
Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls


Abstract
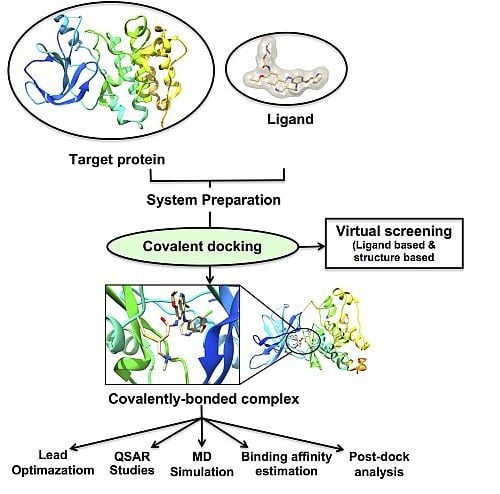
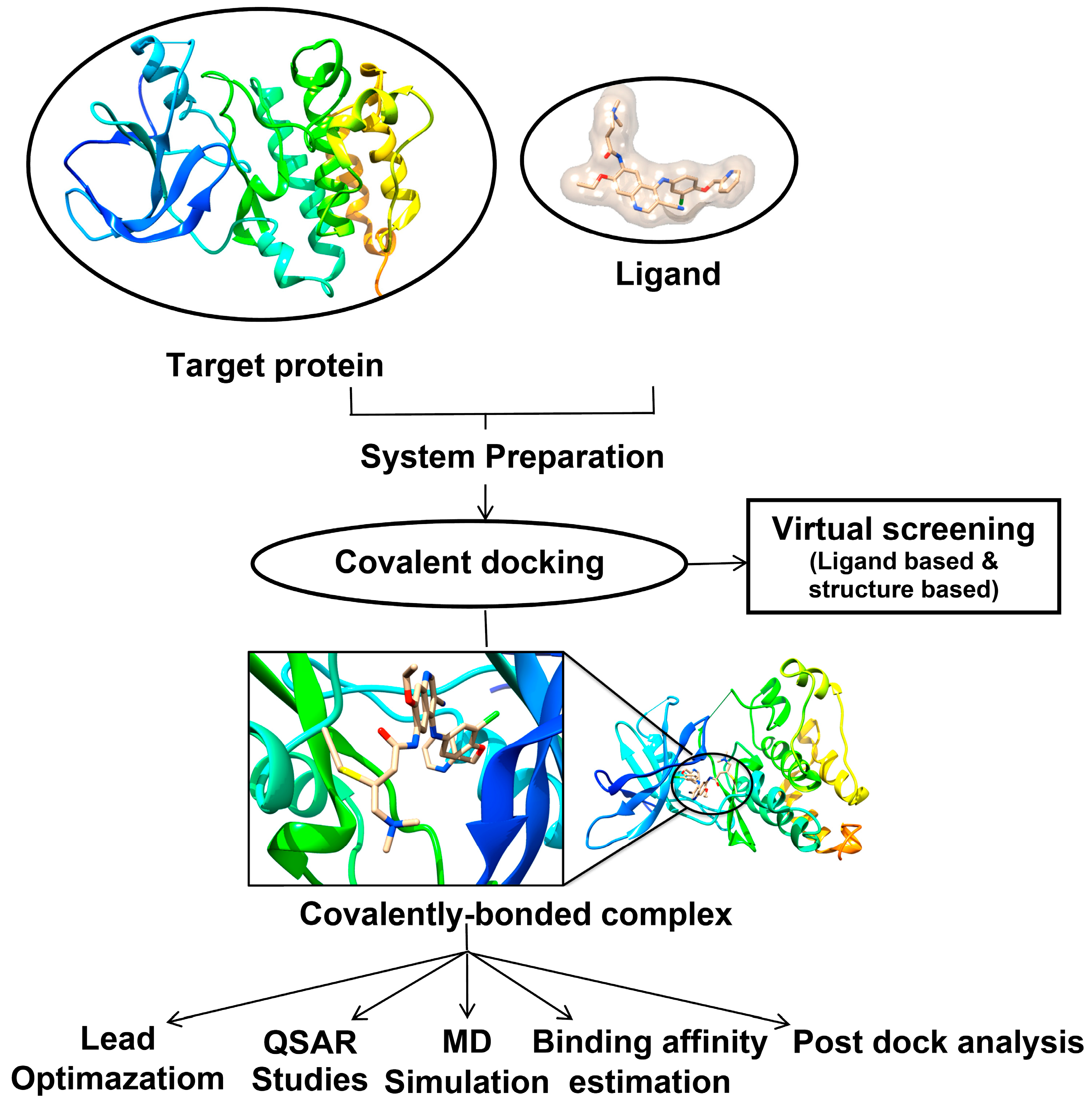
:
1. Introduction
2. Covalent Interactions in Biological Systems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
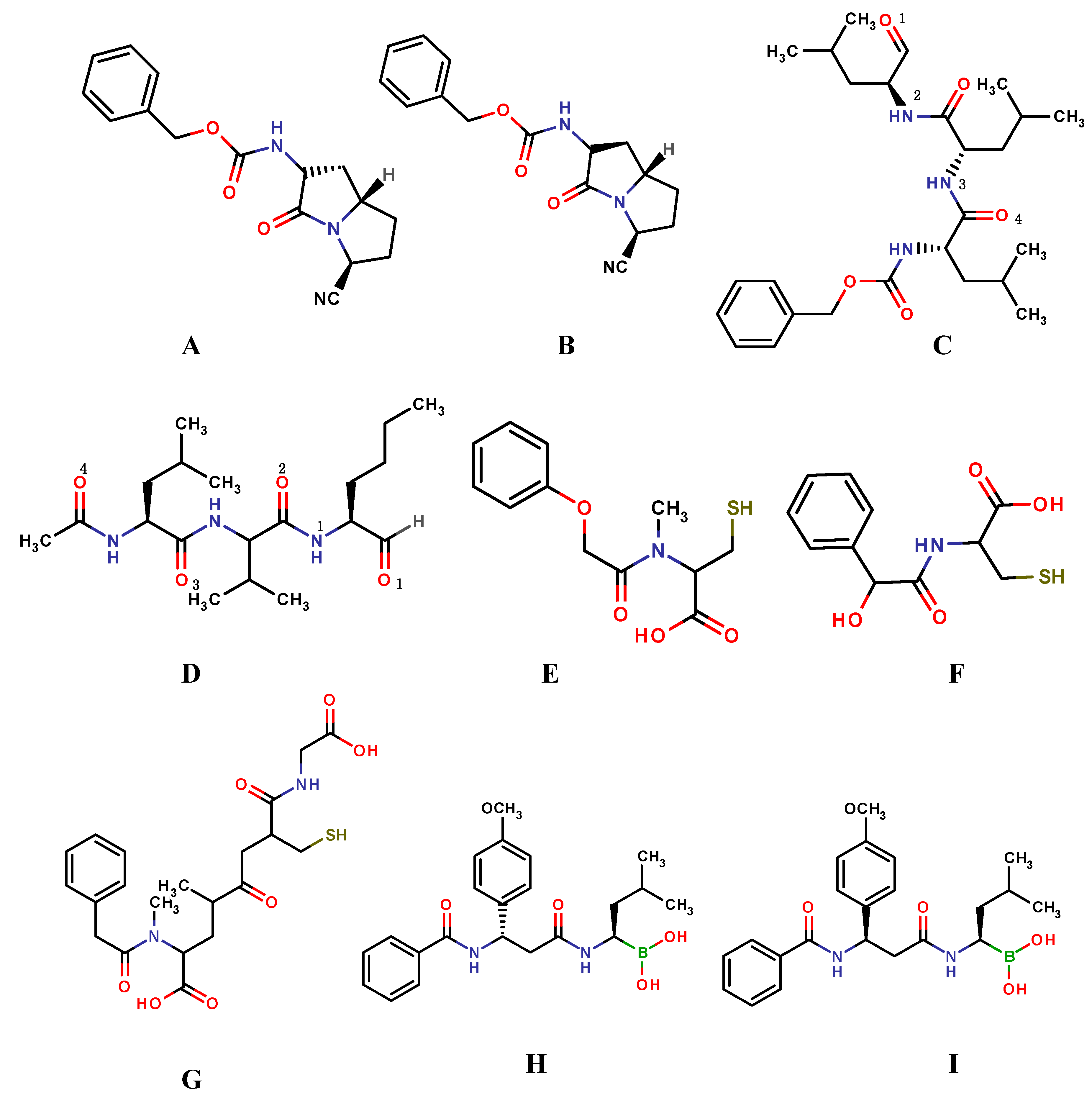{kind=link}
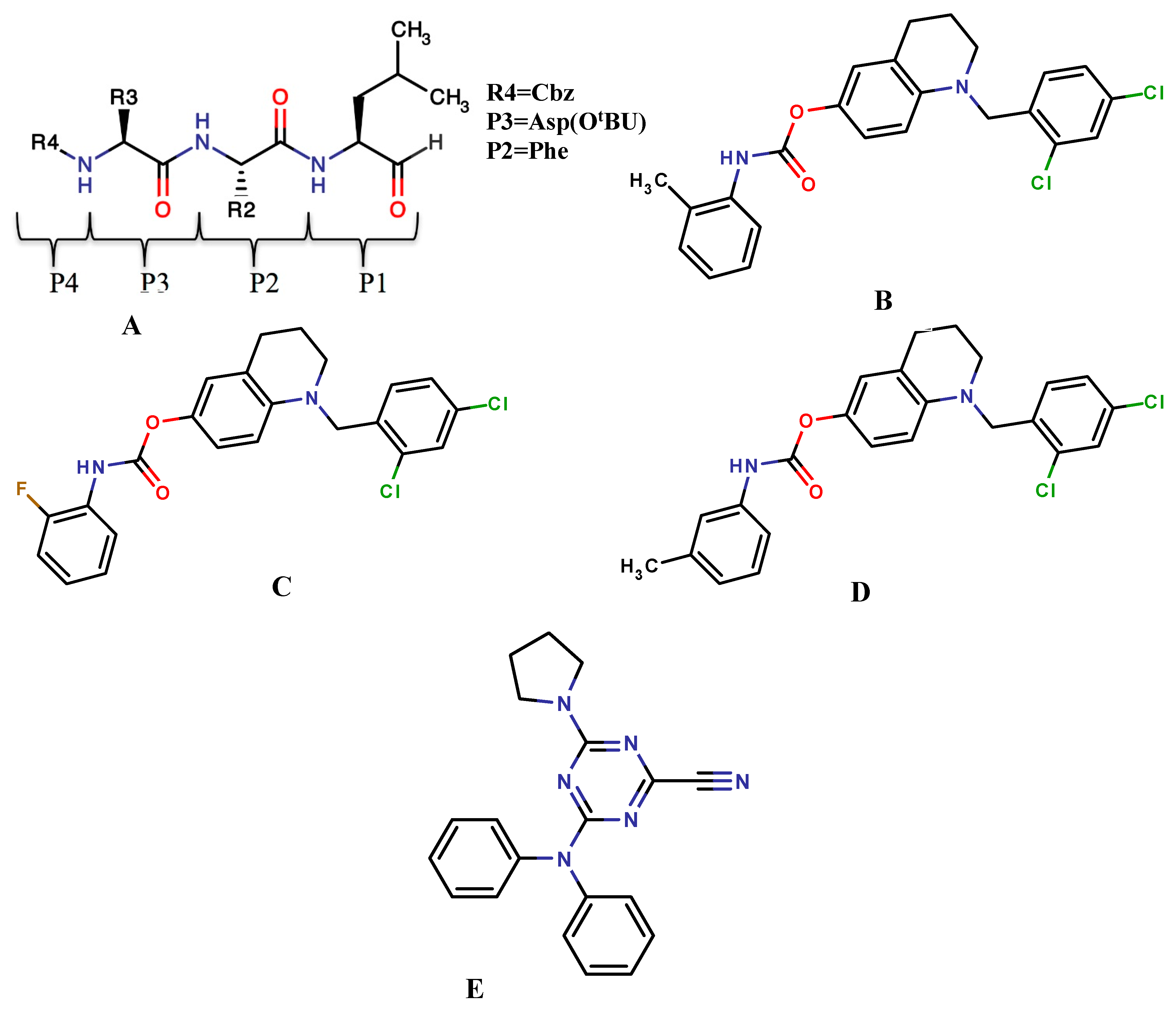{kind=link}
| Drug | Biological Target | Therapeutic Domain |
|---|---|---|
| Amoxicillin | PBP | Anti-infective |
| Cefaclor/Ceclor | PBP | Anti-infective |
| Ceftriaxone/Rocephin | PBP | Anti-infective |
| Cefuroxime axetil/ceftin | PBP | Anti-infective |
| Cephalexin/keflex | PBP | Anti-infective |
| D-cycloserine/seromycin | Alanine racemase | Anti-infective |
| Fosfomycin/monurol | UDP-N-acetylglucosamine-3-enolpyruvyl-transferase | Anti-infective |
| Isoniazid | Enol-acyl carrier protein reductase | Anti-infective |
| Meropenem | PBP | Anti-infective |
| Omnicef | PBP | Anti-infective |
| Penicillin V | PBP | Anti-infective |
| Azacytidine | Methyltranferase | Cancer |
| Bortezomib | Protesome | Cancer |
| Decitabine/azadC | Methyltranferase | Cancer |
| Dutasteride/avodart | 5-α-Reductase | Cancer |
| Exemestane/Aromasin | Aromatase | Cardio-vascular |
| Floxuridine | Thymidylate synthase | Cardio-vascular |
| Gemcitabine/gemzar | Ribonucleoside reductatase | Cardio-vascular |
| Proscar/finasteride | 5-α-Reductase | Cardio-vascular |
| Rasagiline | MAO-B | Parkinson’s disease |
| Selegiline | MAO-B | Parkinson’s disease |
| Warfarin | Vitamin K reductase | Cardio-vascular |
| Vigabatrin/sabril | GABA-Aminotransferase | Anti-epileptic |
| Nexium/esomeprazole | H+/K+ ATPase | Gastro-intestinal |
| Orlistat/ | Lipase | Gastro-intestinal |
| Prevacid/lansoprazole | H+/K+ATPase | Gastro- intestinal |
| Prilosec/omeprazole | H+/K+ATPase | Gastro-intestinal |
| Protonix/pantoprazole | H+/K+ATPase | Gastro-intestinal |
| Aciphex/rabeprazol | H+/K+ATPase | Gastro-intestinal |
| Aspirin | Cyclooxygenase | Inflammation |
| Disulfiram/antabuse | Aldehyde dehydrogenase | Chronic alcoholism |
| Eflornithine | Ornithine decarboxylase | Hirsutism |
| Propylthiouracil/procasil | Thyroxine-5-deiodinase | Hyperthyroidism |
| Saxagliptin/Onglyza | DPP-IV | Anti-diabetic drug |
| Vildagliptin/Eugreas | DPP-IV | Anti-diabetic drug |
| Phenoxy-benzamine hydrochloride | α-Adrenoceptor | Cardio-vascular |
| mercaptopurine/purinthol | Purine-nucleotide synthesis | Cancer |
| Carbidopa/lodosyn | DOPA decarboxylase | CNS |
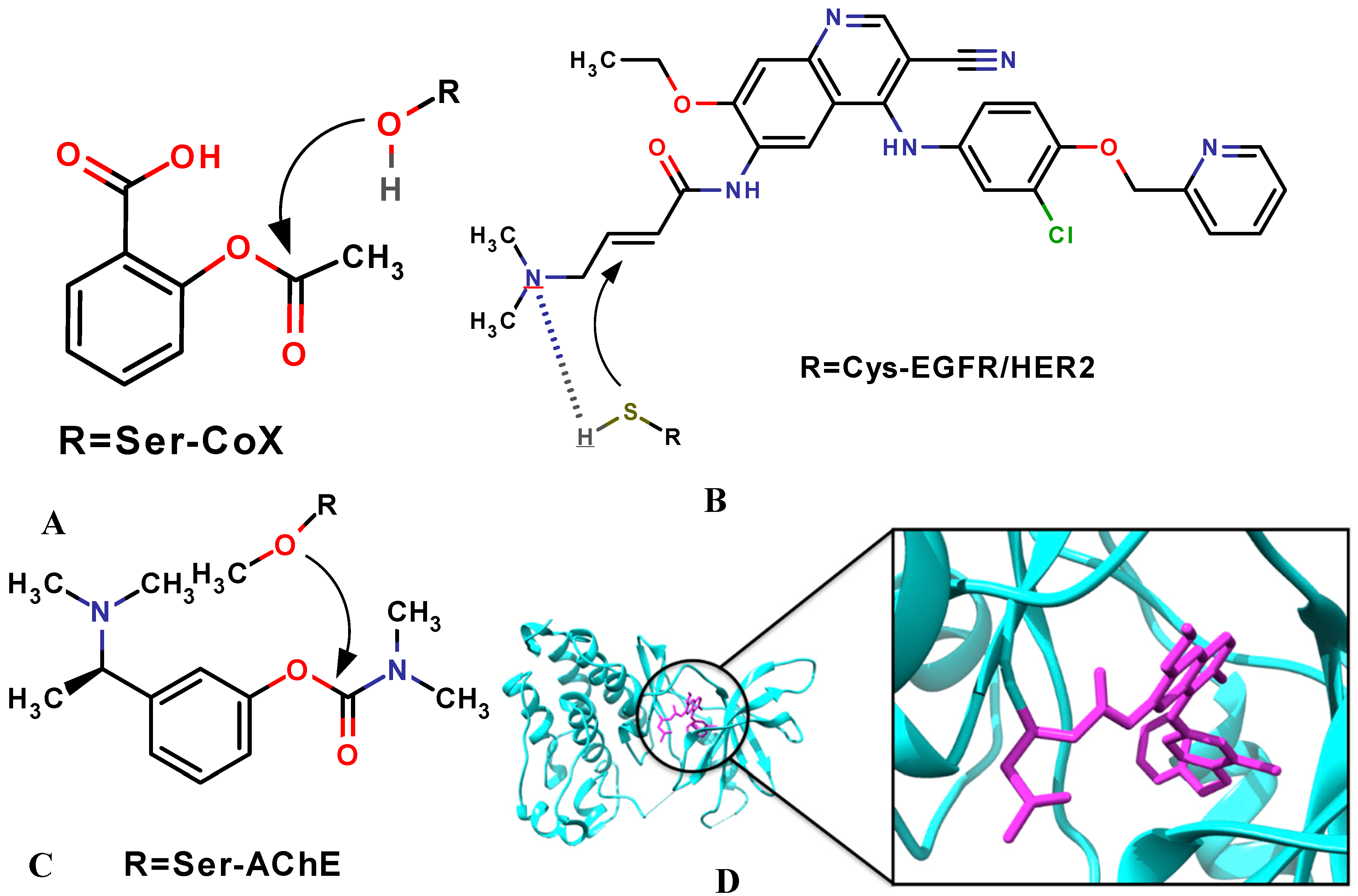
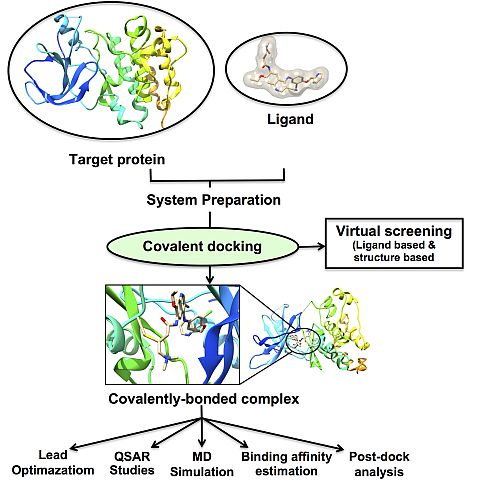
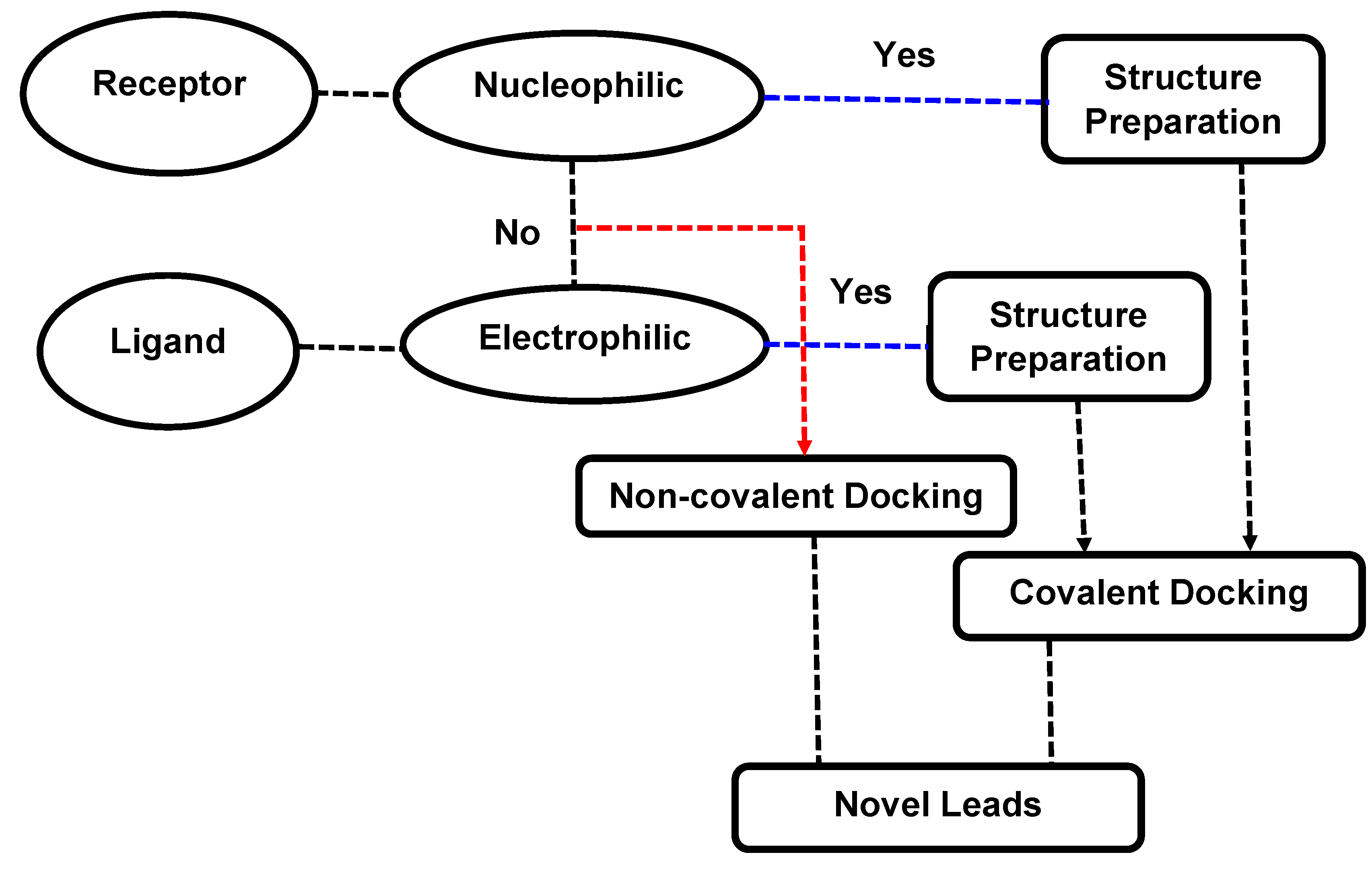
3. Molecular Docking: Non-Covalent and Covalent Docking
3.1. Covalent Docking: Theoretical Background
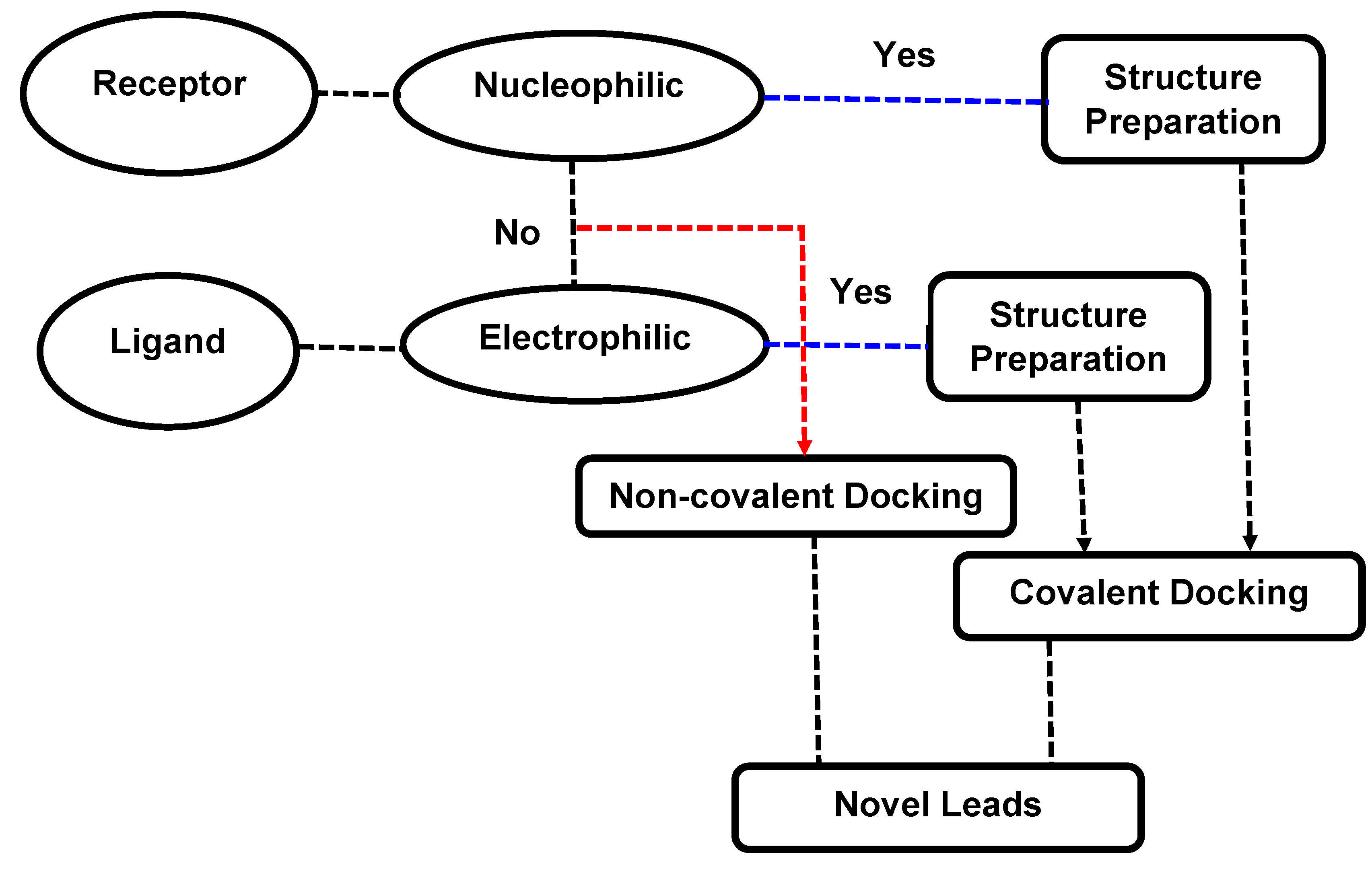
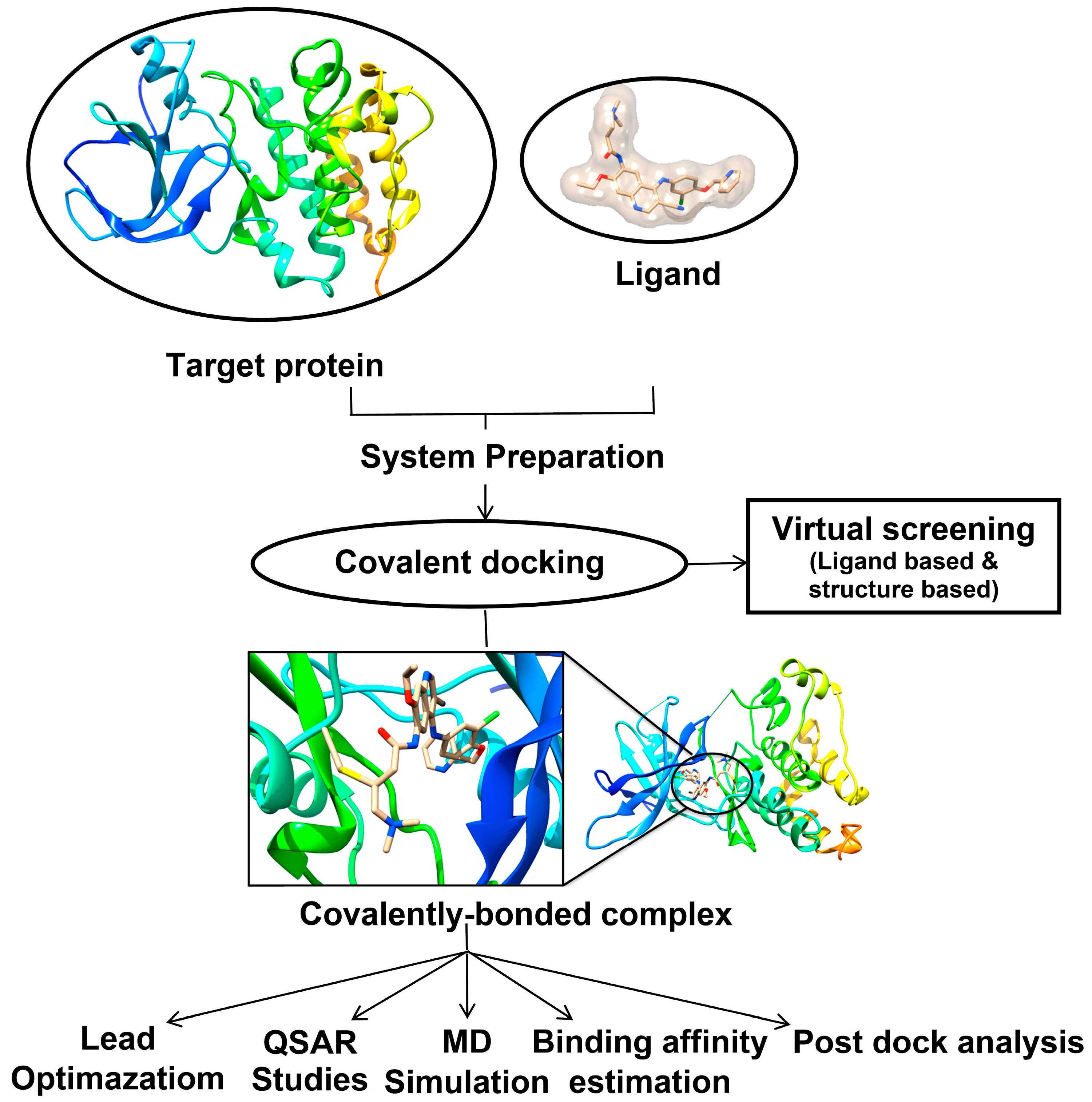
3.2. Implementation of Covalent Docking in Drug Discovery Workflows
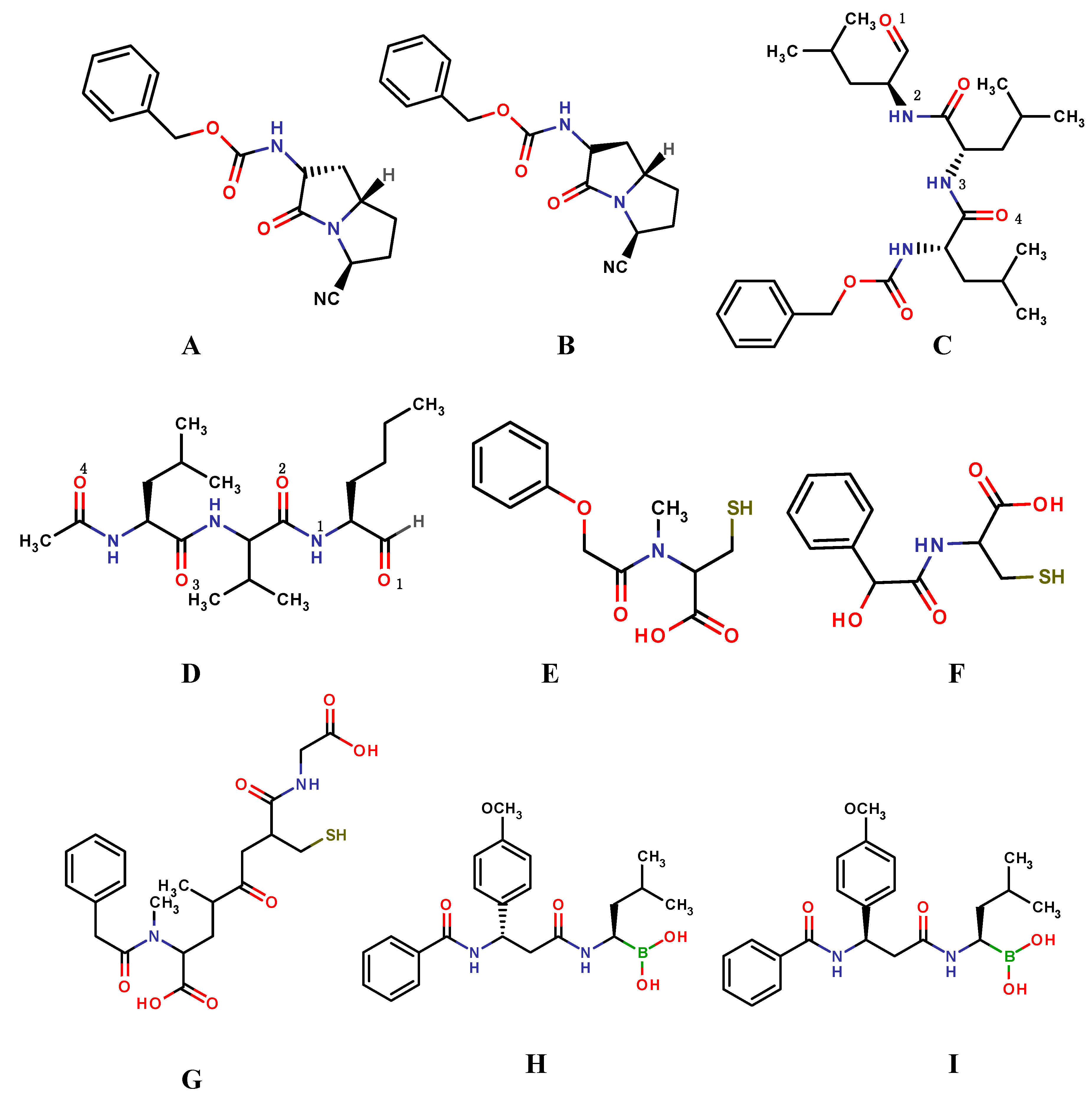
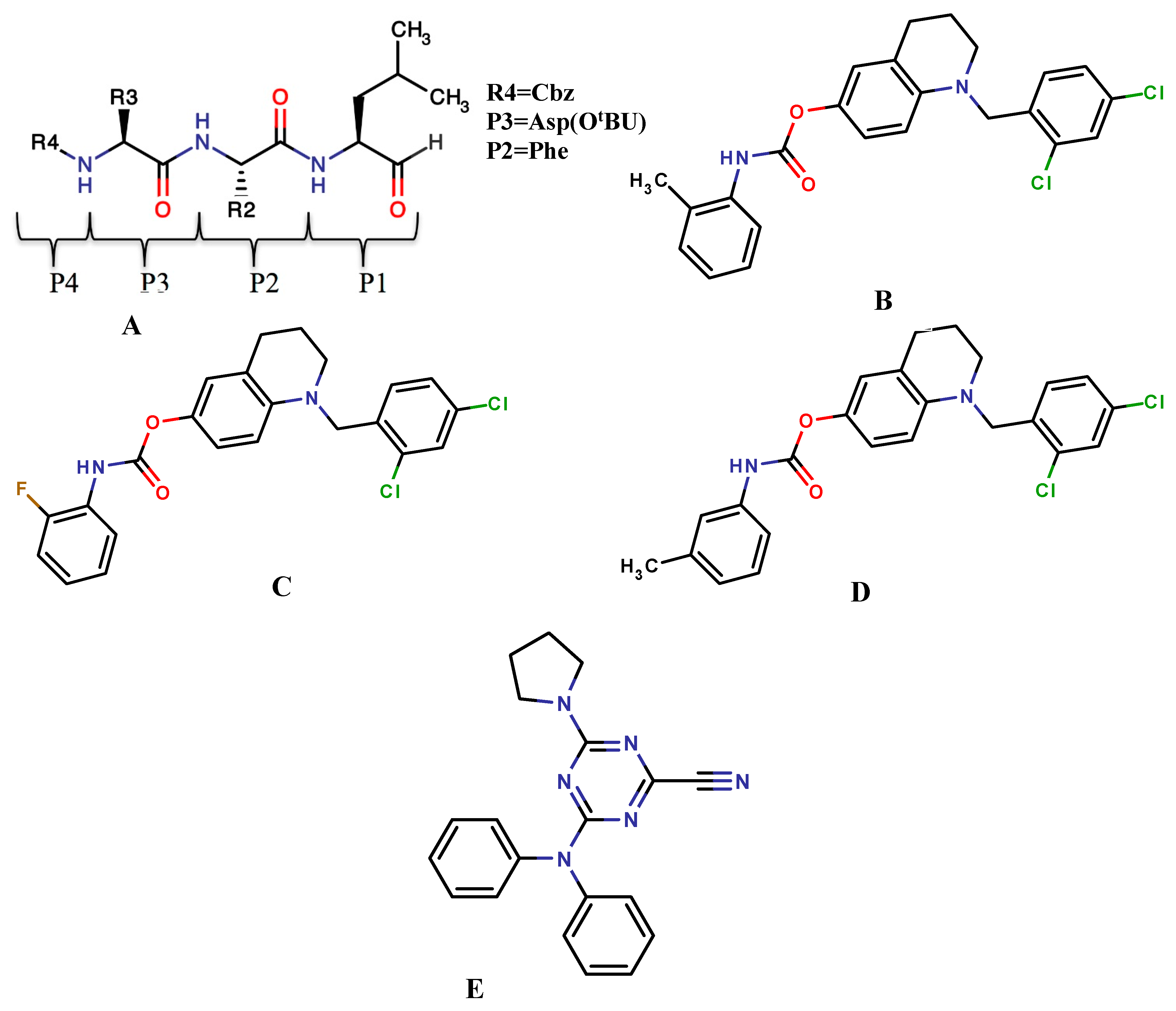
4. Case Studies: Applications of Covalent Docking in Drug Design
5. Software and Web Servers for Covalent Docking
| Standalone Software | Webservers |
|---|---|
| CovalentDock [42] | CovalentDock Cloud [42] http://docking.sce.ntu.edu.sg/ |
| Gold [41] | Dockovalent (Covalent Docking Server) [57] http://covalent.docking.org/ |
| CovDock-VS [58] | DockingServer [59] http://www.dockingserver.com/web/ |
| Autodock [60] | |
| Glide [61] | |
| CovDock [62] |
6. Covalent Docking: Pitfalls and Future Prospectives
7. Conclusions
Acknowledgments
Author Contributions
List of Abbreviations
| AChE | Acetylcholinesterase |
| CNS | Central nervous system |
| Cys | Cysteine |
| DPP-IV | Dipeptidyl peptidase 4 |
| DOPA | 3, 4-Dihydroxyphenethylamine |
| EGFR | Epidermal growth factor receptor |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GABA | γ-Aminobutyric acid |
| H+/K+ ATPase | Hydrogen potassium ATPase |
| MAO-B | Monoamine oxidase B |
| MD | Molecular dynamics |
| PARP | Poly-(ADP-ribose)-polymerase |
| POP | Propyl oligopeptidases |
| PBP | Penicillin-binding protein |
| QSAR | Quantitative structure activity relationships |
| ULP | Ubiquitin-like poxvirus proteinase I7L |
| SBVS | Structure-based virtual screening |
Conflicts of Interest
References
- Ding, H.; Xiong, Y.L. Computational manufacturing. Prog. Nat. Sci. 2002, 12, 641–648. [Google Scholar]
- Lu, X.-G.; Wang, Z.; Cui, Y.; Jin, Z. Computational thermodynamics, computational kinetics, and materials design. Chin. Sci. Bull. 2014, 59, 1662–1671. [Google Scholar] [CrossRef]
- Smith, D.W.; Rubenson, J.; Lloyd, D.; Zheng, M.; Fernandez, J.; Besier, T.; Xu, J.; Gardiner, B.S. A conceptual framework for computational models of Achilles tendon homeostasis. Wiley Interdiscip. Rev.-Syst. Biol. Med. 2013, 5, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Vilela Neto, O.P. Intelligent Computational Nanotechnology: The Role of Computational Intelligence in the Development of Nanoscience and Nanotechnology. J. Comput. Theor. Nanosci. 2014, 11, 928–944. [Google Scholar] [CrossRef]
- Honarparvar, B.; Govender, T.; Maguire, G.E.M.; Soliman, M.E.S.; Kruger, H.G. Integrated Approach to Structure-Based Enzymatic Drug Design: Molecular Modeling, Spectroscopy, and Experimental Bioactivity. Chem. Rev. 2014, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.G. Enzymes as a special class of therapeutic target: Clinical drugs and modes of action. Curr. Opin. Struct. Biol. 2007, 17, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.G. Mechanistic basis of enzyme-targeted drugs. Biochemistry 2005, 44, 8918–8918. [Google Scholar] [CrossRef]
- Doane, T.; Burda, C. Nanoparticle mediated non-covalent drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.K.; Gupta, U. Application of dendrimer-drug complexation in the enhancement of drug solubility and bioavailability. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Dalvie, D.K. Drug discovery for a new generation of covalent drugs. Expert Opin. Drug Discov. 2012, 7, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.E.F.; Lopez-Anaya, A. Strategies for dealing with reactive intermediates in drug discovery and development. Curr. Opin. Drug Discov. Dev. 2004, 7, 126–136. [Google Scholar]
- Pommier, Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Chan, E.; Duan, W.; Huang, M.; Chen, Y.Z. Drug bioactivation, covalent binding to target proteins and toxicity relevance. Drug Metab. Rev. 2005, 37, 41–213. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.P.; Marnett, L.J. Heme prosthetic group required for acetylation of prostaglandin-H synthase by aspirin. FASEB J. 1989, 3, 2294–2297. [Google Scholar] [PubMed]
- Lecomte, M.; Laneuville, O.; Ji, C.; Dewitt, D.L.; Smith, W.L. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J. Biol. Chem. 1994, 269, 13207–13215. [Google Scholar] [PubMed]
- Roth, G.J.; Stanford, N.; Majerus, P.W. Acetylation of prostaglandin synthase by aspirin. Proc. Natl. Acad. Sci. USA 1975, 72, 3073–3076. [Google Scholar] [CrossRef] [PubMed]
- Wells, I.; Marnett, L.J. Acetylation of prostaglandin endoperoxide synthase by n-acetylimidazole—Comparison to acetylation by aspirin. Biochemistry 1992, 31, 9520–9525. [Google Scholar] [CrossRef] [PubMed]
- Guerciolini, R. Mode of action of orlistat. Int. J. Obes. 1997, 21, S12–S23. [Google Scholar]
- Dahal, U.P.; Obach, R.S.; Gilbert, A.M. Benchmarking in Vitro Covalent Binding Burden As a Tool To Assess Potential Toxicity Caused by Nonspecific Covalent Binding of Covalent Drugs. Chem. Res. Toxicol. 2013, 26, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Hadvary, P.; Lengsfeld, H.; Wolfer, H. Inhibition of pancreatic lipase invitro by the covalent inhibitor tetrahydrolipstatin. Biochem. J. 1988, 256, 357–361. [Google Scholar] [PubMed]
- Potashman, M.H.; Duggan, M.E. Covalent Modifiers: An Orthogonal Approach to Drug Design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.M.; Goa, K.L. Rivastigmine—A pharmacoeconomic review of its use in Alzheimer’s disease. Pharmacoeconomics 2001, 19, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.R.; Overbeek-Klumpers, E.G.; Hallett, W.A.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Michalak, R.S.; Nilakantan, R.; Discafani, C.; Golas, J.; et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005, 48, 1107–1131. [Google Scholar] [CrossRef] [PubMed]
- Wissner, A.; Mansour, T.S. The development of HKI-272 and related compounds for the treatment of cancer. Archiv Der Pharmazie 2008, 341, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J. Second-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-small Cell Lung Cancer. J. Thorac. Oncol. 2008, 3, 146–149. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Agostino, M.; Ramsland, P.A. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Mura, C.; McAnany, C.E. An introduction to biomolecular simulations and docking. Mol. Simul. 2014, 40, 732–764. [Google Scholar] [CrossRef]
- Tantar, A.-A.; Conilleau, S.; Parent, B.; Melab, N.; Brillet, L.; Roy, S.; Talbi, E.-G.; Horvath, D. Docking and biomolecular simulations on computer grids: Status and trends. Curr. Comput.-Aided Drug Des. 2008, 4, 235–249. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Ramsland, P.A. Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit. 2013, 26, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.B.; Andersen, T.; McDougal, O.M. Accessible High-Throughput Virtual Screening Molecular Docking Software for Students and Educators. PLoS Comput. Biol. 2012, 8, e1002499. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, Y. Structural ensemble in computational drug screening. Expert Opin. Drug Metab. Toxicol. 2010, 6, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.; Renaud, J.-P.; Horvath, D. Fragment-Based Drug Design: Computational and Experimental State of the Art. Comb. Chem. High Throughput Screen. 2011, 14, 500–520. [Google Scholar] [CrossRef] [PubMed]
- Konteatis, Z.D. In silico fragment-based drug design. Expert Opin. Drug Discov. 2010, 5, 1047–1065. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Docking, virtual high throughput screening and in silico fragment-based drug design. J. Cell. Mol. Med. 2009, 13, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhou, S.; Su, C.T.T.; Ge, Z.; Li, R.; Kwoh, C.K. CovalentDock: Automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J. Computat. Chem. 2013, 34, 326–336. [Google Scholar] [CrossRef]
- Smith, A.J.T.; Zhang, X.; Leach, A.G.; Houk, K.N. Beyond Picomolar Affinities: Quantitative Aspects of Noncovalent and Covalent Binding of Drugs to Proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Byrd, C.M.; Tseitin, V.; Dai, D.; Raush, E.; Totrov, M.; Abagyan, R.; Jordan, R.; Hruby, D.E. Discovery of small molecule inhibitors of ubiquitin-like poxvirus proteinase I7L using homology modeling and covalent docking approaches. J. Comput.-Aided Mol. Des. 2007, 21, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Lawandi, J.; Toumieux, S.; Seyer, V.; Campbell, P.; Thielges, S.; Juillerat-Jeanneret, L.; Moitessier, N. Constrained Peptidomimetics Reveal Detailed Geometric Requirements of Covalent Prolyl Oligopeptidase Inhibitors. J. Med. Chem. 2009, 52, 6672–6684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shi, Y.; Jin, H.; Liu, Z.; Zhang, L.; Zhang, L. Covalent complexes of proteasome model with peptide aldehyde inhibitors MG132 and MG101: Docking and molecular dynamics study. J. Mol. Model. 2009, 15, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mach, R.H.; Reichert, D.E. Docking and 3D-QSAR Studies on Isatin Sulfonamide Analogues as Caspase-3 Inhibitors. J. Chem. Inf. Model. 2009, 49, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Juhl, P.B.; Trodler, P.; Tyagi, S.; Pleiss, J. Modelling substrate specificity and enantioselectivity for lipases and esterases by substrate-imprinted docking. BMC Struct. Biol. 2009, 9. [Google Scholar] [CrossRef]
- Chernorizov, K.A.; Elkina, J.L.; Semenyuk, P.I.; Svedas, V.K.; Muronetz, V.I. Novel inhibitors of glyceraldehyde-3-phosphate dehydrogenase: Covalent modification of NAD-binding site by aromatic thiols. Biochemistry (Moscow) 2010, 75, 1444–1449. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, X.; Wu, G.; Ma, Y.; Li, Y.; Zhao, X.; Yuan, Y.; Yang, J.; Yu, S.; Shao, F.; et al. Synthesis, in Vitro and in Vivo Biological Evaluation, Docking Studies, and Structure-Activity Relationship (SAR) Discussion of Dipeptidyl Boronic Acid Proteasome Inhibitors Composed of β-Amino Acids. J. Med. Chem. 2010, 53, 1990–1999. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xu, B.; Fang, Y.; Yang, Z.; Cui, J.; Zhang, L.; Zhang, L. Synthesis and SAR Study of Novel Peptide Aldehydes as Inhibitors of 20S Proteasome. Molecules 2011, 16, 7551–7564. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.K.; Tota, S.; Tripathi, T.; Chander, S.; Nath, C.; Saxena, A.K. Lead optimization studies towards the discovery of novel carbamates as potent AChE inhibitors for the potential treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 6313–6320. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.; Klinger, A.; Oellien, F.; Marhoefer, R.J.; Duszenko, M.; Selzer, P.M. Docking-Based Virtual Screening of Covalently Binding Ligands: An Orthogonal Lead Discovery Approach. J. Med. Chem. 2013, 56, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Blake, L.; Soliman, M.E.S. Identification of irreversible protein splicing inhibitors as potential anti-TB drugs: insight from hybrid non-covalent/covalent docking virtual screening and molecular dynamics simulations. Med. Chem. Res. 2014, 23, 2312–2323. [Google Scholar] [CrossRef]
- Dong, G.Q.; Calhoun, S.; Fan, H.; Kalyanaraman, C.; Branch, M.C.; Mashiyama, S.T.; London, N.; Jacobson, M.P.; Babbitt, P.C.; Shoichet, B.K.; et al. Prediction of Substrates for Glutathione Transferases by Covalent Docking. J. Chem. Inf. Model. 2014, 54, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins-Struct. Funct. Bioinf. 2014, 82, 1646–1655. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated Docking Screens: A Feasibility Study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminformatics 2009, 1, 15. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discovery 2010, 5, 597–607. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Bonelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M. Biased probability monte-carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Rapid boundary element solvation electrostatics calculations in folding simulations: Successful folding of a 23-residue peptide. Biopolymers 2001, 60, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. Four-Dimensional Docking: A Fast and Accurate Account of Discrete Receptor Flexibility in Ligand Docking. J. Med. Chem. 2009, 52, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, P.L.; Kumar, G.; White, S.W.; Webb, T.R. Recent Advances in Computer-Aided Drug Design as Applied to Anti-Influenza Drug Discovery. Curr. Top. Med. Chem. 2014, 14, 1875–1889. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Marinelli, L.; di Leva, F.S.; la Pietra, V.; de Simone, A.; Mancini, F.; Andrisano, V.; Novellino, E.; Goodsell, D.S.; Olson, A.J. Protein flexibility in virtual screening: the BACE-1 case study. J. Chem. Inf. Model. 2012, 52, 2697–704. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules 2015, 20, 1984-2000. https://doi.org/10.3390/molecules20021984
Kumalo HM, Bhakat S, Soliman MES. Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules. 2015; 20(2):1984-2000. https://doi.org/10.3390/molecules20021984
Chicago/Turabian StyleKumalo, Hezekiel Mathambo, Soumendranath Bhakat, and Mahmoud E. S. Soliman. 2015. "Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls" Molecules 20, no. 2: 1984-2000. https://doi.org/10.3390/molecules20021984
APA StyleKumalo, H. M., Bhakat, S., & Soliman, M. E. S. (2015). Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules, 20(2), 1984-2000. https://doi.org/10.3390/molecules20021984