
Copolymers of Vinyl-Containing Benzoxazine with Vinyl Monomers as Precursors for High Performance Thermosets

Abstract
:1. Introduction
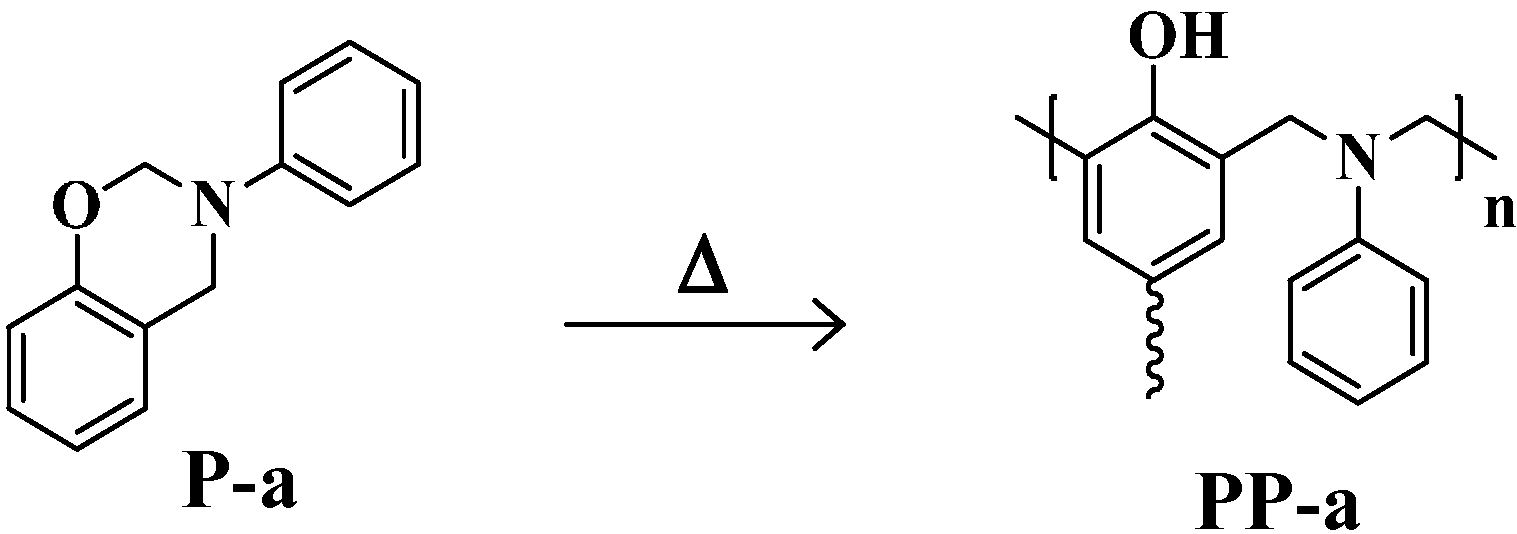
2. Results and Discussion
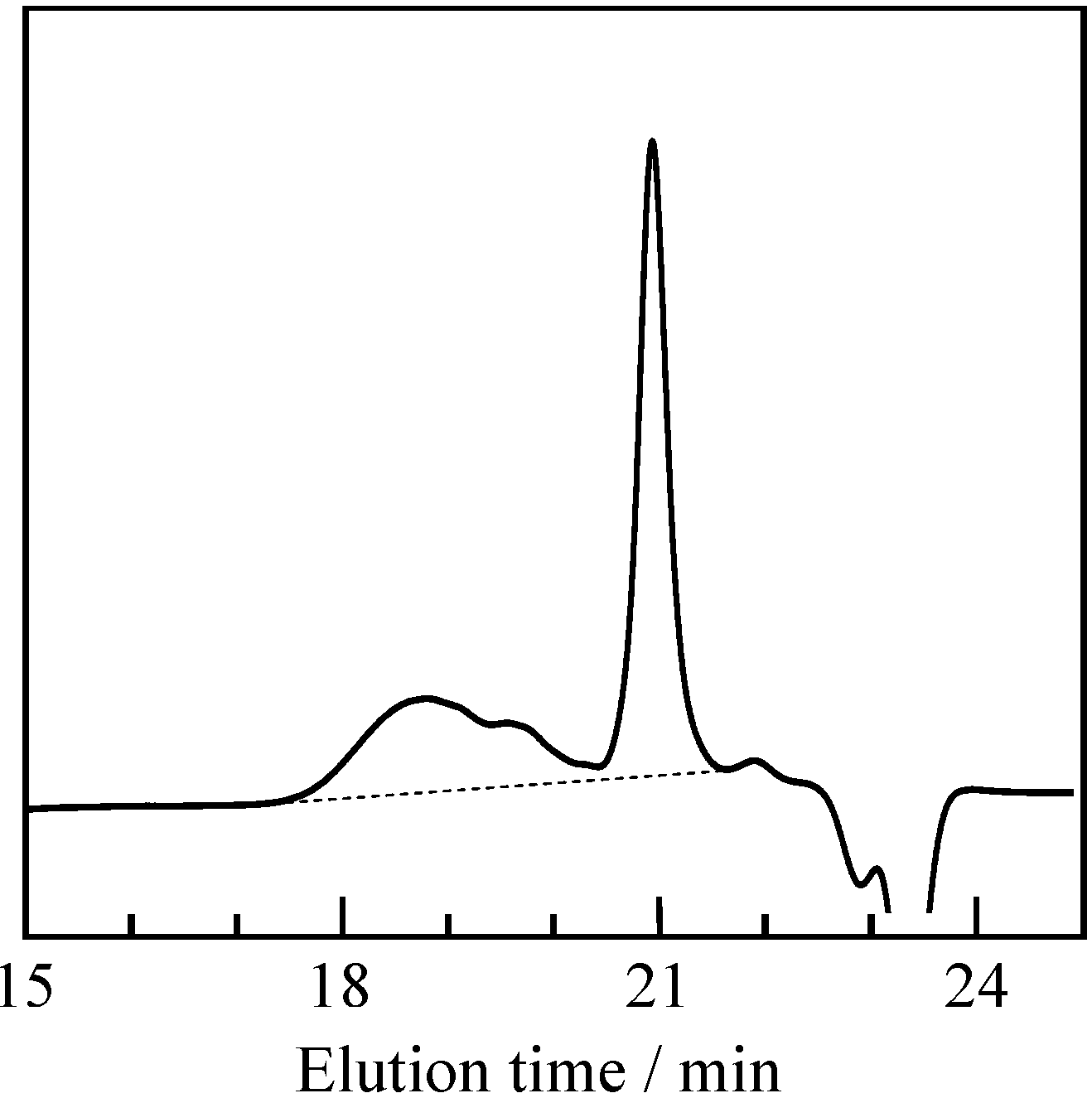
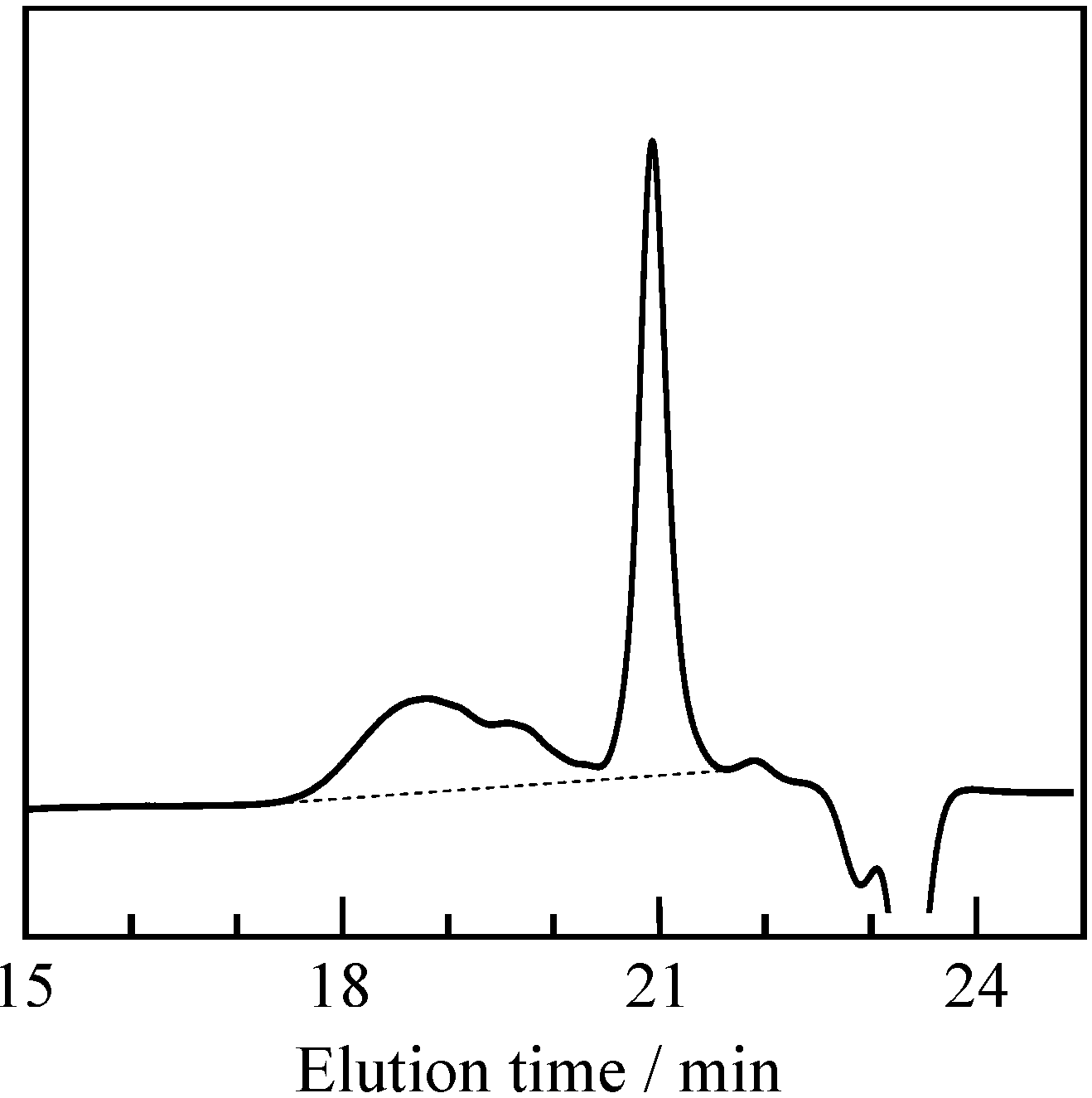
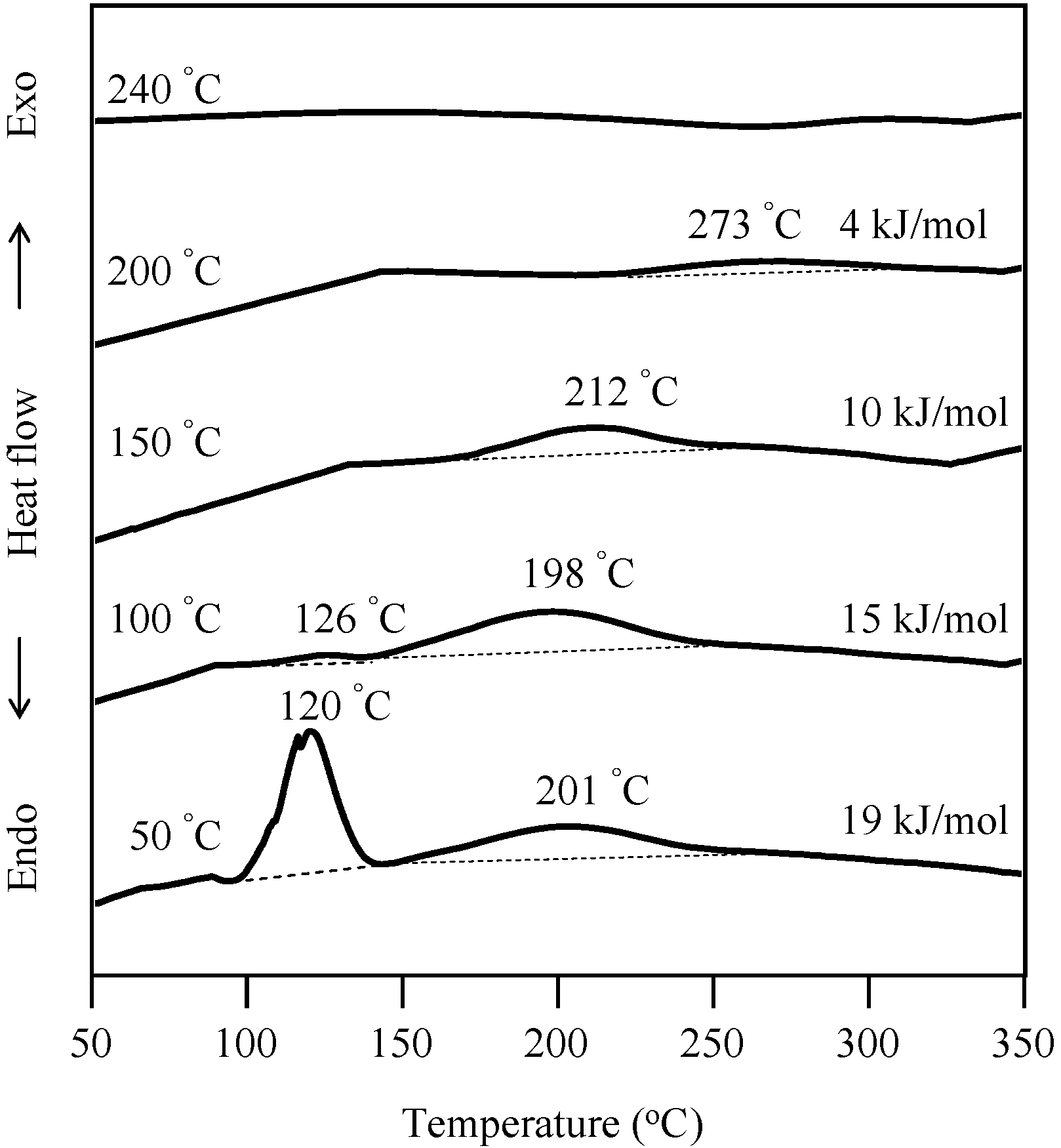
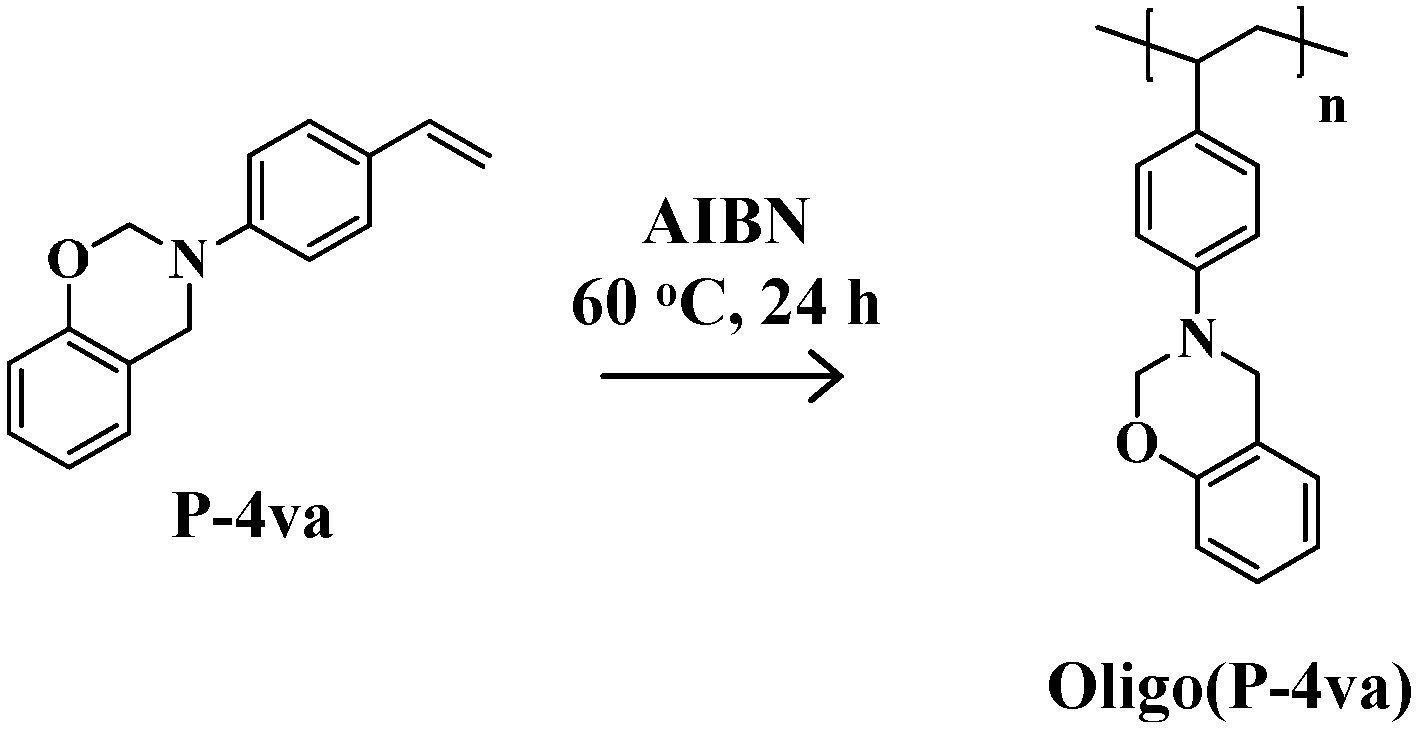
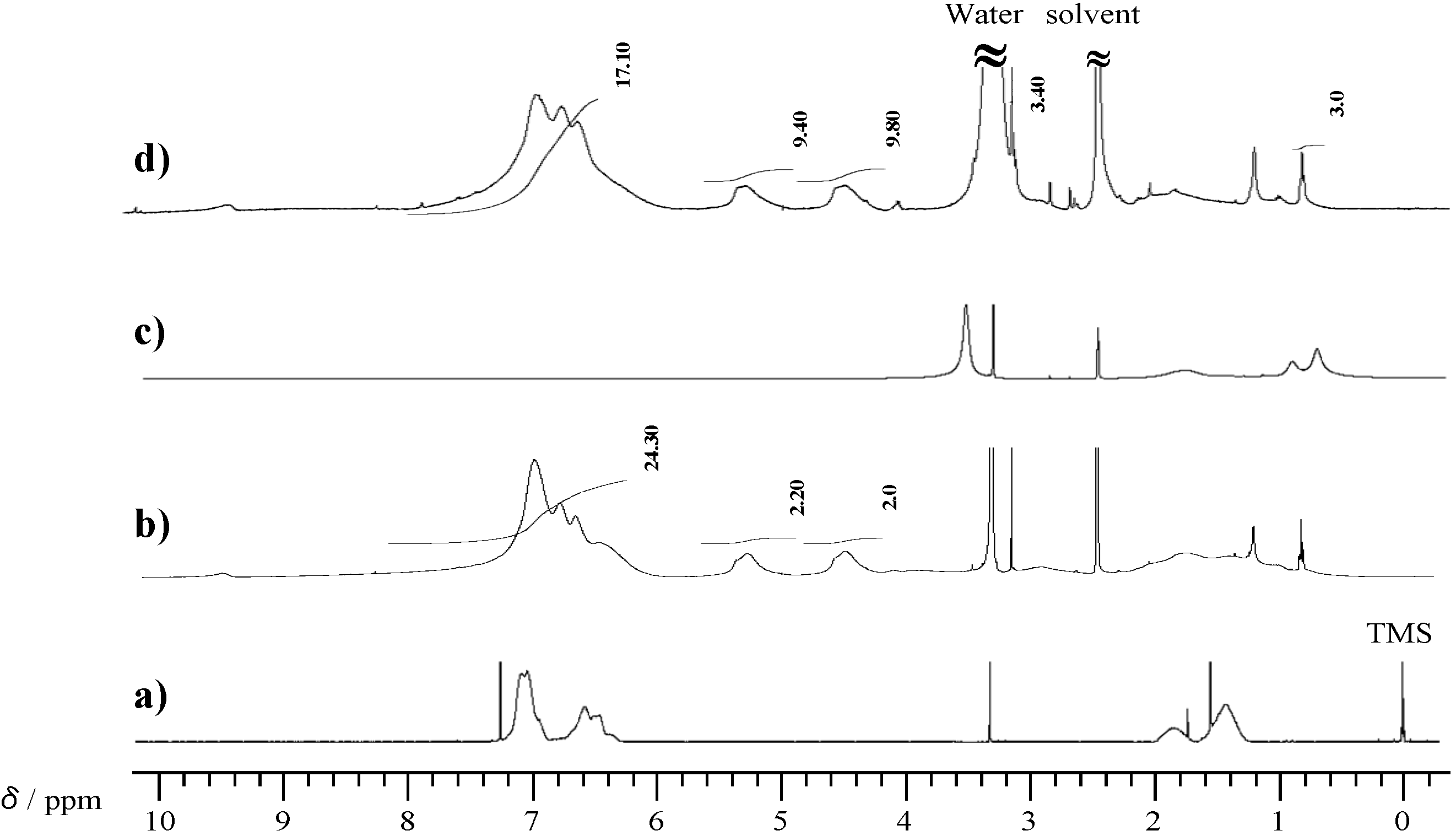
2.1. Radical Homopolymerization of P-4va
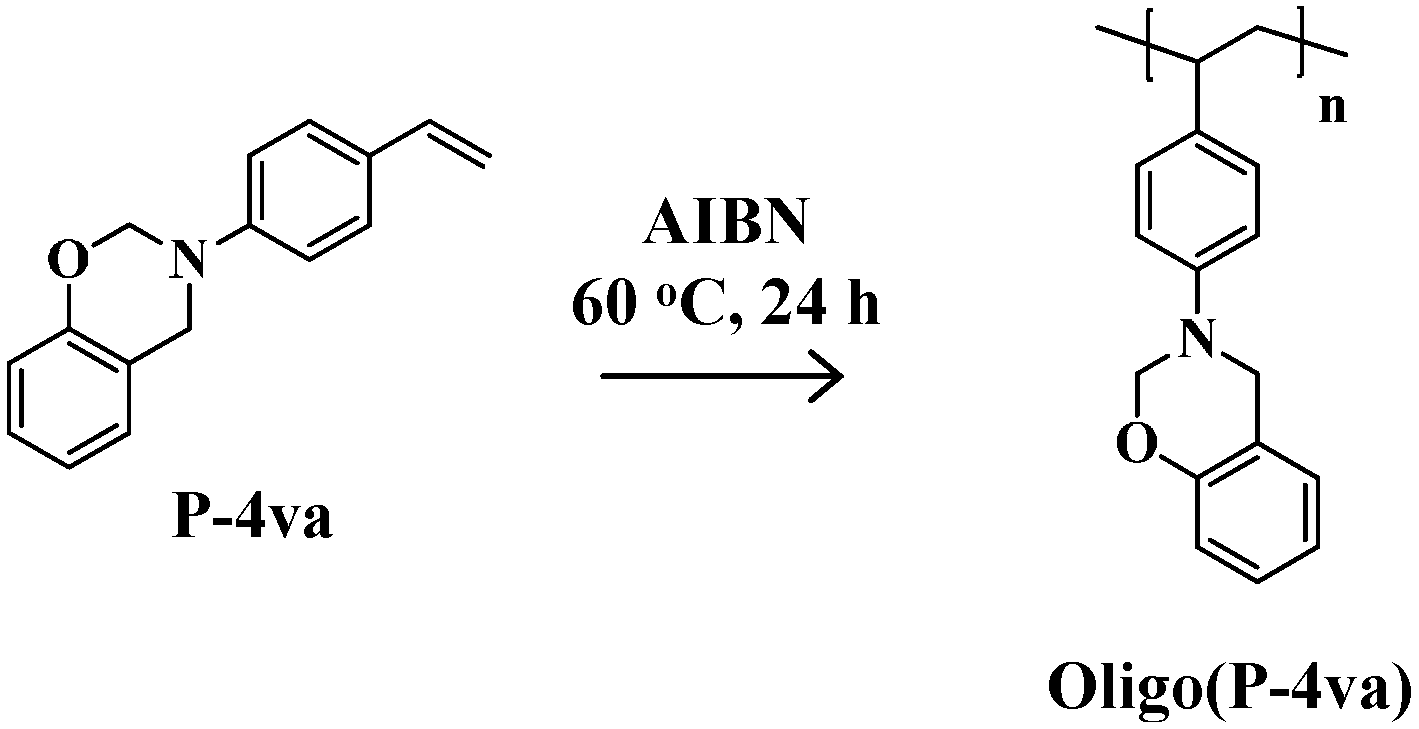
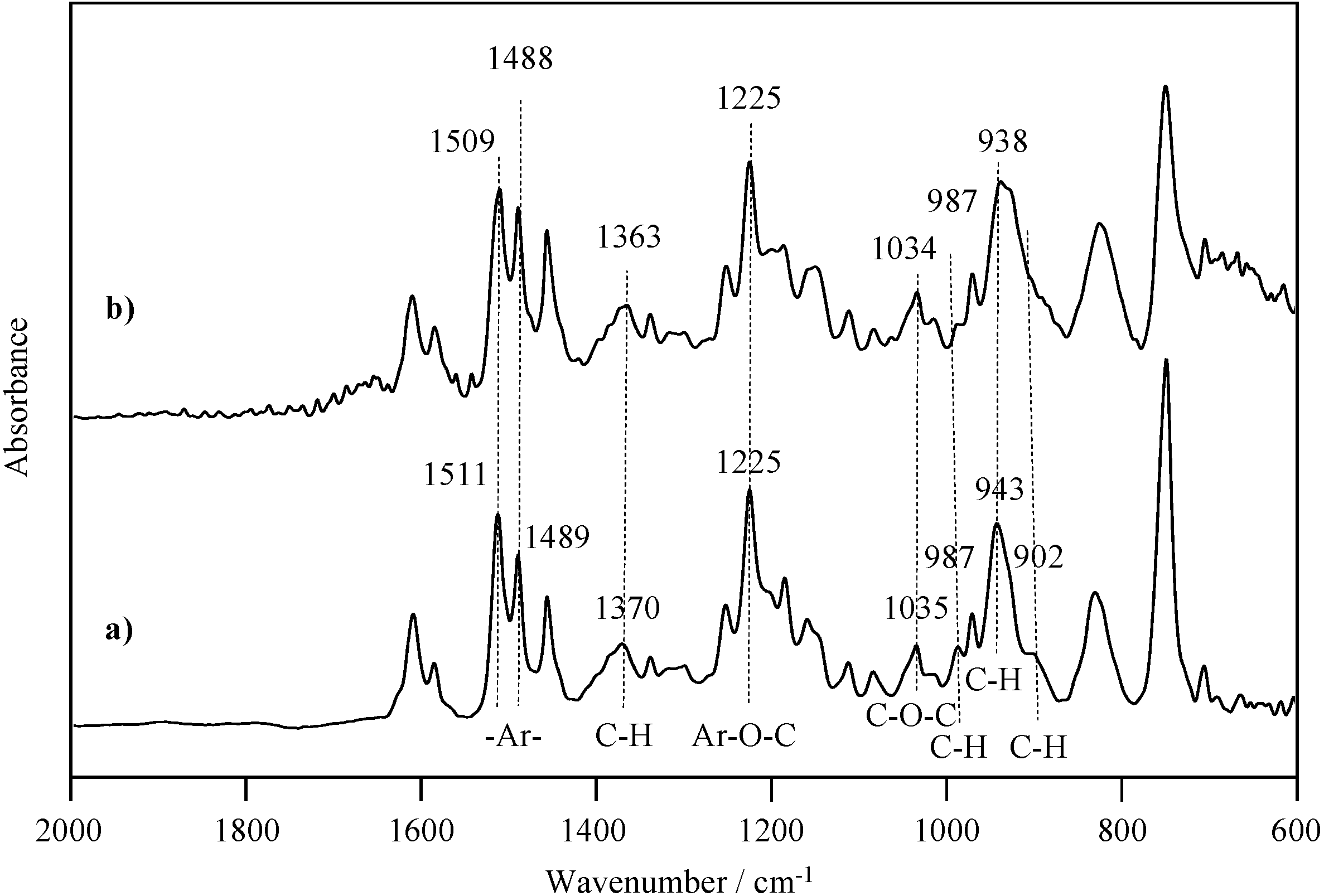
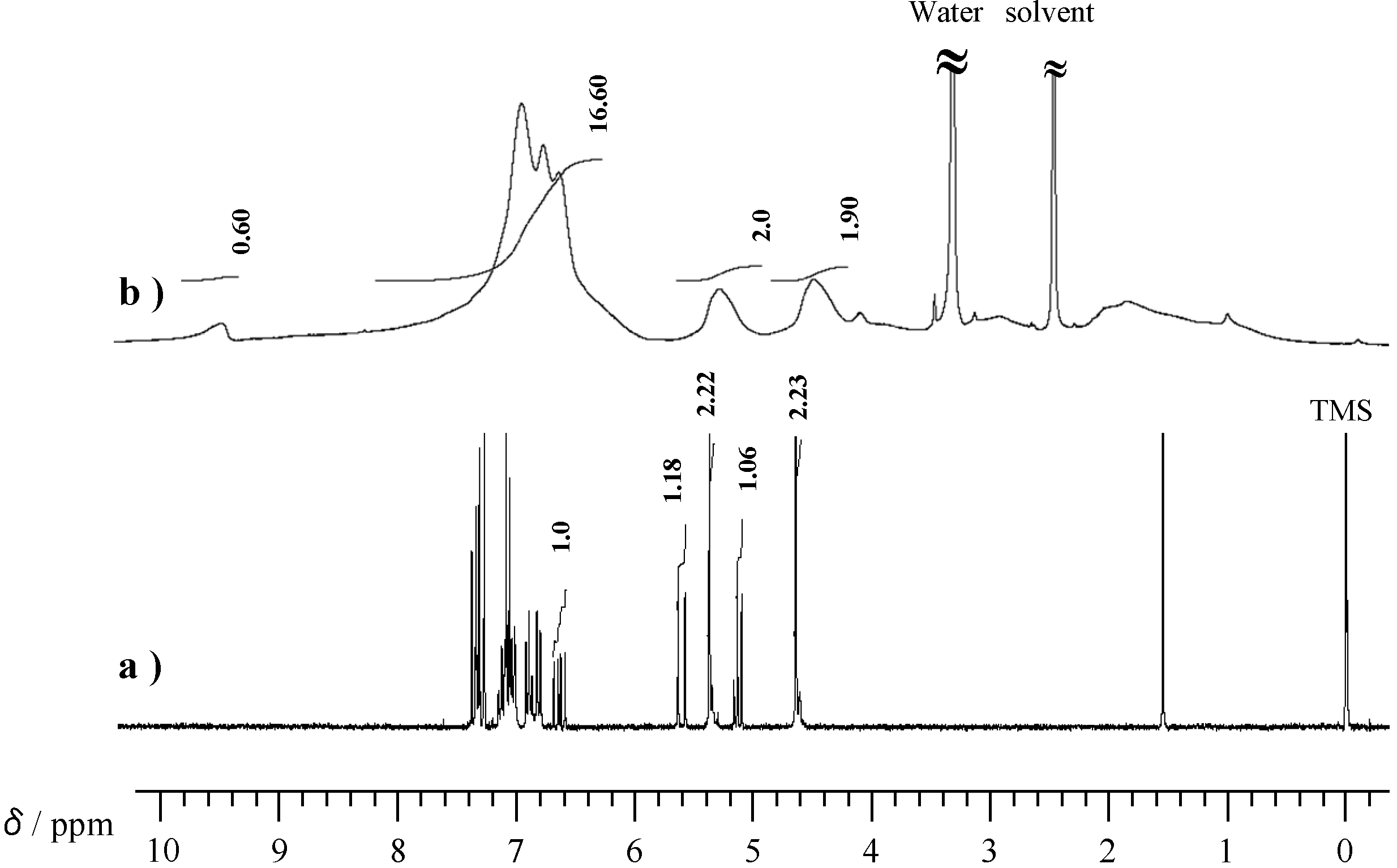
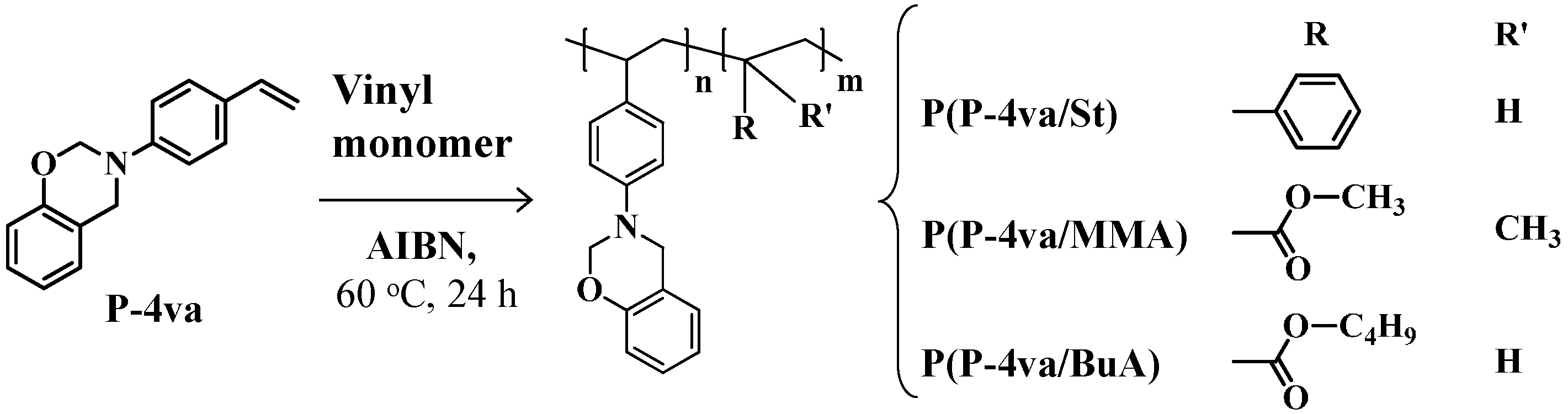
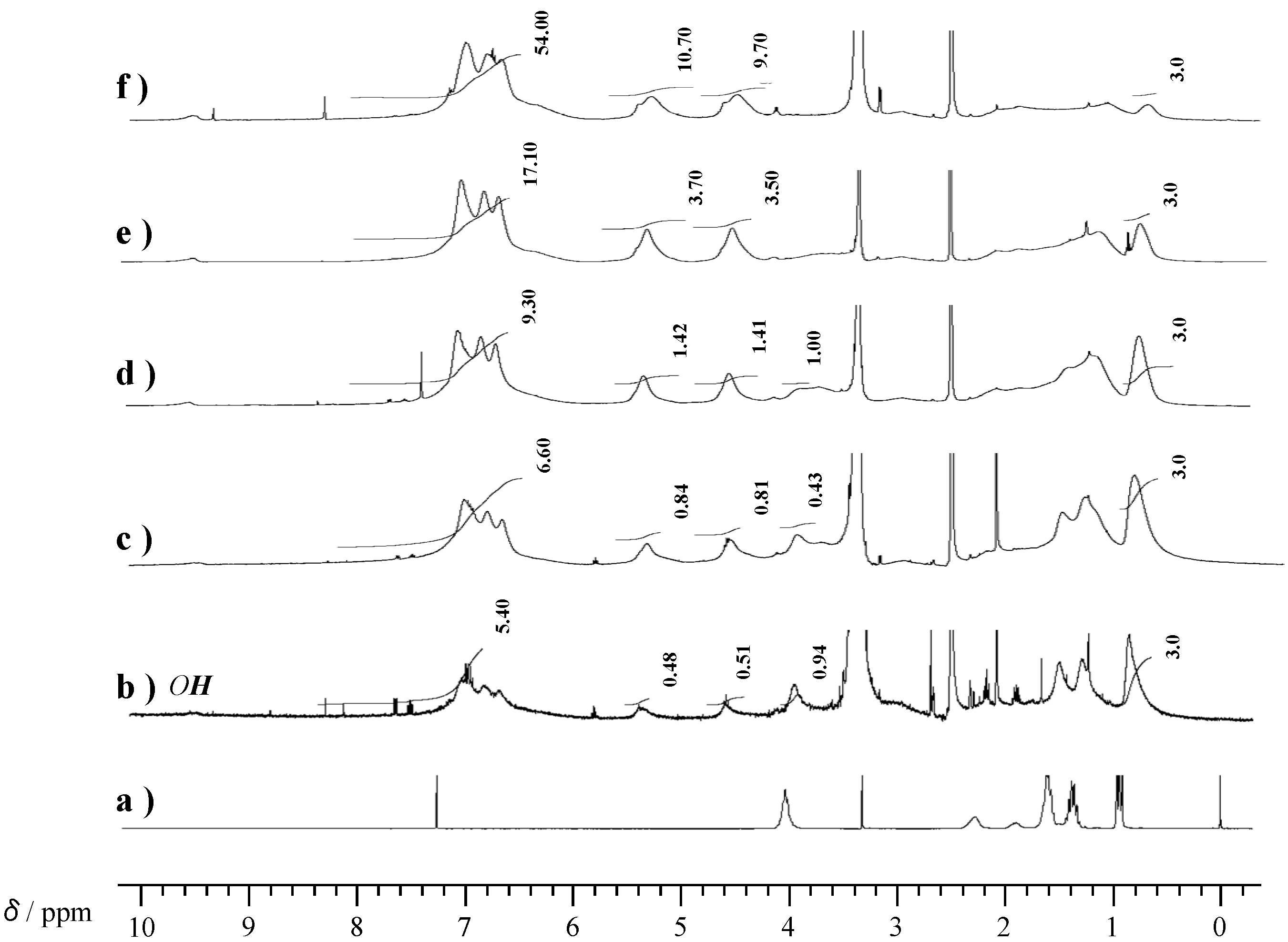
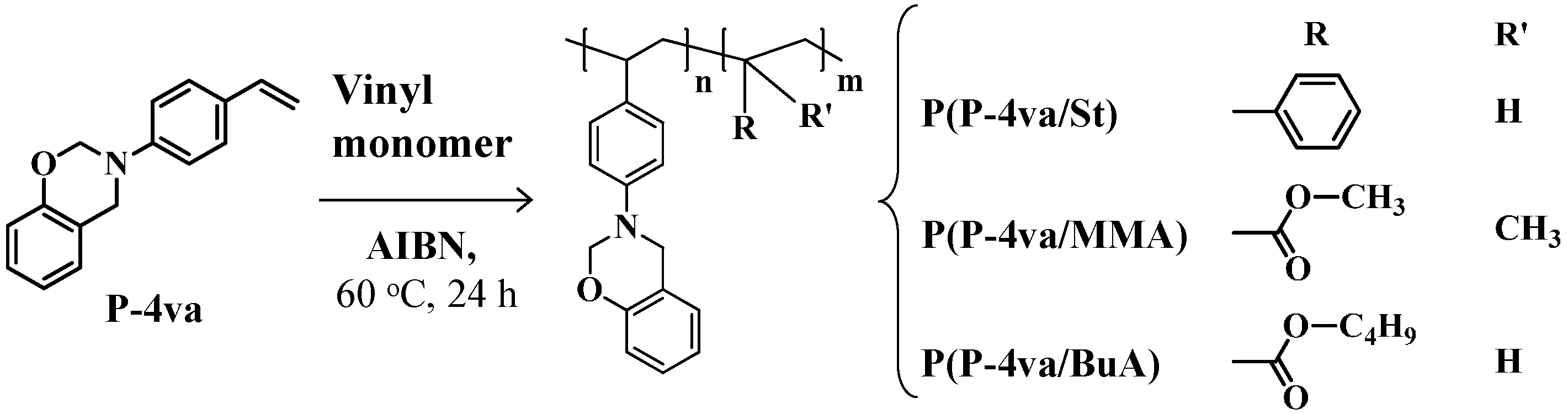
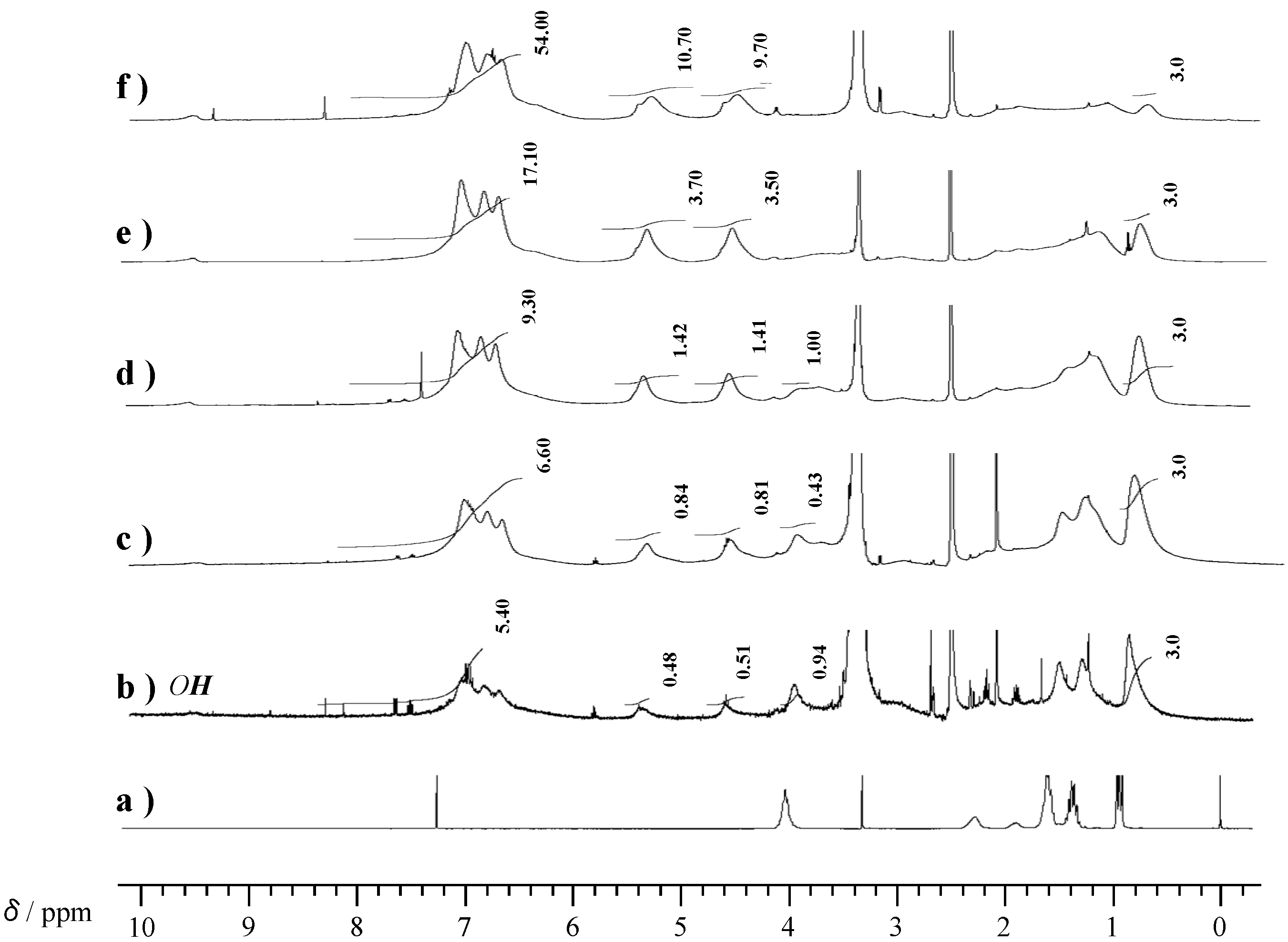
2.2. Radical Copolymerization of P-4va with St, MMA, and BuA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
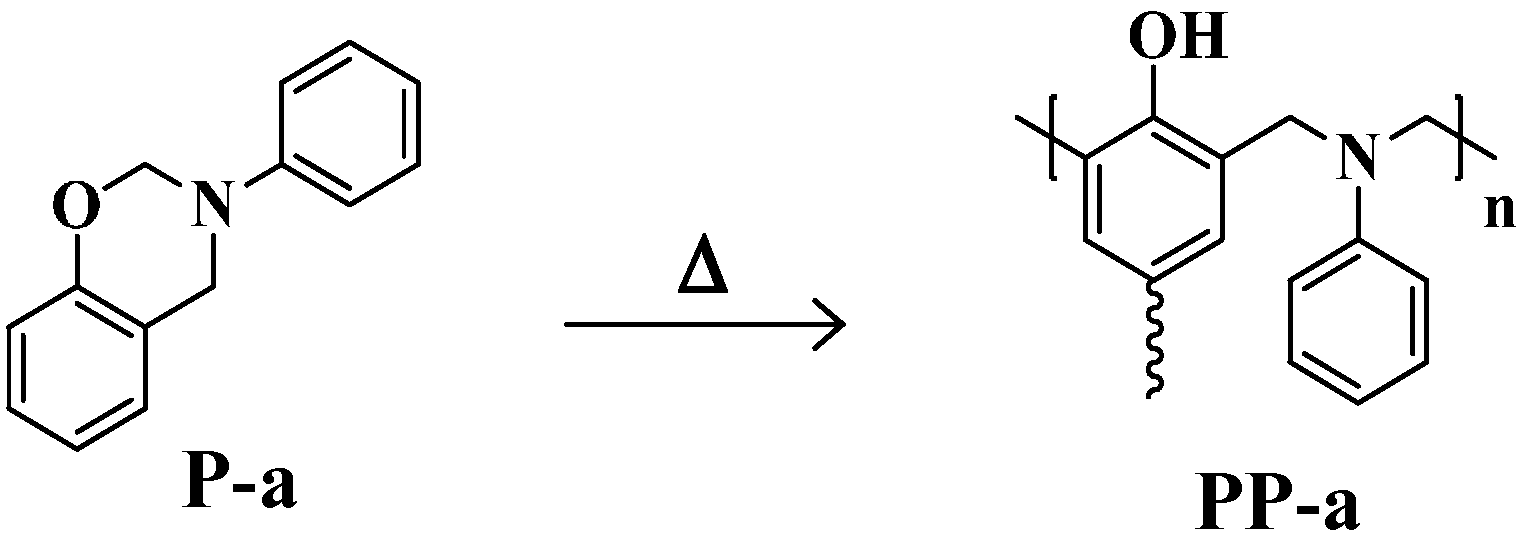{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Monomer feed a (mol %) | Yield b (%) | Composition c (mol %) | Mw d | Mw/Mn d | ||
|---|---|---|---|---|---|---|---|
| P-4va | Comonomer | f1 | f2 | ||||
| oligo(P-4va) | 100 | - | 64 | 100 | - | 1070 | 1.65 |
| PSt | - | 100 | 52 | - | 100 | 33,100 | 1.84 |
| PMMA | - | 100 | 73 | - | 100 | 47,800 | 1.80 |
| PBuA | - | 100 | 80 | - | 100 | 98,800 | 5.40 |
| P(P-4va/St) | 50 | 50 | 60 | 23 | 77 | 8300 | 2.96 |
| P(P-4va/MMA) | 90 | 10 | 57 | 83 | 17 | 1900 | 1.81 |
| P(P-4va/BuA) | 90 | 10 | 98 | 86 | 14 | 3400 | 2.43 |
| P(P-4va/BuA) | 70 | 30 | 92 | 67 | 33 | 13,800 | 5.11 |
| P(P-4va/BuA) | 50 | 50 | 86 | 50 | 50 | 14,000 | 1.28 |
| P(P-4va/BuA) | 30 | 70 | 83 | 42 | 58 | 16,400 | 1.26 |
| P(P-4va/BuA) | 10 | 90 | 64 | 37 | 63 | 51,500 | 1.84 |
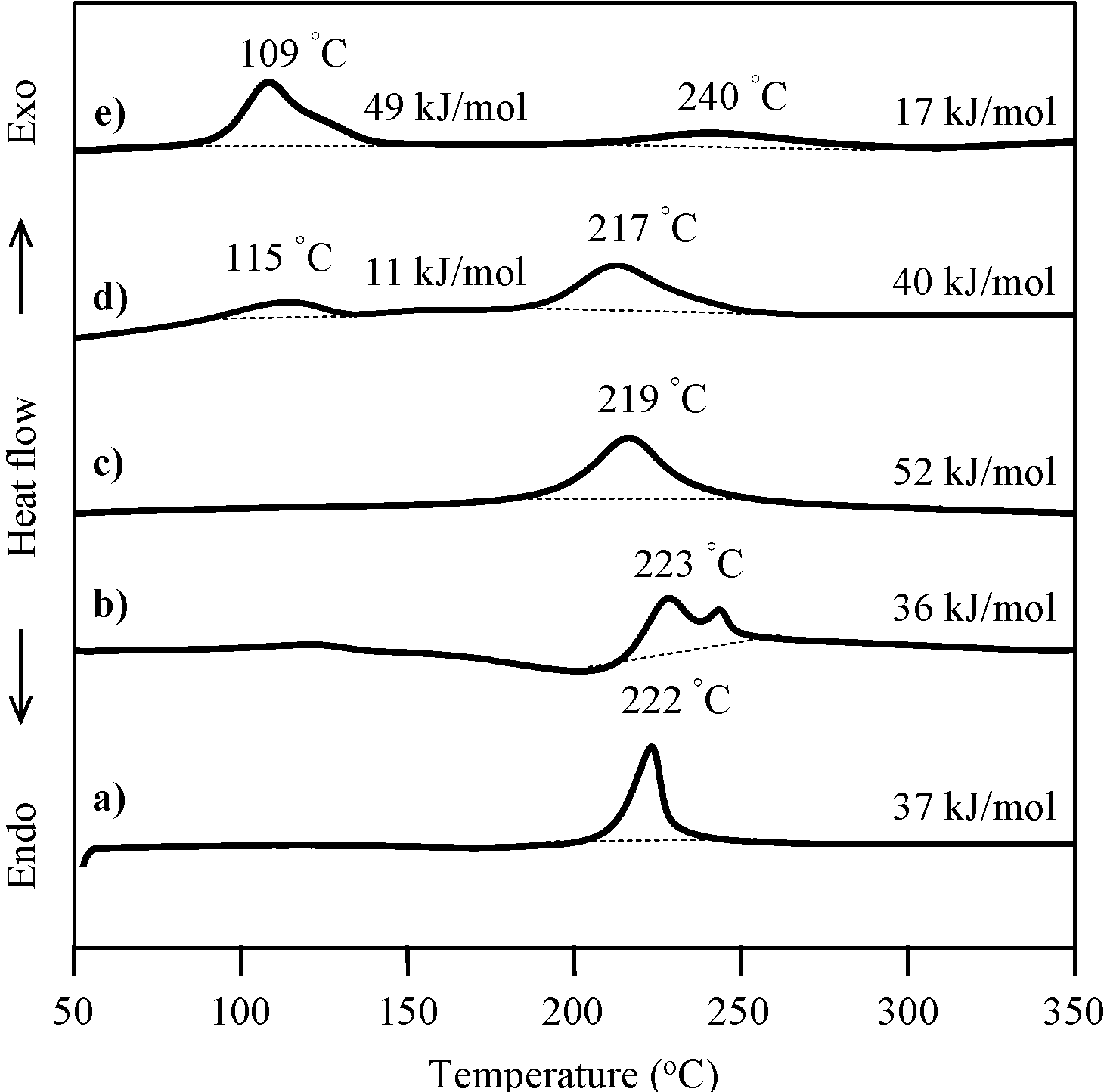
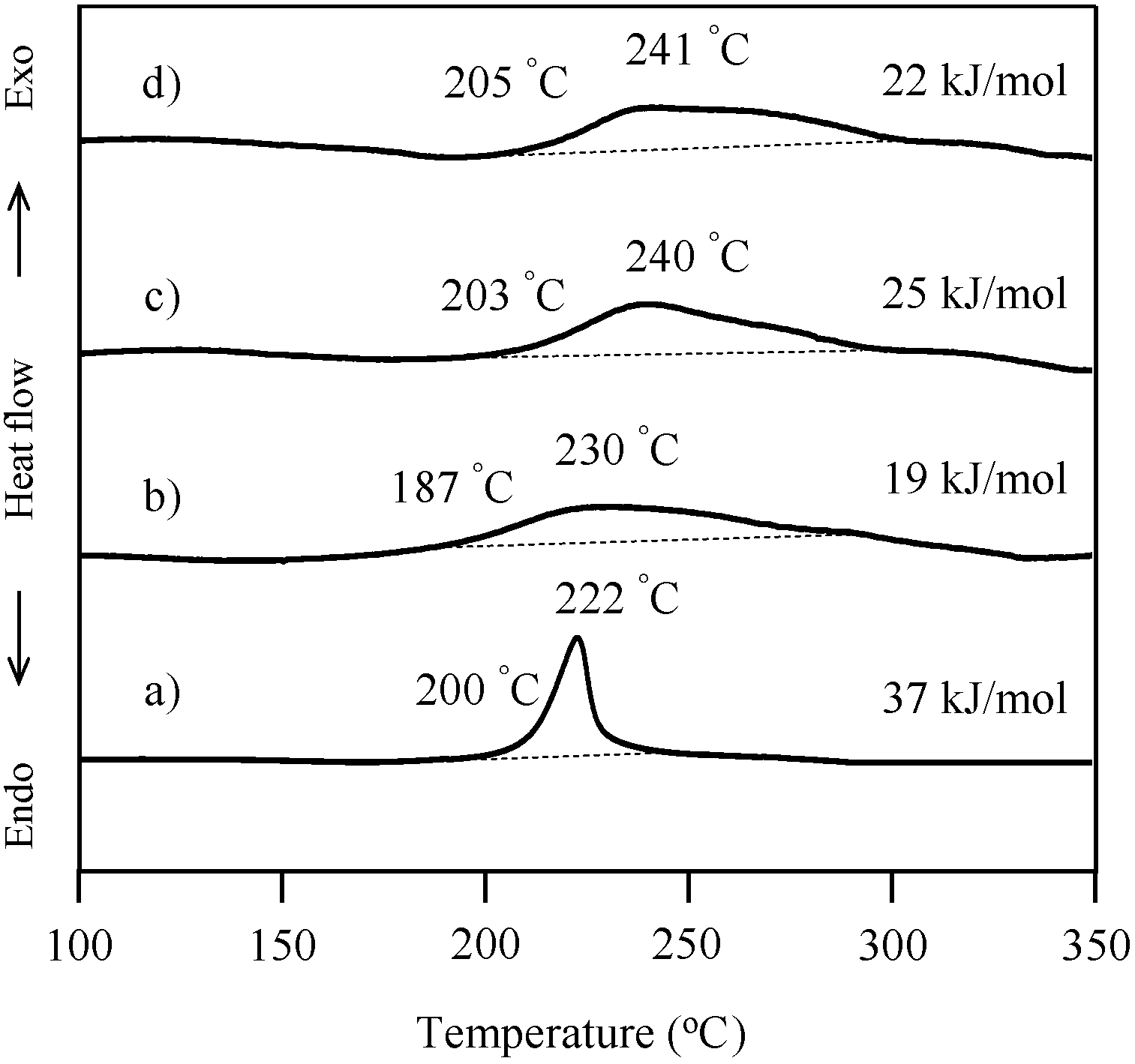
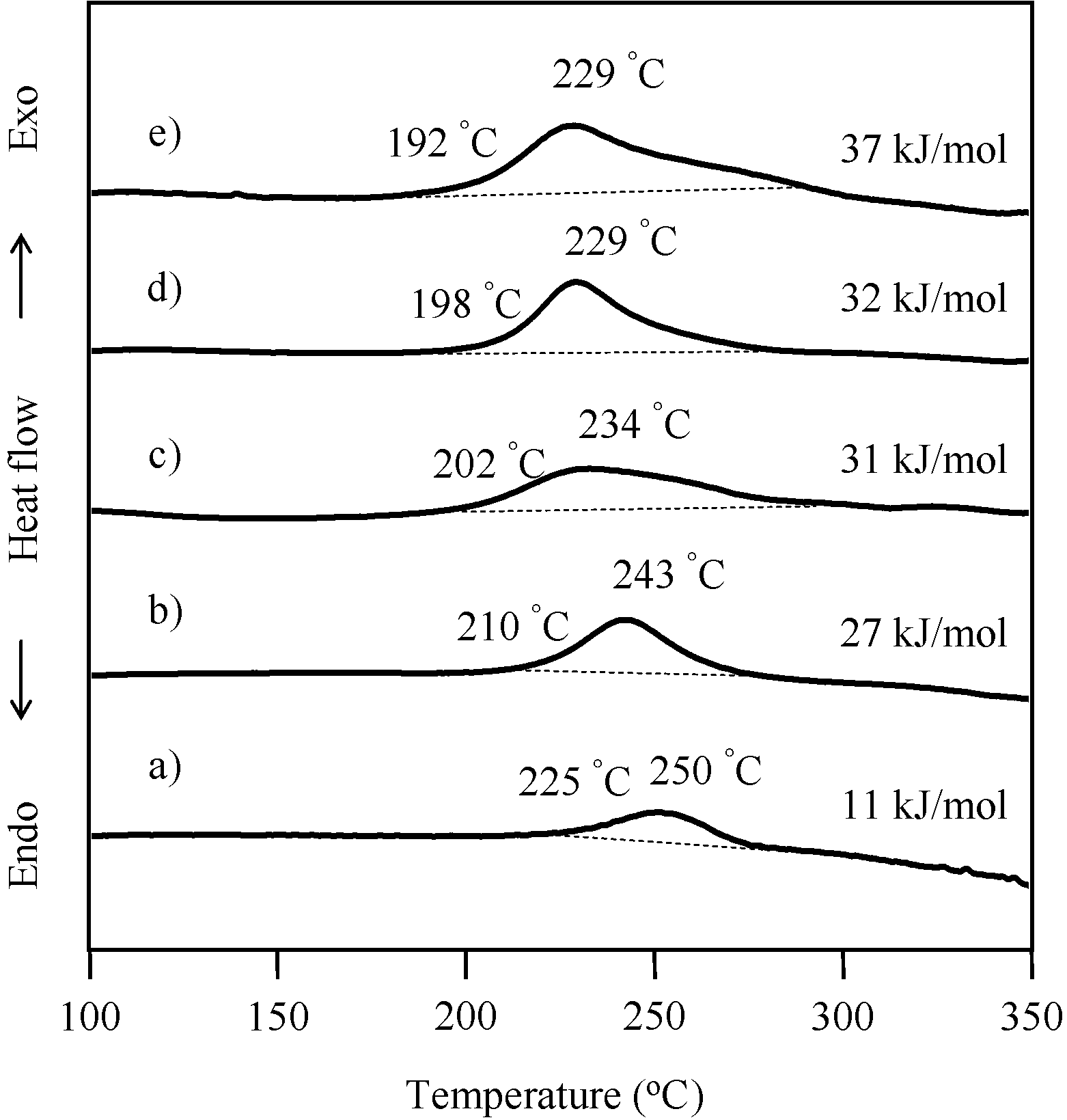
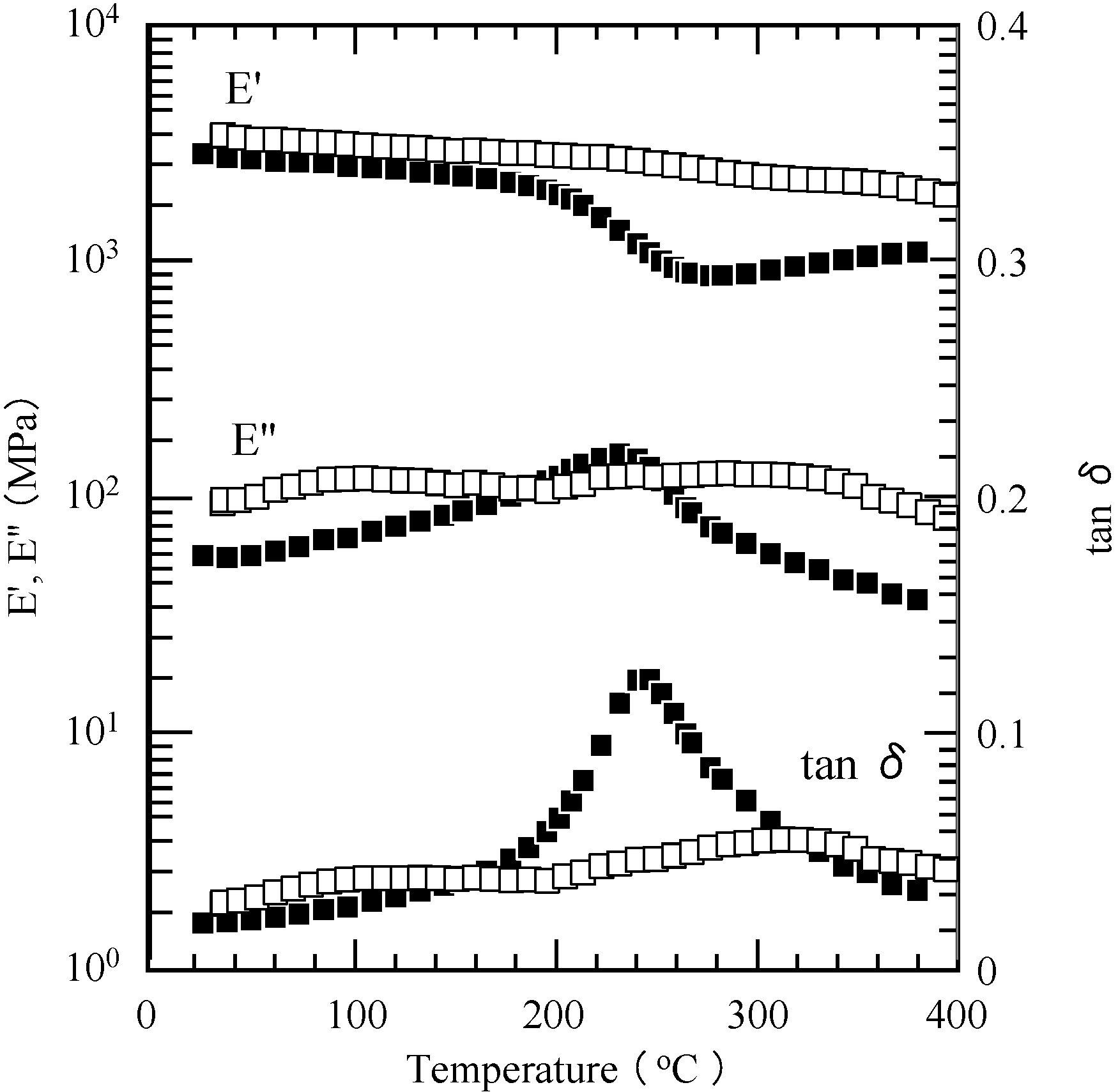
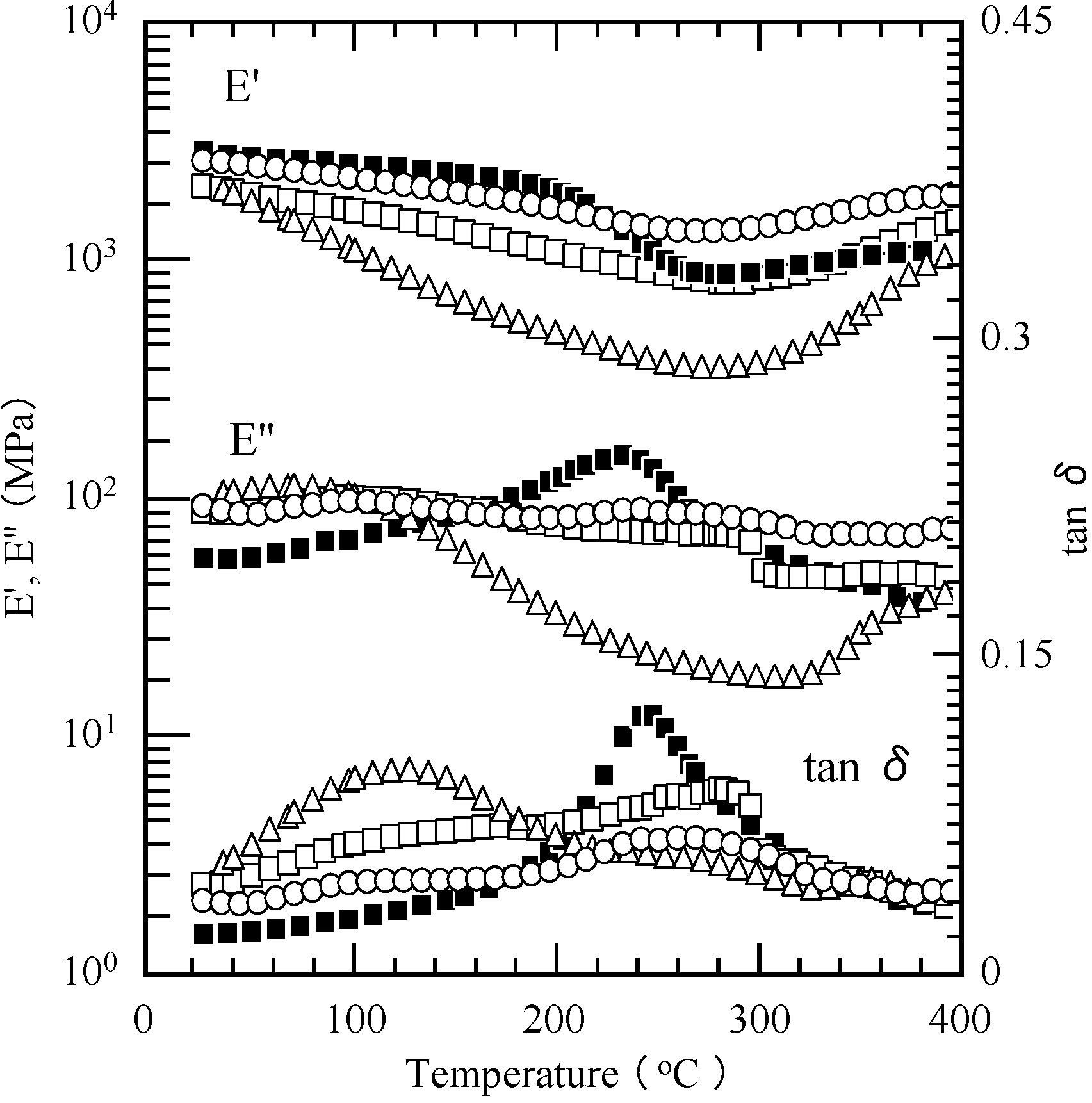
2.3. Thermal Curing Behavior of Oligo(P-4va) and Copolymers
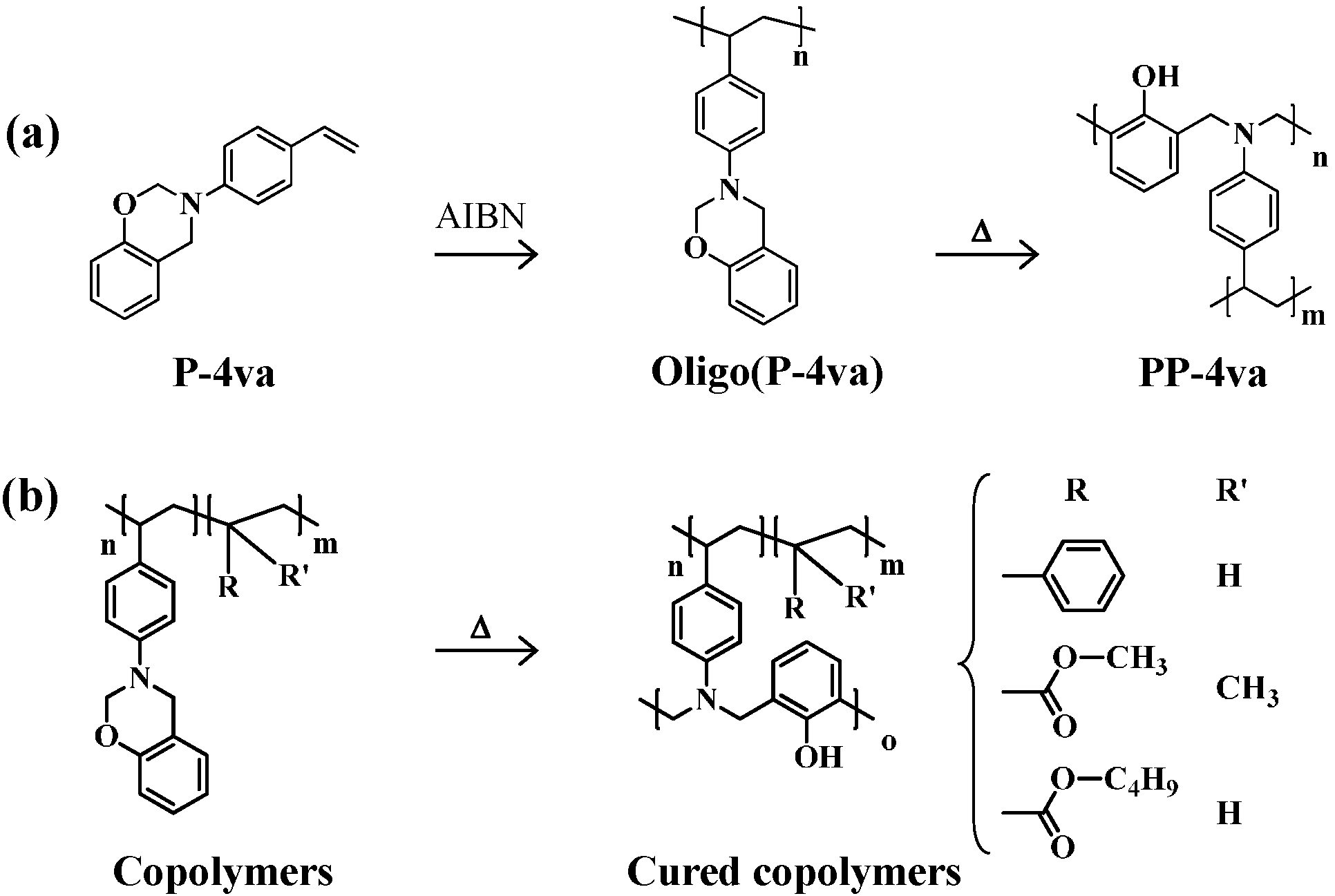
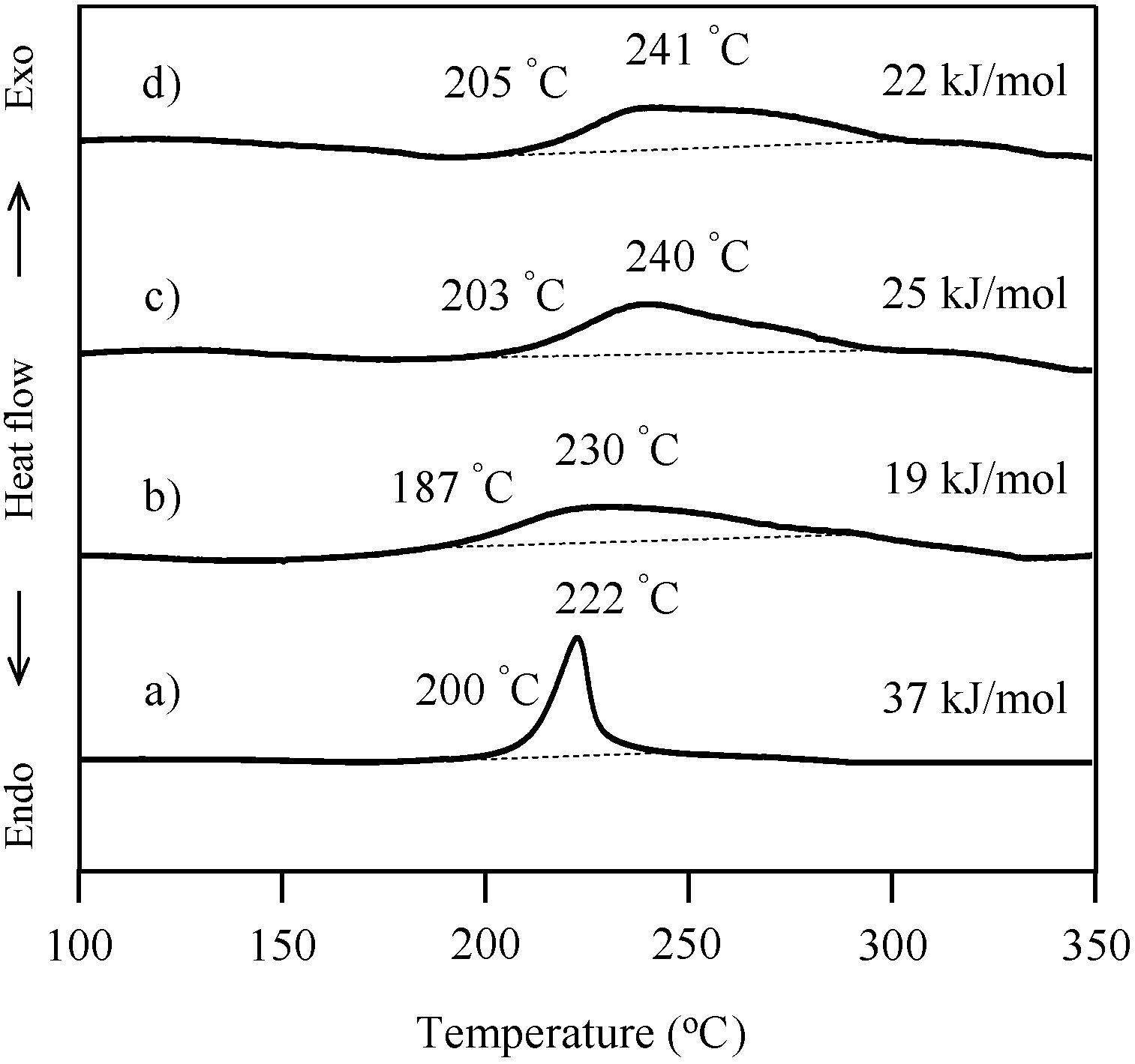
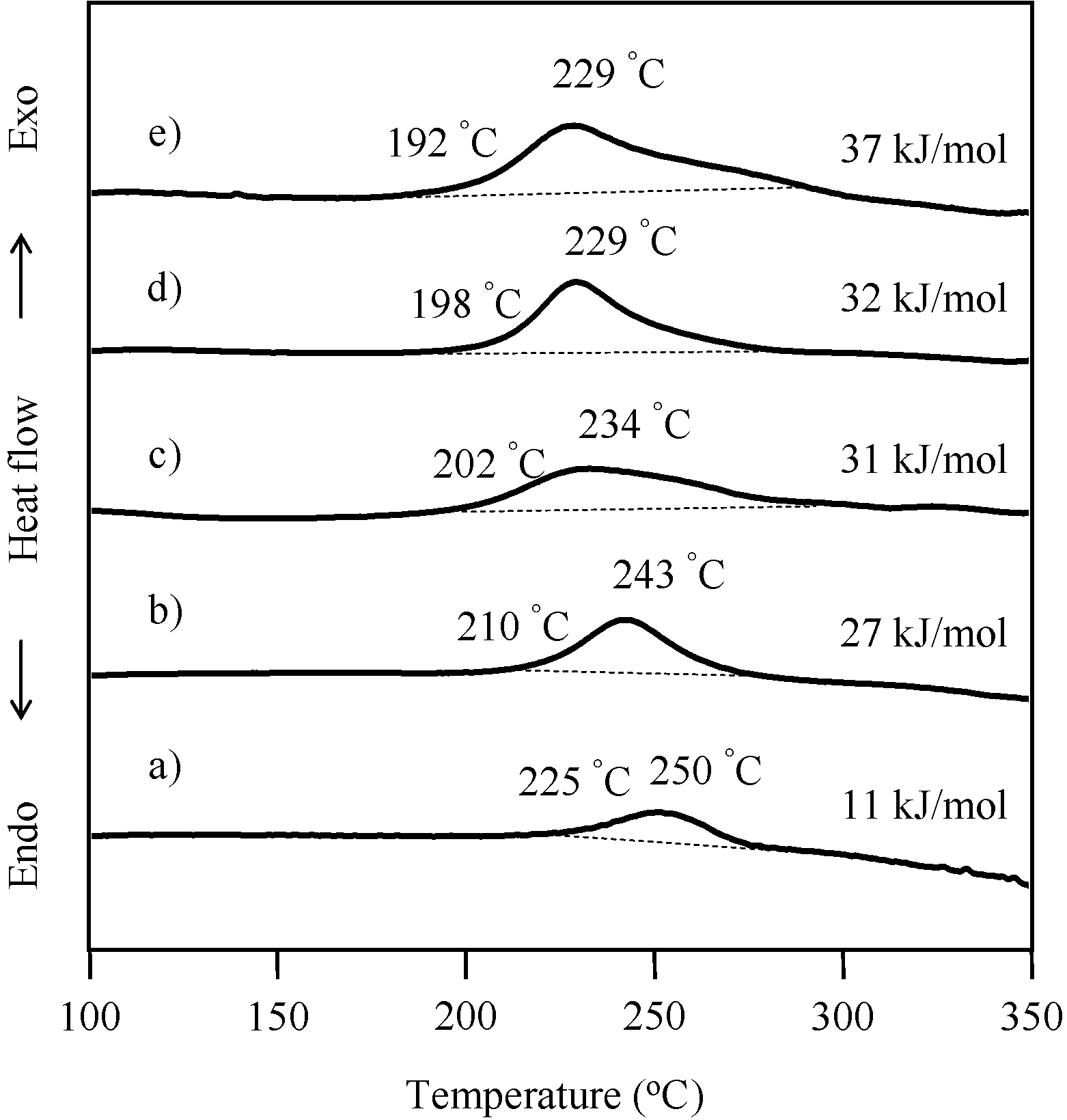
2.4. Thermal Cure of Oligo(P-4va) and the Copolymers
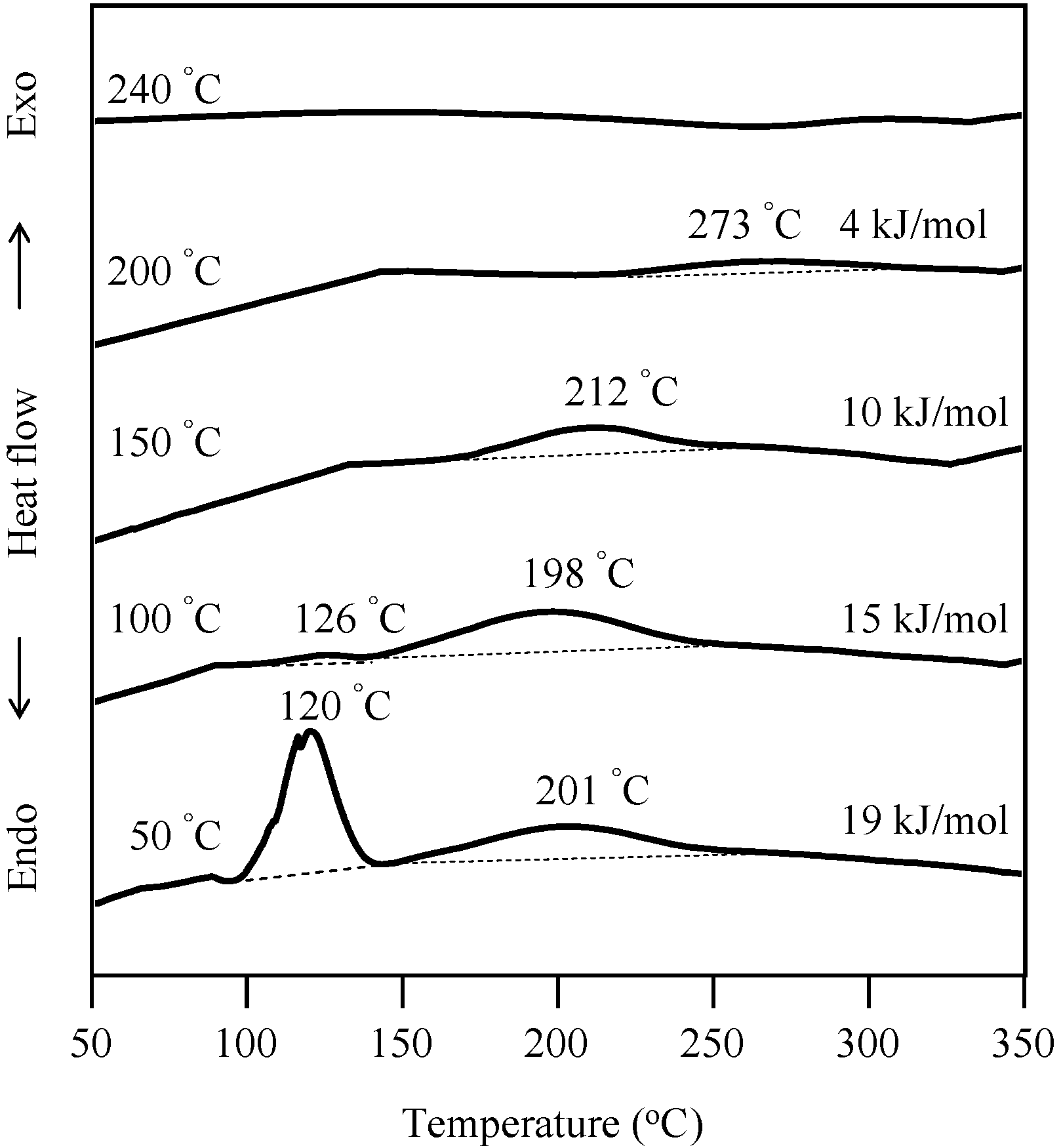
2.5. Properties of the Thermally Cured Films

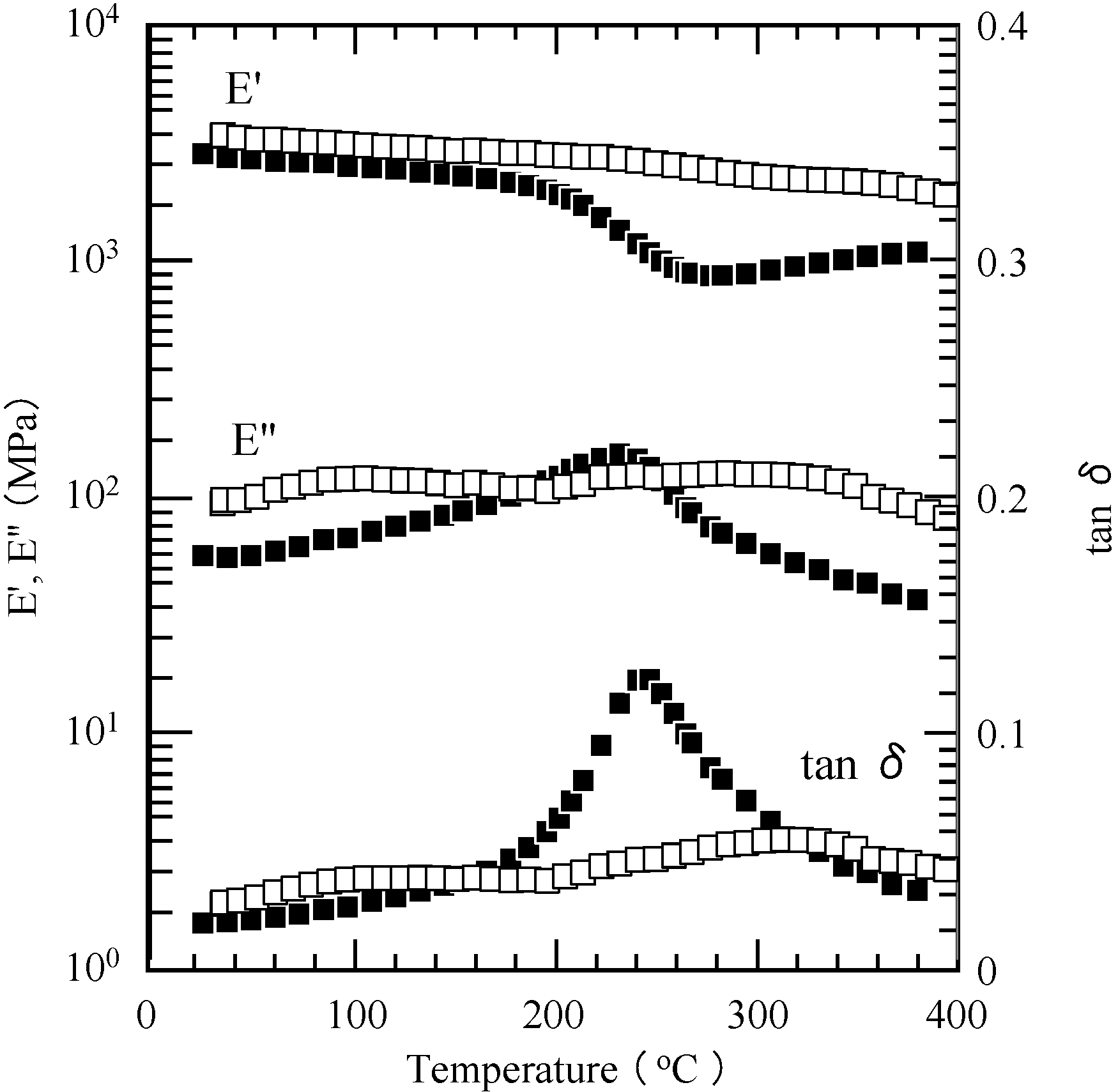
| Code | Composition a (mol %) | Tg (°C) | T5 b (°C) | T10 b (°C) | Char yield at 850 °C b (%) | |||
|---|---|---|---|---|---|---|---|---|
| DMA | ||||||||
| f1 | f2 | E'' | tan δ | DSC | ||||
| PP-4va | 100 | - | 230 c | 244 c | - | 355 c | 378 c | 48 c |
| oligo(P-4va) | 100 | - | - | - | - | 364 | 394 | 53 |
| PSt | - | 100 | - | - | 70 | 346 | 372 | 0 |
| PMMA | - | 100 | 64 | 64 | 73 | 332 | 343 | 0 |
| PBuA | - | 100 | - | - | −62 | 328 | 346 | 0.7 |
| P(P-4va/St) | 23 | 77 | - | - | - | 345 | 378 | 45 |
| P(P-4va/MMA) | 83 | 17 | 286 | 307 | - | 346 | 373 | 44 |
| P(P-4va/BuA) | 86 | 14 | - | - | - | 352 | 385 | 44 |
| P(P-4va/BuA) | 67 | 33 | 238 | 262 | - | 349 | 380 | 43 |
| P(P-4va/BuA) | 50 | 50 | 253 | 283 | 42 | 352 | 380 | 49 |
| P(P-4va/BuA) | 42 | 58 | 235 | 277 | −12 | 339 | 363 | 35 |
| P(P-4va/BuA) | 37 | 63 | - | - | −55 | 346 | 360 | 12 |
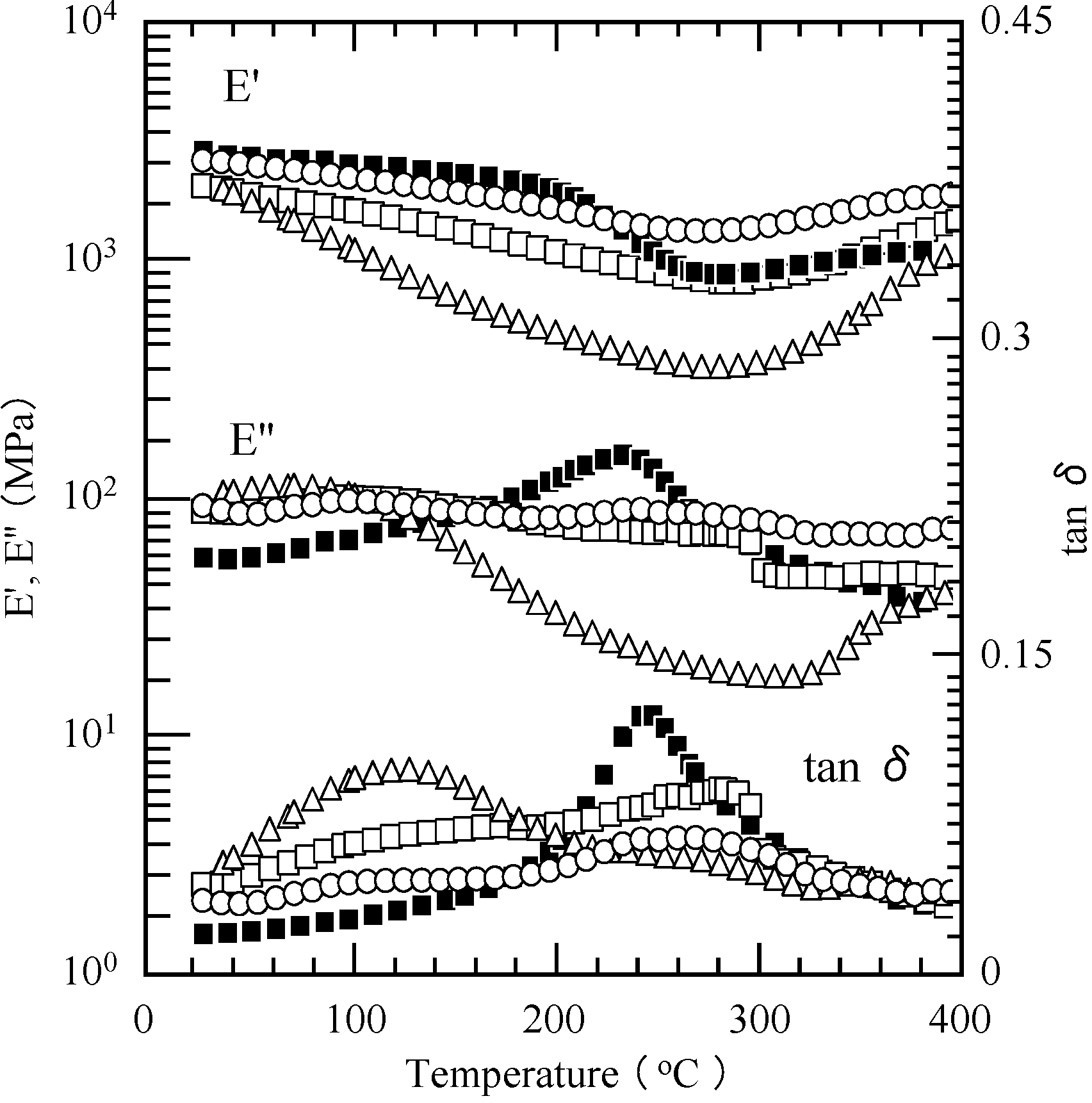
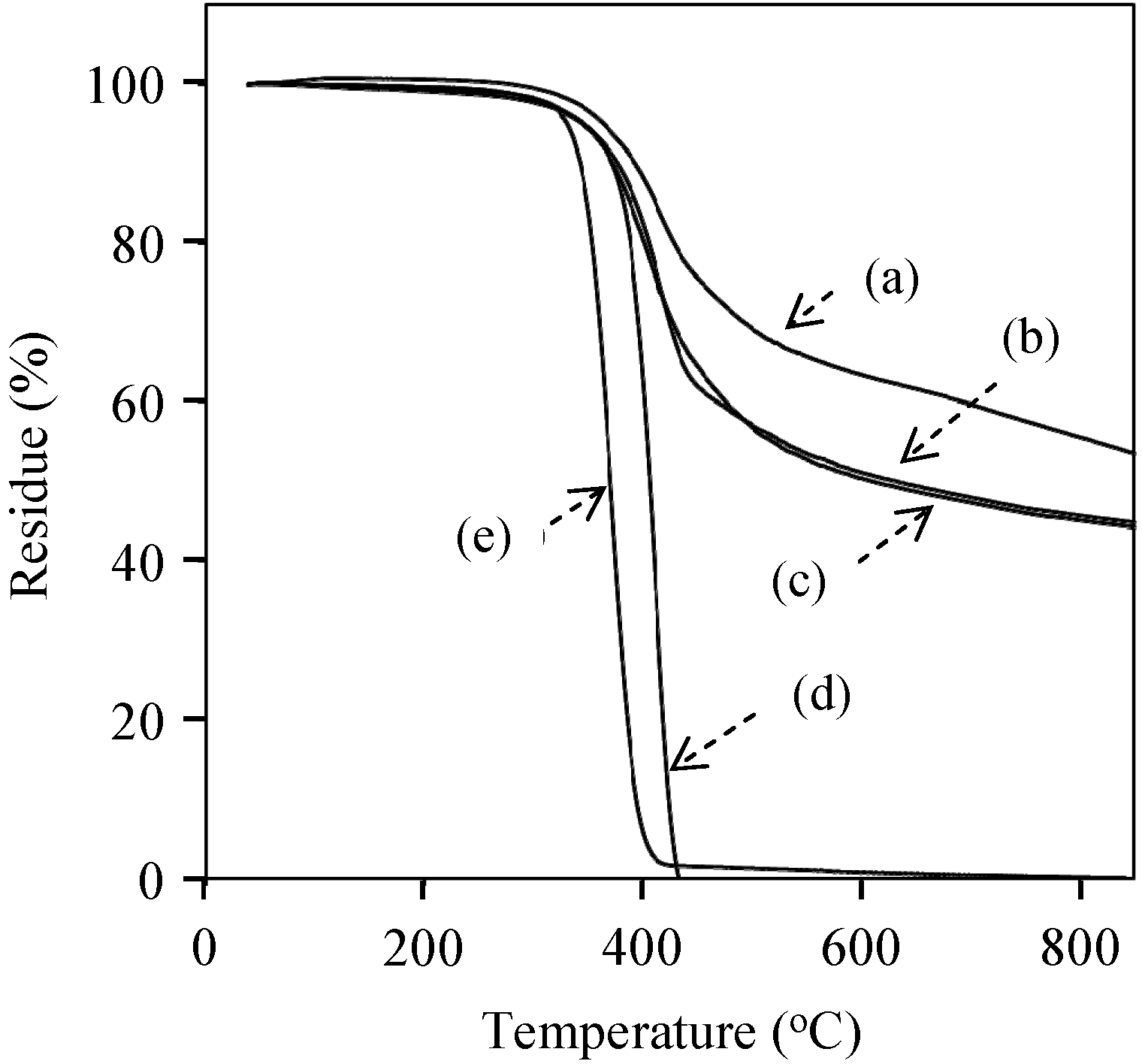
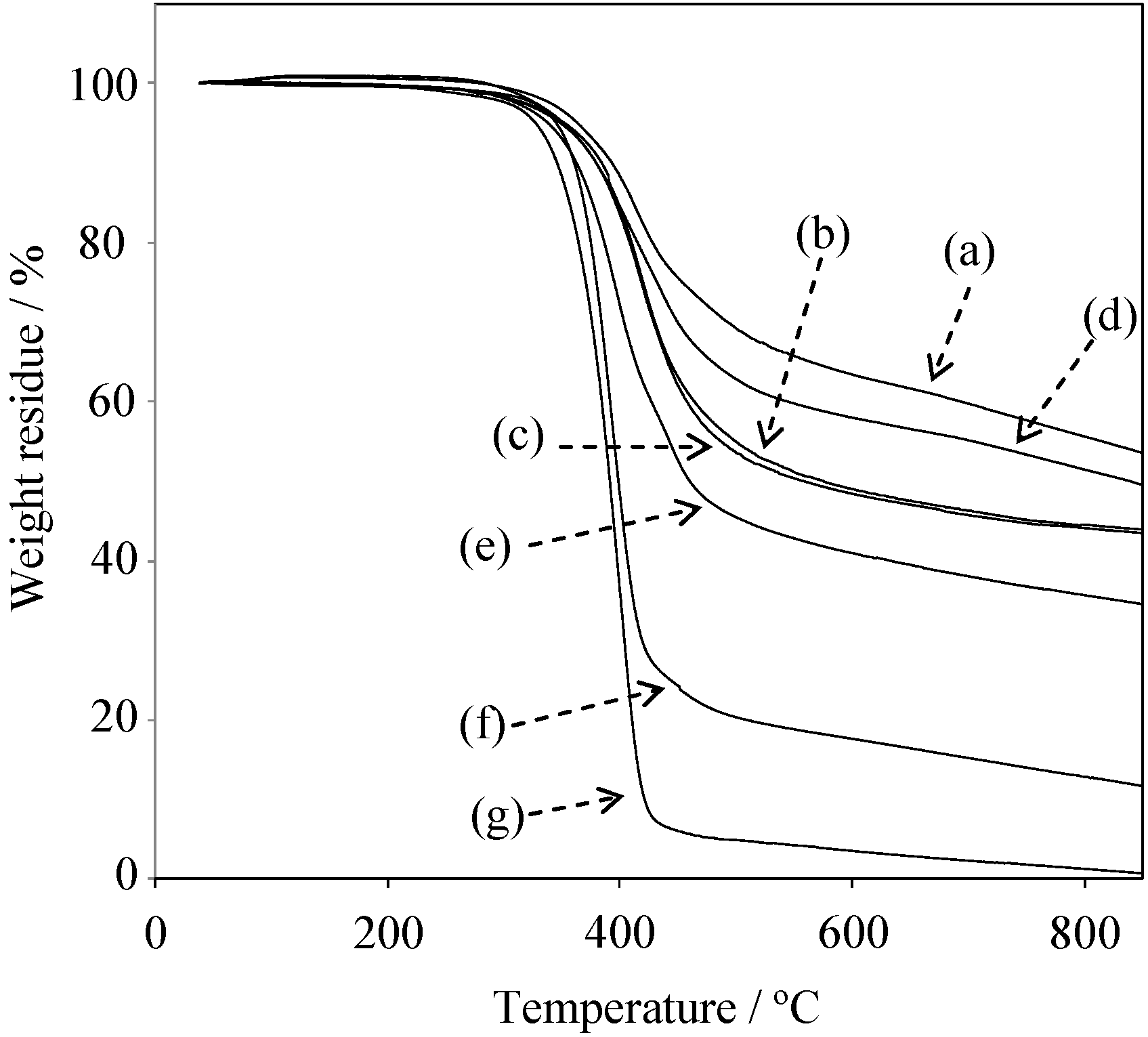
3. Experimental Section
3.1. Reagents
3.2. Radical Polymerization of P-4va and Vinyl Monomers in the Presence of AIBN
3.3. Radical Copolymerization of P-4va with Vinyl Monomers
3.4. Thermal Cure of Oligo(P-4va) and Copolymer Films
3.5. Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ishida, H.; Agag, T. Handbook of Benzoxazine Resins; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Ghosh, N.N.; Kiskan, B.; Yagci, Y. Polybenzoxazines—New high performance thermosetting resins: synthesis and properties. Prog. Polym. Sci. 2007, 32, 1344–1391. [Google Scholar] [CrossRef]
- Takeichi, T.; Kawauchi, T.; Agag, T. High performance polybenzoxazines as a novel type of phenolic resin. Polym. J. 2008, 40, 1121–1131. [Google Scholar]
- Yagci, Y.; Kiskan, B.; Ghosh, N.N. Recent advancement on polybenzoxazine—A newly developed high performance thermoset. J. Polym. Part A: Polym. Chem. 2009, 47, 5565–5576. [Google Scholar] [CrossRef]
- Sarawut Rimdusit, S.; Tiptipakorn, S.; Jubsilp, C.; Takeichi, T. Polybenzoxazine alloys and blends: Some unique properties and applications. React. Funct. Polym. 2013, 73, 369–380. [Google Scholar]
- Ning, X.; Ishida, H. Phenolic materials via ring-opening polymerization: Synthesis and characterization of bisphenol-A based benzoxazines and their polymers. J. Polym. Sci. Part A: Polym. Chem. 1994, 32, 1121–1129. [Google Scholar] [CrossRef]
- Ning, X.; Ishida, H. Phenolic materials via ring-opening polymerization of benzoxazines: Effect of molecular structure on mechanical and dynamic mechanical properties. J. Polym. Sci. Part B: Polym. Phys. 1994, 32, 921–927. [Google Scholar] [CrossRef]
- Ishida, H.; Allen, D.J. Physical and mechanical characterization of near-zero shrinkage polybenzoxazines. J. Polym. Sci. Part B: Polym. Phys. 1996, 34, 1019–1030. [Google Scholar] [CrossRef]
- Ishida, H.; Allen, D.J. Gelation behaviour of near-zero shrinkage polybenzoxazines. J. Appl. Polym. Sci. 2001, 79, 406–417. [Google Scholar] [CrossRef]
- Ishida, H.; Low, H.Y. A study on the volumetric expansion of benzoxazine-based phenolic resin. Macromolecules 1997, 30, 1099–1106. [Google Scholar] [CrossRef]
- Wang, Y.X.; Ishida, H. Development of low-viscosity benzoxazine resins and their polymers. J. Appl. Polym. Sci. 2002, 86, 2953–2966. [Google Scholar] [CrossRef]
- Su, Y.C.; Chang, F.C. Synthesis and characterization of fluorinated polybenzoxazine material with low dielectric constant. Polymer 2003, 44, 7989–7996. [Google Scholar] [CrossRef]
- Wang, C.F.; Su, Y.C.; Kuo, S.W.; Huang, C.F.; Sheen, Y.C.; Chang, F.C. Low-surface-free-energy materials based on polybenzoxazines. Angew. Chem. Int. Ed. 2006, 45, 2248–2251. [Google Scholar] [CrossRef]
- Brunovska, Z.; Ishida, H. Thermal study on the copolymers of phthalonitrile and phenylnitrile-functional benzoxazines. J. Appl. Polym. Sci. 1999, 73, 2937–2949. [Google Scholar] [CrossRef]
- Kim, H.J.; Brunovska, Z.; Ishida, H. Molecular characterization of the polymerization of acetylene-functional benzoxazine resins. Polymer 1999, 40, 1815–1822. [Google Scholar] [CrossRef]
- Agag, T.; Takeichi, T. Novel benzoxazine monomers containing p-phenyl propargyl ether: Polymerization of monomers and properties of polybenzoxazines. Macromolecules 2001, 34, 7257–7263. [Google Scholar] [CrossRef]
- Agag, T.; Takeichi, T. Synthesis and characterization of novel benzoxazine monomers containing allyl groups and their high performance thermosets. Molecromolecules 2003, 36, 6010–6017. [Google Scholar] [CrossRef]
- Takeichi, T.; Kano, T.; Agag, T. Synthesis and thermal cure of high molecular weight polybenzoxazine precursors and the properties of the thermosets. Polymer 2005, 46, 12172–12180. [Google Scholar] [CrossRef]
- Agag, T.; Takeichi, T. High-molecular-weight AB-type benzoxazines as new precursors for high-performance thermosets. J. Polym. Sci. Part A: Polym. Chem. 2007, 45, 1878–1888. [Google Scholar] [CrossRef]
- Takeichi, T.; Kano, T.; Agag, T.; Kawauchi, T.; Furukawa, N. Preparation of high molecular weight polybenzoxazine prepolymers containing siloxane unites and properties of their thermosets. J. Polym. Sci. Part A: Polym. Chem. 2010, 48, 5945–5952. [Google Scholar] [CrossRef]
- Agag, T.; Takeichi, T. Synthesis, characterization and clay-reinforcement of epoxy cured with benzoxazine. High Perform. Polym. 2002, 14, 115–132. [Google Scholar]
- Takeichi, T.; Guo, Y. Synthesis and characterization of poly(urethane-benzoxazine)/clay hybrid nanocomposites. J. Appl. Polym. Sci. 2003, 90, 4075–4083. [Google Scholar] [CrossRef]
- Takeichi, T.; Guo, Y.; Rimdusit, S. Performance improvement of polybenzoxazine by alloying with polyimide: Effect of preparation method on the properties. Polymer 2005, 46, 4909–4916. [Google Scholar] [CrossRef]
- Takeichi, T.; Saito, Y.; Agag, T.; Muto, H.; Kawauchi, T. High performance polymer alloys of polybenzoxazine and bismaleimide. Polymer 2008, 49, 1173–1179. [Google Scholar] [CrossRef]
- Ardhyananta, H.; Wahid, M.H.; Sasaki, M.; Agag, T.; Kawauchi, T.; Ismail, H.; Takeichi, T. Performance enhancement of polybenzoxazine by hybridization with polysiloxane. Polymer 2008, 49, 4585–4591. [Google Scholar] [CrossRef]
- Ardhyananta, H.; Kawauchi, T.; Ismail, H.; Takeichi, T. Effect of pendant group of polysiloxanes on the thermal and mechanical properties of polybenzoxazine hybrids. Polymer 2009, 50, 5959–5969. [Google Scholar] [CrossRef]
- Ardhyananta, H.; Kawauchi, T.; Takeichi, T.; Ismail, H. Preparation and properties of polybenzoxazine-poly(dimethylsiloxane-co-diphenylsiloxane) hybrids as high performance polymers. High Perform. Polym. 2010, 22, 609–632. [Google Scholar] [CrossRef]
- Kiskan, B.; Yagci, Y. Synthetic strategies to combine high performance benzoxazine thermosets with polymers. Macromol. Symp. 2010, 298, 145–153. [Google Scholar] [CrossRef]
- Jubsilp, C.; Takeichi, T.; Rimdusit, S. Property enhancement of polybenzoxazine modified with dianhydride. Polym. Degrad. Stab. 2011, 96, 1047–1053. [Google Scholar] [CrossRef]
- Rimdusit, S.; Bangsen, W.; Kasemsiri, P. Chemorheology and thermomechanical characteristics of benzoxazine-urethane copolymers. J. Appl. Polym. Sci. 2011, 121, 3669–3678. [Google Scholar] [CrossRef]
- Takeichi, T.; Uchida, S.; Inoue, Y.; Kawauchi, T.; Furukawa, N. Preparation and properties of polymer alloys consisting of high-molecularweight-benzoxazine and bismaleimide. High Perform. Polym. 2014, 26, 265–273. [Google Scholar] [CrossRef]
- Uchida, S.; Kawauchi, T.; Furukawa, N.; Takeichi, T. Polymer alloys of high-molecular-weight benzoxazine and epoxy resin. High Perform. Polym. 2014, 26, 846–855. [Google Scholar] [CrossRef]
- Jang, J.; Seo, D. Performance improvement of rubber-modified polybenzoxazine. J. Appl. Polym. Sci. 1998, 67, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ishida, H. Probing the properties of particle-matrix interphase in reactive rubber-grafted polybenzoxazine resins by atomic force microscopy. Compos. Interfaces 2005, 12, 481–499. [Google Scholar] [CrossRef]
- Lee, Y.H.; Allen, D.J.; Ishida, H. Effect of rubber reactivity on the morphology of polybenzoxazine blends investigated by atomic force microscopy and dynamic mechanical analysis. J. Appl. Polym. Sci. 2006, 100, 2443–2454. [Google Scholar] [CrossRef]
- Agag, T.; Takeichi, T. Effect of hydroxyphenylmaleimide on the curing behavior and thermomechanical properties of rubber-modified polybenzoxazine. High Perform. Polym. 2001, 13, S327–S342. [Google Scholar] [CrossRef]
- Kiskan, B.; Colak, D.; Muftuoglu, A.E.; Cianga, I.; Yagci, Y. Synthesis and characterization of thermally curable benzoxazine-functionalized polystyrene macromonomers. Macromol. Rapid Commun. 2005, 26, 819–824. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Kiskan, B.; Yagci, Y. Photoinitiated free radical polymerization using benzoxazines as hydrogen donors. Macromol. Rapid Commun. 2006, 27, 1539–1544. [Google Scholar] [CrossRef]
- Takeichi, T.; Thongpradith, S.; Hirai, S.; Takiguchi, T.; Kawauchi, T. Syntheses of novel benzoxazines having vinyl groups and thermal properties of the thermosets. High Perform. Polym. 2012, 24, 765–774. [Google Scholar] [CrossRef]
- Sample Availability: Samples are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeichi, T.; Thongpradith, S.; Kawauchi, T. Copolymers of Vinyl-Containing Benzoxazine with Vinyl Monomers as Precursors for High Performance Thermosets. Molecules 2015, 20, 6488-6503. https://doi.org/10.3390/molecules20046488
Takeichi T, Thongpradith S, Kawauchi T. Copolymers of Vinyl-Containing Benzoxazine with Vinyl Monomers as Precursors for High Performance Thermosets. Molecules. 2015; 20(4):6488-6503. https://doi.org/10.3390/molecules20046488
Chicago/Turabian StyleTakeichi, Tsutomu, Soulideth Thongpradith, and Takehiro Kawauchi. 2015. "Copolymers of Vinyl-Containing Benzoxazine with Vinyl Monomers as Precursors for High Performance Thermosets" Molecules 20, no. 4: 6488-6503. https://doi.org/10.3390/molecules20046488
APA StyleTakeichi, T., Thongpradith, S., & Kawauchi, T. (2015). Copolymers of Vinyl-Containing Benzoxazine with Vinyl Monomers as Precursors for High Performance Thermosets. Molecules, 20(4), 6488-6503. https://doi.org/10.3390/molecules20046488