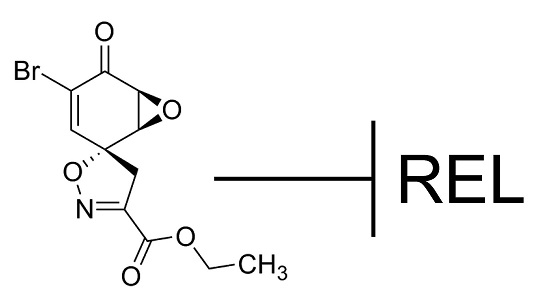
2.1. Calafianin Monomer (CM101) Preferentially Inhibits REL and p65 DNA-Binding Activity
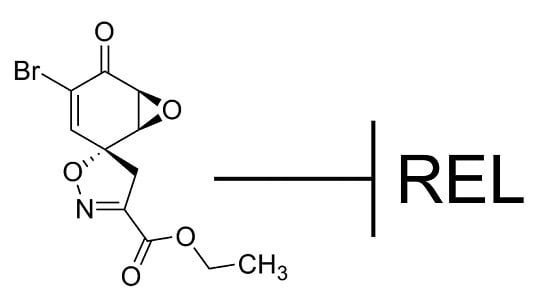
While screening for compounds that inhibit NF-κB signaling, we identified calafianin monomer (CM101) as a promising hit. CM101, the monomer unit of the spiroisoxazoline natural product [
16] calafianin [
17], shares chemical moieties (
i.e., epoxy ketone, α,β-unsaturated carbonyl) with other known NF-κB inhibitors such as parthenolide, DHMEQ, and epoxyquinone A monomer, but substantially differs in its overall chemical structure (
Figure 1).
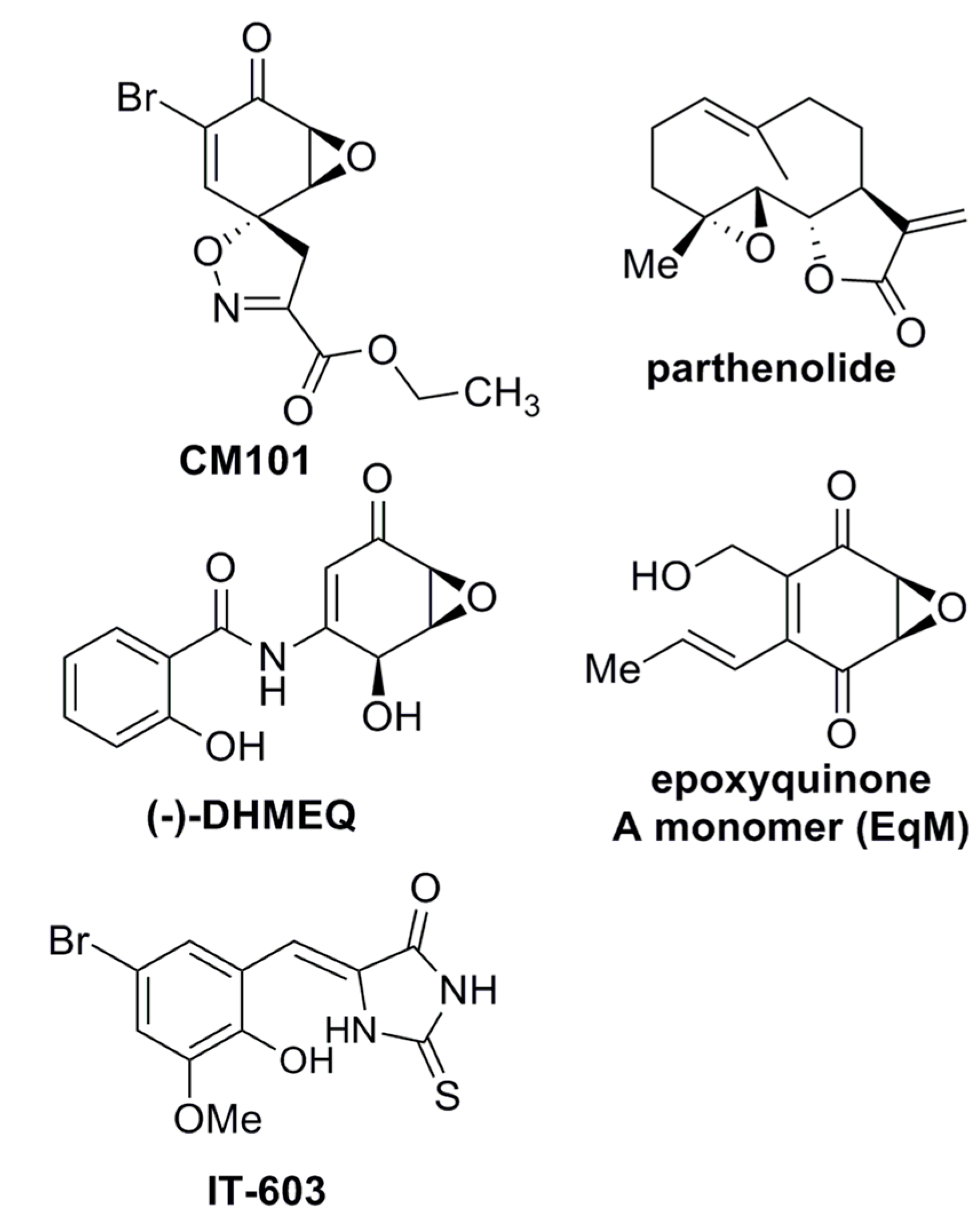
Figure 1.
Structures of compounds with anti-REL activity. Shown are the chemical structures of CM101, parthenolide [
18,
19], DHMEQ [(−)-dehydroxymethylepoxquinomicin] [
20], epoxyquinone A monomer [
21], and IT-603 [
22].
Figure 1.
Structures of compounds with anti-REL activity. Shown are the chemical structures of CM101, parthenolide [
18,
19], DHMEQ [(−)-dehydroxymethylepoxquinomicin] [
20], epoxyquinone A monomer [
21], and IT-603 [
22].
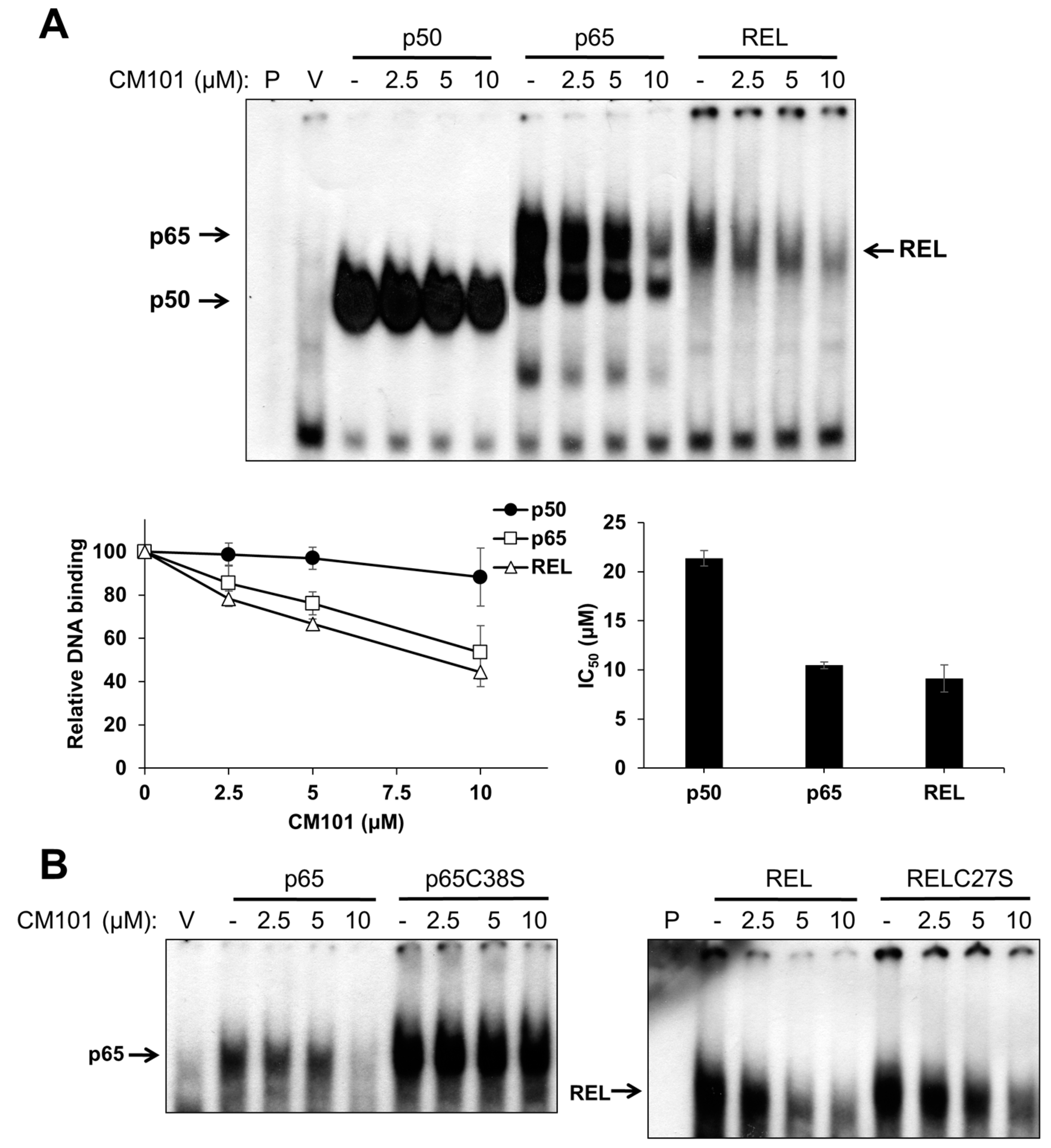
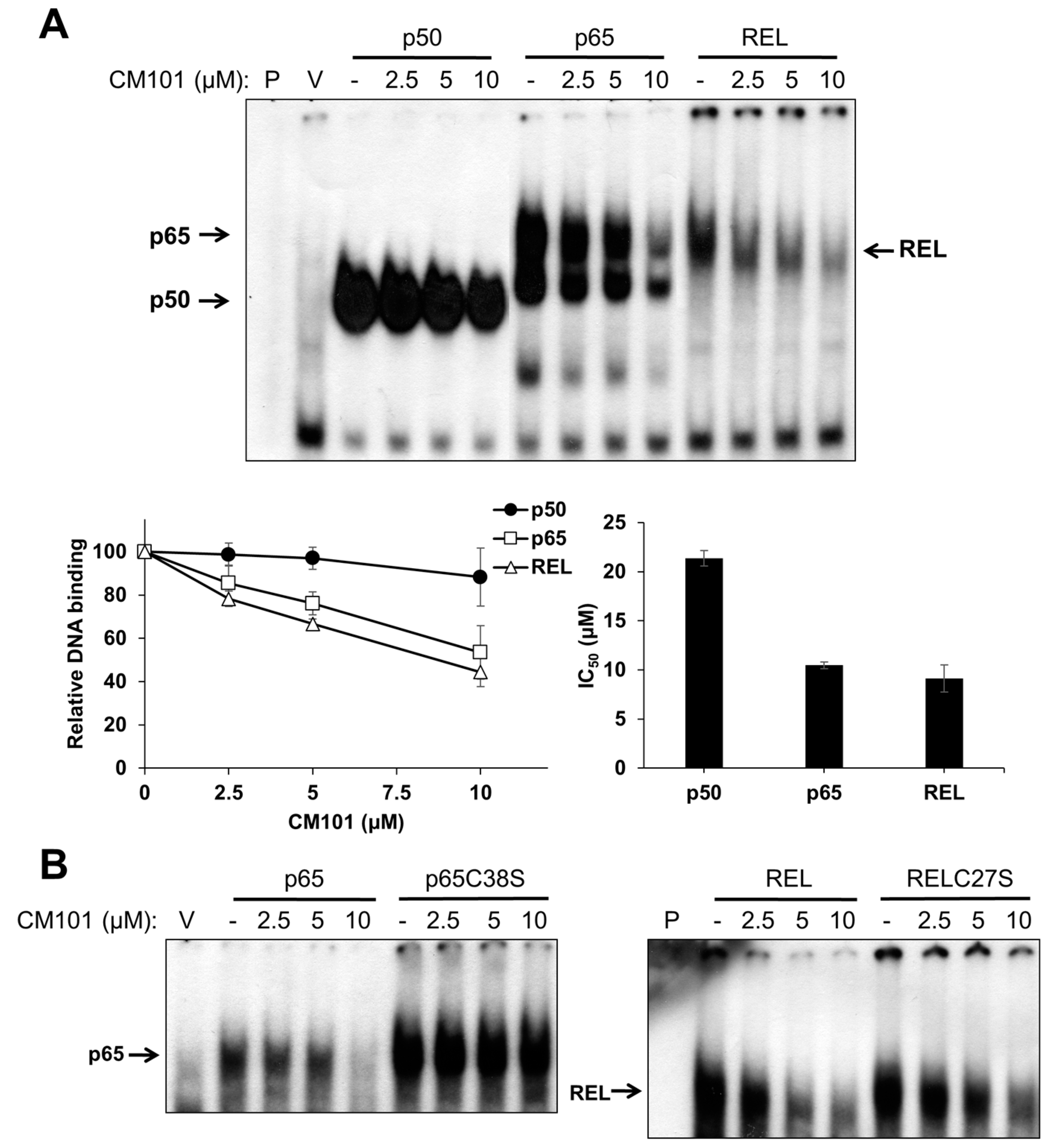
To assess the effects of CM101 on the DNA-binding activity of canonical NF-κB subunits, human 293 cells were transfected with expression vectors for FLAG-tagged p50, p65, and REL. Two days later, transfected cells were treated with increasing concentrations of CM101 for 2 h, and then extracts were prepared and analyzed by electrophoretic mobility shift assay (EMSA) using a consensus NF-κB probe. The DNA-binding activities of p65 and REL were inhibited by CM101 at doses as low as 2.5 µM, whereas inhibition of p50 began at 10 µM (
Figure 2A) The IC
50 values for CM101 inhibition of p65 and REL DNA-binding activity were approximately 10 µM, whereas the IC
50 for inhibition of p50 was greater than 20 µM.
Figure 2.
CM101 selectively inhibits REL and p65 but not p50 through covalent modification of cysteine residues. (A) 293 cells were transfected with expression vectors encoding FLAG-tagged human p50, p65 and REL, or with the empty vector (V). Transfected cells were treated with the indicated concentrations of calafianin monomer (CM101) for 2 h, extracts were made, and analyzed by EMSA with a κB-site probe. EMSA bands were detected by phosphorimaging and quantified. IC50 values were determined by plotting the band intensities at each concentration of CM101, using the control (no CM101) band intensity as 100%. All samples were treated with up to 20 µM CM101 (not shown), which was required to calculate the IC50 value for p50. Error bars indicate standard errors (three independent experiments); (B) 293 cells transfected with expression vectors encoding p65, p65C38S, REL or RELC27S were treated with CM101 and analyzed by EMSA as in (A). P, EMSA lane containing the probe alone.
Figure 2.
CM101 selectively inhibits REL and p65 but not p50 through covalent modification of cysteine residues. (A) 293 cells were transfected with expression vectors encoding FLAG-tagged human p50, p65 and REL, or with the empty vector (V). Transfected cells were treated with the indicated concentrations of calafianin monomer (CM101) for 2 h, extracts were made, and analyzed by EMSA with a κB-site probe. EMSA bands were detected by phosphorimaging and quantified. IC50 values were determined by plotting the band intensities at each concentration of CM101, using the control (no CM101) band intensity as 100%. All samples were treated with up to 20 µM CM101 (not shown), which was required to calculate the IC50 value for p50. Error bars indicate standard errors (three independent experiments); (B) 293 cells transfected with expression vectors encoding p65, p65C38S, REL or RELC27S were treated with CM101 and analyzed by EMSA as in (A). P, EMSA lane containing the probe alone.
Some NF-κB subunit inhibitors require a conserved Cys residue that is present in p65 (Cys38) and REL (Cys27) for maximal inhibition [
6,
18,
19,
20,
21,
23]. To determine whether inhibition by CM101 also requires this Cys residue, 293 cells transfected with expression vectors for wild-type or C38S mutant p65 and wild-type or C27S mutant REL were treated with increasing concentrations of CM101, and extracts were analyzed by EMSA. For both p65 and REL, the Cys mutants showed reduced inhibition of DNA binding by CM101 as compared to the respective wild-type proteins (
Figure 2B). Western blotting of the same extracts under standard reducing SDS-PAGE conditions showed that treatment with CM101 converted a portion of wild-type p65 and REL to high molecular weight, covalently modified forms, whereas p50 cross-linking appeared to be restricted to a dimer (
Figure S1A). Migration of the Cys-to-Ser mutant of REL was not affected by the same concentrations of CM101 (
Figure S1B). Therefore, CM101-mediated inhibition of p65 and REL DNA binding may involve covalent modification at Cys38 and Cys27, respectively.
2.2. CM101 Inhibits Proliferation and Induces Apoptosis in B-Lymphoma Cell Lines
REL has been previously shown to be required for the proliferation of several B-lymphoma cell lines [
2,
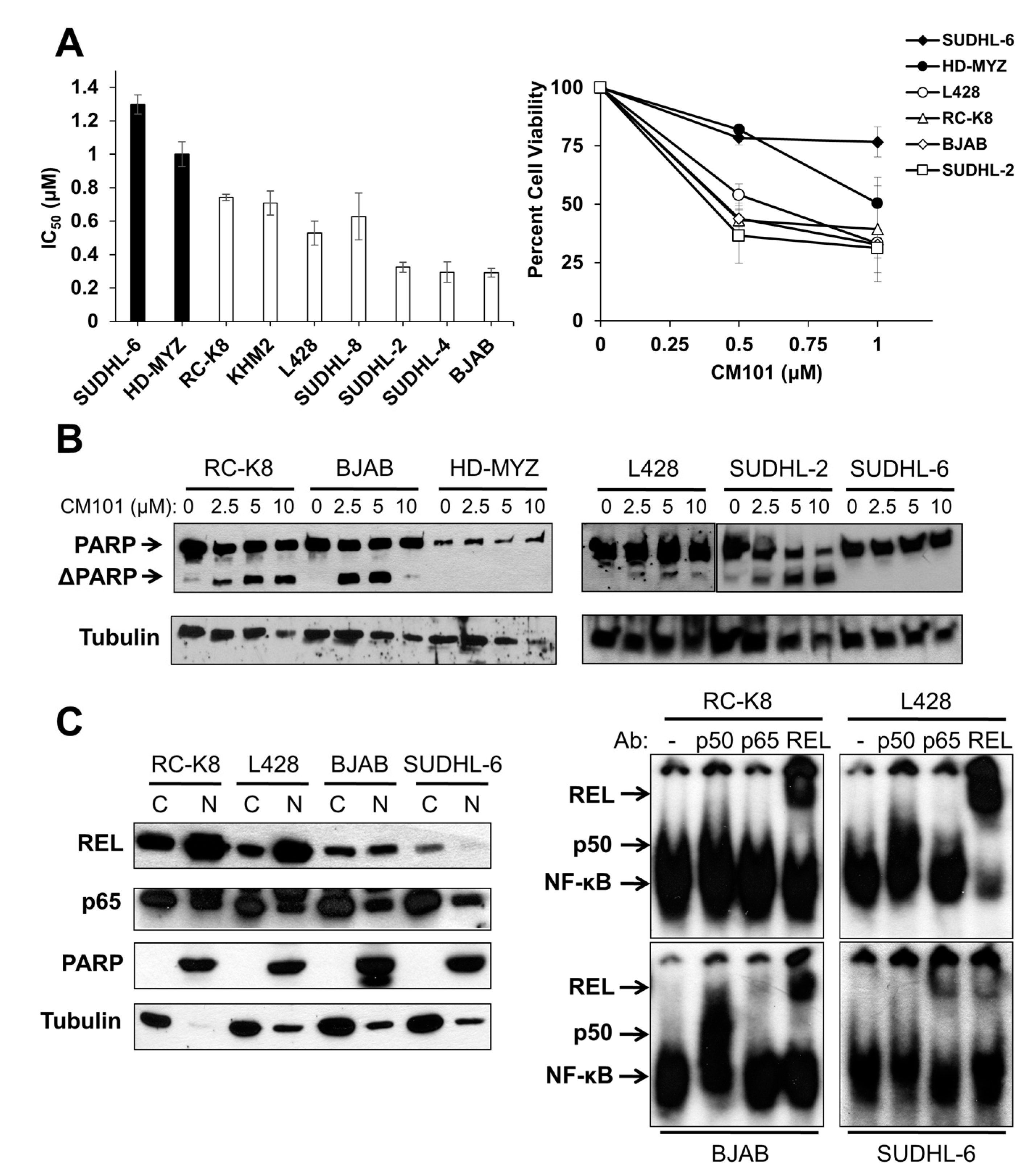
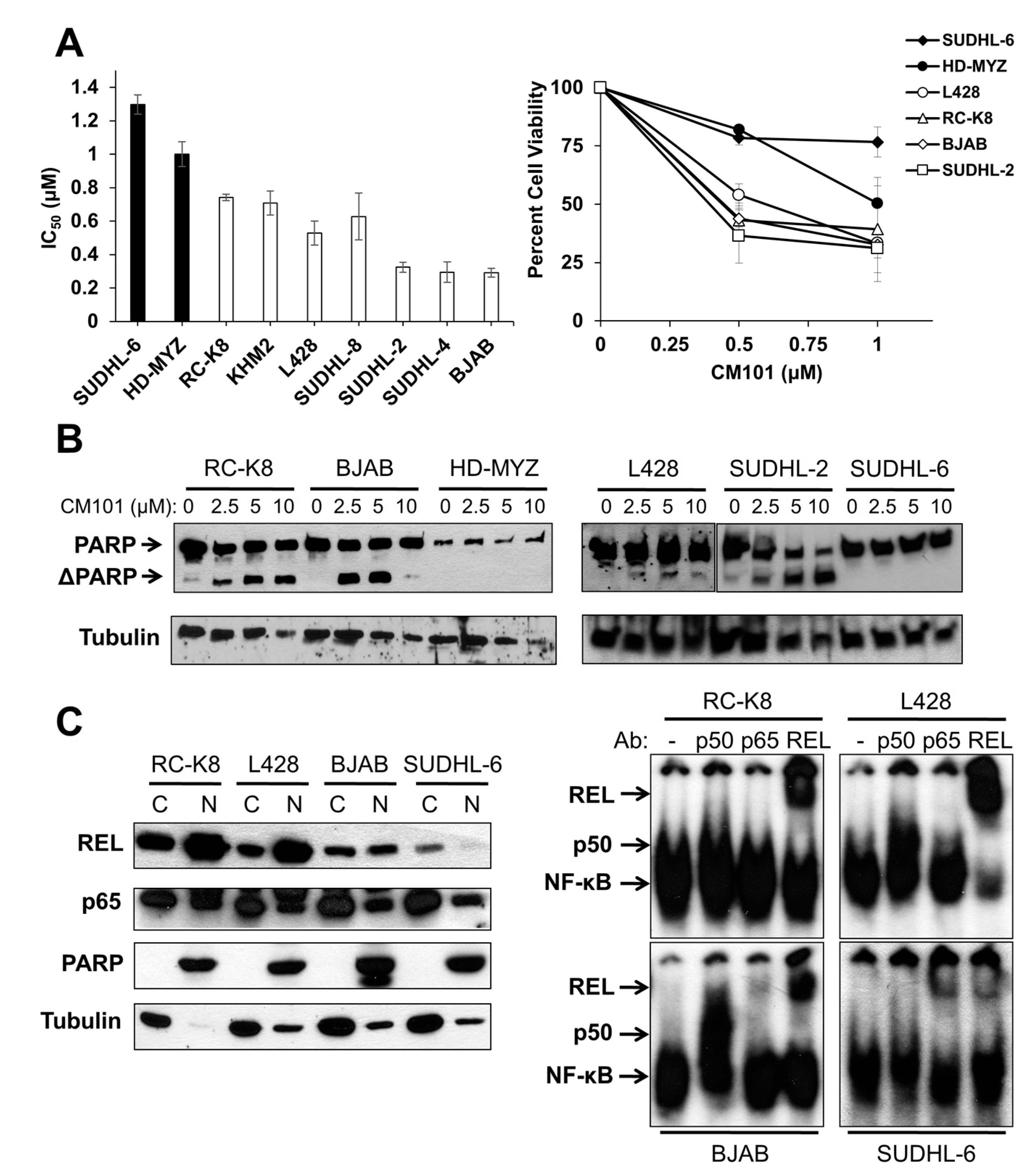
14]. Because of CM101’s ability to inhibit REL DNA binding, we assessed the effect of CM101 on the proliferation and survival of human B-lymphoma cell lines. To do this, we treated nine human B-lymphoma cell lines with increasing concentrations of CM101 for 72 h and then counted cells at this time point. CM101 reduced cell proliferation in the different cell lines to varying degrees (
Figure S2A). Based on the IC
50 values for inhibition of cell proliferation by CM101 (
Figure 3A), the nine B-lymphoma cell lines were separated into relatively resistant cell lines (defined as ones with IC
50 ≥; 0.9 µM; SUDHL-6, HD-MYZ) and relatively sensitive cell lines (with the IC
50 < 0.9 µM; RC-K8, KMH2, L428, SUDHL-8, SUDHL-2, SUDHL-4, BJAB). As a second measure of the effect of CM101 on lymphoma cells, we demonstrated that CM101 also had lesser effects on the cell viability of resistant SUDHL-6 and HD-MYZ cells as judged by an MTT assay (
Figure 3A), which measures the activity of mitochondrial oxidoreductases that are associated with cell viability. In contrast, CM101 greatly reduced cell viability in all of the sensitive B-lymphoma cell lines.
We have previously shown that cleavage of the caspase-3 substrate poly(ADP-ribose) polymerase (PARP) is a reliable indicator of apoptosis in many of these same B-lymphoma cell lines [
6,
21,
24]. To determine whether CM101 can induce apoptosis in B-lymphoma cell lines, four sensitive and two resistant B-lymphoma cell lines were treated with increasing concentrations of CM101 for 24 h, and cleavage of PARP was monitored by western blotting (
Figure 3B). CM101 induced cleavage of PARP in the sensitive SUDHL-2, BJAB, and RC-K8 cell lines and to a lesser extent in L428 cells, but CM101 did not induce detectable PARP cleavage in resistant SUDHL-6 and HD-MYZ cell lines. In all six cell lines, CM101 induced apoptosis with potencies that had the same relative order as the IC
50 values measured in the cell proliferation and MTT assays (
Figure 3B). These results indicate that CM101 can selectively induce both cell growth arrest and apoptosis in certain B-lymphoma cell lines.
Figure 3.
Sensitivity of human B-lymphoma cell lines to CM101 is correlated with active REL nuclear complexes. (
A) A panel of B-lymphoma cell lines were seeded in triplicate at a cell number of 10
5. Approximately 18 h later, cells were treated with increasing concentrations of CM101, incubated for an additional 72 h and cells were then counted. Cell numbers are relative to methanol-treated control cells (left panel). IC
50 values were calculated by plotting the relative cell numbers at each concentration of CM101, which were normalized to cells incubated with no CM101 (100%) (see
Figure S2A for the graph that was used to calculate IC
50 values). Error bars indicate standard error. Right panel, MTT assays were performed and cell numbers were normalized to methanol-treated control cells; (
B) The indicated B-lymphoma cell lines treated with various concentrations of CM101 for 24 h and cell extracts were analyzed for PARP cleavage by western blotting. The full-length and cleaved PARP (ΔPARP) bands are indicated. β-Tubulin western blotting was performed as a loading control; (
C) Nuclear levels of REL were assessed from cytosolic and nuclear extracts prepared from RC-K8, L428, BJAB and SUDHL-6 cells. Extracts were analyzed by western blotting for REL and p65 (left panel). PARP and β-tubulin western blotting was performed to assess the purity of the nuclear and cytoplasmic fractions, respectively. EMSAs were performed on nuclear extracts with no antibody or antibodies for p50, p65 or REL for supershift analyses (right panel).
Figure 3.
Sensitivity of human B-lymphoma cell lines to CM101 is correlated with active REL nuclear complexes. (
A) A panel of B-lymphoma cell lines were seeded in triplicate at a cell number of 10
5. Approximately 18 h later, cells were treated with increasing concentrations of CM101, incubated for an additional 72 h and cells were then counted. Cell numbers are relative to methanol-treated control cells (left panel). IC
50 values were calculated by plotting the relative cell numbers at each concentration of CM101, which were normalized to cells incubated with no CM101 (100%) (see
Figure S2A for the graph that was used to calculate IC
50 values). Error bars indicate standard error. Right panel, MTT assays were performed and cell numbers were normalized to methanol-treated control cells; (
B) The indicated B-lymphoma cell lines treated with various concentrations of CM101 for 24 h and cell extracts were analyzed for PARP cleavage by western blotting. The full-length and cleaved PARP (ΔPARP) bands are indicated. β-Tubulin western blotting was performed as a loading control; (
C) Nuclear levels of REL were assessed from cytosolic and nuclear extracts prepared from RC-K8, L428, BJAB and SUDHL-6 cells. Extracts were analyzed by western blotting for REL and p65 (left panel). PARP and β-tubulin western blotting was performed to assess the purity of the nuclear and cytoplasmic fractions, respectively. EMSAs were performed on nuclear extracts with no antibody or antibodies for p50, p65 or REL for supershift analyses (right panel).
![]()
2.3. CM101 Inhibits REL DNA-Binding Activity in B-Lymphoma Cell Lines
To determine whether REL plays a role in the sensitivity of B-lymphoma cells to CM101, we first compared nuclear levels of REL in sensitive cell lines RC-K8, L428, and BJAB to levels in resistant SUDHL-6 cells. As a control for the integrity of the fractions, the blots were also probed for PARP (nuclear marker) and β-tubulin (cytosolic marker). Sensitive cell lines RC-K8 and L428 displayed high levels of nuclear REL, and BJAB had moderate levels of nuclear REL. Resistant SUDHL-6 cells had low levels of nuclear REL. In contrast, the levels of nuclear p65 did not differ among the four cell lines (
Figure 3C). These results suggest that nuclear levels of REL, but not p65, correlate with the sensitivity of these cells to CM101.
To determine whether nuclear REL is active in these same four cell lines, an EMSA was performed on nuclear extracts. Constitutive κB site-binding activity was detected in all four cell lines, with the highest levels being in RC-K8 and L428 cells, lower levels in BJAB cells, and much lower levels in SUDHL-6 cells (
Figure 3C). Using subunit-specific antibodies for supershifts, we found that RC-K8, L428 and BJAB EMSA complexes contained mostly REL, whereas SUDHL-6 complexes contained primarily p65 with lesser amounts of REL (
Figure 3C). These results indicate that the sensitivity of B-lymphoma cells to CM101 correlates with the presence of nuclear REL DNA-binding activity, and not with the level or activity of p65.
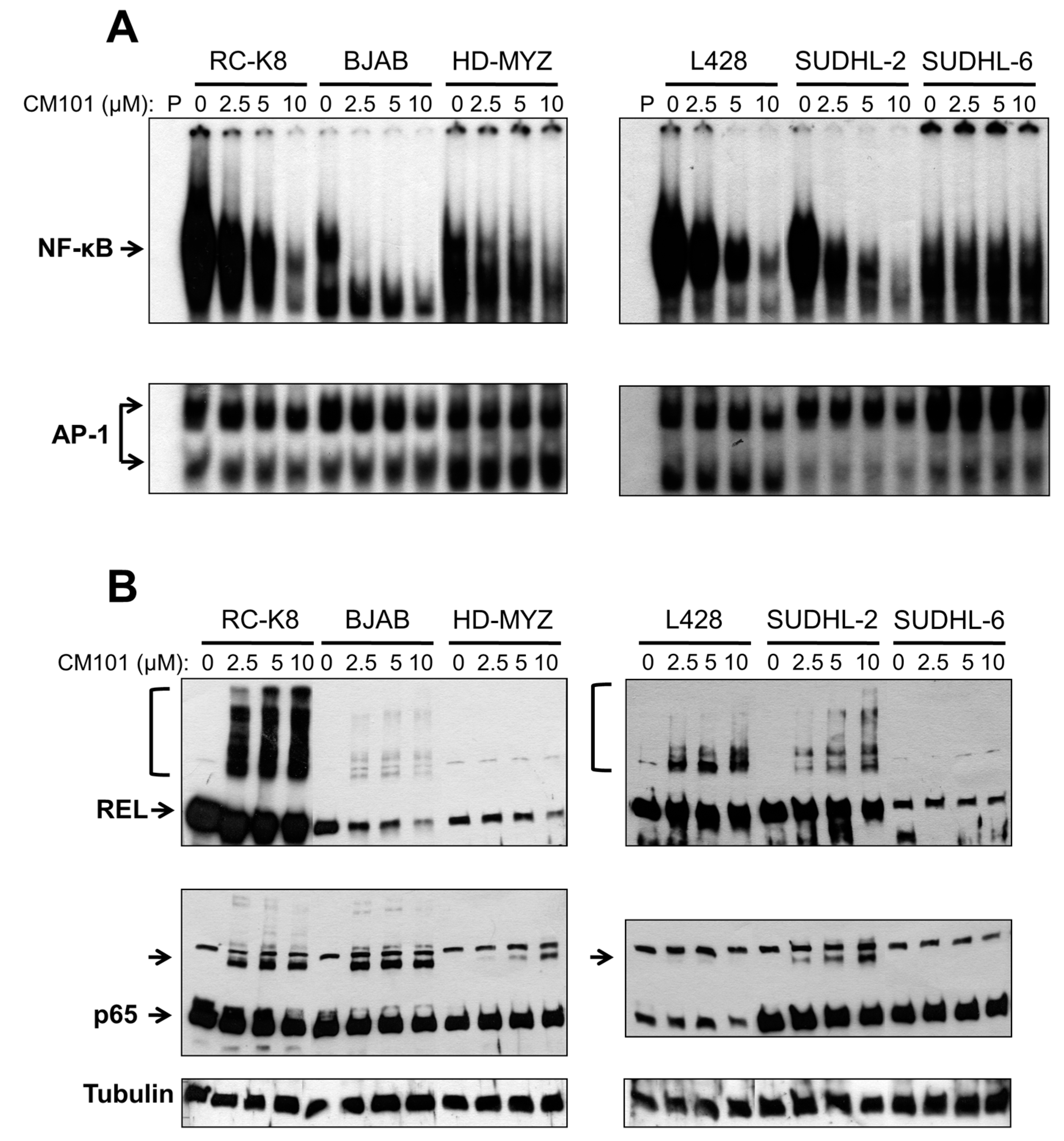
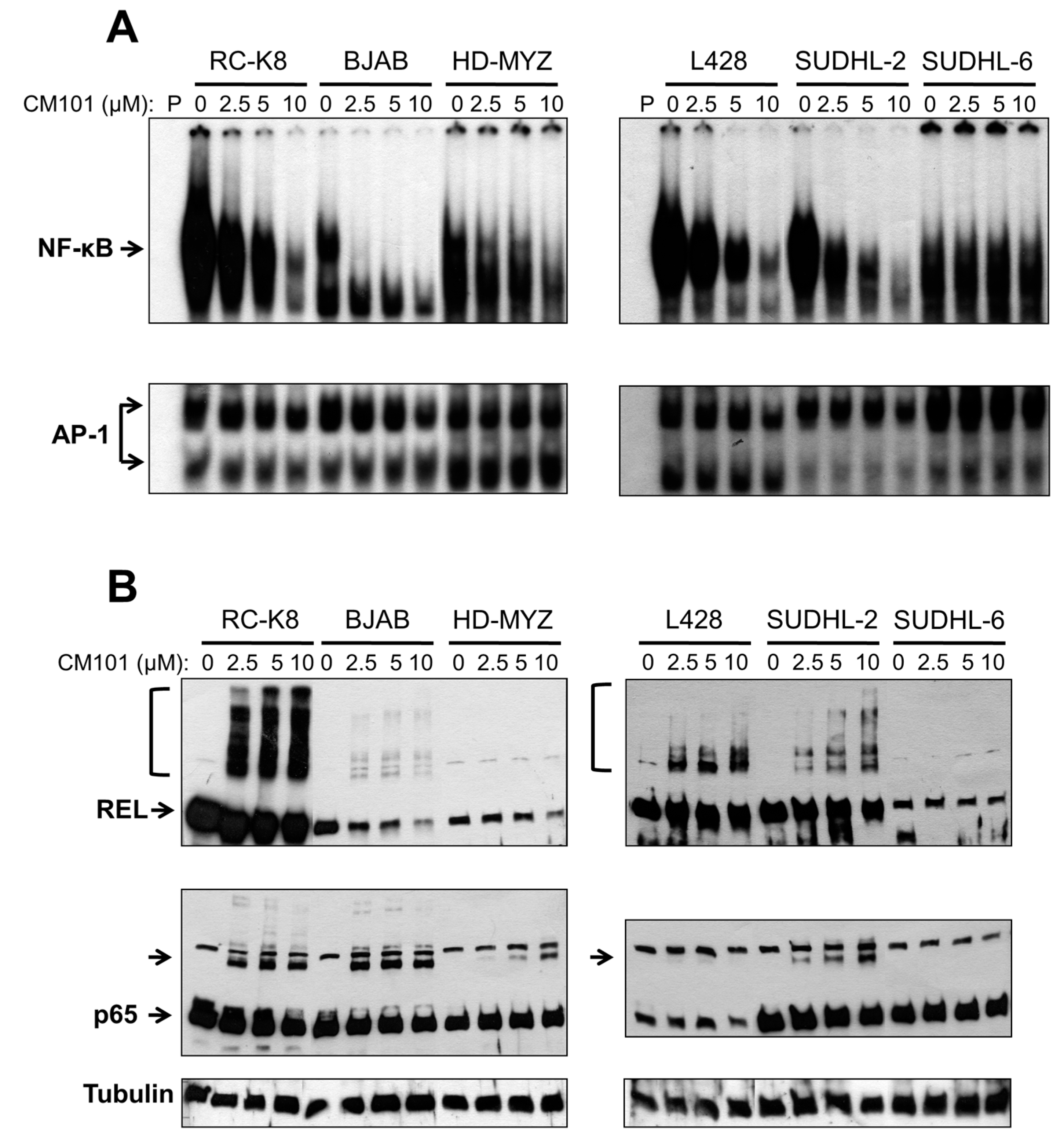
We next investigated whether CM101 could inhibit constitutive NF-κB DNA binding in these lymphoma cells, and if so, whether CM101-induced inhibition of nuclear κB-site DNA binding correlated with the ability of CM101 to induce growth arrest and apoptosis. Sensitive (RC-K8 BJAB, L428 and SUDHL-2) and resistant (HD-MYZ, SUDHL-6) cell lines were treated with increasing concentrations of CM101 for 2 h, and extracts were analyzed by EMSA using a κB-site probe. The amount of cell extract used in the EMSAs was adjusted to make the amount of κB-site DNA-binding activity approximately equal for the various cell lines (see legend to
Figure 4A). CM101 efficiently inhibited NF-κB DNA-binding activity in sensitive cell lines (BJAB, SUDHL-2, RC-K8, L428), but showed much less inhibition of NF-κB DNA-binding activity in resistant cells (HD-MYZ, SUDHL-6 cells), which had considerably less overall nuclear κB-site binding activity (
Figure 4A). These results suggest that inhibition of NF-κB DNA binding by CM101 is responsible for its ability to induce cell growth arrest and apoptosis. Of note, CM101 did not inhibit transcription factor AP-1 DNA-binding activity at similar concentrations in these cell lines (
Figure 4A). Thus, CM101 has a selective ability to inhibit NF-κB DNA binding among B-lymphoma cell lines, and B-lymphoma cell lines with high κB-site DNA-binding activity are sensitive to CM101-based inhibition.
To determine whether CM101 converts REL to higher molecular weight forms in B-lymphoma cells, as seen in 293 cells (
Figure S1), western blotting was performed on the same lymphoma cell extracts used for the EMSAs in
Figure 4A. REL was converted to high molecular weight forms in a dose-dependent manner in sensitive cell lines (RC-K8, L428, BJAB, SUDHL-2), but not in resistant cell lines (HD-MYZ, SUDHL-6) (
Figure 4B). p65 had a lesser ability than REL to be converted to higher molecular weight forms. Additionally, the low basal κB-site DNA-binding activity in CM101-resistant SUDHL-6 and HD-MYZ cells may explain why they are not dependent on NF-κB signaling for survival.
Figure 4.
CM101 inhibits REL/NF-κB DNA binding in sensitive B-lymphoma cell lines. The named B-lymphoma cell lines were treated with the indicated concentrations of CM101 for 2 h, and extracts were made. (A) EMSAs were performed using a κB-site probe (top panels) or an AP-1 site probe (bottom panels). To make the amount of κB-site DNA-binding activity approximately equal for the various cell lines, the following amounts of cell extract were used: RC-K8, 5 µg; BJAB, 10 µg; HD-MYZ, 40 µg; L428, 10 µg; SUDHL-2, 15 µg; and SUDHL-6, 60 µg; (B) To assess the extent of cross-linking induced by CM101, extracts prepared from (A) were subjected to anti-REL or anti-p65 western blotting as indicated. REL and p65 indicate the monomer forms of the proteins. Brackets (for REL, top panels) or unlabeled arrow (p65 blots) designate higher molecular weight forms of the proteins. β-Tubulin western blotting was performed as a loading control (bottom panel).
Figure 4.
CM101 inhibits REL/NF-κB DNA binding in sensitive B-lymphoma cell lines. The named B-lymphoma cell lines were treated with the indicated concentrations of CM101 for 2 h, and extracts were made. (A) EMSAs were performed using a κB-site probe (top panels) or an AP-1 site probe (bottom panels). To make the amount of κB-site DNA-binding activity approximately equal for the various cell lines, the following amounts of cell extract were used: RC-K8, 5 µg; BJAB, 10 µg; HD-MYZ, 40 µg; L428, 10 µg; SUDHL-2, 15 µg; and SUDHL-6, 60 µg; (B) To assess the extent of cross-linking induced by CM101, extracts prepared from (A) were subjected to anti-REL or anti-p65 western blotting as indicated. REL and p65 indicate the monomer forms of the proteins. Brackets (for REL, top panels) or unlabeled arrow (p65 blots) designate higher molecular weight forms of the proteins. β-Tubulin western blotting was performed as a loading control (bottom panel).
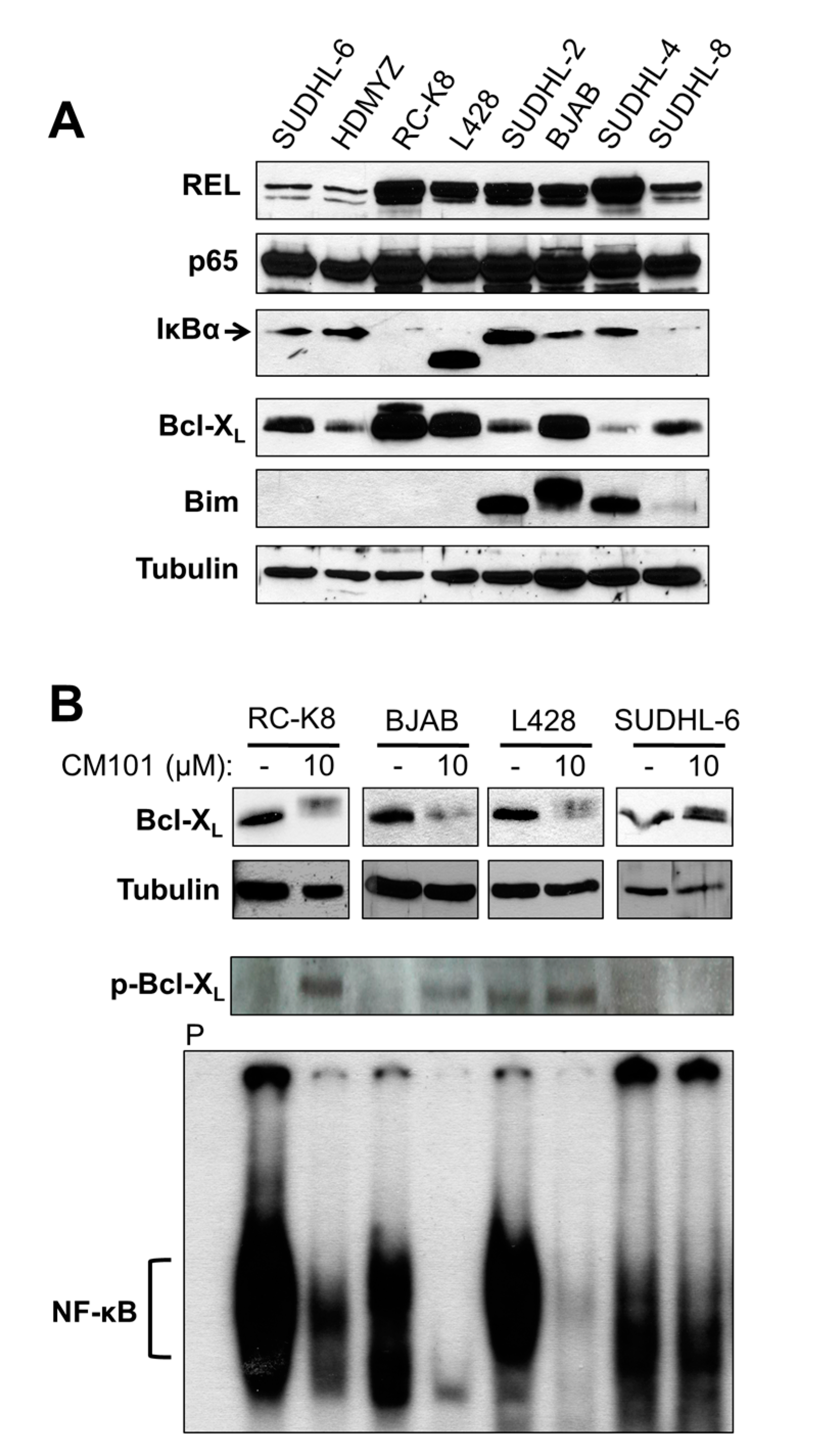
2.4. Cellular Levels of REL Predict the Sensitivity of B-Lymphoma Cells to CM101, Which May Involve a REL-to-Bcl-XL Pathway
We have previously shown that the sensitivity of B-lymphoma cell lines to apoptosis-inducing compounds depends on levels of Bcl-2 family proteins [
23,
24]. Moreover, anti-apoptotic protein Bcl-X
L is the product of a REL target gene [
25]. To identify proteins in the NF-κB and Bcl-2 families that could be important for the sensitivity of B-lymphoma cell lines to CM101, whole-cell extracts from the eight B-lymphoma cell lines were analyzed by western blotting for expression of NF-κB signaling proteins (REL, p65, IκBα) and of two major apoptotic regulatory proteins (anti-apoptotic Bcl-X
L, pro-apoptotic Bim) (
Figure 5A). Consistent with the EMSA results (
Figure 4A), the total cellular levels of REL were low in CM101-resistant cell lines SUDHL-6 and HD-MYZ, whereas REL levels were high in all six CM101-sensitive cell lines. The p65 levels were similar in all eight cell lines. Moreover, all cell lines that were sensitive to CM101 had high levels of Bcl-X
L and/or high levels of Bim, whereas the two resistant cell lines, SUDHL-6 and HD-MYZ, had comparatively low levels of Bcl-X
L and no detectable Bim.
Figure 5.
Total cellular levels of REL generally correlate with the sensitivity of B-lymphoma cells to CM101-induced apoptosis. (A) Extracts from the indicated B-lymphoma cell lines were probed by western blotting for REL, p65, IκBα, Bcl-XL, and Bim. β-Tubulin western blotting was performed as a loading control. (B) B-lymphoma cell lines RC-K8, L428, BJAB and SUDHL-6 were treated for 24 h with either the solvent methanol (−) or 10 μM CM101 (+). Anti-Bcl-XL, anti-phospho-Bcl-XL and β-tubulin (loading control) western blotting was then performed on whole-cell extracts (upper three panels). In the lower panel, the indicated cell extracts (RC-K8, 10 µg; BJAB, 10 µg; L428, 15 µg, SUDHL-6, 80 µg) were analyzed by EMSA with a κB site-containing probe. P designates an EMSA lane containing the probe alone.
Figure 5.
Total cellular levels of REL generally correlate with the sensitivity of B-lymphoma cells to CM101-induced apoptosis. (A) Extracts from the indicated B-lymphoma cell lines were probed by western blotting for REL, p65, IκBα, Bcl-XL, and Bim. β-Tubulin western blotting was performed as a loading control. (B) B-lymphoma cell lines RC-K8, L428, BJAB and SUDHL-6 were treated for 24 h with either the solvent methanol (−) or 10 μM CM101 (+). Anti-Bcl-XL, anti-phospho-Bcl-XL and β-tubulin (loading control) western blotting was then performed on whole-cell extracts (upper three panels). In the lower panel, the indicated cell extracts (RC-K8, 10 µg; BJAB, 10 µg; L428, 15 µg, SUDHL-6, 80 µg) were analyzed by EMSA with a κB site-containing probe. P designates an EMSA lane containing the probe alone.
To determine if Bcl-X
L plays a role in CM101-induced apoptosis, we analyzed the effect of CM101 treatment on expression of Bcl-X
L in sensitive (RC-K8, L428, BJAB) and resistant (SUDHL-6) cells. Treatment with 10 µM CM101 for 24 h led to a reduction in Bcl-X
L levels in sensitive cell lines, whereas CM101 treatment did not affect the Bcl-X
L level in SUDHL-6 cells (
Figure 5B). Moreover, CM101-sensitive cells showed the appearance of a slightly higher molecular weight form of Bcl-X
L, which we hypothesized to be a phosphorylated form of Bcl-X
L seen in cells treated with other cytotoxic agents [
26,
27,
28,
29,
30]. Indeed, anti-phospho-Bcl-X
L western blotting showed that treatment with CM101 led to increased levels of phosophorylated Bcl-X
L in sensitive (RC-K8, L428, BJAB) but not resistant (SUDHL-6) cells (
Figure 5B). Furthermore, REL DNA-binding activity was inhibited by treatment with 10 µM CM101 for 24 h in sensitive cell lines, but κB-site binding was not inhibited in resistant SUDHL-6 cells (
Figure 5B). These results show that down-regulation of both REL DNA-binding activity and Bcl-X
L expression are correlated with sensitivity to CM101-induced apoptosis in certain B-lymphoma cell lines.
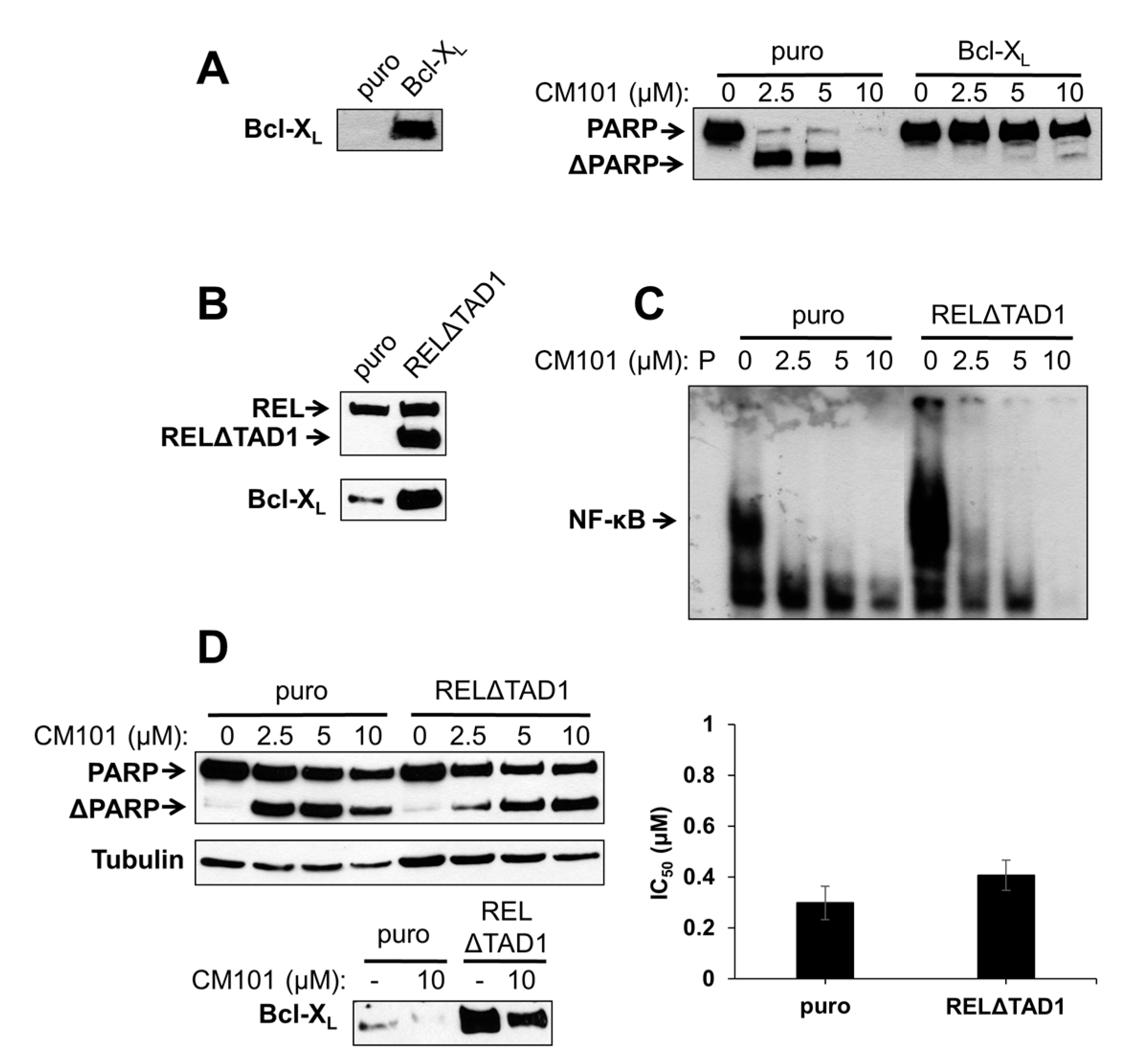
2.5. Overexpression of Bcl-XL or an Activated REL Mutant Can Protect B-Lymphoma Cells from CM101-Induced Apoptosis
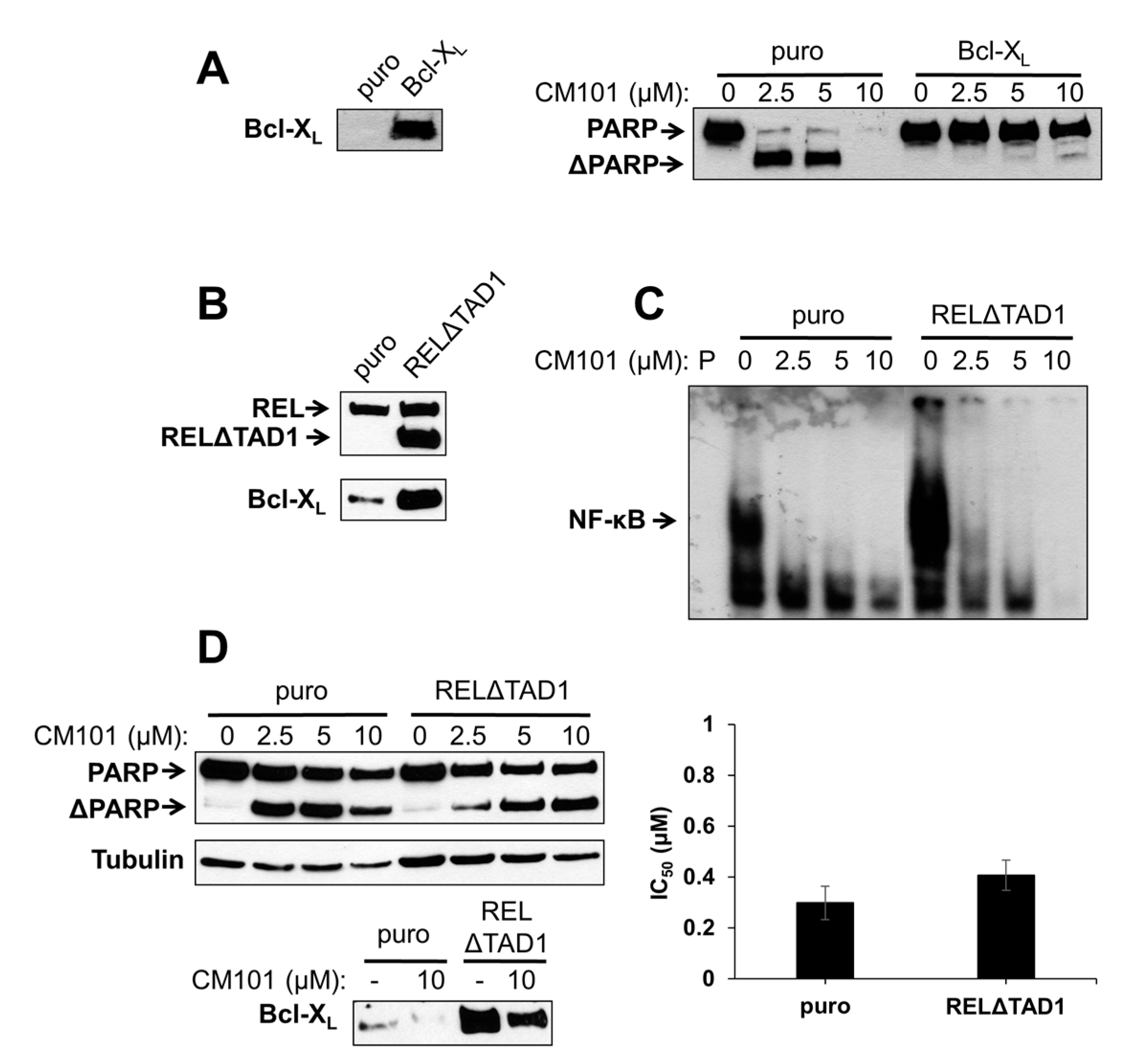
To test directly whether Bcl-X
L can protect B-lymphoma cells from CM101-induced apoptosis, SUDHL-2 cells, which are sensitive to CM101-induced apoptosis and have low levels of Bcl-X
L, were transduced with a retroviral vector containing the human Bcl-X
L gene or the parallel control vector. Pools of infected cells were selected with puromycin. Western blotting confirmed that Bcl-X
L was overexpressed in SUDHL-2 cells transduced with the Bcl-X
L vector as compared to control cells (
Figure 6A). Control SUDHL-2-puro and experimental SUDHL-2-Bcl-X
L cells were then treated with increasing concentrations of CM101 for 24 h, and apoptosis was assessed by the extent of PARP cleavage. PARP cleavage was greatly reduced in SUDHL-2-Bcl-X
L cells as compared to control SUDHL-2-puro cells (
Figure 6A). Thus, Bcl-X
L can protect SUDHL-2 B-lymphoma cells from CM101-induced apoptosis.
We have previously shown that overexpression of an activated REL mutant (RELΔTAD1) can enhance the transformed properties of BJAB cells and decrease their sensitivity to doxorubicin-induced apoptosis [
4]. BJAB-RELΔTAD1 cells have increased levels of nuclear REL DNA-binding activity and consequently increased levels of Bcl-X
L [
4,
23] (
Figure 6B). Therefore, we hypothesized that BJAB-RELΔTAD1 cells would be less sensitive to CM101-induced apoptosis than control BJAB-puro cells. Consistent with our previous findings [
4], BJAB-RELΔTAD1 cells displayed higher levels of κB-site DNA binding than control BJAB-puro cells. CM101 inhibited κB-site DNA-binding activity in both BJAB-RELΔTAD1 and BJAB-puro cells (
Figure 6C); however, because of the increased levels of κB site-binding activity in BJAB-RELΔTAD1 cells, the residual levels of DNA-binding activity after treatment with CM101 were higher in these cells than in control BJAB-puro cells (e.g., compare 2.5 µM and 5 µM lanes in
Figure 6C).
To determine whether activated REL can protect cells from CM101-induced apoptosis, BJAB-puro and BJAB-RELΔTAD1 cells were treated with increasing concentrations of CM101, and PARP cleavage was monitored. Although PARP cleavage was observed in both cell types following treatment with CM101, the control cells were reproducibly more sensitive to CM101 (
i.e., PARP cleavage was seen at lower concentrations, e.g., 2.5 µM, of CM101 in BJAB-puro compared to BJAB-RELΔTAD1 cells) (
Figure 6D). These results further suggest that enhanced REL activity can decrease sensitivity to CM101-induced apoptosis, possibly through up-regulation of Bcl-X
L. Nevertheless, CM101 inhibited cell proliferation to a similar extent in both control BJAB-puro and BJAB-RELΔTAD1 (
Figure 6D). Taken together, these results indicate that RELΔTAD1 protects BJAB cells from CM101-induced apoptosis, but not CM101-induced proliferation arrest.
Figure 6.
Ectopic expression of Bcl-X
L or an activated REL mutant can protect B-lymphoma cells from CM101-induced apoptosis. (
A) SUDHL-2 cells were infected with a pMSCV-puro empty vector or the same vector encoding Bcl-X
L. Stable pools were selected with puromycin. The left panel is a western blot confirming stable overexpression of retrovirally transduced Bcl-X
L. In the right panel, SUDHL-2-puro and SUDHL-2-Bcl-X
L cells were treated with the indicated concentrations of CM101 for 24 h, extracts were prepared, and PARP cleavage was monitored by western blotting; (
B) BJAB cells stably infected with pMSCV-puro empty vector or the same vector encoding RELΔTAD1 [
4] were analyzed by western blotting for REL and Bcl-X
L; (
C) An EMSA with a κB-site probe using extracts of BJAB-puro and BJAB-RELΔTAD1 cells treated for 2 h with the indicated concentrations of CM101. P indicates probe alone; (
D) BJAB-puro and BJAB-RELΔTAD1 cells were treated with the indicated concentrations of CM101 for 24 h, extracts were made, and PARP cleavage was monitored by western blotting. β-Tubulin western blotting was performed as a loading control. In the bottom left panel, Bcl-X
L levels were monitored by western blotting on extracts of BJAB-puro and BJAB-RELΔTAD1 cells treated for 24 h with either the solvent methanol (−) or 10 μM CM101 (+). In the right graph, BJAB-puro or BJAB-RELΔTAD1 cells were treated with increasing concentrations of CM101, and cells were counted 72 h later. IC
50 values were calculated and plotted, as in
Figure 3A.
Figure 6.
Ectopic expression of Bcl-X
L or an activated REL mutant can protect B-lymphoma cells from CM101-induced apoptosis. (
A) SUDHL-2 cells were infected with a pMSCV-puro empty vector or the same vector encoding Bcl-X
L. Stable pools were selected with puromycin. The left panel is a western blot confirming stable overexpression of retrovirally transduced Bcl-X
L. In the right panel, SUDHL-2-puro and SUDHL-2-Bcl-X
L cells were treated with the indicated concentrations of CM101 for 24 h, extracts were prepared, and PARP cleavage was monitored by western blotting; (
B) BJAB cells stably infected with pMSCV-puro empty vector or the same vector encoding RELΔTAD1 [
4] were analyzed by western blotting for REL and Bcl-X
L; (
C) An EMSA with a κB-site probe using extracts of BJAB-puro and BJAB-RELΔTAD1 cells treated for 2 h with the indicated concentrations of CM101. P indicates probe alone; (
D) BJAB-puro and BJAB-RELΔTAD1 cells were treated with the indicated concentrations of CM101 for 24 h, extracts were made, and PARP cleavage was monitored by western blotting. β-Tubulin western blotting was performed as a loading control. In the bottom left panel, Bcl-X
L levels were monitored by western blotting on extracts of BJAB-puro and BJAB-RELΔTAD1 cells treated for 24 h with either the solvent methanol (−) or 10 μM CM101 (+). In the right graph, BJAB-puro or BJAB-RELΔTAD1 cells were treated with increasing concentrations of CM101, and cells were counted 72 h later. IC
50 values were calculated and plotted, as in
Figure 3A.
![]()
To determine whether inhibition of REL could affect Bcl-X
L levels, control BJAB-puro and BJAB-RELΔTAD1 cells were treated with 10 µM CM101 for 24 h and cellular levels of Bcl-X
L were assessed. CM101 treatment resulted in decreased levels of Bcl-X
L in both BJAB-puro and BJAB-RELΔTAD1 cells (
Figure 6D). However, the Bcl-X
L levels following CM101 treatment were higher in BJAB-RELΔTAD1 than BJAB-puro cells. These results suggest that the PARP cleavage observed in CM101-treated BJAB-RELΔTAD1 cells is due to down-regulation of Bcl-X
L; however, apoptosis occurs to a lesser extent in BJAB-RELΔTAD1 cells compared to BJAB-puro cells due to the higher levels of Bcl-X
L in BJAB-RELΔTAD1 cells. These results indicate that up-regulation of Bcl-X
L can play a role in protecting B-lymphoma cells from CM101-induced apoptosis.
2.7. Discussion
In this report, we have characterized a compound that directly and selectively inhibits the DNA-binding activity of cellular oncoprotein REL to cause B-lymphoma cell growth arrest and apoptosis. We show that the ability of CM101 to induce apoptosis in B-lymphoma cell lines correlated with the levels of constitutive REL DNA-binding activity and REL-controlled expression of anti-apoptotic Bcl-XL. In contrast, LPS-induced NF-κB pathway activation still occurs in the presence of CM101, supporting the conclusion that CM101 inhibits NF-κB pathway function by acting selectively at the level of the transcription factors. Therefore, CM101 belongs to a class of molecules that may be valuable for treatment of certain types of B-cell lymphoma.
CM101 has multiple electrophilic sites (indicated by bold arrows,
Figure S3) that could enable it act as a covalent inhibitor [
31]. Epoxy ketone pharmacophores have been shown to serve as irreversible covalent inhibitors by thiophilic ring opening with Cys residues on target proteins [
32,
33]. A probable mechanism includes initial attack of a cysteine thiol to the carbonyl carbon followed by epoxide ring opening via intramolecular 1,2 hydride shift to afford a dehydrated α-addition product (
Figure S3). We have previously shown that other epoxyquinoids can cross-link REL to generate high molecular weight forms, and can inhibit REL DNA-binding activity [
6]. CM101 acts on REL and p65, at least in part, through modification of a conserved Cys residue in a DNA-binding loop within the RHD. CM101 does not effectively inhibit p50 DNA-binding activity at concentrations where it inhibits REL and p65 DNA-binding activity, even though Cys is present at the analogous position in p50 and CM101 can crosslink p50 to a dimer. Flanking residues may affect the nature of the interaction of CM101-like compounds with p65 or REL vs p50, and the selective inhibitory activity of CM101 towards REL and p65 may depend on its ability to crosslink these NF-κB subunits to forms of higher molecular weight than the dimer. Of note, the specific inhibitory effect of DHMEQ on p65 can be reversed by changing five residues near to this Cys to those residues found in p50, which is not affected by DHMEQ [
34]. Moreover, alterations in the DHMEQ structure can convert it from a p65 inhibitor to primarily an IKK inhibitor [
35].
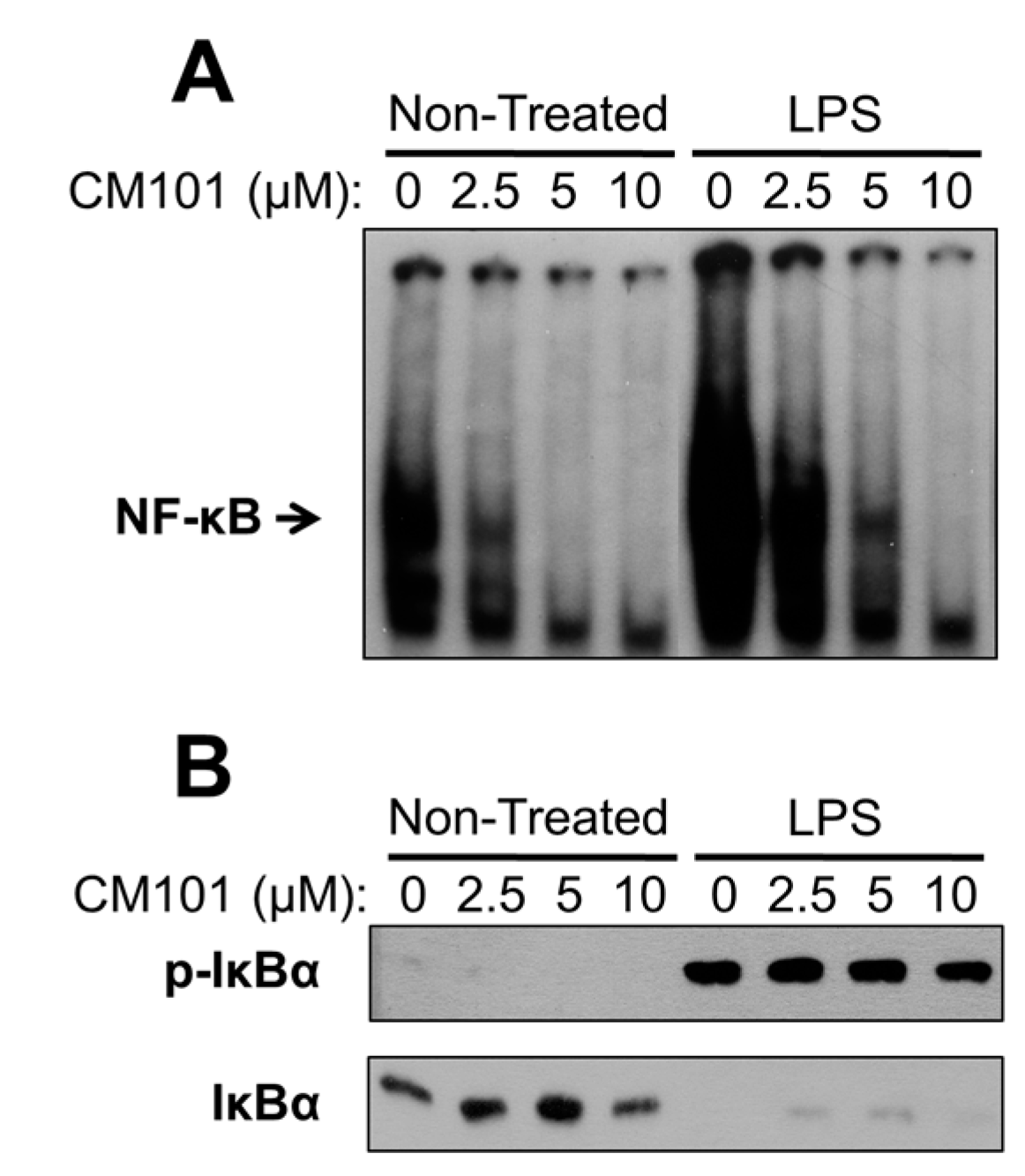
Cys-reactive inhibitors of NF-κB that act on multiple steps in the signaling pathway have been characterized by several groups [
6,
11,
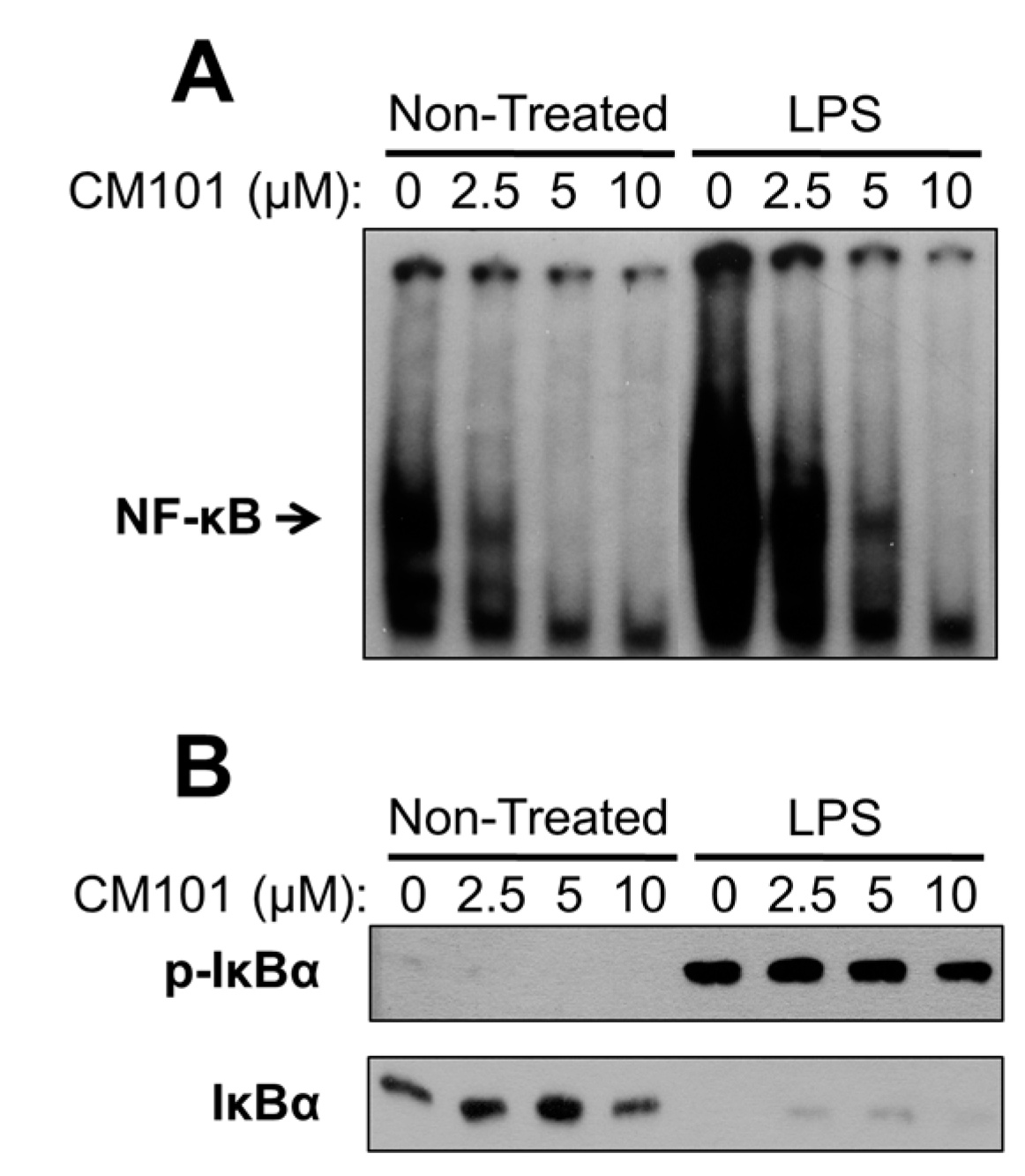
21]. CM101 is different because of its selectivity for inhibiting REL and p65 DNA-binding activity at concentrations at which upstream NF-κB signaling is unaffected. Namely, in many B-lymphoma cell lines CM101 substantially inhibits REL DNA-binding activity, arrests proliferation, and induces apoptosis at concentrations of 2.5–10 µM. However, in a macrophage cell line 10 µM CM101 had no detectable effect on upstream IKK activation as judged by LPS-induced phosphorylation and degradation of IκBα (
Figure 7B). These results suggest that low concentrations of CM101 act primarily on REL and p65 rather than on p50 or the upstream IKK complex. Thus, CM101 and related compounds may have clinical relevance for inhibition of B-cell lymphomas that are dependent on constitutively high levels of nuclear REL DNA-binding activity. Moreover, REL and p65 have been shown to be required for optimal transformation by certain oncoproteins, including Ras [
36,
37], and
REL was identified as a synthetic lethal gene in K-RAS mutant cancers [
38]. Therefore, REL and p65 could also be drug targets in the many types of cancer that have activated Ras. Consistent with these observations, CM101 showed greater cell killing of Ras-transformed 3T3 cells than of control non-transformed 3T3 cells (
Figure S2B).
NF-κB signaling has been commonly proposed as an anti-leukemia/lymphoma drug target. In particular, IKK inhibitors have been shown to be selectively toxic for the ABC subtype of diffuse large B-cell lymphoma (DLBCL), which has high levels of NF-κB activity and target gene expression [
39,
40,
41]. However, in our study the sensitivity of lymphoma cell lines to inhibition by CM101 did not correlate with DLBCL subtype. That is, CM101 was effective at killing both ABC (RC-K8, SUDHL-2) and GCB (SUDHL-4, SUDHL-8, BJAB) cells, all of which have high levels of REL. Therefore, REL may be required for the growth/survival of certain lymphoma cell lines regardless of DLBCL subtype. In addition, nuclear REL staining has recently been shown to occur in both ABC and GCB tumors [
42]. Thus, oncogenic NF-κB addiction cannot be solely identified through gene expression profiling, and may require NF-κB subunit profiling.
The RC-K8 and L428 cell lines both have inactivating mutations in the gene encoding IκBα [
5,
43,
44]. Consequently, RC-K8 and L428 cells have abundant nuclear REL DNA-binding activity, and notably, undergo growth arrest upon ectopic expression of normal or super-repressor forms of IκBα, but not upon expression of dominant-negative IKK [
5,
39]. We show here that RC-K8 and L428 cells are both sensitive to inhibition of growth and REL DNA binding by CM101. Thus, direct inhibitors of REL, such as CM101, may be preferable to IKK inhibitors for those lymphomas that are refractory to IKK inhibitors due to mutations that activate the NF-κB pathway downstream of IKK.
Among the CM101-sensitive and -resistant B-lymphoma cell lines that we have characterized herein, only sensitive cell lines have high levels of REL and some of these cells also have high levels of Bcl-X
L. The Bcl-X
L gene has been shown to be a direct target gene of REL [
25]. Consistent with these observations, expression of the oncogenic RELΔTAD1 protein increased expression of Bcl-X
L in BJAB cells and made them slightly less sensitive to CM101-induced apoptosis. In addition, overexpression of Bcl-X
L dramatically protected SUDHL-2 cells from CM101-induced apoptosis. Taken together, these results confirm that Bcl-X
L can play an important role in the survival of B-lymphoma cells and that Bcl-X
L expression is dependent on transcription factor REL in some B-cell lymphomas. Thus, REL and/or Bcl-X
L should be suitable drug targets in some B-cell lymphomas.
SUDHL-6 and, to a lesser extent, HD-MYZ cells were both relatively resistant to CM101-induced growth arrest and apoptosis. Also, the total amount of constitutive NF-κB DNA-binding activity is low in both of these resistant cell lines (
Figure 4A). In particular, SUDHL-6 cells have low amounts of active REL and its levels of Bcl-X
L were not affected by CM101. Therefore, Bcl-X
L is almost certainly not regulated by REL in SUDHL-6 cells, and SUDHL-6 does not appear to be an NF-κB/REL-addicted cell line. Moreover, phosphorylation of Bcl-X
L, which is associated with sensitivity of several cell types to cytotoxic agents [
26,
27,
28,
29,
30], did not occur in SUDHL-6 cells in response to CM101 treatment (
Figure 5B). Although the SUDHL-6 cell line has been shown to be resistant to certain other toxic compounds, such as IKK and HDAC inhibitors [
24,
39], SUDHL-6 cells are not more resistant than other B-lymphoma cell lines to the cytotoxic effects of all compounds [
45,
46,
47].
Inflammation is a key process for activation of the innate immune system [
48]. Many kinase inhibitors used in targeted cancer therapies can compromise the responsiveness of the innate immune system [
49], which may lessen their utility in clinical settings. NF-κB is a critical pathway for both inflammation and innate immune responses. Therefore, one concern for the use of NF-κB pathway inhibitors for cancer therapy is that such inhibitors would lessen immune system anti-tumor activity. CM101 appears promising in this regard because it selectively inhibits REL and p65 DNA-binding activity but not the activity of IKK. Thus, low concentrations of CM101-like compounds may still permit NF-κB DNA binding to be sufficiently induced in normal anti-tumor immune cells, while still inhibiting basal REL DNA-binding activity required for lymphoma cell growth. As such, compounds with CM101-like activity may be more effective anti-tumor agents because normal anti-tumor immune responses can still occur in the presence of lower concentrations of CM101-like compounds, in addition to the direct action of CM101 on tumor cell growth.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}