
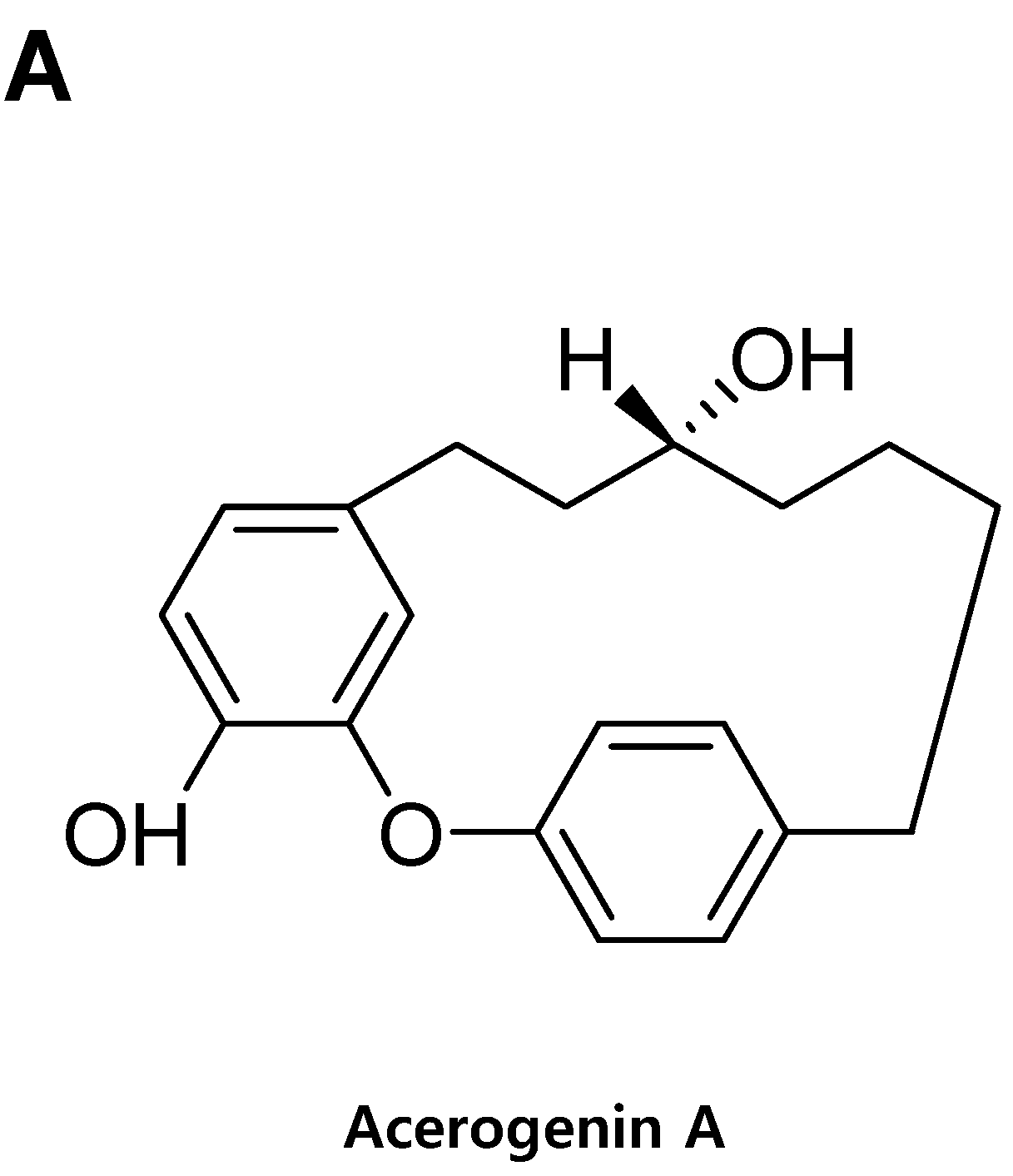
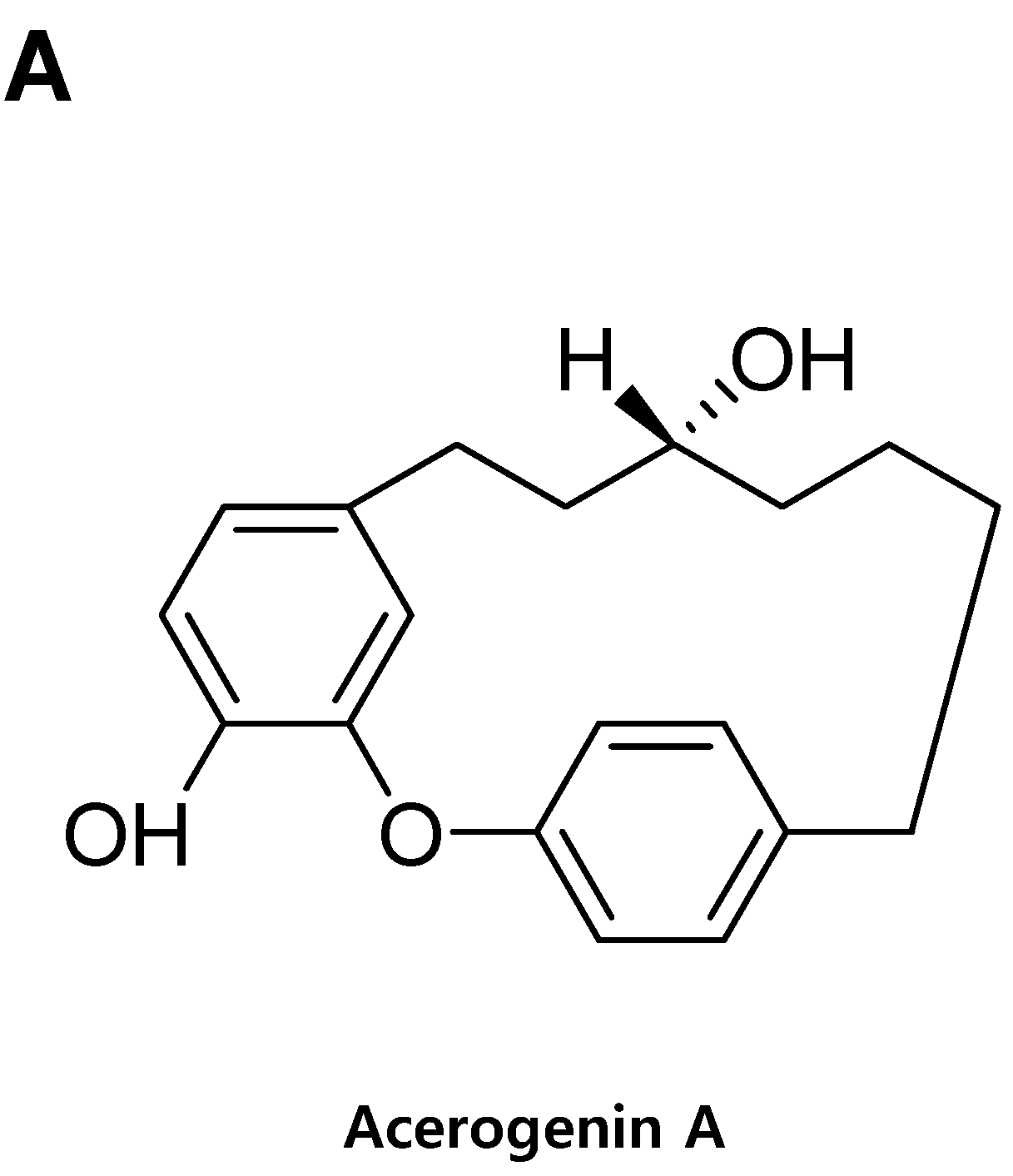
Acerogenin A from Acer nikoense Maxim Prevents Oxidative Stress-Induced Neuronal Cell Death through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse Hippocampal HT22 Cell Line



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
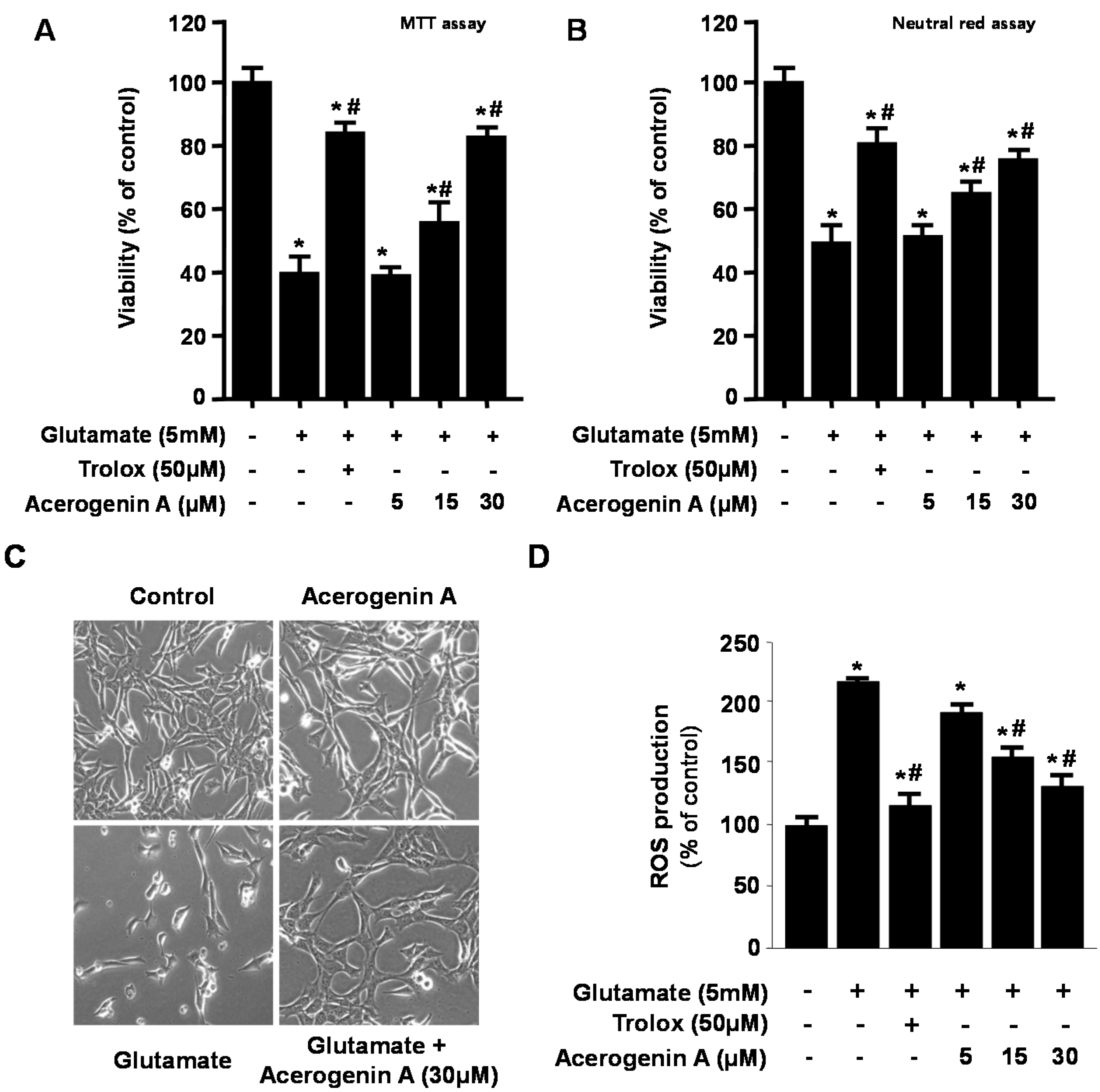
2.1. Effects of Acerogenin A on Glutamate-Induced Cytotoxicity and Inhibition of ROS Generation in HT22 Cells
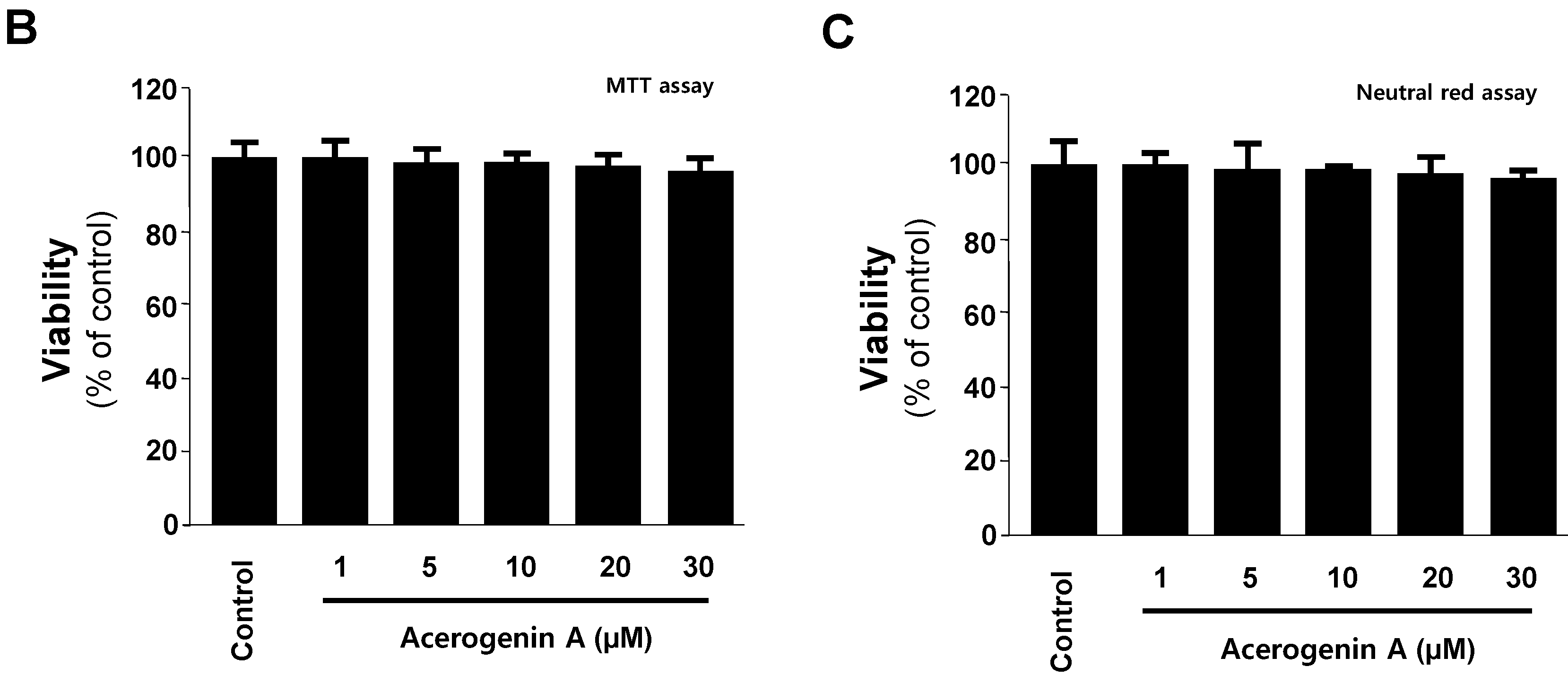
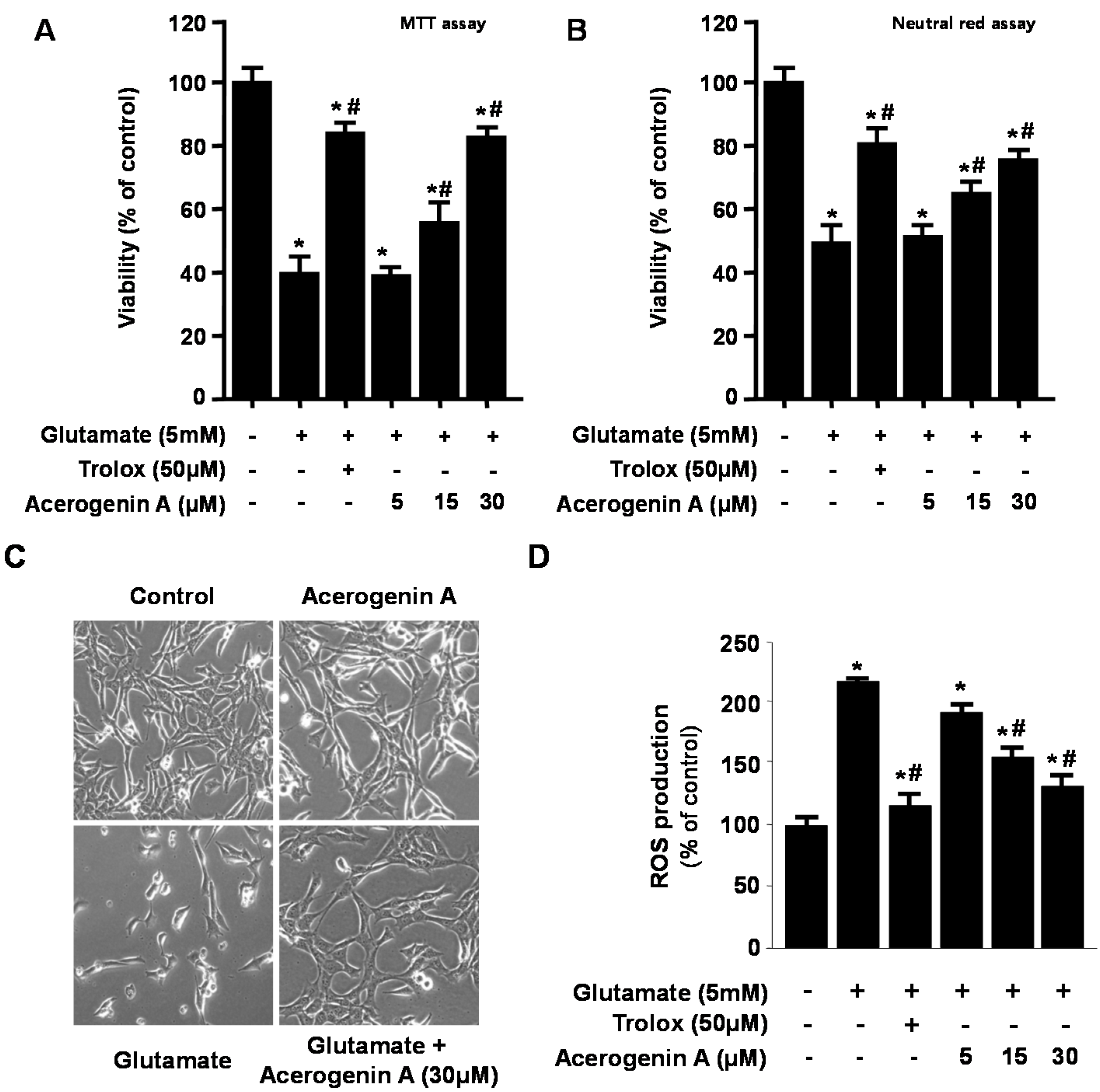
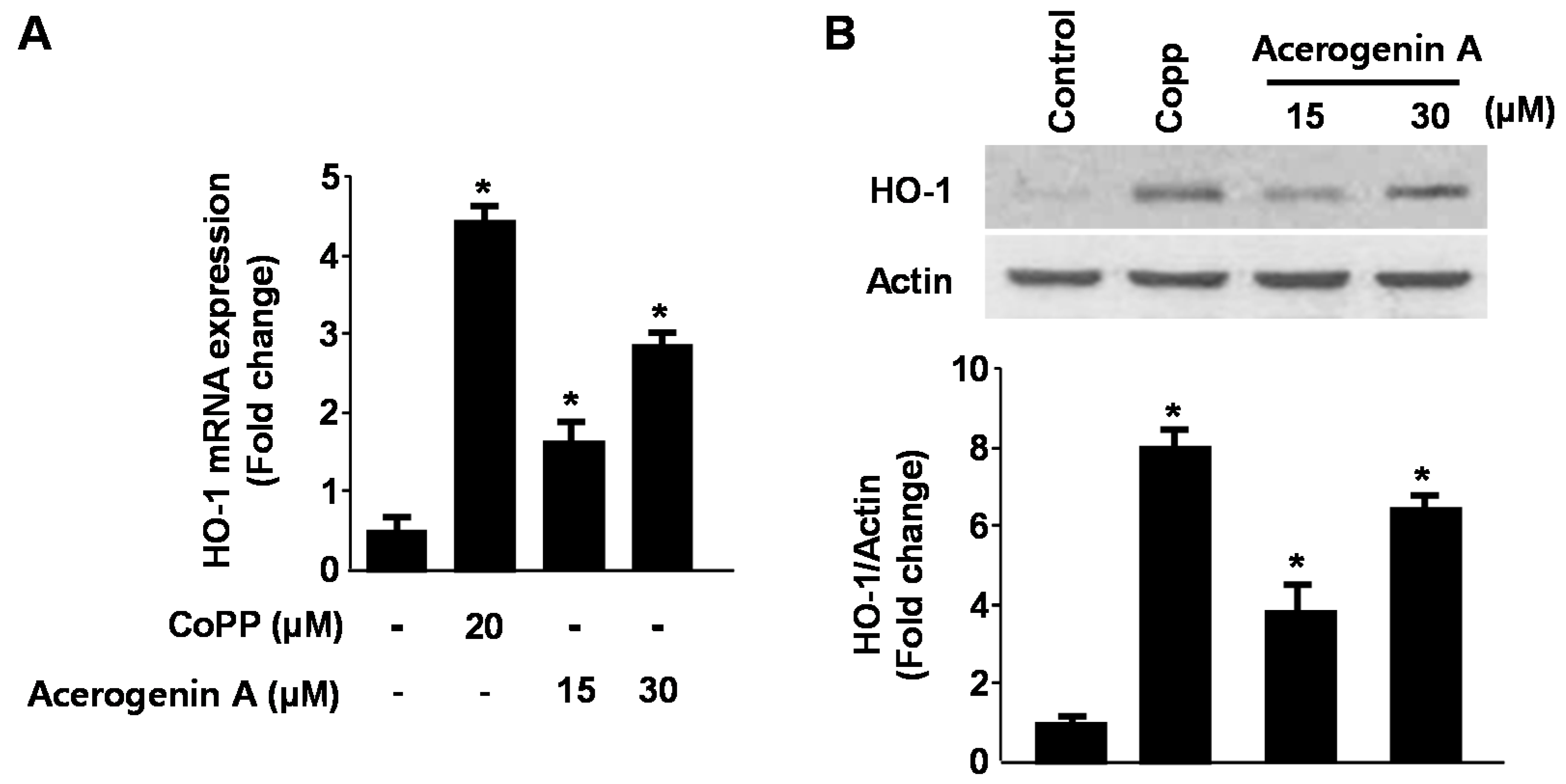
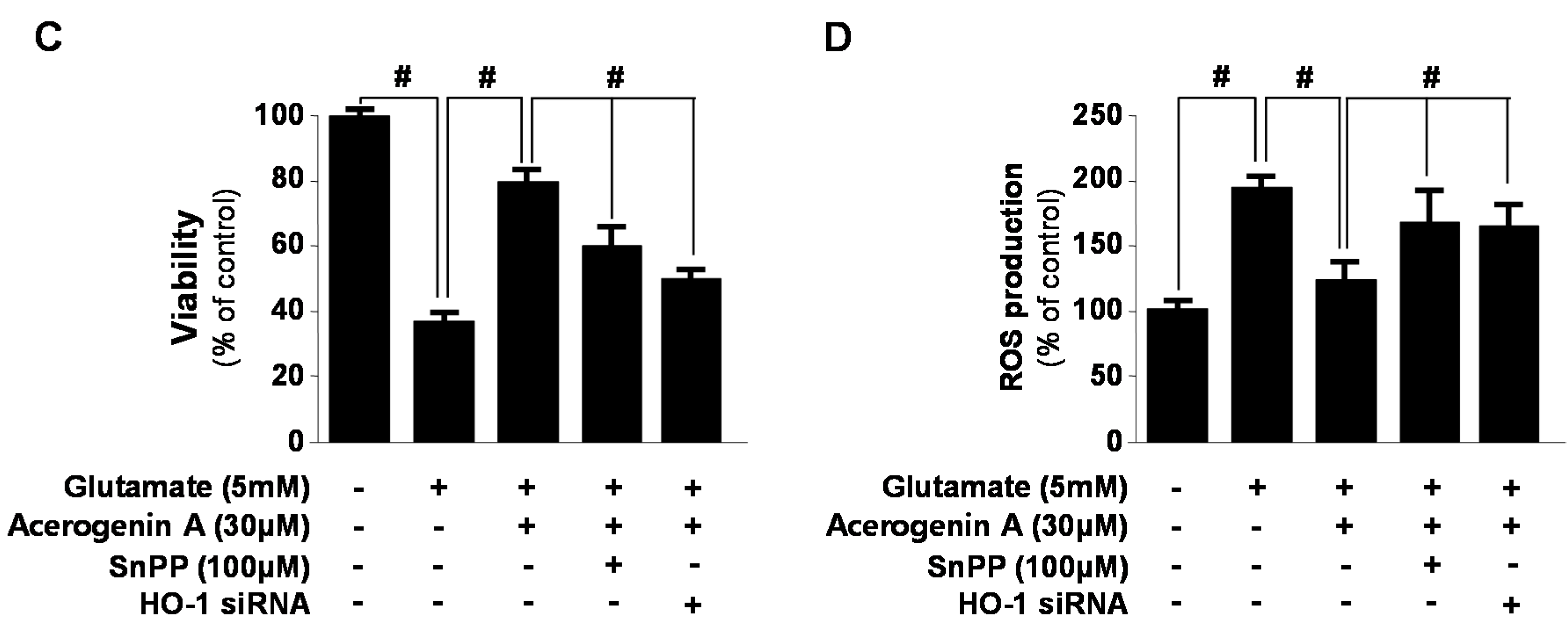
2.2. Effects of Acerogenin A on Glutamate-Induced Oxidative Neurotoxicity through HO-1 Expression Pathway in HT22 Cells
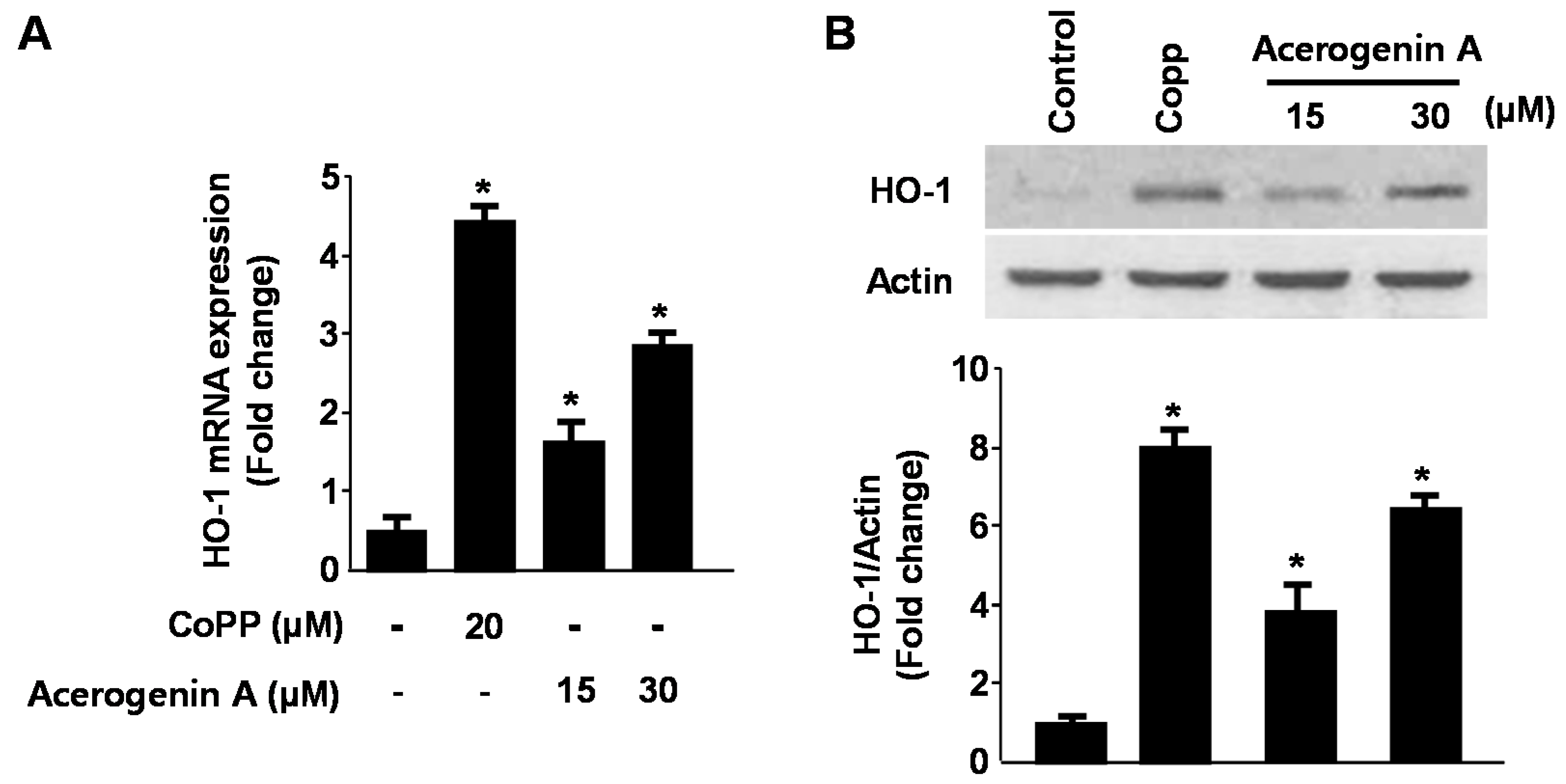
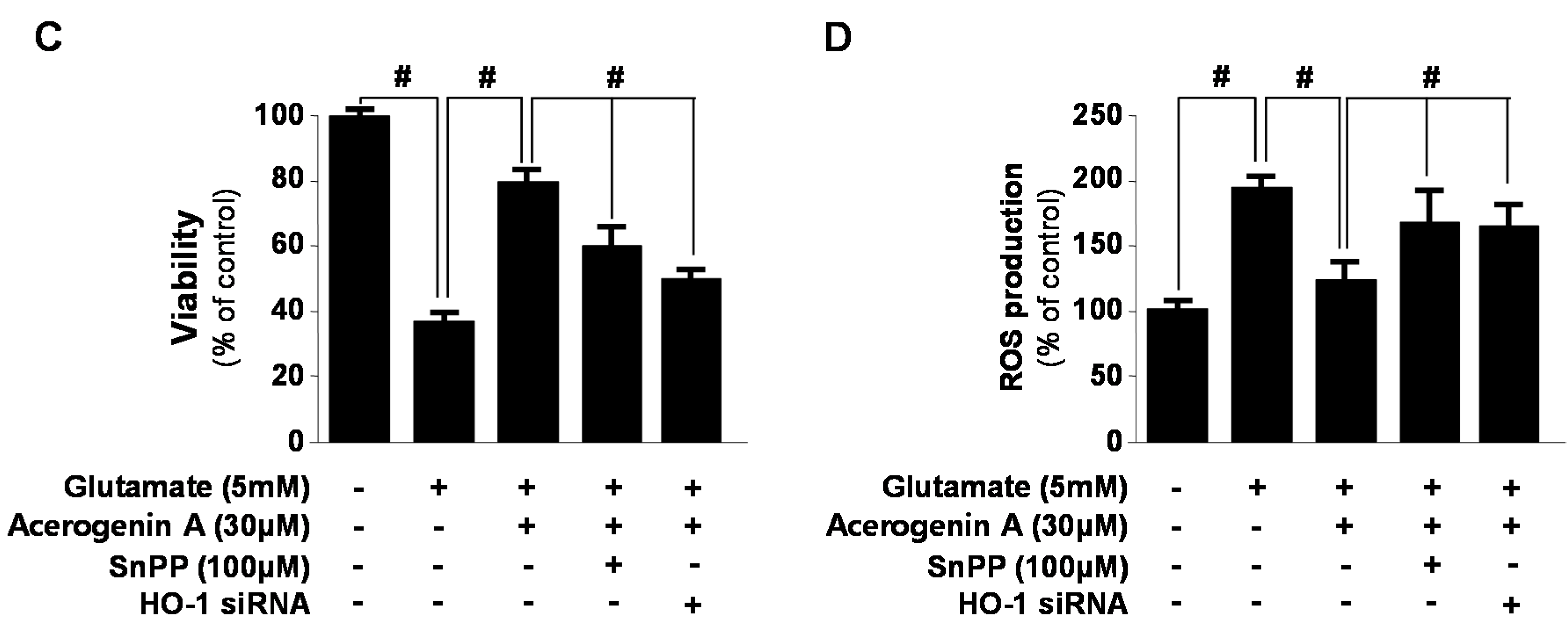
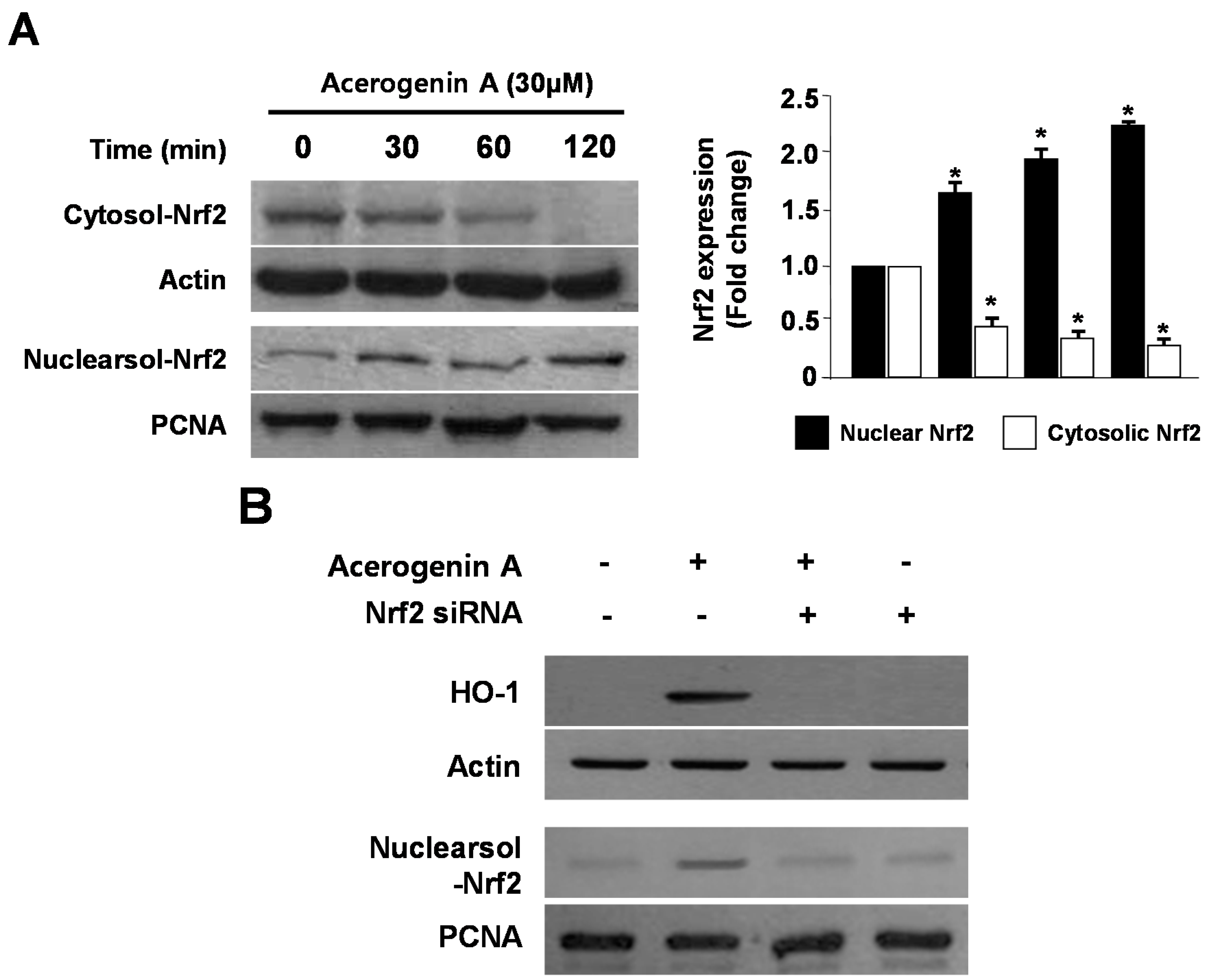
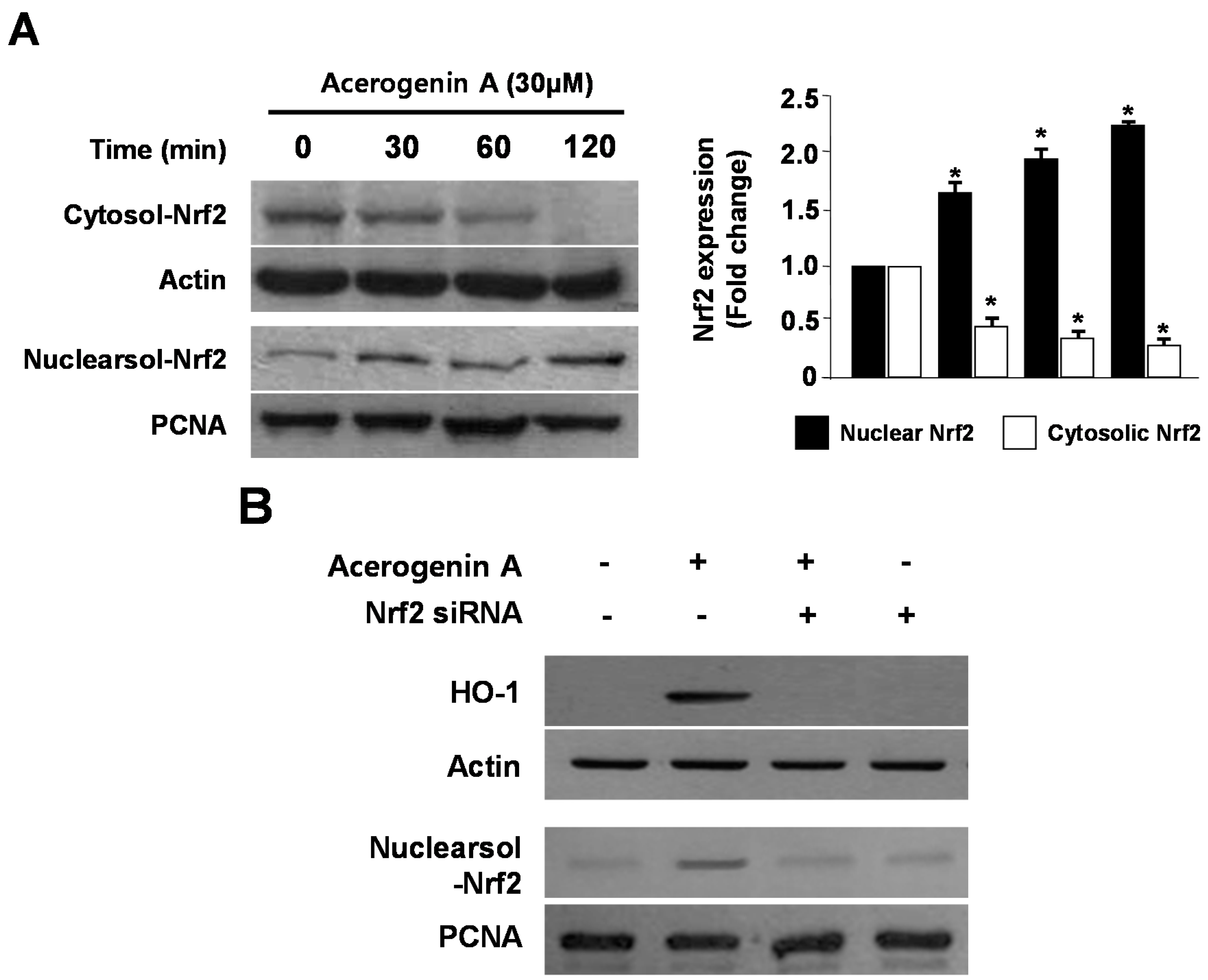
2.3. Effects of Acerogenin A on Nrf2 Nuclear Translocation and Nrf2-Mediated HO-1 Expression in HT22 Cells
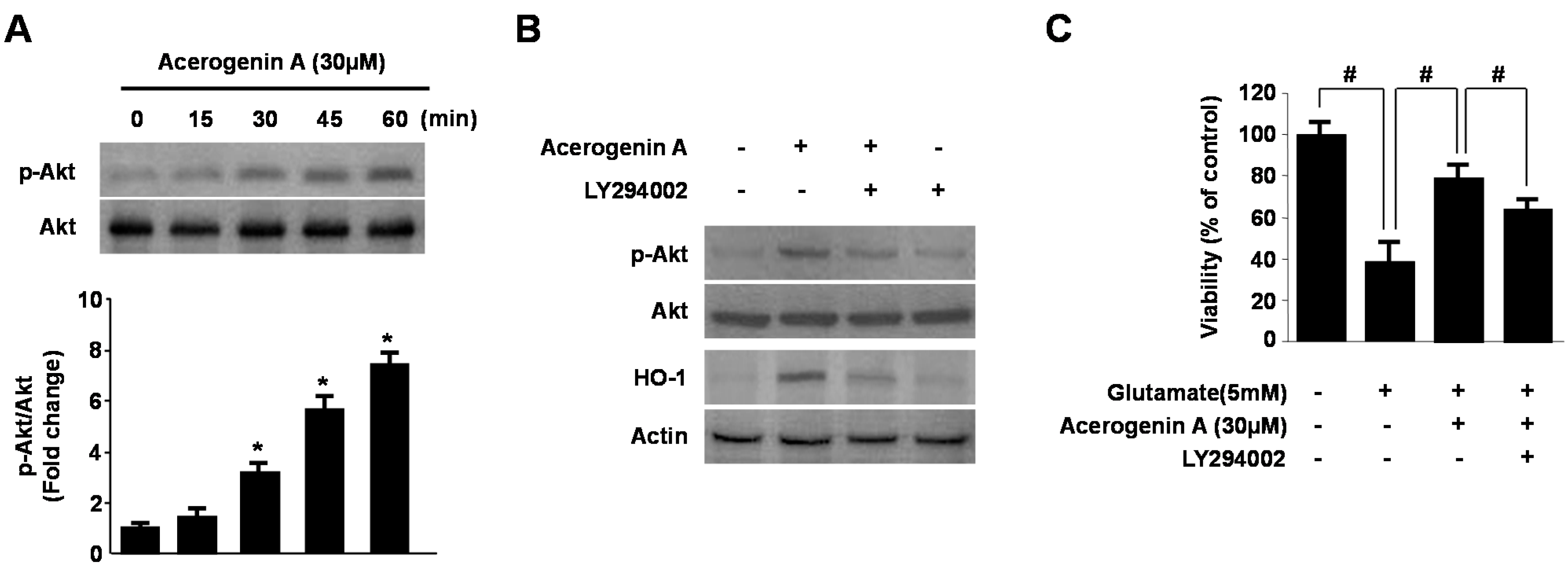
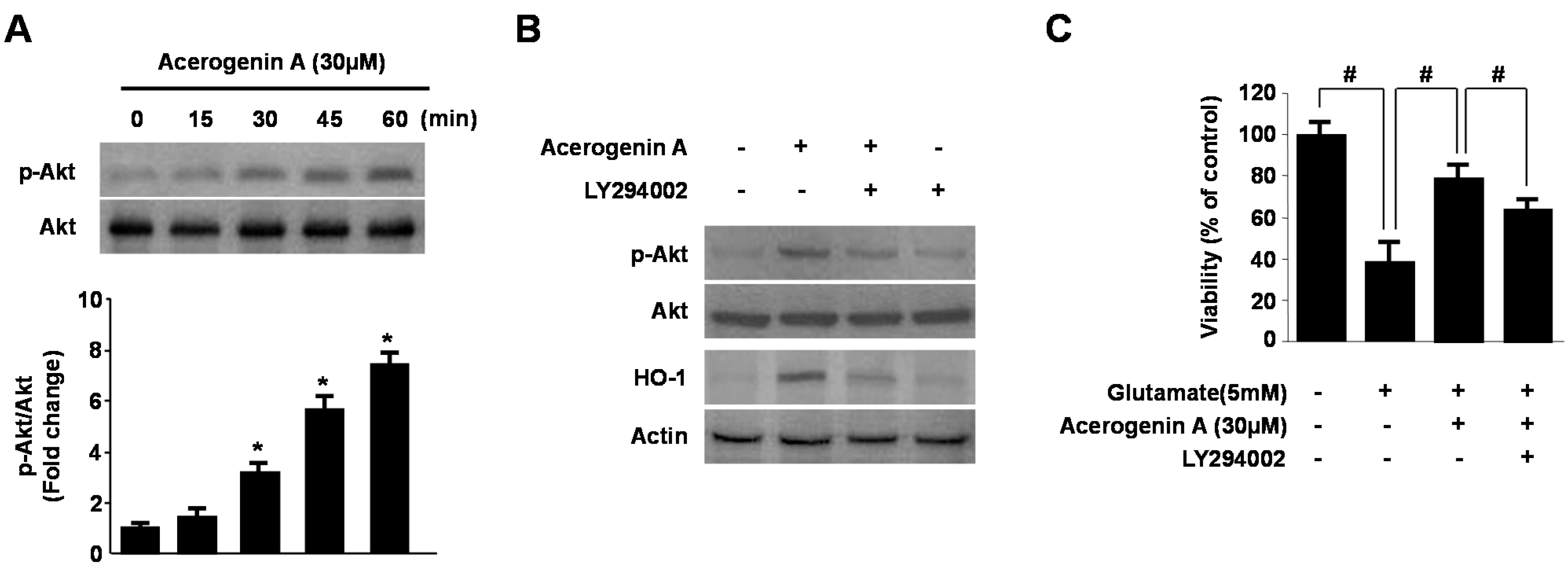
2.4. Involvement of the PI3K/Akt Pathway in Acerogenin A-Induced HO-1 Expression
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cell Culture
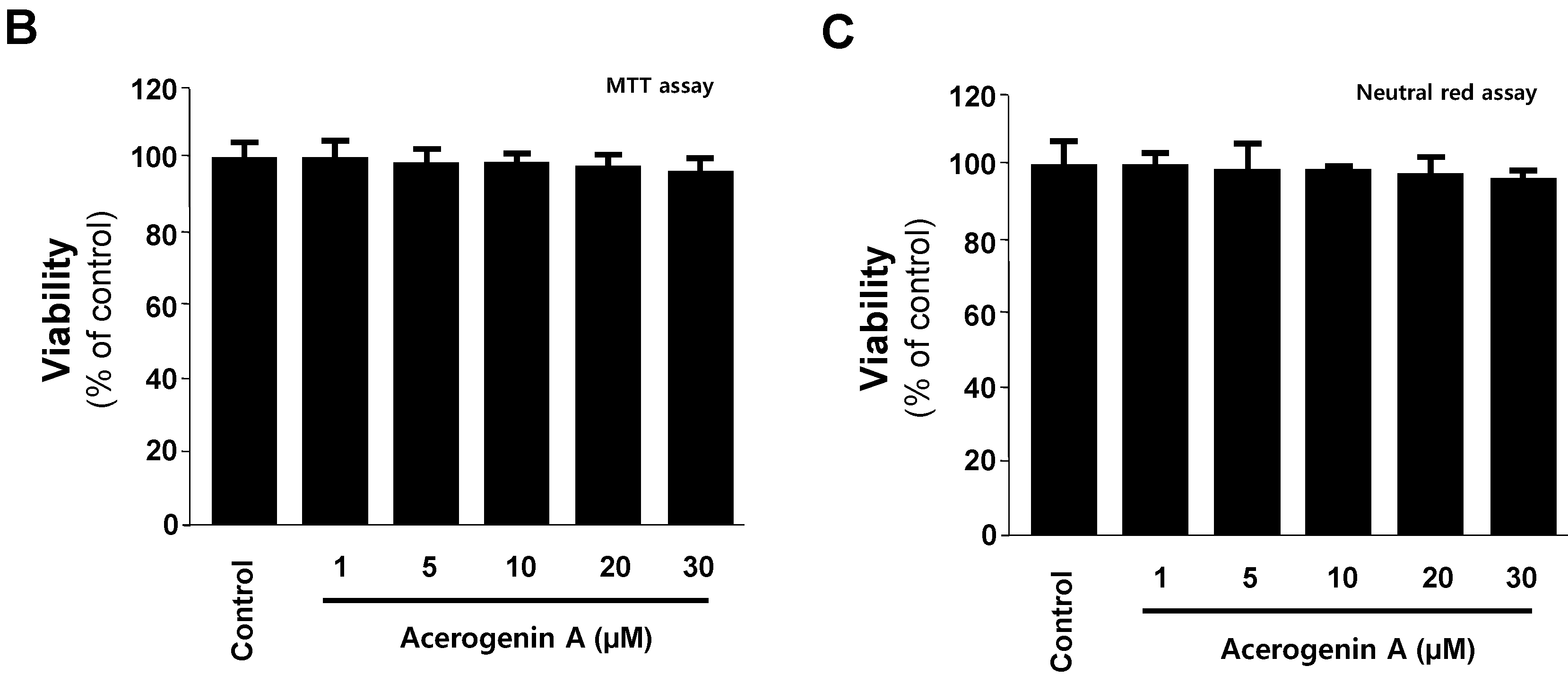
3.3. MTT Assay and Neutral Red Assay
3.4. ROS Measurement
3.5. Preparation of Nuclear and Cytosolic Fractions
3.6. Western Blot Analysis
3.7. Transfection
3.8. RNA Quantification
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hald, A.; Lotharius, J. Oxidative stress and inflammation in Parkinson’s disease: Is there a causal link? Exp. Neurol. 2005, 193, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Mattson, M.P. Roles of lipid peroxidation in modulation of cellular signaling pathways, cell dysfunction, and death in the nervous system. Rev. Neurosci. 1998, 9, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Simonian, N.A.; Coyle, J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Boutten, A.; Goven, D.; Boczkowski, J.; Bonay, M. Oxidative stress targets in pulmonary emphysema: Focus on the NRF2 pathway. Expert Opin. Ther. Targets 2010, 14, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Tuder, R.M. Pathobiology of cigarette smoke induced chronic obstructive pulmonary disease. Physiol. Rev. 2007, 87, 1047–1082. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Otterbein, L.E.; Morse, D.; Choi, A.M.K. Heme oxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol. Cell. Biochem. 2002, 37, 249–263. [Google Scholar] [CrossRef]
- Rőssler, O.G.; Bauer, I.; Chung, H.Y.; Thiel, G. Glutamate-induced cell death of immortalized murine hippocampal neurons: Neuroprotective activity of heme oxygenase-1, heat shock protein 70, and sodium selenite. Neurosci. Lett. 2004, 362, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lee, D.S.; Jeong, G.S.; Kim, Y.C. Involvement of heme oxygenase-1 induction in the cytoprotective and immunomodulatory activities of 6,4′-dihydroxy-7-methoxyflavanone in murine hippocampal and microglia cells. Eur. J. Pharmacol. 2012, 674, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Kubo, M.; Fujita, M.; Inoue, T.; Matsuo, M. Studies on the constituents of Aceraceae Plants. II. Structure of aceroside I, a glucose of a novel cyclic diarylheptanoids from Acer nikoense Maxim. Chem. Pharm. Bull. (Tokyo) 1978, 26, 2805–2810. [Google Scholar] [CrossRef]
- Inoue, T. Constituents of Acer nikoense and Myrica rubra. On diarylheptanoids. Yakugaku Zasshi 1993, 113, 181–197. [Google Scholar] [PubMed]
- Morikawa, T.; Tao, J.; Toguchida, I.; Matsuda, H.; Yoshikawa, M. Structures of new cyclic diarylheptanoids and inhibitors of nitric oxide production from Japanese folk medicine Acer nikoense. J. Nat. Prod. 2003, 66, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Nagumo, S.; Yotsumoto, H.; Moriyama, H.; Nagai, M. Vasorelaxant effects of Acer nikoense extract and isolated coumarinolignans on rat aortic rings. Biol. Pharm. Bull. 2007, 30, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Tao, J.; Ueda, K.; Matsuda, H.; Yoshikawa, M. Medicinal foodstuffs. XXXI. Structures of new aromatic constituents and inhibitors of degranulation in RBL-2H3 cells from Japanese folk medicine, the stem bark of Acer nikoense. Chem. Pharm. Bull. (Tokyo) 2003, 51, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, M.; Ohta, S.; Kumasaka, M.; Fujita, M.; Nagai, M.; Inoue, T. Protective effect of the bark of Acer nikoense on hepatic injury induced by carbon. Shoyakugaku Zasshi 1986, 40, 177–181. [Google Scholar]
- Nagai, M.; Kubo, M.; Fujita, M.; Inoue, T.; Matsuo, M. Acerogenin A, a novel cyclic diarylheptanoid. J. Chem. Soc. Chem. Commun. 1976, 338–339. [Google Scholar] [CrossRef]
- Maioli, E.; Torricelli, C.; Fortino, V.; Carlucci, F.; Tommassini, V.; Pacini, A. Evaluation of viability assays for anthocyanins in cultured cells. Phytochem. Anal. 2008, 1, 479–486. [Google Scholar]
- Elisia, I.; Popovich, D.G.; Hu, C.; Kitts, D.D. Accurate assessment of the bioactivities of redox-active polyphenolics in cell culture. J. Agric. Food Chem. 2008, 56, 7831–7837. [Google Scholar]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Jeong, G.S. Arylbenzofuran isolated from Dalbergia odorifera suppresses lipopolysaccharide-induced mouse BV2 microglial cell activation, which protects mouse hippocampal HT22 cells death from neuroinflammation-mediated toxicity. Eur. J. Pharmacol. 2014, 728, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Ko, W.; Kim, D.C.; Kim, Y.C.; Jeong, G.S. Cudarflavone B provides neuroprotection against glutamate-induced mouse hippocampal HT22 cell damage through the Nrf2 and PI3K/Akt signaling pathways. Molecules 2014, 19, 10818–10831. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Jang, J.H.; Ko, W.; Kim, K.S.; Sohn, J.H.; Kang, M.S.; Ahn, J.S.; Kim, Y.C.; Oh, H. PTP1B inhibitory and anti-inflammatory effects of secondary metabolites isolated from the marine-derived fungus Penicillium sp. JF-55. Mar. Drugs 2013, 11, 1409–1426. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Li, B.; Kim, K.S.; Jeong, G.S.; Kim, E.C.; Kim, Y.C. Butein protects human dental pulp cells from hydrogen peroxide-induced oxidative toxicity via Nrf2 pathway-dependent heme oxygenase-1 expressions. Toxicol. In Vitro 2013, 27, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Cahill, J.M.; Liu, J.; Kuang, X.; Liu, N.; Scofield, V.L.; Voorhees, J.R.; Reid, A.J.; Yan, M.; Lynn, W.S.; et al. Activation of transcription factor Nrf-2 and its downstream targets in response to moloney murine leukemia virus ts1-induced thiol depletion and oxidative stress in astrocytes. J. Virol. 2004, 78, 11926–11938. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bnnai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; de Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3′-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hsu, M.C.; Hsieh, C.W.; Lin, J.B.; Lai, P.H.; Wung, B.S. Upregulation of heme oxygenase-1 by Epigallocatechin-3-gallate via the phosphatidylinositol 3-kinase/Akt and ERK pathways. Life Sci. 2006, 78, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the acerogenin A are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-S.; Cha, B.-Y.; Woo, J.-T.; Kim, Y.-C.; Jang, J.-H. Acerogenin A from Acer nikoense Maxim Prevents Oxidative Stress-Induced Neuronal Cell Death through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse Hippocampal HT22 Cell Line. Molecules 2015, 20, 12545-12557. https://doi.org/10.3390/molecules200712545
Lee D-S, Cha B-Y, Woo J-T, Kim Y-C, Jang J-H. Acerogenin A from Acer nikoense Maxim Prevents Oxidative Stress-Induced Neuronal Cell Death through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse Hippocampal HT22 Cell Line. Molecules. 2015; 20(7):12545-12557. https://doi.org/10.3390/molecules200712545
Chicago/Turabian StyleLee, Dong-Sung, Byung-Yoon Cha, Je-Tae Woo, Youn-Chul Kim, and Jun-Hyeog Jang. 2015. "Acerogenin A from Acer nikoense Maxim Prevents Oxidative Stress-Induced Neuronal Cell Death through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse Hippocampal HT22 Cell Line" Molecules 20, no. 7: 12545-12557. https://doi.org/10.3390/molecules200712545
APA StyleLee, D.-S., Cha, B.-Y., Woo, J.-T., Kim, Y.-C., & Jang, J.-H. (2015). Acerogenin A from Acer nikoense Maxim Prevents Oxidative Stress-Induced Neuronal Cell Death through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse Hippocampal HT22 Cell Line. Molecules, 20(7), 12545-12557. https://doi.org/10.3390/molecules200712545