N.; Ishihara, K.; Narain, R. Well-controlled cationic water-soluble phospholipid polymer-DNA nanocomplexes for gene delivery. Bioconjug. Chem. 2011, 22, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, B.; Jiang, H.; Wang, B.; Ma, B. Cationic lipids and polymers mediated vectors for delivery of siRNA. J. Control. Release 2007, 123, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Piest, M.; Engbersen, J.F.J. Effects of charge density and hydrophobicity of poly(amido amine)s for non-viral gene delivery. J. Control. Release 2010, 148, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Yi, J.W.; Bang, E.K.; Jeon, E.M.; Kim, B.H. Synthesis and efficient siRNA delivery of polyamine-conjugated cationic nucleoside lipids. Med. Chem. Commun. 2011, 2, 505–508. [Google Scholar] [CrossRef]
- Geihe, E.I.; Cooley, C.B.; Simon, J.R.; Kiesewetter, M.K.; Edward, J.A.; Hickerson, R.P.; Kaspar, R.L.; Hedrick, J.L.; Waymouth, R.M.; Wender, P.A. Designed guanidinium-rich amphipathic oligocarbonate molecular transporters complex, deliver and release siRNA in cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13171–13176. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, P.L.; Arya, D.P. Natural product DNA major groove binders. Nat. Prod. Rep. 2012, 29, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Davis, E.; Ranjan, N.; Xue, L.; Hyde-Volpe, D.; Arya, D.P. Thermodynamics of nucleic acid “shape readout” by an aminosugar. Biochemistry 2011, 50, 9088–9113. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Lollo, B.; Freier, S.; Esau, C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006, 34, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Behlke, M.A.; Rose, S.D.; Chang, M.S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Cekaite, L.; Furset, G.; Hovig, E.; Sioud, M. Gene Expression Analysis in Blood Cells in Response to Unmodified and 2′-Modified siRNAs Reveals TLR-dependent and Independent Effects. J. Mol. Biol. 2007, 365, 90–108. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, S.; Mueller-Ibeler, J.; Kluge, J.; Wäss, L.; Burdinski, G.; Havens, J.R.; Onofrey, T.J.; Wang, D.; Schweitzer, M. Hydrazide oligonucleotides: New chemical modification for chip array attachment and conjugation. Nucleic Acids Res. 2002, 30, 4793–4802. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.C.; Park, H.G.; Chang, H.N.; Cho, D.Y. Array-based mutation detection of BRCA1 using direct probe/target hybridization. Anal. Biochem. 2005, 337, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Wengel, J. Nucleic acid nanotechnology-towards Angstrom-scale engineering. Org. Biomol. Chem. 2004, 2, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Liu, D.; Watari, M.; Riener, C.K.; Strunz, T.; Welland, M.E.; Balasubramanian, S.; McKendry, R.A. DNA molecular motor driven micromechanical cantilever arrays. J. Am. Chem. Soc. 2005, 127, 17054–17060. [Google Scholar] [CrossRef] [PubMed]
- Mangraviti, A.; Tzeng, S.Y.; Kozielski, K.L.; Wang, Y.; Jin, Y.; Gullotti, D.; Pedone, M.; Buaron, N.; Liu, A.; Wilson, D.R.; et al. Polymeric Nanoparticles for Nonviral Gene Therapy Extend Brain Tumor Survival in Vivo. ACS Nano 2015, 9, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Saneyoshi, H.; Tamaki, K.; Ohkubo, A.; Seio, K.; Sekine, M. Synthesis and hybridization properties of 2′-O-(tetrazol-5-yl)ethyl-modified oligonucleotides. Tetrahedron 2008, 64, 4370–4376. [Google Scholar] [CrossRef]
- Urban, E.; Noe, C.R. Structural modifications of antisense oligonucleotides. Il Farmaco 2003, 58, 243–258. [Google Scholar] [CrossRef]
- Debart, F.; Abes, S.; Deglane, G.; Moulton, H. M.; Clair, P.; Gait, M.J.; Vasseur, J.J.; Lebleu, B. Chemical modifications to improve the cellular uptake of oligonucleotides. Curr. Top. Med. Chem. 2007, 7, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Saadat, K.; Díaz-Gavilán, M.; Urban, E.; Noe, C.R. Oligonucleotide-polyamine conjugates: Influence of length and position of 2′-attached polyamines on duplex stability and antisense effect. Eur. J. Med. Chem. 2009, 44, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.A.; Baba, T.; Kodama, T.; Islam, M.A.; Obika, S. Hybridizing ability and nuclease resistance profile of backbone modified cationic phosphorothioate oligonucleotides. Bioorg. Med. Chem. 2012, 20, 4098–5102. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, U. Polyamines and cancer: Minireview article. Amino Acids 2004, 26, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.C.; Mehta, D.J.; Gerner, E.W. Polyamine-dependent gene expression. Cell. Mol. Life Sci. 2003, 60, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Venkiteswaran, S.; Vijayanathan, V.; Shirahata, A.; Thomas, T.; Thomas, T.J. Antisense recognition of the HER-2 mRNA: Effects of phosphorothioate substitution and polyamines on DNA·RNA, RNA·RNA, and DNA·DNA duplex stability. Biochemistry 2005, 44, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Antony, T.; Thomas, T.; Shirahata, A.; Thomas, T.J. Selectivity of polyamines on the stability of RNA-DNA hybrids containing phosphodiester and phosphorothioate oligodeoxyribonucleotides. Biochemistry 1999, 38, 10775–10784. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.H.; Lin, S.B.; Yuann, J.M.; Lin, W.C.; Wang, A.H.J.; Kan, L. Effects of polyamines on the thermal stability and formation kinetics of DNA duplexes with abnormal structure. Nucleic Acids Res. 2001, 29, 5121–5128. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, J.; Gdaniec, Z.; Popenda, L.; Markiewicz, W.T. Polyaminooligonucleotide: NMR structure of duplex DNA containing a nucleoside with spermine residue, N-[4,9,13-triazatridecan-1-yl]-2′-deoxycytidine. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Malkoch, M.; Schleicher, K.; Drockenmuller, E.; Hawker, C.J.; Russell, T.P.; Wu, P.; Fokin, V.V. Structurally Diverse Dendritic Libraries: A Highly Efficient Functionalization Approach Using Click Chemistry. Macromolecules 2005, 38, 3663–3678. [Google Scholar] [CrossRef]
- Asanuma, H.; Liang, X.; Komiyama, M. meta-Aminoazobenzene as a thermo-insensitive photo-regulator of DNA-duplex formation. Tetrahedron Lett. 2000, 41, 1055–1058. [Google Scholar] [CrossRef]
- Ihre, H.; Padilla De Jesús, O.L.; Fréchet, J.M.J. Fast and convenient divergent synthesis of aliphatic ester dendrimers by anhydride coupling. J. Am. Chem. Soc. 2001, 123, 5908–5917. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, W.T.; Godzina, P.; Markiewicz, M.; Astriab, A. Synthesis of A Polyaminooligonucleotide Combinatorial Library. Nucleos. Nucleot. 1998, 17, 1871–1880. [Google Scholar] [CrossRef]
- Godzina, P.; Adrych-Rozek, K.; Markiewicz, W.T. Synthetic Oligonucleotide Combinatorial Libraries. 3. Synthesis of Polyaminonucleosides. Nucleos. Nucleot. 1999, 18, 2397–2414. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Sund, C.; Puri, N.; Chattopadhyaya, J. The Chemistry of C-Branched Spermine Tethered Oligo-DNAs and Their Properties in Forming Duplexes and Triplexes. Nucleos. Nucleot. 1997, 16, 755–760. [Google Scholar] [CrossRef]
- Sund, C.; Puri, N.; Chattopadhyaya, J. Synthesis of C-branched spermine tethered oligo-DNA and the thermal stability of the duplexes and triplexes. Tetrahedron 1996, 52, 12275–12290. [Google Scholar] [CrossRef]
- Moriguchi, T.; Sakai, H.; Suzuki, H.; Shinozuka, K. Spermine moiety attached to the C-5 position of deoxyuridine enhances the duplex stability of the phosphorothioate DNA/complementary DNA and shows the susceptibility of the substrate to RNase H. Chem. Pharm. Bull. (Tokyo) 2008, 56, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Breslauer, K.J.; Frank, R.; Blöcker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [Google Scholar] [CrossRef] [PubMed]
- Bommarito, S.; Peyret, N.; SantaLucia, J. Thermodynamic parameters for DNA sequences with dangling ends. Nucleic Acids Res. 2000, 28, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Longfellow, C.E.; Kierzek, R.; Turner, D.H. Thermodynamic and spectroscopic study of bulge loops in oligoribonucleotides. Biochemistry 1990, 29, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Minetti, C.A.; Remeta, D.P.; Dickstein, R.; Breslauer, K.J. Energetic signatures of single base bulges: Thermodynamic consequences and biological implications. Nucleic Acids Res. 2009, 38, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Dauben, W.G.; Hendricks, R.T.; Pandy, B.; Wu, S.C.; Zhang, Xiaoming; Luzzio, M.J. Stereoselective intramolecular cyclopropanations: Enantioselective syntheses of 1α,25-dihydroxyvitamin D3 A-ring precursors. Tetrahedron Lett. 1995, 36, 2385–2388. [Google Scholar] [CrossRef]
- Markiewicz, W.T.; Biała, E.; Adamiak, R.W.; Grześkowiak, K.; Kierzek, R.; Kraszewski, A.; Stawiński, J.; Wiewiórowski, M. Further studies on oligoribonucleotide synthesis. Nucleic Acids Symp. Ser. 1980, 115–127. [Google Scholar]
- Markiewicz, W.T.; Biała, E.; Kierzek, R. Application of the Tetraisopropyldisiloxane-1,3-diyl Group in the Chemical Synthesis of Oligoribonucleotides. Bull. Pol. Acad. Sci. Chem. 1984, 32, 433–451. [Google Scholar]
- Borer, P.N.; Dengler, B.; Tinoco, I.; Uhlenbeck, O.C. Stability of ribonucleic acid double-stranded helices. J. Mol. Biol. 1974, 86, 843–853. [Google Scholar] [CrossRef]
- McDowell, J.A.; Turner, D.H. Investigation of the structural basis for thermodynamic stabilities of tandem GU mismatches: Solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing. Biochemistry 1996, 35, 14077–14089. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are not available from authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzezinska, J.; Markiewicz, W.T. Non-Nucleosidic Analogues of Polyaminonucleosides and Their Influence on Thermodynamic Properties of Derived Oligonucleotides. Molecules 2015, 20, 12652-12669. https://doi.org/10.3390/molecules200712652
Brzezinska J, Markiewicz WT. Non-Nucleosidic Analogues of Polyaminonucleosides and Their Influence on Thermodynamic Properties of Derived Oligonucleotides. Molecules. 2015; 20(7):12652-12669. https://doi.org/10.3390/molecules200712652
Chicago/Turabian StyleBrzezinska, Jolanta, and Wojciech T. Markiewicz. 2015. "Non-Nucleosidic Analogues of Polyaminonucleosides and Their Influence on Thermodynamic Properties of Derived Oligonucleotides" Molecules 20, no. 7: 12652-12669. https://doi.org/10.3390/molecules200712652
APA StyleBrzezinska, J., & Markiewicz, W. T. (2015). Non-Nucleosidic Analogues of Polyaminonucleosides and Their Influence on Thermodynamic Properties of Derived Oligonucleotides. Molecules, 20(7), 12652-12669. https://doi.org/10.3390/molecules200712652