
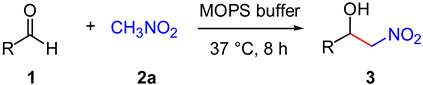
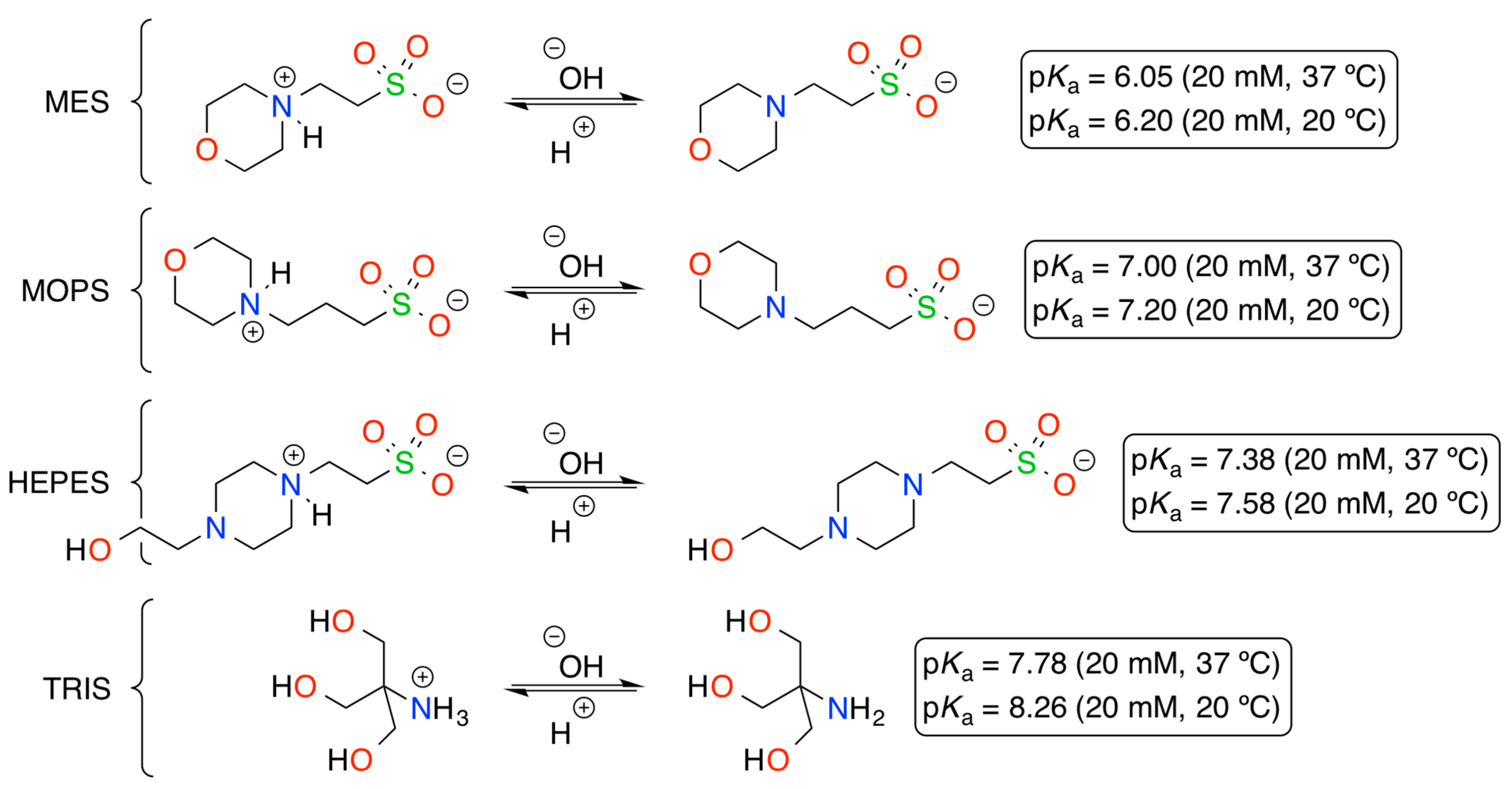
DNA-Catalyzed Henry Reaction in Pure Water and the Striking Influence of Organic Buffer Systems


Abstract
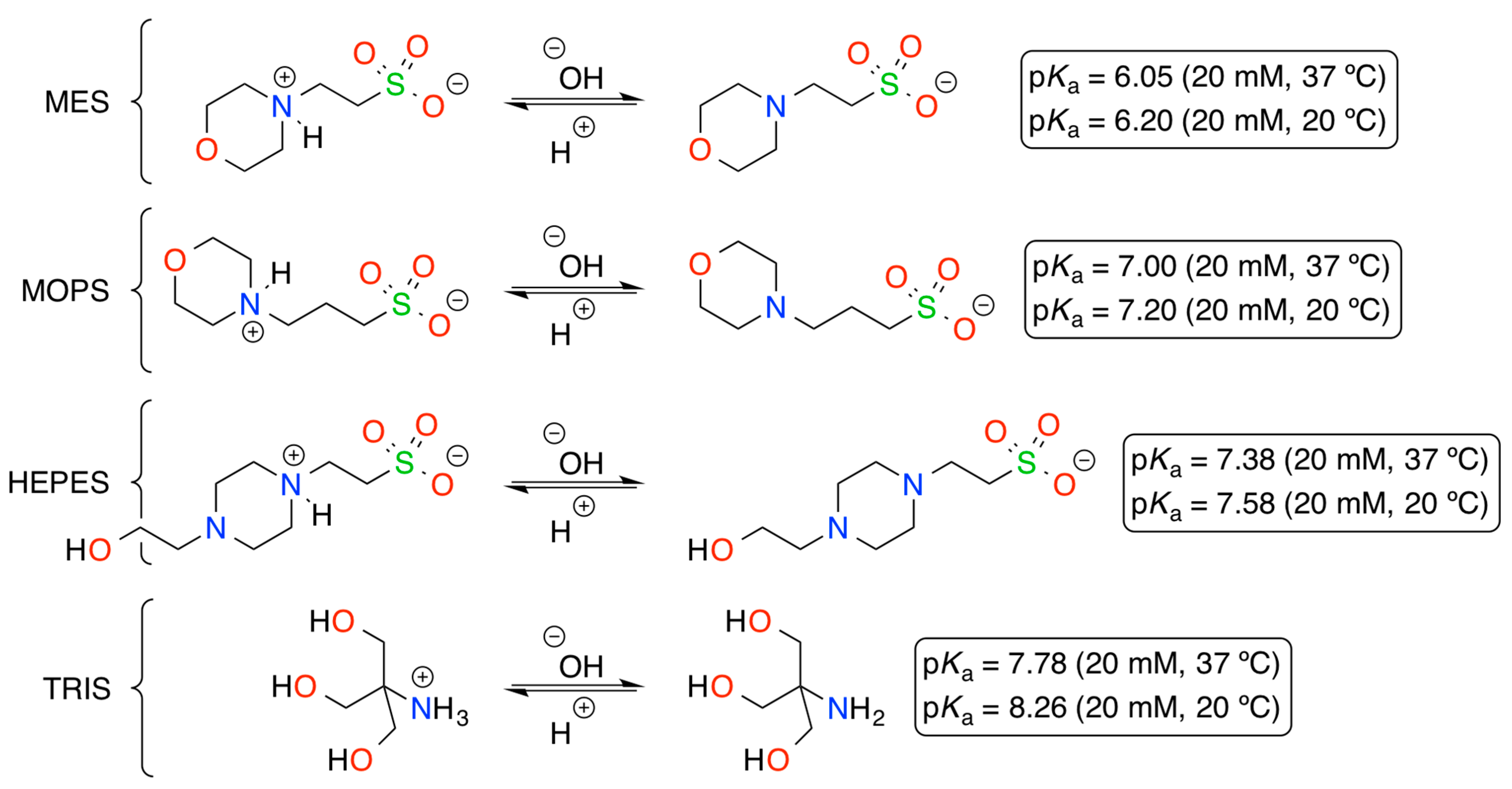
:
1. Introduction
2. Results and Discussion


| Entry | Solvent System | Yield e of 3a (%) |
|---|---|---|
| 1 | DMSO, EtOH, THF, toluene | 0 f |
| 2 | MES (pH 5.5) | 70 |
| 3 | MOPS (pH 6.5) | 86 |
| 4 | HEPES (pH 7.5) | 71 |
| 5 | TRIS (pH 7.5) | 83 |
| 6 | H2O (pH 6.5) b | ≤2 (4) g |
| 7 | H2O + MgCl2 (pH 6.5) c | 5 |
| 8 | H2O + NaCl (pH 6.5) d | ≤2 |
| Entry | R | 1 | Yield b of 3 (%) |
|---|---|---|---|
| 1 | (4-NO2)-C6H4 | 1a | 86 |
| 2 | (2-NO2)-C6H4 | 1b | 87 |
| 3 | Pyrid-2-yl | 1c | 90 |
| 4 | (4-Cl)-C6H4 | 1d | 21 |
| 5 | Furfur-2-yl | 1e | 27 |
{kind=link}
{kind=link}
{kind=link}
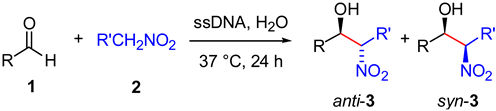
| Entry | R (1) | 1 | R' | 2 | Yield b of 3 (%) | dr d (anti/syn) |
|---|---|---|---|---|---|---|
| 1 | (4-NO2)-C6H4 | 1a | H | 2a | 80 | - |
| 2 | (4-NO2)-C6H4 | 1a | CH3 | 2b | 18 (46) c | 1:1.7 |
| 3 | (3-NO2)-C6H4 | 1f | H | 2a | 57 | - |
| 4 | (2-NO2)-C6H4 | 1b | H | 2a | 51 | - |
| 5 | (2-NO2)-C6H4 | 1b | CH3 | 2b | 12 | 1:2.1 |
| 6 | Pyrid-2-yl | 1c | H | 2a | 84 | - |
| 7 | Pyrid-2-yl | 1c | CH3 | 2b | 64 | 1:1.3 |
| 8 | (4-Cl)-C6H4 | 1d | H | 2a | 19 c | - |
| 9 | Furfur-2-yl | 1e | H | 2a | 37 c | - |
| 10 | C6H5 | 1g | H | 2a | 4 c | - |
| 11 | (4-OH)-C6H4 | 1h | H | 2a | 6 c | - |
| 12 | (4-NC)-C6H4 | 1i | H | 2a | 61 | - |
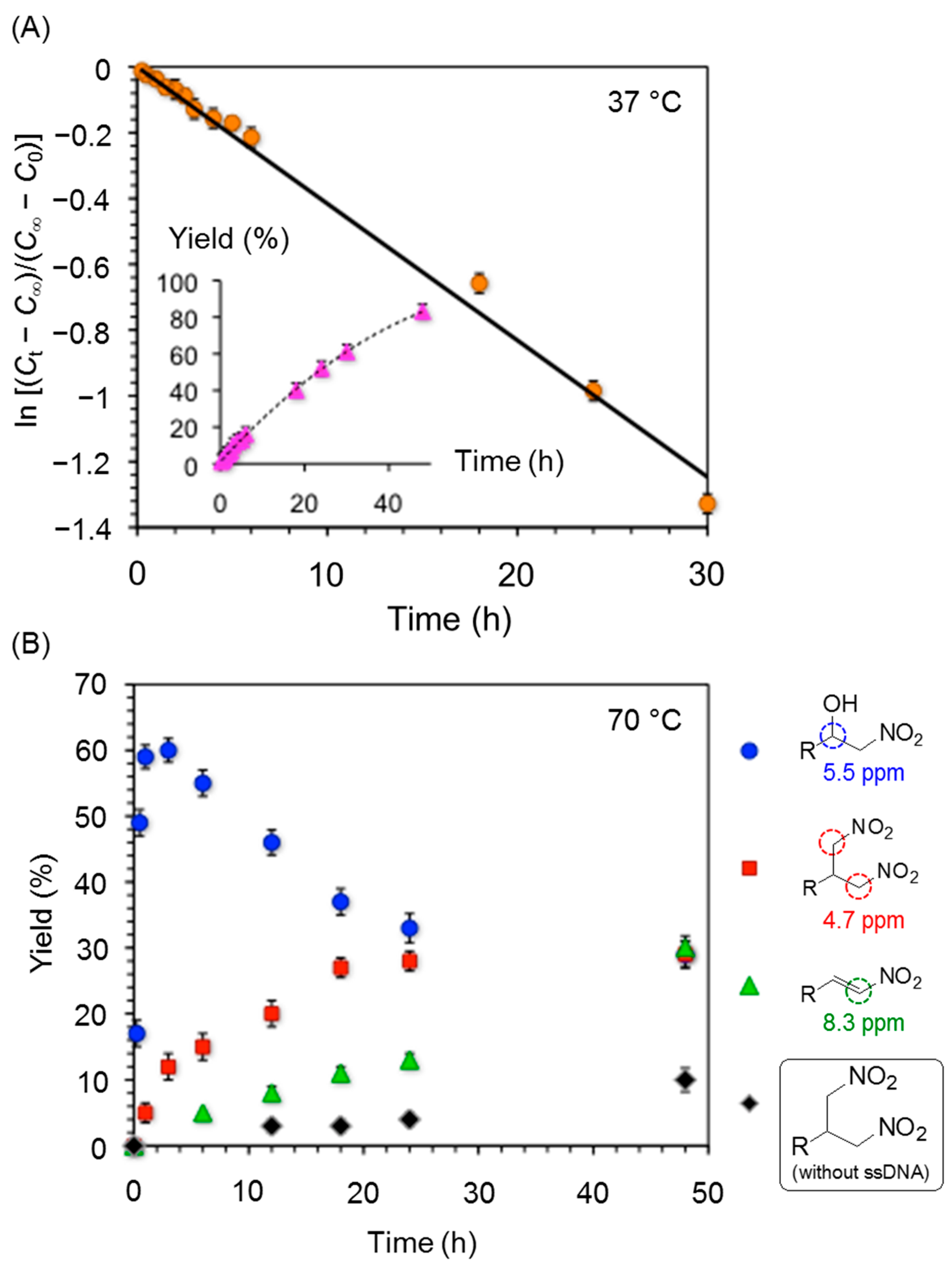
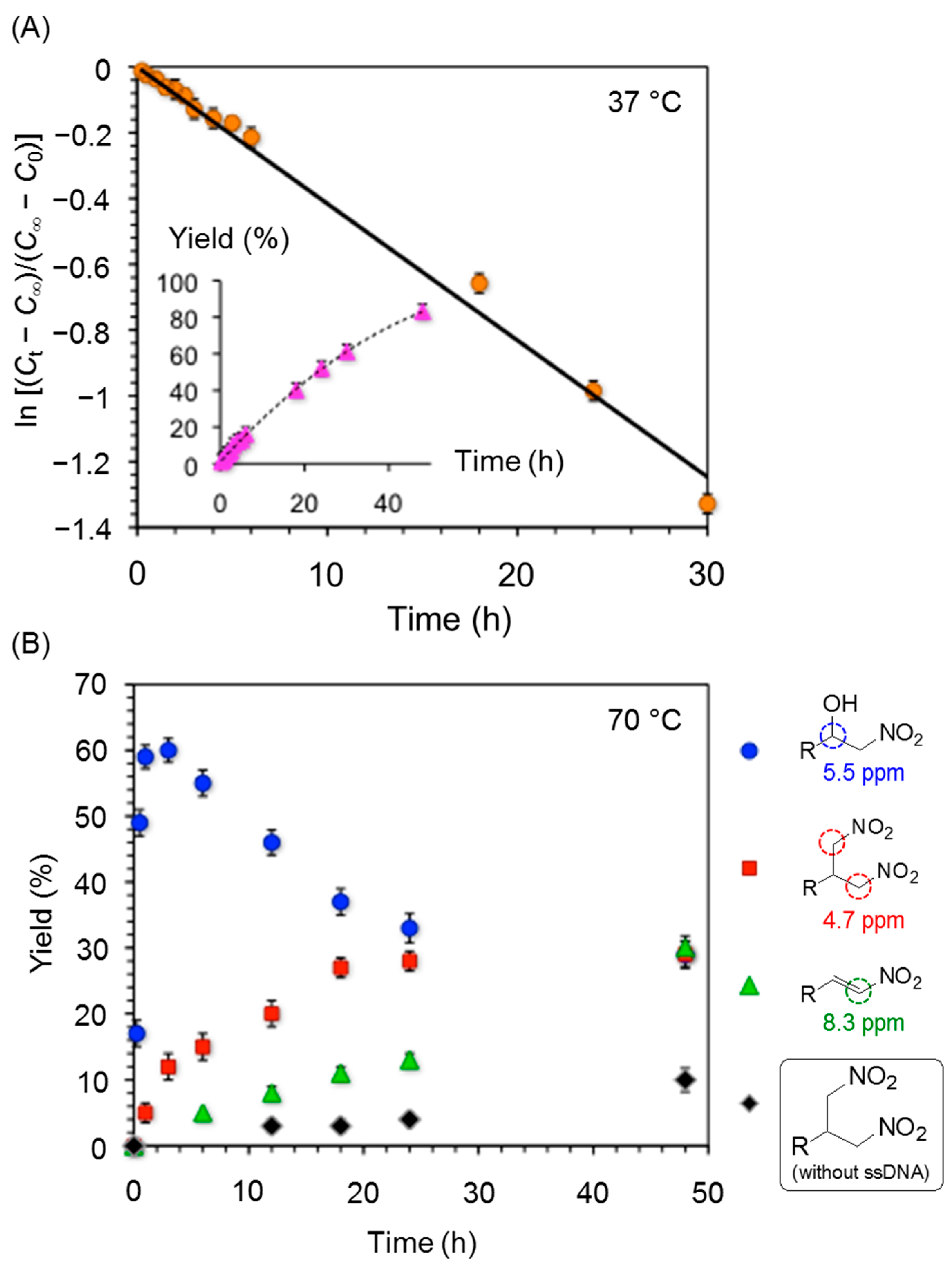
3. Experimental Section
3.1. Materials
3.2. Methods
3.3. Preliminary Optimization Experiments
| Entry | Catalyst | Solvent | Time (h) | Catalyst Loading (mg) | Conditions | Yield c of 3a (%) |
|---|---|---|---|---|---|---|
| 1 | hsDNA | H2O | 8 | 10 | A | 0 |
| 2 | ssDNA | H2O | 8 | 10 | A | 25 |
| 3 | ssDNA | H2O | 24 | 10 | A | 38 |
| 4 | ssDNA | H2O | 48 | 10 | A | 77 |
| 5 | ssDNA | H2O | 24 | 2 | B | 80 |
| 6 | ssDNA | H2O | 48 | 2 | B | 93 |
| 7 | hsDNA | H2O | 24 | 40 | B | 0 |
| 8 | Na+-free ssDNA | H2O | 24 | 2 | B | 77 |
| 9 | - | H2O | 24 | - | B | 4 |
| 10 | - | H2O | 48 | - | B | 15 |
| 11 | - | DMSO | 24 | - | B | 31 |
| 12 | ctDNA b | DMSO | 24 | 2 | B | 76 |
3.4. General Procedure for ssDNA-catalyzed Henry Reaction
3.5. Typical Recycling Procedure
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H. A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Gavanji, S. Application of recombinant DNA technology—A review. App. Sci. Rep. 2013, 2, 29–31. [Google Scholar]
- Endo, M.; Sugiyama, H. Chemical approaches to DNA nanotechnology. ChemBioChem 2009, 10, 2420–2443. [Google Scholar] [CrossRef] [PubMed]
- Ono, A. DNA-synthetic polymer conjugates. Macromol. Chem. Phys. 2006, 207, 1629–1632, and references therein. [Google Scholar] [CrossRef]
- Silverman, S.K. DNA as a versatile chemical component for catalysis, encoding, and stereocontrol. Angew. Chem. Int. Ed. 2010, 49, 7180–7201. [Google Scholar] [CrossRef]
- Silverman, S.K. Deoxyribozymes: Selection design and serendipity in the development of DNA catalysts. Acc. Chem. Res. 2009, 42, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Jäschke, A.; Seeling, B. Evolution of DNA/RNA as catalysts for chemical reaction. Curr. Opin. Chem. Biol. 2000, 4, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, T.N.; Strohbach, A. Achieving turnover in DNA-templated reactions. ChemBioChem 2008, 9, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.R.; Li, X. DNA-templated organic synthesis: Nature’s strategy for controlling chemical reactivity applied to synthetic molecules. Angew. Chem. Int. Ed. 2004, 43, 4848–4870. [Google Scholar] [CrossRef]
- Chandra, M.; Silverman, S.K. DNA and RNA can be equally efficient catalysts for carbon–carbon bond formation. J. Am. Chem. Soc. 2008, 130, 2936–2937. [Google Scholar] [CrossRef] [PubMed]
- Oelerich, J.; Roelfes, G. DNA-based asymmetric organometallic catalysis in water. Chem. Sci. 2013, 4, 2013–2017. [Google Scholar] [CrossRef]
- Rioz-Martínez, A.; Roelfes, G. DNA-based hybrid catalysis. Curr. Opin. Chem. Biol. 2015, 25, 80–87, and references therein. [Google Scholar] [CrossRef]
- Park, S.; Sugiyama, H. DNA-based hybrid catalysts for asymmetric organic synthesis. Angew. Chem. Int. Ed. 2010, 49, 3870–3878. [Google Scholar] [CrossRef]
- Coquire, D.; Feringa, B.L.; Roelfes, G. DNA-based catalytic enantioselective Michael reactions in water. Angew. Chem. Int. Ed. 2007, 46, 9308–9311. [Google Scholar] [CrossRef]
- Roelfes, G.; Feringa, B.L. DNA-based asymmetric catalysis. Angew. Chem. Int. Ed. 2005, 44, 3230–3232. [Google Scholar] [CrossRef]
- Fan, J.; Sun, G.; Wan, C.; Wang, Z.; Li, Y. Investigation of DNA as a catalyst for Henry reaction in water. Chem. Commun. 2008, 3792–3794. [Google Scholar] [CrossRef]
- Izquierdo, C.; Luis-Barrera, J.; Fraile, A.; Alemán, J. 1,4-Michael additions of cyclic-β-ketoesters catalyzed by DNA in aqueous media. Catal. Commun. 2014, 44, 10–14. [Google Scholar] [CrossRef]
- De Rosa, M.; di Marino, S.; D’Ursi, A.M.; Strianese, M.; Soriente, A. Genomic salmon testes DNA as a catalyst for Michael reactions in water. Tetrahedron 2012, 68, 3086–3091. [Google Scholar] [CrossRef]
- Kühbeck, D.; Ghosh, M.; Gupta, S.S.; Díaz, D.D. Investigation of C-C bond formation Mediated by Bombyx Mori silk fibroin materials. ACS Sustain. Chem. Eng. 2014, 2, 1510–1517. [Google Scholar] [CrossRef]
- Kühbeck, D.; Schön, E.-M.; Bachl, J.; Gotor-Fernández, V.; Díaz, D.D. Gelatin protein-mediated direct aldol reaction. Helv. Chim. Acta 2014, 97, 574–580. [Google Scholar] [CrossRef]
- Kühbeck, D.; Dhar, B.B.; Schön, E.-M.; Cativiela, C.; Gotor-Fernández, V.; Díaz, D.D. C-C bond formation catalyzed by natural gelatin and collagen proteins. Beilstein J. Org. Chem. 2013, 9, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Kühbeck, D.; Saidulu, G.; Reddy, K.R.; Díaz, D.D. Critical assessment of the efficiency of chitosan biohydrogel beads as recyclable and heterogenous organocatalyst for C-C bond formation. Green Chem. 2012, 14, 378–392. [Google Scholar] [CrossRef]
- Cielecka-Piontek, J.; Jelinska, A. Catalytic effect of buffers on the degradation of doripenem in aqueous solutions. React. Kinet. Mech. Catal. 2011, 102, 37–47. [Google Scholar] [CrossRef]
- Elabprotocol—Laboratory Protocol Management. Available online: https://www.elabprotocols.com (accessed on 2 March 2015).
- Biological Buffers. Available online: http://www.reachdevices.com/Protein/BiologicalBuffers.html (accessed on 2 March 2015).
- Li, C.H. Catalytic effect of acetate and phosphate buffers on the iodination of tyrosine. J. Am. Chem. Soc. 1944, 66, 228–230. [Google Scholar] [CrossRef]
- Alvarez-Casao, Y.; Marques-Lopez, E.; Herrera, R.P. Organocatalytic enantioselective Henry Reactions. Symmetry 2011, 3, 220–245. [Google Scholar] [CrossRef]
- Rueping, M.; Antonchick, A.P. Brønsted-acid-catalyzed activation of nitroalkanes: A direct enantioselective aza-Henry reaction. Org. Lett. 2008, 10, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Oiarbide, M.; Laso, A. Enantioselective Henry reactions under dual Lewis acid/amine catalysis using chiral amino alcohol ligands. Angew. Chem. Int. Ed. 2005, 44, 3881–3884. [Google Scholar] [CrossRef]
- Seayad, J.; List, B. Asymmetric organocatalysis. Org. Biomol. Chem. 2005, 3, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.P.; Bez, G. Henry reaction in aqueous media at neutral pH. Eur. J. Org. Chem. 2013, 2922–2929. [Google Scholar] [CrossRef]
- Stephenson, R.M. Mutual solubilities: Water-ketones, water-ethers, and water-gasoline-alcohols. J. Chem. Eng. Data 1992, 37, 80–95. [Google Scholar] [CrossRef]
- Gruber-Khadjawi, M.; Purkarthofer, T.; Skranc, W.; Griengla, H. Hydroxynitrile lyase-catalyzed enzymatic nitroaldol (Henry) reaction. Adv. Synth. Catal. 2007, 349, 1445–1450. [Google Scholar] [CrossRef]
- Matsumoto, K.; Asakura, S. Albumin-mediated asymmetric nitroaldol reaction of aromatic aldehydes with nitromethane in water. Tetrahedron Lett. 2014, 55, 6919–6921. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernández, V.; Gotor, V. Protein-mediated nitroaldol addition in aqueous media. Catalytic promiscuity or unspecific catalysis? Org. Process Res. Rev. 2011, 15, 236–240. [Google Scholar] [CrossRef]
- Blay, G.; Domingo, L.R.; Hernández-Olmos, V.; Pedro, J.R. New highly asymmetric Henry reaction catalyzed by CuII and a C1-symmetric aminopyridine Ligand, and its application to the synthesis of miconazole. Chem. Eur. J. 2008, 14, 4725–4730. [Google Scholar] [CrossRef] [PubMed]
- Purkarthofer, T.; Gruber, K.; Gruber-Khadjawi, M.; Waich, K.; Skranc, W.; Mink, D.; Griengl, H.A. A biocatalytic Henry Reaction—The hydroxynitrile Lyase from Hevea brasiliensis also catalyzes nitroaldol reactions. Angew. Chem. Int. Ed. 2006, 45, 3454–3456. [Google Scholar] [CrossRef]
- Liang, C.; Jiaxing, D.; Jingsong, Y.; Ge, G.; Jingbo, L. A Highly syn-selective nitroaldol reaction catalyzed by CuII–bisimidazoline. Chem. Eur. J. 2010, 16, 6761–6765. [Google Scholar] [CrossRef] [PubMed]
- Nitabaru, T.; Nojiri, A.; Kobayashi, M.; Kumagai, N.; Shibasaki, M. anti-Selective catalytic asymmetric nitroaldol reaction via a heterobimetallic heterogeneous catalyst. J. Am. Chem. Soc. 2009, 131, 13860–13869. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not apply.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Häring, M.; Pérez-Madrigal, M.M.; Kühbeck, D.; Pettignano, A.; Quignard, F.; Díaz, D.D. DNA-Catalyzed Henry Reaction in Pure Water and the Striking Influence of Organic Buffer Systems. Molecules 2015, 20, 4136-4147. https://doi.org/10.3390/molecules20034136
Häring M, Pérez-Madrigal MM, Kühbeck D, Pettignano A, Quignard F, Díaz DD. DNA-Catalyzed Henry Reaction in Pure Water and the Striking Influence of Organic Buffer Systems. Molecules. 2015; 20(3):4136-4147. https://doi.org/10.3390/molecules20034136
Chicago/Turabian StyleHäring, Marleen, Maria M. Pérez-Madrigal, Dennis Kühbeck, Asja Pettignano, Françoise Quignard, and David Díaz Díaz. 2015. "DNA-Catalyzed Henry Reaction in Pure Water and the Striking Influence of Organic Buffer Systems" Molecules 20, no. 3: 4136-4147. https://doi.org/10.3390/molecules20034136
APA StyleHäring, M., Pérez-Madrigal, M. M., Kühbeck, D., Pettignano, A., Quignard, F., & Díaz, D. D. (2015). DNA-Catalyzed Henry Reaction in Pure Water and the Striking Influence of Organic Buffer Systems. Molecules, 20(3), 4136-4147. https://doi.org/10.3390/molecules20034136