2. Results and Discussion
Cairicoside I (1) obtained as a white, amorphous powder, which gave a quasi-molecular ion at
m/z 1321.7212 [M+Na]
+ in HR-TOF-MS, which suggested the molecular formula C
65H
102O
26 (calcd. for C
65H
102O
26Na, 1321.6557). Its IR spectrum gave a hydroxyl group absorption band at 3442 cm
−1, carbonyl group at 1724 cm
−1, and aromatic group at 1641 cm
−1. Alkaline hydrolysis of
1 afforded a glycosidic acid (compound
7) and organic acids. The organic acids were identified as 2-methylbutyric, and Cna (
trans-cinnamic acids), with a ratio of 2:1 by GC-MS. Mba
a (2-Methylbutyric acid) was found to have the
S-configuration by comparison of its optical rotation value with that of an authentic sample [
3]. Acid hydrolysis the glycosidic acid afforded Rha (
l-rhamnose) and Glu (
d-glucose), with a ratio of 4:1. The monosaccharides was derivatized and identified as Rha and Glu by GC-MS by comparison with authentic samples. Taken together the hydrolysis information and NMR data identified the glycosidic acid as simonic acid A (
7).
The
1H-NMR spectrum of
1 (
Figure 1) exhibited two
trans-coupled olefinic protons at δ
H 6.53 (1H, d) and 7.81 (1H, d), a multiplet due to five protons at δ
H 7.27–7.45 (m) suggesting the presence of a Cna. The protons at δ
H 0.82 (1H, t), 1.13 (1H, d), and 2.38 (m) were assignable to another Mba group. The
13C-NMR spectrum data of
1 (
Table 1) exhibited four ester carbonyl carbons at δ
C 175.7, 175.2, 173.7, and 166.1 and five anomeric carbon signals at δ
C 104.6, 104.3, 103.6, 99.2 and 98.3. The anomeric protons were assigned to the peaks at 5.66 (1H, br s), 4.91 (1H, d), 5.94 (1H, br s), 6.08 (1H, br s) and 5.58 (1H, br s), respectively, by the HSQC data. All protons in each saccharide system and carbon signals were assigned by
1H-,
13C-NMR, TOCSY, HMBC and HSQC experiments, leading to the identification of one glucopyranosyl unit and four rhamnopyranosyl units as the monosaccharides present in
1. The anomeric configuration for the sugar moieties were defined as β for the glucopyranosyl moieties from their coupling constants of 7.2 Hz, and α for the rhamnopyranosyl from the C-5 chemical shift [
9].
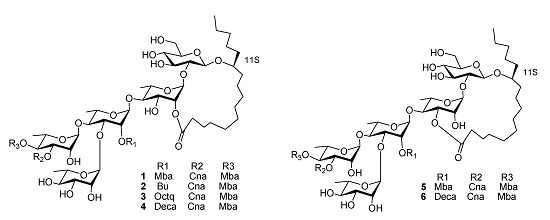
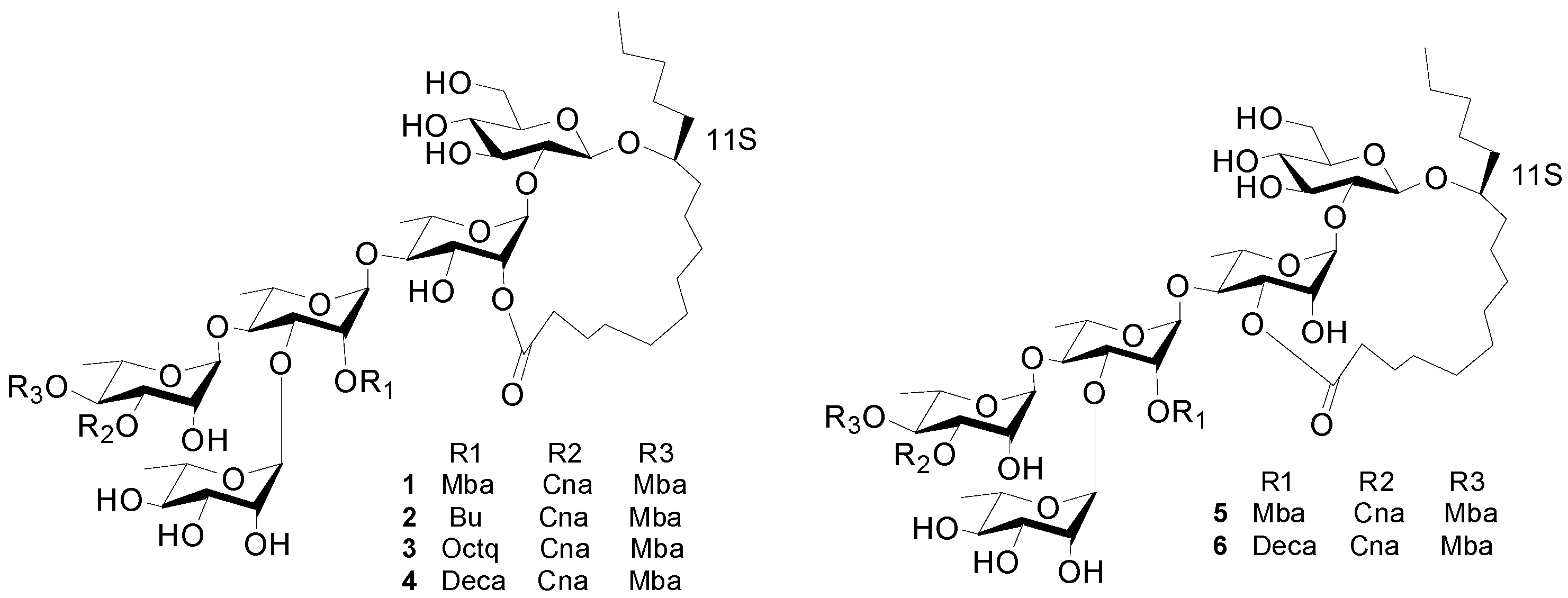
Figure 1.
The structures of compounds 1–6.
Figure 1.
The structures of compounds 1–6.
Table 1.
NMR Data for Compounds 1–4 in pyridine-d5.
Table 1.
NMR Data for Compounds 1–4 in pyridine-d5.
| Position b | 1 a | 2 a | 3 a | 4 a |
|---|
| 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H |
|---|
| Glc-1 | 104.3 | 4.91 d (7.2) | 104.3 | 4.97 d (7.2) | 102.7 | 4.95 d (7.2) | 104.3 | 4.91 d (7.2) |
| 2 | 81.8 | 3.87 * | 81.8 | 3.95 * | 80.3 | 3.92 * | 81.8 | 3.89 * |
| 3 | 76.3 | 4.17 dd (9.0, 9.0) | 76.1 | 4.24 dd (9.0, 9.0) | 74.8 | 4.21 dd (9.0, 9.0) | 76.3 | 4.21 dd (9.0, 9.0) |
| 4 | 71.7 | 4.12 dd (9.0, 9.0) | 71.7 | 4.12 dd (9.0, 9.0) | 70.2 | 4.15 dd (9.0, 9.0) | 71.7 | 4.12 dd (9.0, 9.0) |
| 5 | 77.8 | 3.83 * | 77.8 | 4.18 * | 76.3 | 3.90 * | 77.8 | 3.84 * |
| 6a | 62.6 | 4.44–4.46 * | 62.6 | 4.52 * | 61.1 | 4.48 * | 62.6 | 4.48 * |
| 6b | | 4.29 * | | 4.34 * | | 4.30 * | | 4.30 * |
| Rha-1 | 98.3 | 5.58 br s | 98.5 | 5.64 br s | 97.0 | 5.61 br s | 98.6 | 5.58 br s |
| 2 | 71.2 | 6.04 br s | 71.2 | 6.16 br s | 69.7 | 6.15 br s | 71.2 | 6.15 br s |
| 3 | 69.6 | 5.05 dd (9.6, 3.0) | 69.6 | 5.12 dd (9.6, 2.0) | 67.9 | 5.10 dd (9.6, 3.0) | 69.5 | 5.08 dd (9.6, 2.0) |
| 4 | 79.8 | 4.18 dd (9.6, 9.6) | 79.4 | 4.24 dd (9.6, 9.6) | 77.8 | 4.23 dd (9.6, 9.6) | 79.3 | 4.20 dd (9.6, 9.6) |
| 5 | 68.2 | 4.24 * | 68.1 | 4.27 * | 66.6 | 4.28 * | 68.1 | 4.30 * |
| 6 | 19.2 | 1.56 d (6.0) | 18.6 | 1.60 d (6.0) | 18.6 | 1.59 d (6.0) | 18.6 | 1.59 d (6.0) |
| Rha'-1 | 99.2 | 6.08 br s | 99.3 | 6.12 br s | 97.6 | 6.08 br s | 99.0 | 6.05 br s |
| 2 | 72.8 | 5.98 br s | 72.9 | 6.06 br s | 71.3 | 6.05 br s | 73.0 | 6.02 br s |
| 3 | 79.4 | 4.56 dd (9.0, 2.4) | 79.9 | 4.56 dd (9.0, 2.4) | 78.3 | 4.66 dd (10.0, 2.4) | 79.8 | 4.64 dd (9.0, 2.4) |
| 4 | 80.2 | 4.29 dd (9.0, 9.0) | 79.4 | 4.20 dd (9.0, 9.0) | 78.8 | 4.20 dd (10.0, 10.0) | 80.2 | 4.20 dd (9.0, 9.0) |
| 5 | 68.1 | 4.34 dd (9.0, 6.0) | 68.1 | 4.54 * | 66.6 | 4.53 * | 68.0 | 4.50 * |
| 6 | 18.4 | 1.64 d (6.0) | 18.3 | 1.67 d (5.5) | 16.1 | 1.64 d (5.4) | 17.6 | 1.64 d (5.4) |
| Rha"-1 | 103.6 | 5.94 br s | 103.1 | 5.94 br s | 101.8 | 5.99 br s | 103.4 | 5.96 br s |
| 2 | 72.4 | 4.78 br s | 72.4 | 4.84 br s | 70.9 | 4.81 br s | 72.4 | 4.77 br s |
| 3 | 73.2 | 5.91 dd (3.0, 10.0) | 73.1 | 5.96 dd (3.0, 10.0) | 71.4 | 5.93 dd (2.9, 10.2) | 73.1 | 5.90 dd (3.0, 10.0) |
| 4 | 73.5 | 6.06 t (10.0) | 73.5 | 6.11 t (10.0) | 71.8 | 6.10 dd (10.2, 10.2) | 73.4 | 6.05 dd (10.0, 10.0) |
| 5 | 68.4 | 4.49 dd (10.0, 6.5) | 68.4 | 4.30 * | 66.9 | 4.30 * | 68.4 | 4.21 * |
| 6 | 17.6 | 1.36 d (6.5) | 19.2 | 1.46 d (6.0) | 17.7 | 1.43 d (6.0) | 19.2 | 1.42 d (5.5) |
| Rha"'-1 | 104.6 | 5.66 br s | 104.3 | 5.69 br s | 102.7 | 5.65 br s | 104.3 | 5.62 br s |
| 2 | 69.8 | 4.97 br s | 69.8 | 4.97 br s | 68.3 | 4.98 br s | 69.8 | 4.94 br s |
| 3 | 72.3 | 4.39 * | 72.3 | 4.37 * | 70.9 | 4.38 * | 72.3 | 4.35 * |
| 4 | 73.2 | 4.23 * | 73.4 | 4.24 * | 71.8 | 4.23 * | 73.4 | 4.21 * |
| 5 | 70.4 | 4.41 * | 70.6 | 4.45 * | 69.1 | 4.43 * | 70.6 | 4.40 * |
| 6 | 18.6 | 1.58 d (6.0) | 18.5 | 1.58 d (6.0) | 16.9 | 1.61 d (6.0) | 18.4 | 1.59 d (6.0) |
| Ag-1 | 173.7 | | 173.5 | | 171.6 | | 173.1 | |
| 2 | 33.0 | 2.40 m, 2.24 m | 34.2 | 2.30 m, 2.24 m | 32.6 | 2.40 m, 2.29 m | 33.0 | 2.40 m 2.26 m |
| Ag-1 | 173.7 | | 173.5 | | 171.6 | | 173.1 | |
| 2 | 33.0 | 2.40 m, 2.24 m | 34.2 | 2.30 m, 2.24 m | 32.6 | 2.40 m, 2.29 m | 33.0 | 2.40 m 2.26 m |
| 11 | 82.5 | 3.89 * | 82.6 | 3.96 * | 81.1 | 3.90 * | 82.6 | 3.90 * |
| 16 | 14.0 | 0.84 t (7.0) | 14.0 | 0.85 t (7.0) | 12.6 | 0.82 t (7.0) | 14.1 | 0.84 t (7.0) |
| Cna-1 | 166.1 | | 166.0 | | 164.8 | | 166.2 | |
| 2 | 118.1 | 6.53 d (16.0) | 118.0 | 6.60 d (16.0) | 116.8 | 6.57 d (16.0) | 118.1 | 6.54 d (16.0) |
| 3 | 145.4 | 7.81 d (16.0) | 145.3 | 7.86 d (16.0) | 143.9 | 7.84 d (16.0) | 145.3 | 7.75 (16.0) |
| 1' | 134.3 | | 134.4 | | 133.0 | | 134.4 | |
| 2' and 6' | 128.4 | 7.43 m | 128.3 | 7.49 m | 126.8 | 7.46 m | 128.3 | 7.44 m |
| 3' and 5' | 129.0 | 7.34 m | 129.1 | 7.38 m | 127.6 | 7.36 m | 129.1 | 7.34 m |
| 4' | 130.7 | 7.34 m | 130.4 | 7.38 m | 129.0 | 7.36 m | 130.5 | 7.34 m |
| Mba-1 | 175.2 | | 175.6 | | 175.7 | | 175.7 | |
| 2 | 41.2 | 2.38 m | 41.3 | 2.45 m | 39.8 | 2.49 m | 41.3 | 2.47 m |
| 2-CH3 | 16.6 | 1.06 d (7.0) | 16.7 | 1.17 d (7.0) | 15.2 | 1.13 d (7.0) | 16.7 | 1.13 d (7.0) |
| 3 | 27.5 | 1.41 m * | 27.5 | 1.41 m * | 27.4 | 1.38 m * | 27.1 | 1.44 m * |
| 4 | 11.6 | 0.81 t (7.0) | 11.7 | 0.79 t (7.0) | 10.1 | 0.79 t (7.0) | 11.6 | 0.79 t (7.0) |
| Mba'-1 | 175.7 | | | | | | | |
| 2 | 41.3 | 2.47 m | | | | | | |
| 2-CH3 | 16.7 | 1.13 d (7.0) | | | | | | |
| 3 | 27.0 | 1.22 m * | | | | | | |
| 4 | 11.6 | 0.82 t (7.0) | | | | | | |
| Bu-1 | | | 172.5 | | | | | |
| 2 | | | 17.6 | 2.50 tq (7.0, 7.0) | | | | |
| 4 | | | 13.4 | 0.84 t (7.0) | | | | |
| Oct-1 | | | | | 171.3 | | | |
| 2 | | | | | 32.6 | 2.30 m | | |
| 8 | | | | | 12.5 | 0.81 t (7.0) | | |
| Deca-1 | | | | | | | 172.7 | |
| 2 | | | | | | | 34.2 | 2.32 tq (7.0, 7.0) |
| 10 | | | | | | | 14.0 | 0.83 t (7.0) |
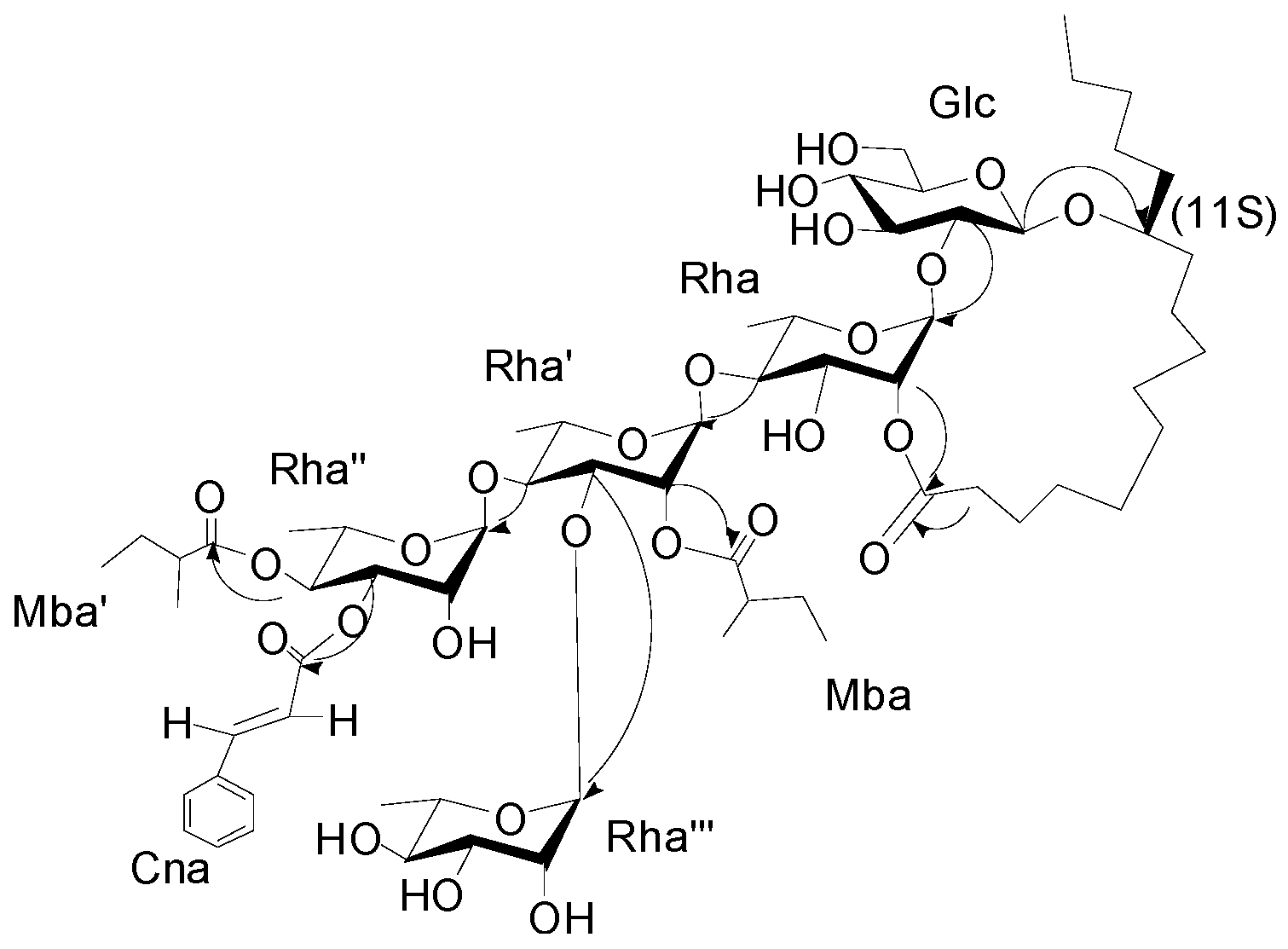
The interglycosidic connectivities were determined from the following HMBC (
Figure 2) correlations from Rha-H-1 (δ
H 5.58) to Glu-C-2 (δ
C 81.8), Rha'-H-1 (6.08) to Rha-C-4 (79.8), Rha''-H-1 (δ
H 5.94) to Rha'-C-4 (80.2), Rha'''-H-1 (δ
H 5.66) to Rha'-C-3 (79.4). Esterification positions were also determined by HMBC data, between Rha''-H-4 (δ
H 6.06) to Mba-C-1(δ
C 175.7); Rha''-H-3 of rhamnose'' (δ
H 5.91) to Cna-C-1 (δ
C 166.1); Rha'-H-2 (δ
H 5.98) to Mba-C-1 (δ
C 175.2); and Rha-H-2 (δ
H 6.04) and aglycone-C-1 (δ
C 173.7), respectively. The position of the jalapinolic acid unit was finally determined by HMBC to be between aglycone-H-11 (δ
H 3.89) to Glu-C-1 (δ
C 104.3), which correlations established the structure of
1 as (
S)-jalapinolic acid 11-
O-α-
l-rhamnopyranosyl-(1→3)-
O-[3-O-trans-cinnamoyl-4-O-(
S)-2-methyl-butyryl-α-
l-rhamnopyranosyl-(1→4)]-
O-[2-O-(
S)-2-methylbutyryl]-α-
l-rhamnopyranosyl-(1→4)-
O-α-
l-rhamnopyranosyl-(1→2)-
O-β-
d-glucopyranoside, intramolecular 1,2''-ester (
Figure 1). This new compound was named cairicoside I.
Figure 2.
Key HMBC correlations from H to C for cairicoside I (1).
Figure 2.
Key HMBC correlations from H to C for cairicoside I (1).
Cairicosides II–IV (compounds
2–
4) afforded white, amorphous powders, and gave quasi-molecular ions at
m/z 1307.6437 [M+Na]
+, 1363.7032 [M+Na]
+ and 1391.7395 [M+Na]
+ in HR-TOF-MS, which suggested the molecular formulas C
64H
100O
26 (calcd. for C
64H
100O
26Na: 1391.7340), C
65H
101O
26 (calcd. for C
68H
108O
26Na: 1363.7027) and C
70H
112O
26 (calcd. for C
70H
112O
26Na: 1391.7340). The IR spectra gave absorption bands of hydroxyl groups at 3418, 3443 and 3444 cm
−1, carbonyl groups at 1721, 1740 and 1740 cm
−1, and aromatic groups at 1641, 1638 and 1638 cm
−1. Independent alkaline hydrolysis of
2–
4 afforded a mixture of organic acids and a glycosidic acid, respectively. 2
S-Methylbutanooic acid and a
trans-cinnamoic acid were found in
2–
4 by GC-MS experiments. Additionally, a butyl acid in
2, a
n-octanoyl acid in
3 and a
n-decanoyl acid were found in
4 by GC-MS expriments. The glycosidic acid obtained also proved to be simonic acid A (
7) from the NMR and MS data. The
1H-NMR spectrum of
2 exhibited two
trans-coupled olefinic protons at δ
H 6.60 (1H, d,
J = 16.0 Hz, Cna-2) and 7.86 (1H, d,
J = 16.0 Hz Cna-3), a multiplet due to five protons at δ
H 7.38–7.49 (m, Cna-2-6), suggesting the presence of a
trans-cinnamoyl moiety. The same
trans-cinnamoyl moiety appeared in
3–
4, two
trans-coupled olefinic protons at δ
H 6.57 (1H, d,
J = 16.0 Hz, Cna-2) and 7.84 (1H, d,
J = 16.0 Hz Cna-3), a multiplet due to five protons at δ
H 7.36–7.46 (m, Cna-2-6) in
3, and two
trans-coupled olefinic protons at δ
H 6.54 (1H, d,
J = 16.0Hz, Cna-2 ) and 7.75 (1H, d,
J = 16.0 Hz, Cna-3), a multiplet due to five protons at δ
H 7.34–7.44 (m, Cna-2-6) in
4. The protons at δ
H 0.79 (1H, t,
J = 7.0 Hz, Mba-4), 1.17 (1H, d,
J = 7.0 Hz, Mba-2-Me), and 2.45 (m, Mba-2) were assignable to a 2-methylbutanoyl (Mba) group in
2 and the protons at δ
H 0.79 (1H, t,
J = 7.0 Hz, Mba-4), 1.13 (1H, d,
J = 7.0 Hz, Mba-2-Me), and 2.49 (m, Mba-2) suggested an Mba group in
3, and δ
H 0.79 (1H, t,
J = 7.0 Hz, Mba-4), 1.13 (1H, d,
J = 7.0 Hz, Mba-2- CH
3), and 2.47 (m, Mba-2) were also ive of an Mba group in
4. Alkaline hydrolysis
2–
4 gave an organic acid mixture and one glycosidic acid, which were identified as 2-methylbutyric and
trans-cinnamic acids in
2–
4, butyl acid in
2,
n-octanoyl acid in
3 and
n-decanoyl acid in
4 by GC-MS expriments. The
13C-NMR spectrum of
2–
4 were highly similar from δ
C 180 to δ
C 40 with
1, so the three compounds should have very similar connection positions, just differing in the substituents. The key HMBC correlations confirmed the esterification positions of the acyl residues in the oligosaccharide core, thus a
2S-methylbutanoyl group and a
trans-cinnamoyl group were located at C-4 of Rha" in
2–
4. A butyl,
n-octanoyl and
n-decanoyl group were located at C-2 of Rha', in
2,
3 and
4, respectively. The lactonization position of the aglycone was C-2 of Rha for
2–
4. Accordingly, the structures of
2–
4 were as shown in
Figure 1.
Cairicoside A (
5) and cairicoside C (
6) appeared as white, amorphous powders, and gave quasi-molecular ions at
m/z 1333 [M+Cl]
−, and 1403 [M+Cl]
− in ESI-MS, which suggested molecular weights of 1298 and 1368, so these two compounds were isomeric with
1 and
4, respectively. Alkaline hydrolysis of
5 afforded the same substituent groups as
1, and alkaline hydrolysis of
6 afforded the same substituent groups as
4. The groups’ esterfication sites were suggested by HMBC data, and just the lactone sites were different between
1 and
4, which were bonded at C-3 of Rha. The structures of
5–
6 are shown in
Figure 1.
Some plants from the Convolvulaceae family have been reported to exert anti-diabetes activities or potent α-glucosidase inhibitory activities, so compounds
1–
4 have been evaluated for inhibitory activities against α-glucosidase. As shown in
Table 2, new compounds
1–
4 exhibited more potent α-glucosidase inhibitory activities compared to acarbose, a widely used clinically useful drug, used as a positive control (IC
50 = 385.0 ± 9.3 μM). To our knowledge, these are the first examples of resin glycosides with α-glucosidase inhibitory activities. The IC
50 values of the four compounds are almost the same, probably due to the similarity of their structures, and every compound with strong inhibitory activity has four α-anomeric monosaccharides in the structure, which is the same relative configuration as α-glucosidase.
Table 2.
α-Glucosidase inhibition of compounds 1–4.
Table 2.
α-Glucosidase inhibition of compounds 1–4.
| Compound | α-Glucosidase Inhibition Contstant a |
|---|
| (IC50) [μM] |
|---|
| 1 | 21.4 ± 2.9 |
| 2 | 26.2 ± 4.6 |
| 3 | 30.4 ± 3.9 |
| 4 | 28.9 ± 1.4 |
| Acarbose b | 385.0 ± 9.3 |
3. Experimental Section
3.2. Plant Material
The aerial parts of I. cairica were collected at Guangzhou City, Guangdong Province, China, in August 2012, and identified by Prof. Ji-Zhu Liu. A voucher specimen (No. 2012-8) was deposited at Department of Traditional Chinese Medicinal Chemistry, Guangdong Pharmaceutical University.
3.3. Extraction and Isolation
The aerial parts of I. cairica (5 kg) were extracted two times with 95% EtOH (20 L) under reflux for 2 h and then evaporated in vacuo. The extract was divided into CHCl3 and H2O-soluble fractions. The CHCl3 extract (107 g) was subjected to column chromatography on silica gel (200–300 mesh, 2 kg, 10 × 80 cm) and eluted with CH2Cl2–MeOH (v/v, 100:0, 20:1, 5:1) to give three fractions (Fr.1-3). Fr.2 (13 g) was purified by Sephadex LH-20 (75–150 μm, 120 g, 1.5 × 160 cm) column chromatography with MeOH as eluent, fractions of 10–19 (3.8 g) were put together as Fr. A1 and fractions of 3–9 were combined as Fr. A2. Fr. A1 was purified by a reverse-phase HPLC system (10 mL/min, 280 nm), eluted with MeOH/H2O (v/v, 95:5) to furnish 1 (52 mg), 2 (9.3 mg), 5 (34 mg) and 6 (17 mg). Fr. A2 was purified by the same system, eluted with MeOH/H2O (v/v, 98:2) to yield 3 (32 mg), and 4 (40 mg).
3.4. Product Identification
Cairicoside I (
1), white amorphous powder,
+ 53.8° (
c 0.3, MeOH). UV (MeOH) λ
max: (log ε) 278 (0.657), 216 (0.402) nm; IR (KBr) ν
max: 3442, 2931, 2859, 2334, 1724, 1641, 1134, 1058 cm
−1;
1H- and
13C-NMR (C
5D
5N) data see
Table 1; HR-TOF-MS
m/z 1321.6542 [M+Na]
+ (calcd for C
65H
102O
26Na: 1321.6557).
Cairicoside II (
2), white amorphous powder,
+ 30.5° (
c 0.1, MeOH). UV (MeOH) λ
max: (log ε) 280 (0.26), 226 (0.54) nm; IR (KBr) ν
max: 3418, 2938, 1721, 1641, 1082 cm
−1;
1H- and
13C-NMR (C
5D
5N) data see
Table 1; HR-TOF-MS
m/z 1307.6437 [M+Na]
+ (calcd for C
64H
100O
26Na: 1307.6401).
Cairicoside III (
3), white amorphous powder,
+ 8.7° (
c 0.2, MeOH). UV (MeOH) λ
max: (log ε) 278 (0.695), 220 (0.485) nm; IR (KBr) ν
max: 3444, 2931, 2858, 1739, 1638, 1054 cm
−1;
1H- and
13C-NMR (C
5D
5N) data see
Table 1; HR-TOF-MS
m/z 1363.7032 [M+Na]
+ (calcd for C
68H
108O
26Na: 1363.7027).
Cairicoside IV (
4), white amorphous powder,
+ 88.3° (
c 0.10, MeOH); UV (MeOH) λ
max 220 (0.34), 278 (0.56) nm; IR (KBr) ν
max: 3444, 2931, 2858, 1740, 1638, 1054 cm
−1;
1H- and
13C-NMR (C
5D
5N) data see
Table 1; HR-TOF-MS
m/z 1391.7395 [M+Na]
+ (calcd for C
70H
112O
26Na:1391.7340).
Cairicoside A (5), white amorphous powder, − 54.0° (c 0.10, MeOH); UV (MeOH) λmax 222 (0.33), 278 (0.58) nm; IR (KBr) νmax: 3439, 2933, 2863, 1727, 1637, 1053 cm−1; ESIMS m/z 1333 [M+Cl]−.
Cairicoside C (6), white amorphous powder, − 58.1° (c 0.11, MeOH); UV (MeOH) λmax (log ε) 216 (0.4), 278 (0.66) nm; IR (KBr) νmax: 3442, 2931, 2859, 1724, 1641, 1058 cm−1; ESIMS m/z 1403 [M+Cl]−.
3.5. Alkaline Hydrolysis of 1–6
Compounds
1–
6 (7 mg each) in 5% KOH (3 mL) were refluxed at 90 °C for 2 h, respectively. The reaction mixture was acidified to pH 4.0 with 2 mol/L HCl and extracted with hexane (3 mL × 2) and
n-BuOH (3 mL × 2). The organic layer was washed with H
2O, dried over anhydrous Na
2SO
4, then methylated following [
10]. The hexane extract, was combined with 0.1 mL 0.5 M CH
3ONa solution, then shaken for 5 min at room temperature, before adding 5 μL CH
3COOH and 1 g anhydrous CaCl
2 powder, heating for 1 h, followed by centrifugation for 2–3 min at 2000–3000 rpm.min
−1. The supernatant was analyzed by GC-MS on a TRACE GC ULTRA DSQ II intrument under the following conditions: 30 m × 0.25 mm × 0.25 μm, TG-5MS (Thermo) column; He, 0.8 mL/min; 40 °C, 3min; 50–310 °C, ∆10 °C/min, 70 eV. 2-Methylbutyric acid methyl ester (
tR 4.39 min)
m/z [M+H]
+ 117 (5), 101 (23), 88 (96), 57 (100), 41 (55), 29 (45), 27 (19), and
trans-cinnamic acid methyl ester (
tR 13.29 min)
m/z [M]
+ 162 (40), 131 (100), 103 (66), 77 (32), from
1–
6 was identified.
n-Butyric acid methyl ester (
tR 4.37 min)
m/z [M]
+ 101 (33), 88 (100), 57 (70), 41 (35) from
2.
n-Octanoic acid methyl ester (
tR 10.82 min):
m/z [M]
+ 158 (4), 127 (18), 87 (45), 74 (100), 43 (22) from
3 was identified.
n-Decanoic acid methyl ester (
tR 12.37 min):
m/z 172 [M]
+ (4), 155 (5), 143 (30), 129 (5), 87 (59), 74 (100), 55 (18) from
4 and
6 was identified. The 2-methylbutanoic acid was proved to have an
S configuration by comparing the specific rotation with that of authentic 2
S-methylbutanoic acid [
11].
3.6. Acid Hydrolysis and Sugar Analysis
The glycosidic acid (
7, 4 mg, from alkaline hydrolysis) was methylated with MeOH catalyzed by 1.0 mol/L H
2SO
4 to give simonic acid A methyl ester (
8). Compound
7 was hydrolyzed with 2 mol/L H
2SO
4 and extracted with ether to obtain 11-hydroxyhexadecanoic acid methyl ester (
9) [
12]. The aqueous layer of acidic hydrolysis was concentrated under reduced pressure to give a sugar residue. The protocols described in [
13] were applied to determine the stereochemistry of sugars, which allowed the identification of the components of the mixture of sugars as
l-rhamnose and
d-glucose by comparison their derivatives with those of authentic samples. GC-MS was performed on a TRACE GC ULTRA DSQ II intrument under the following conditions: 30 m × 0.25 mm × 0.25 μm, TG-5MS (Thermo)column; He, 0.8 mL/min; 60 °C, 3 min; 60–180 °C, ∆10 °C/min keep 3 min, 180–205 °C, ∆3 °C/min keep 5 min, 205–300 °C, ∆20 °C/min keep 5 min, 70 eV. In the acid hydrolysate of operculinic acid A methyl ester
l-rhamnose, and
d-glucose were confirmed by comparison of their retention times of their derivatives with those of authentic
l-rhamnose (
tR 30.14 min) and
d-glucose (
tR 31.65 min) derivatives prepared in the same way, respectively.
3.7. Preparation of Mosher’s Esters
The procedures for preparation of Mosher’s esters to determination of absolute configuration of 11
S of the aglycone were same as described previously for resin glycosides from
Ipomoea batatas [
10]. According to the ∆δ of the derivative of
9 with
R-MPA and
S-MPA, so compound
9 was assigned to 11
S.
3.8. Enzyme Inhibition Assay
The a-glucosidase inhibition assay was performed according to a slightly modified method of Pierre
et al. [
14]. α-Glucosidase was purchased from Sigma (San Francisco, CA, USA, No. M0035-15). The inhibition was measured spectrophotometrically at pH 6.8 and at 37 °C for 10 min, using 0.01 M
p-nitrophenyl a-
d-glucopyranoside (PNPG) from Sigma (No. M0103) as a substrate and 1 U/mL of enzyme, in 0.067 M KH
2PO
4-Na
2HPO
4 buffer. Acarbose (Aladdin Company, Shanghai, China, No. l1424006) was used as positive control. The increment in absorption at 410 nm due to the hydrolysis of PNPG by α-glucosidase was monitored continuously with an automatic multi-functional microplate reader.



{kind=link}
{kind=link}
{kind=link}