
Electron Transfer-Induced Coupling of Haloarenes to Styrenes and 1,1-Diphenylethenes Triggered by Diketopiperazines and Potassium tert-Butoxide

Abstract
:1. Introduction
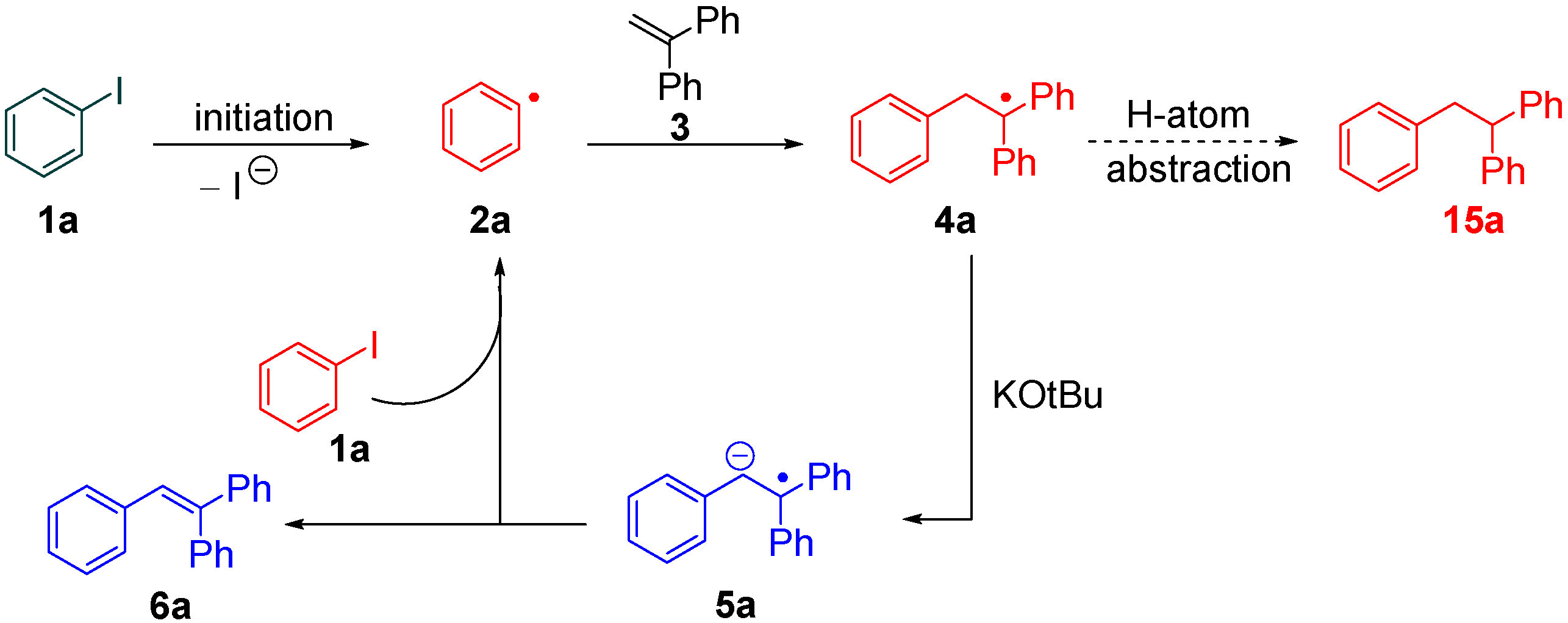
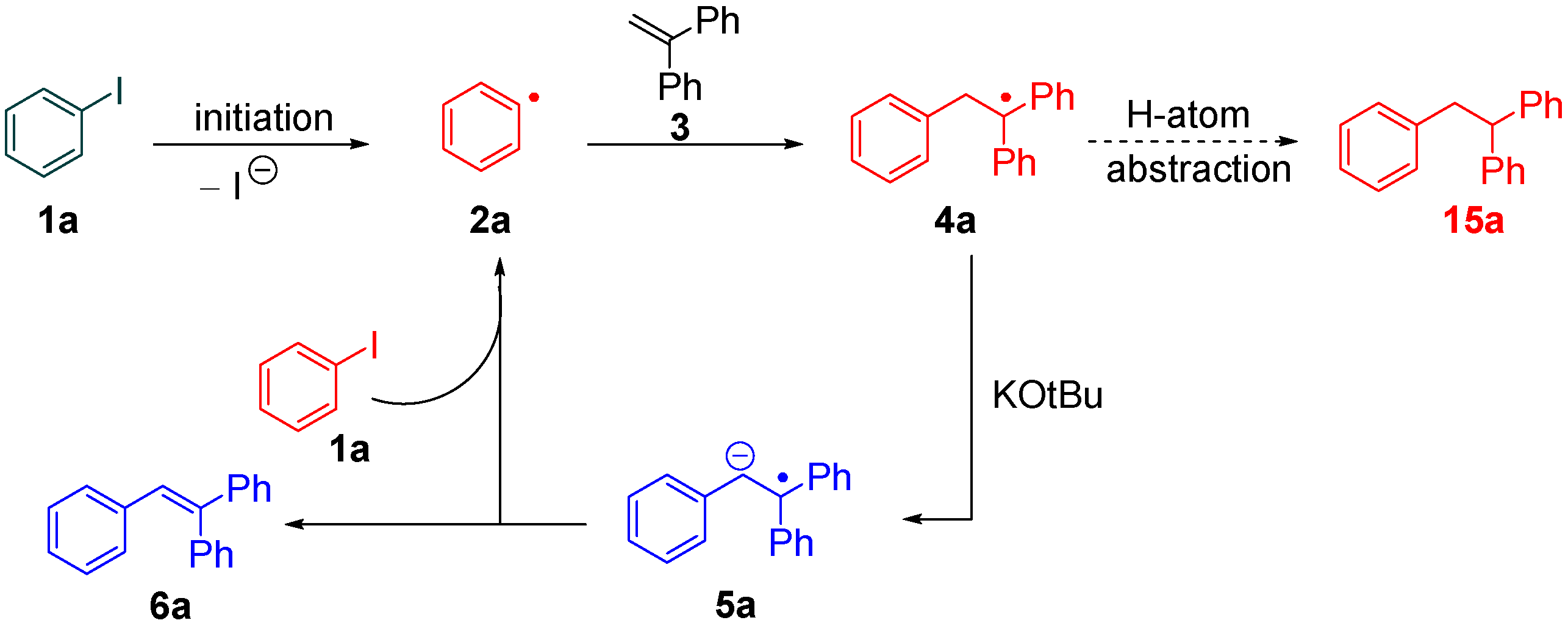
2. Results and Discussion

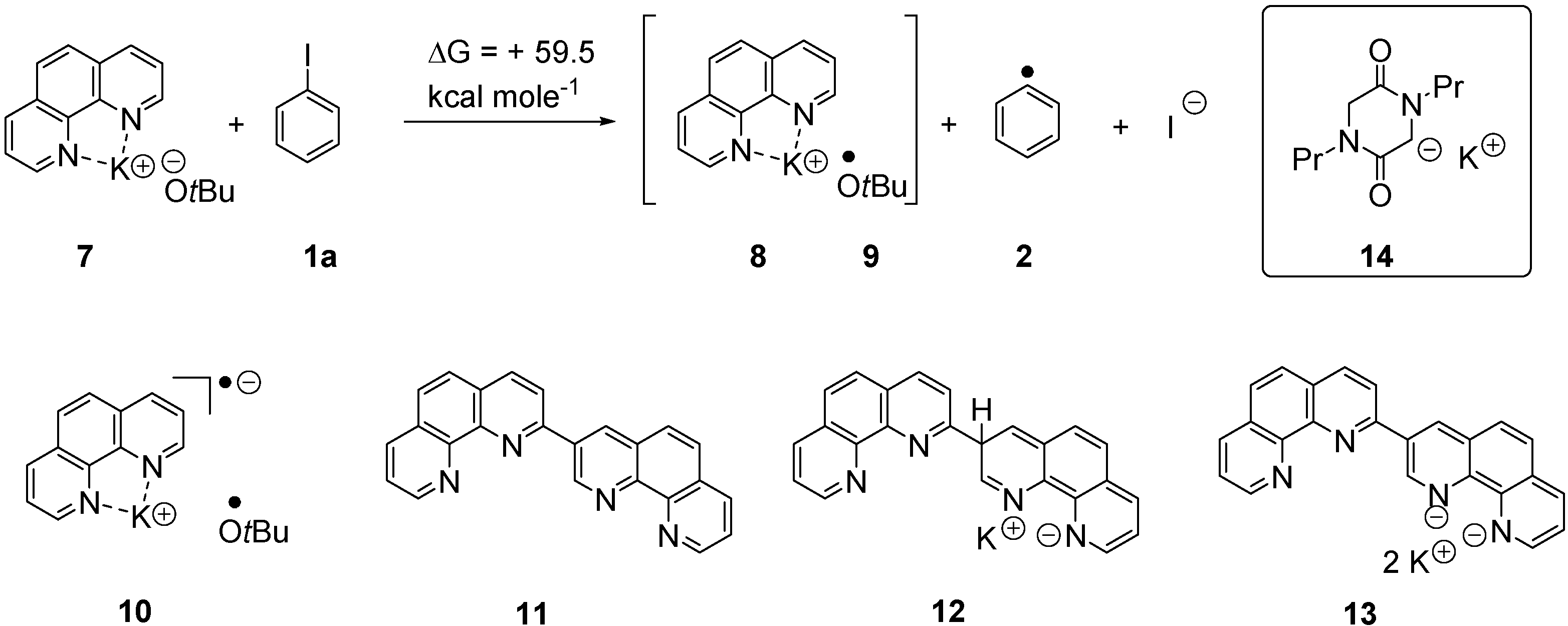
{kind=link}
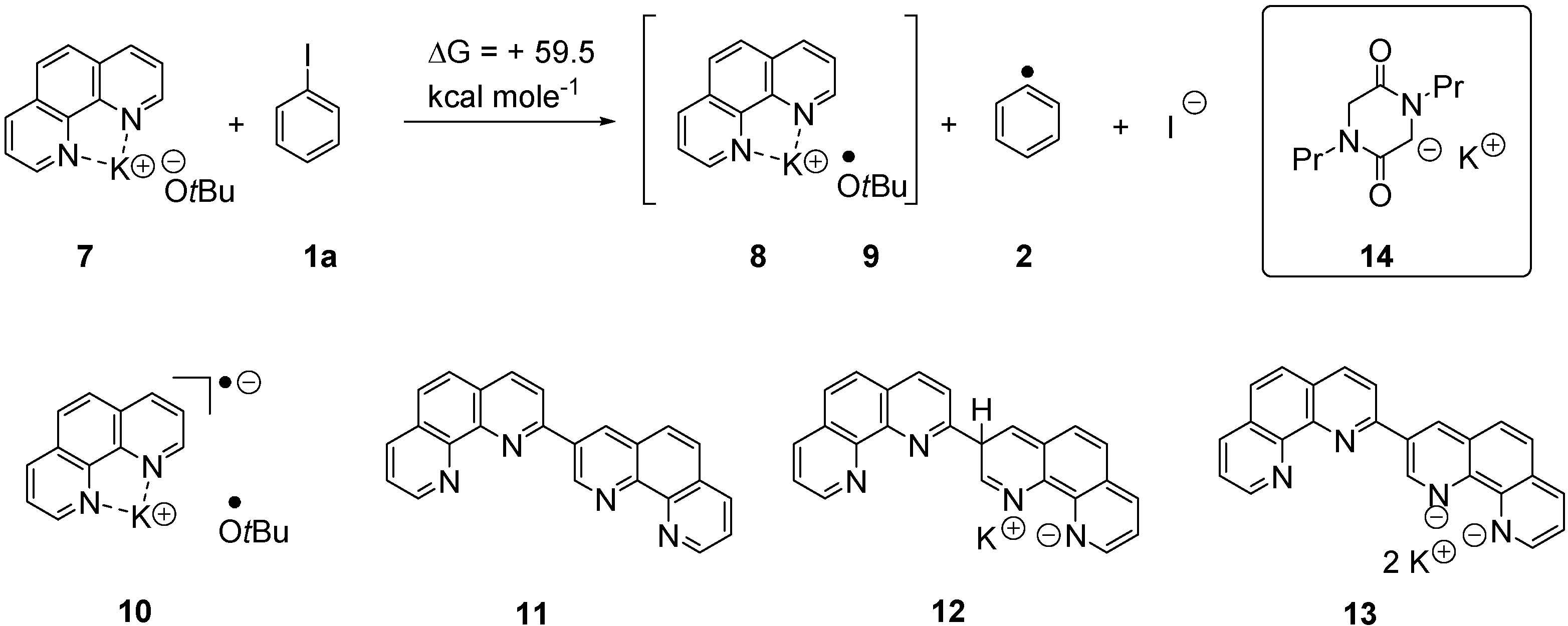{kind=link}
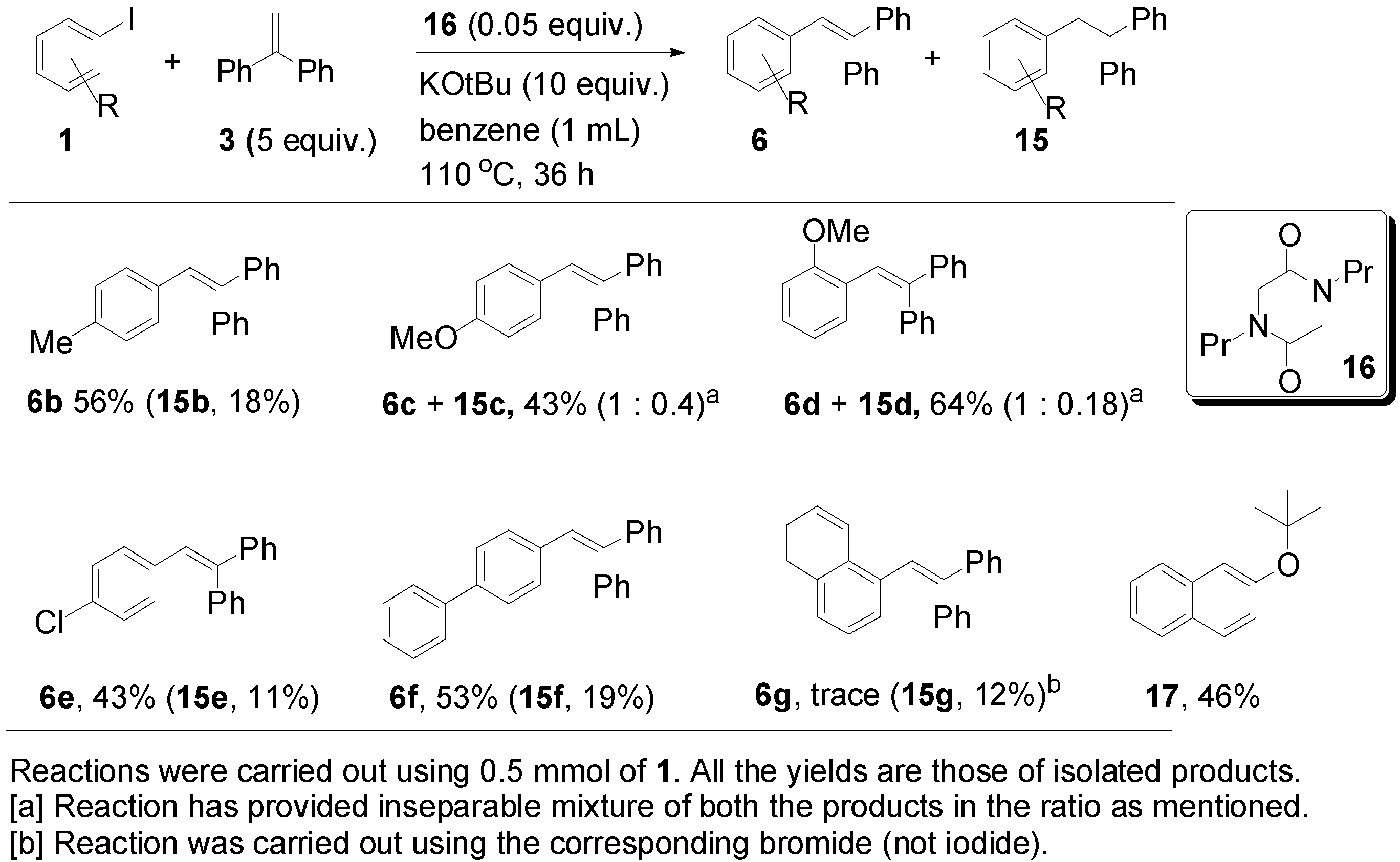{kind=link}
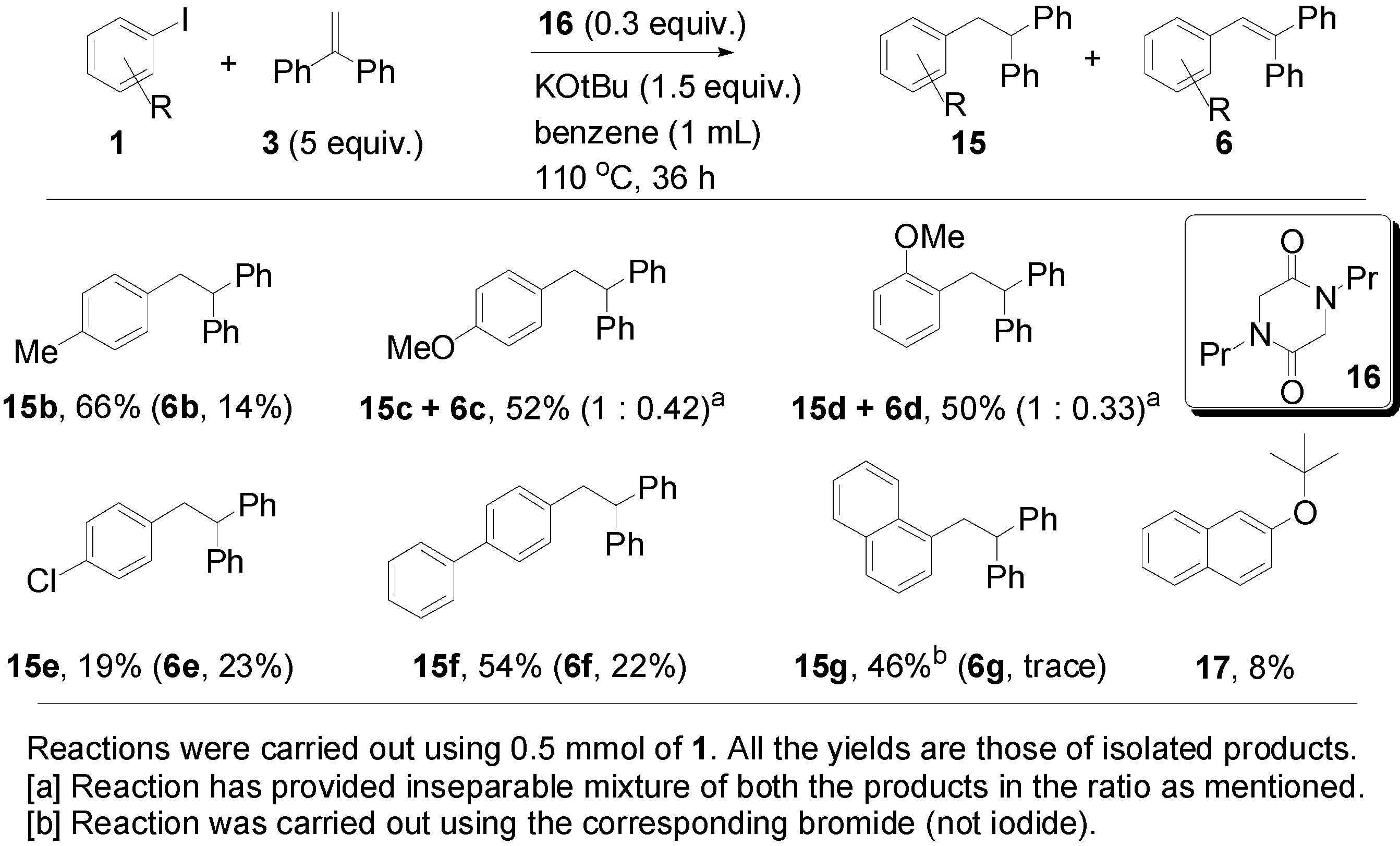{kind=link}
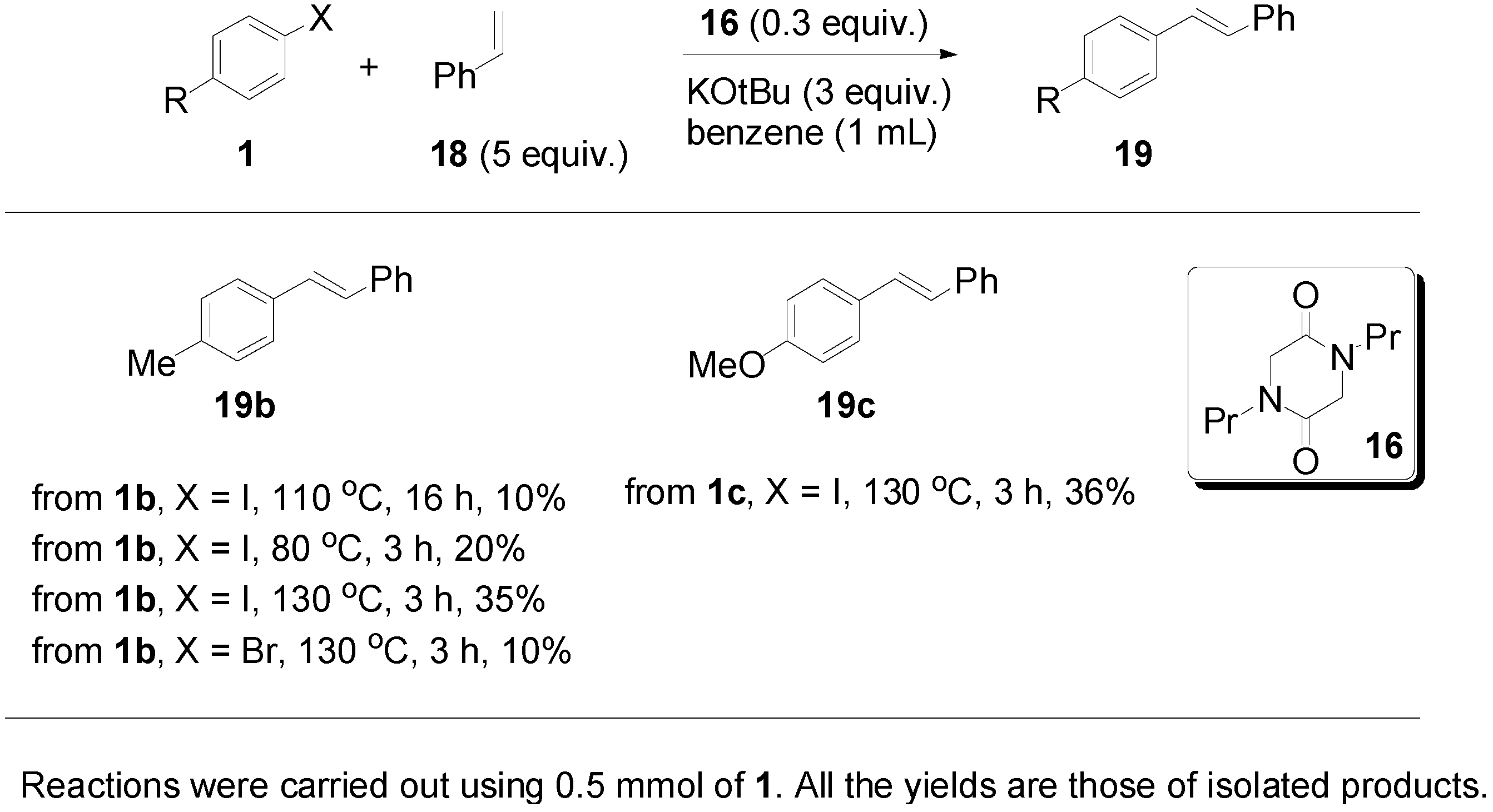{kind=link}
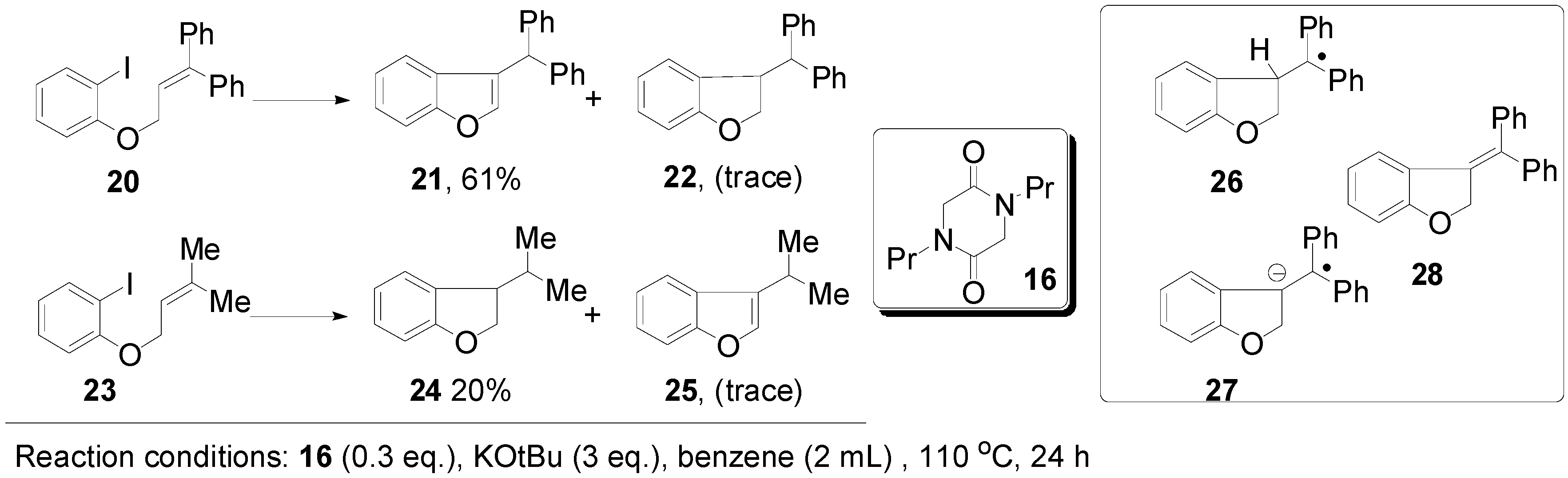{kind=link}
{kind=link}
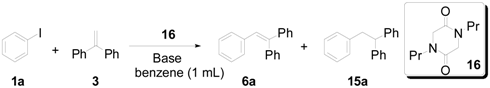
| Entry | 3 (Equiv.) | 16 (Equiv.) | Base (Equiv.) | Solvent | T (°C) | Reaction Time (h) | 6a (%) a | 15a (%) a |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 0.5 | KOtBu (3) | benzene | 100 | 16 | 11 | 18 |
| 2 | 1 | 0.1 | KOtBu (5) | benzene | 110 | 16 | 22 | 13 |
| 3 | 1 | 0.1 | KOtBu (5) | benzene | 160 | 16 | 25 | 8 |
| 4 | 1 | 0.1 | KOtBu (5) | DMSO | 160 | 16 | -- b | -- b |
| 5 | 3 | 0.1 | KOtBu (5) | DMF | 160 | 16 | 6 | 13 |
| 6 | 3 | 0.3 | KOtBu (3) | benzene | 110 | 36 | 25 | 43 |
| 7 | 5 | 0.1 | KOtBu (6) | benzene | 110 | 36 | 36 | 26 |
| 8 | 5 | 0.1 | K2CO3 (6) | benzene | 110 | 36 | 0 | 0 |
| 9 | 5 | 0.2 | KOtBu (6) | benzene | 110 | 36 | 25 | 42 |
| 10 | 5 | 0.05 | KOtBu (10) | benzene | 110 | 36 | 58 | 14 |
| 11 | 5 | 0.3 | KOtBu (1.5) | benzene | 110 | 36 | 20 | 63 |
| 12 | 5 | - | KOtBu (6) | benzene | 110 | 36 | trace | trace |
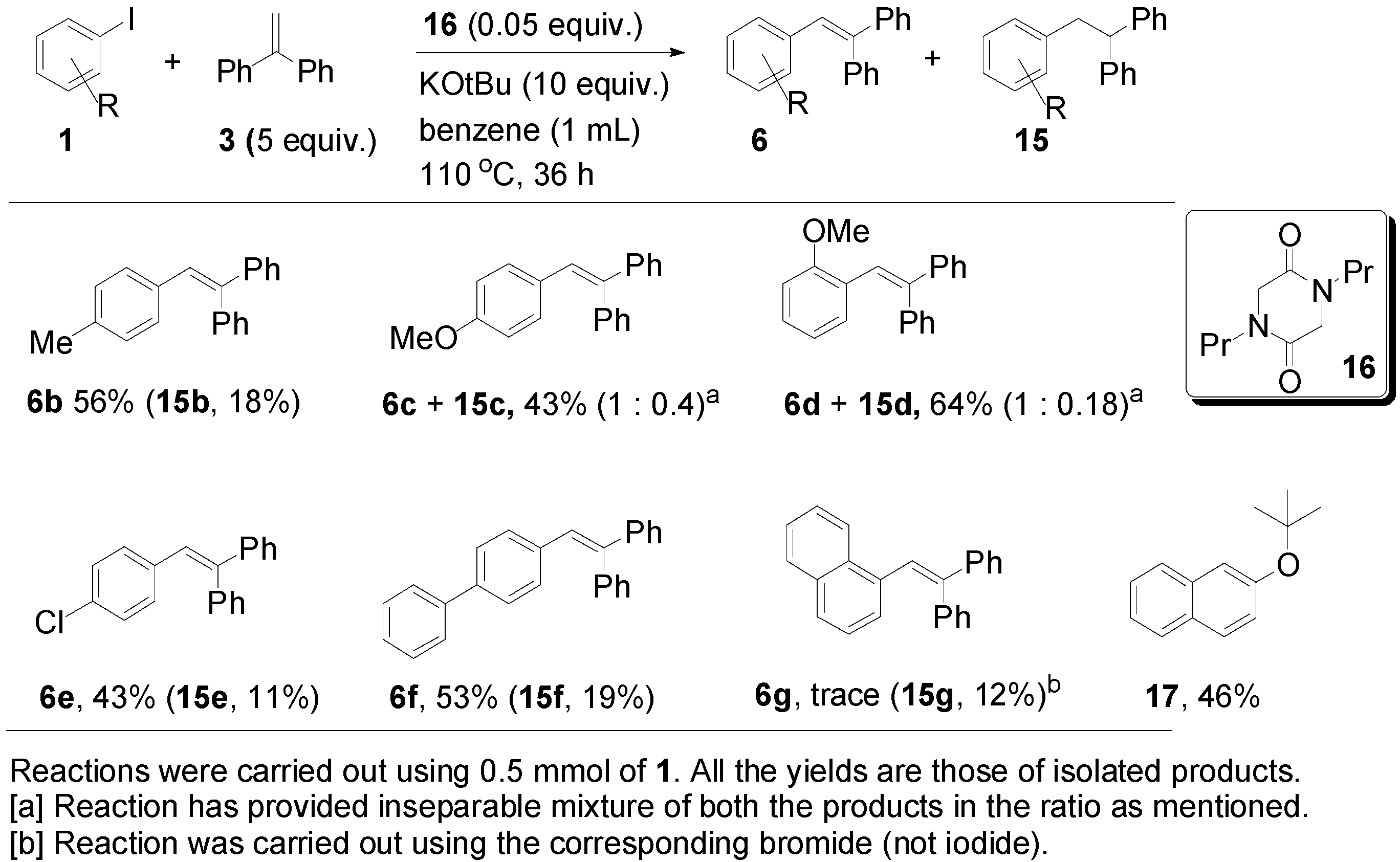
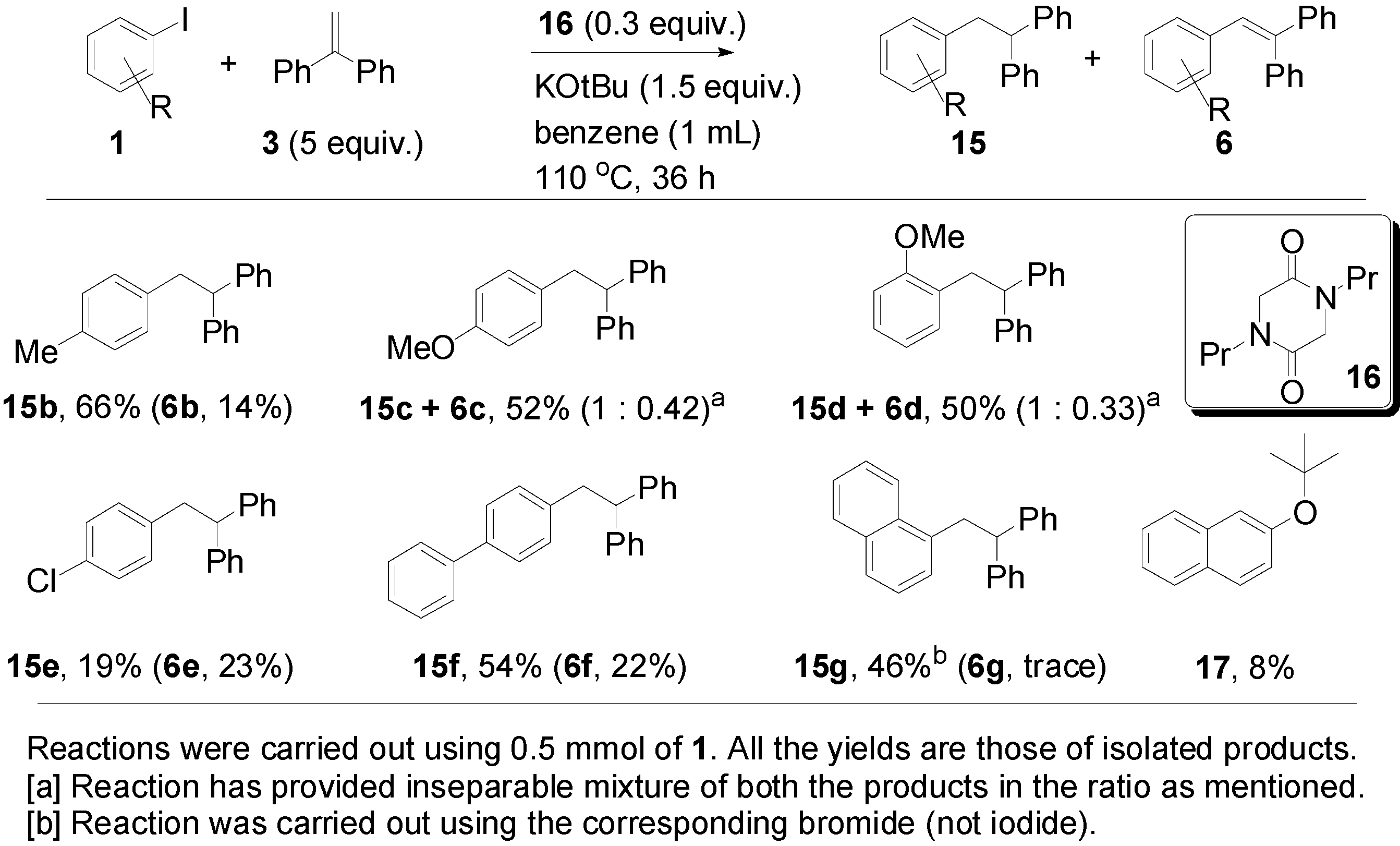
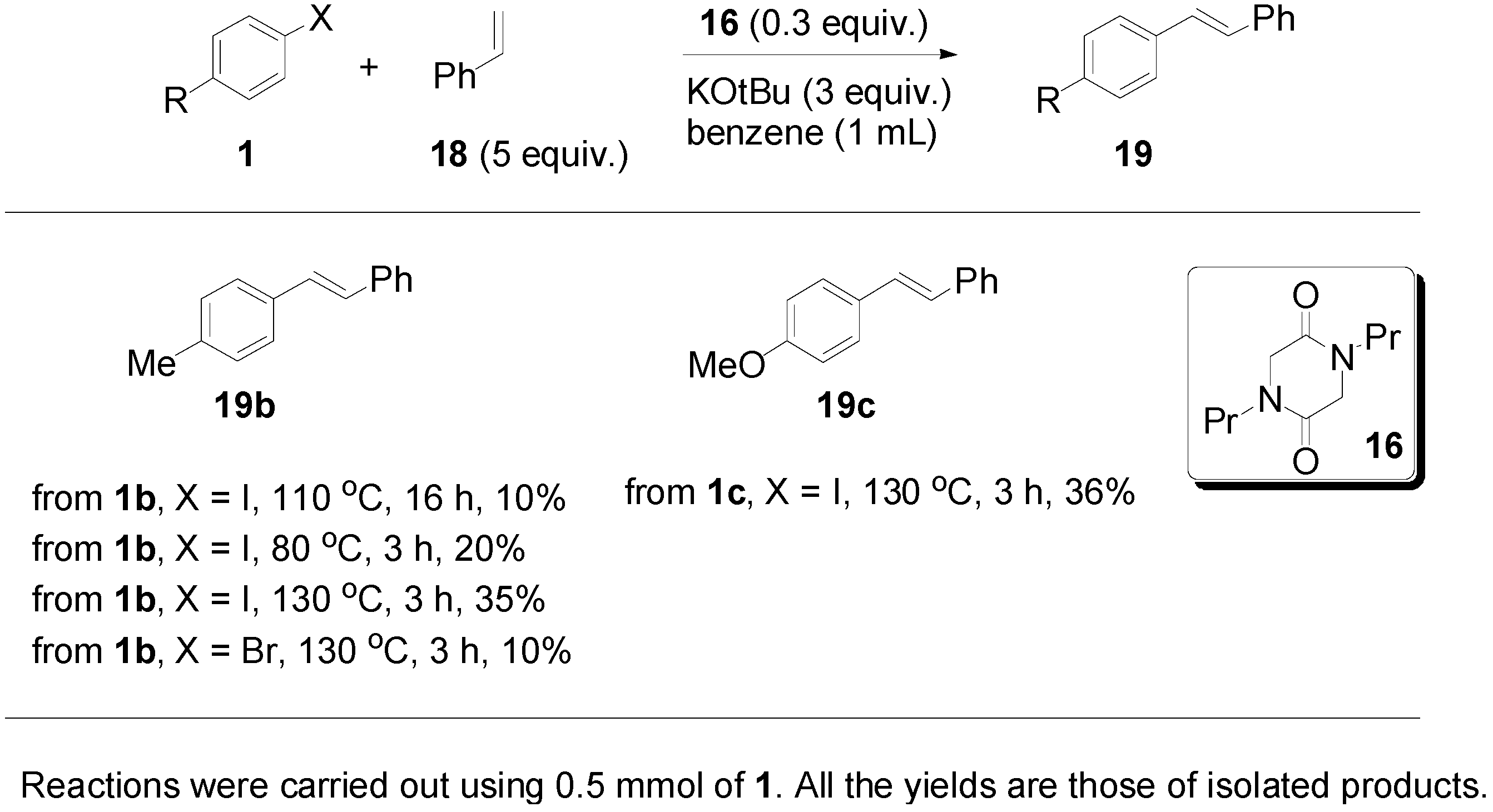
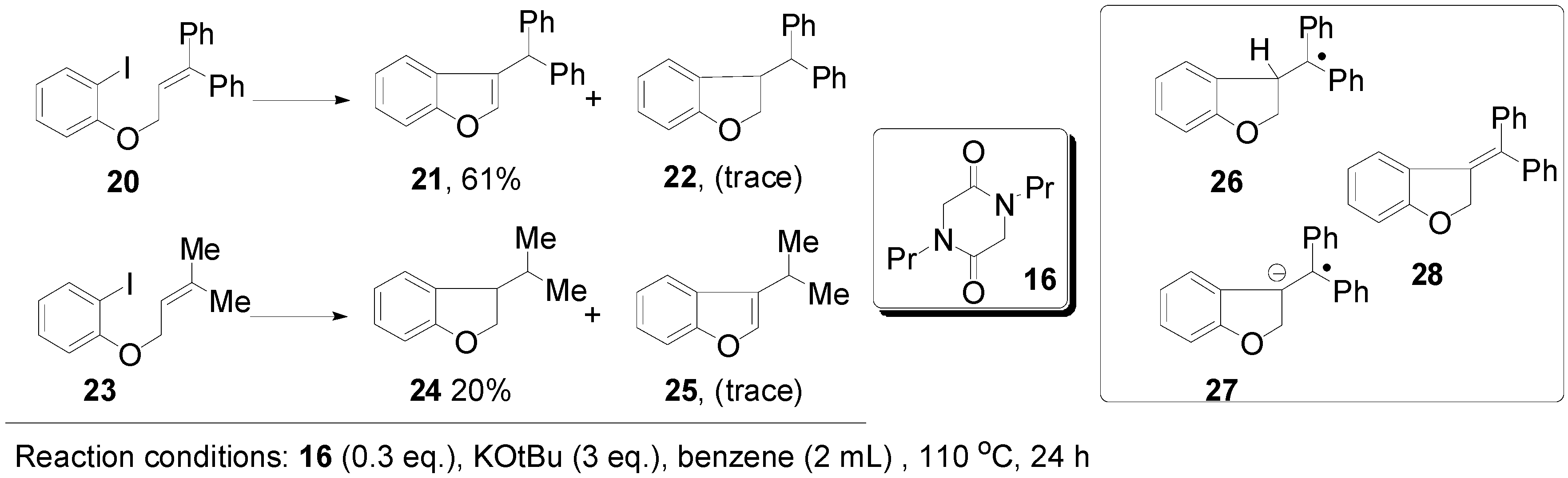
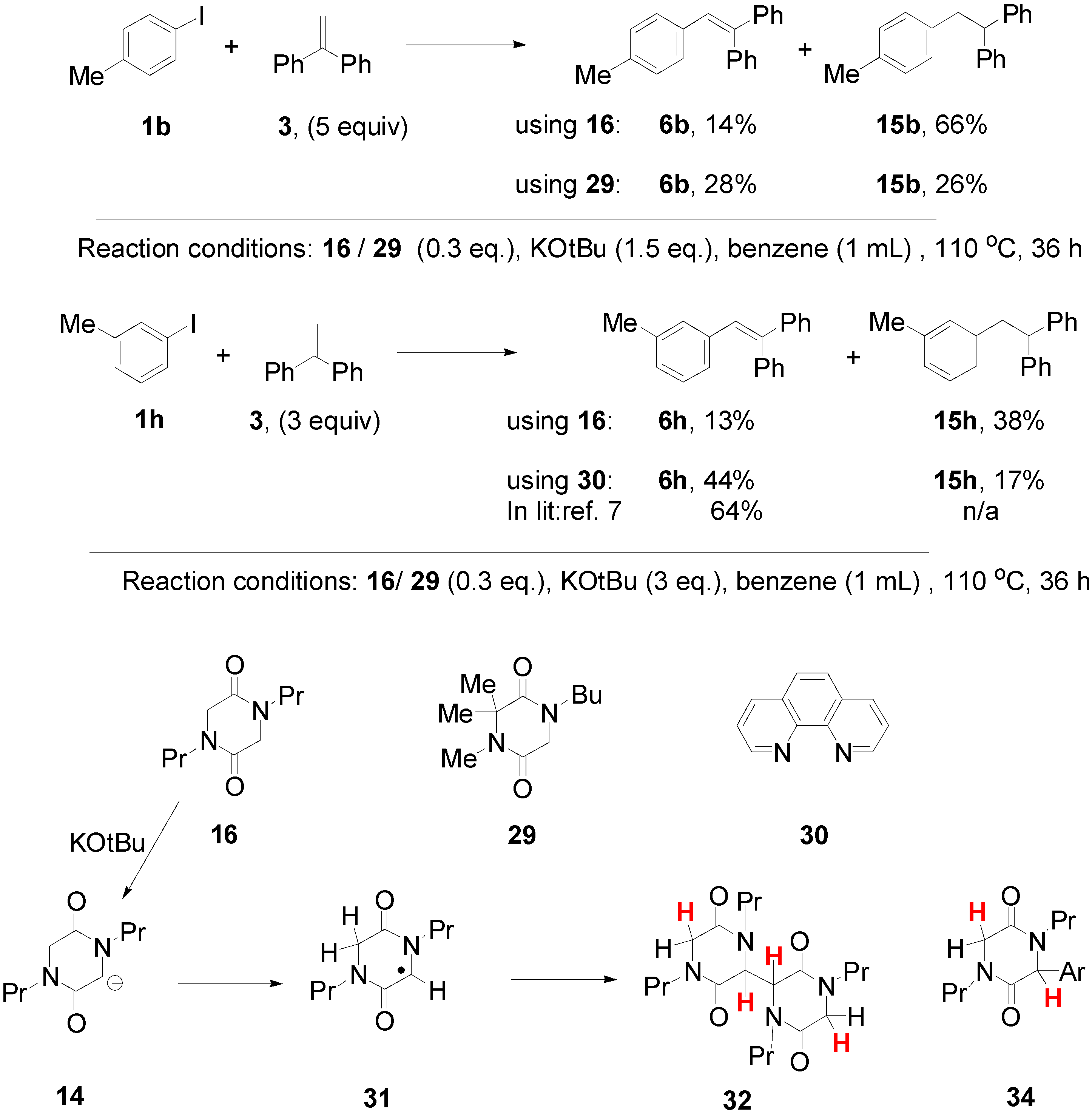
3. Experimental Section
3.1. General Information
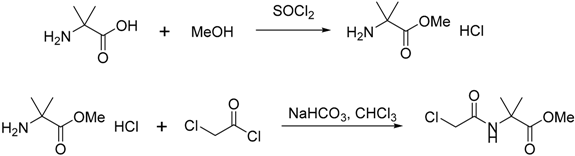
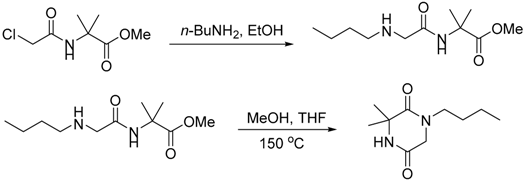
3.2. Preparation of Piperazinediones
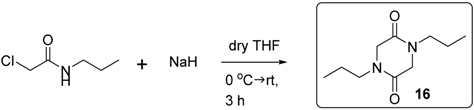
3.2.1. Synthesis of Compound 16
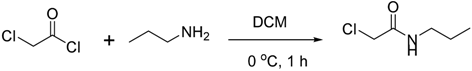
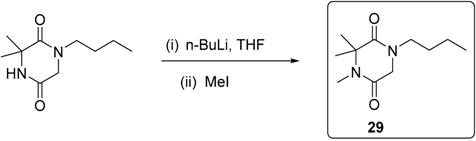
3.2.2. Synthesis of Compound 29
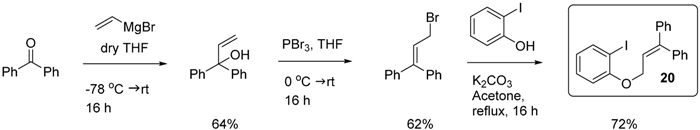
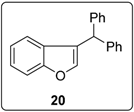
3.3. Synthesis of (3-(2-Iodophenoxy)prop-1-ene-1,1-diyl)dibenzene (20)
3.4. Synthesis of 1-Iodo-2-((3-methylbut-2-en-1-yl)oxy)benzene (23)
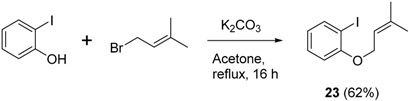
3.5. General Reaction Procedure
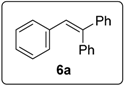
3.5.1. Data for Compound 6a
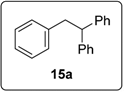
3.5.2. Data for Compound 15a
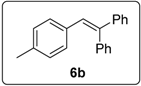
3.5.3. Data for Compound 6b
3.5.4. Data for Compound 15b
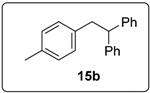
3.5.5. Data for Compound 6e
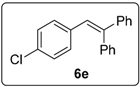
3.5.6. Data for Compound 15e
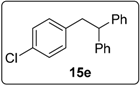
3.5.7. Data for Compound 6f
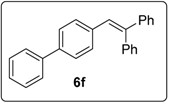
3.5.8. Data for Compound 15f
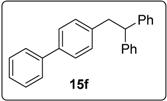
3.5.9. Data for Compound 17
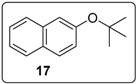
3.5.10. Data for Compound 15g
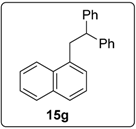
3.5.11. Data for Compound 18b
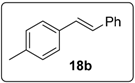
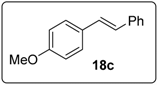
3.5.12. Data for Compound 18c
3.5.13. Data for Compound 20
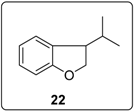
3.5.14. Data for Compound 22
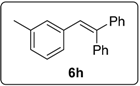
3.5.15. Data for Compound 6h
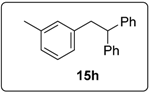
3.5.16. Data for Compound 15h
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guastavino, J.F.; Buden, M.E.; Rossi, R.A. Room-temperature and transition-metal-free Mizoroki-Heck-type Reaction. Synthesis of E-stilbenes by photoinduced C-H functionalization. J. Org. Chem. 2014, 79, 9104–9111. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Cheprakov, A.V. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- Braese, S.; de Meijere, A. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A., Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 217–315. [Google Scholar]
- Larhed, M.; Hallberg, A. Handbook of OrganopalladiumChemistry for Organic Synthesis; Negishi, E., de Meijere, A., Eds.; Wiley-Interscience: New York, NY, USA, 2002; Volume 1, Chapter IV.2; pp. 1133–1178. [Google Scholar]
- Ackermann, L.; Born, R. Mizoroki-Heck Reaction; Oestreich, M., Ed.; Wiley: Chichester, UK, 2009; pp. 383–403. [Google Scholar]
- Shirakawa, E.; Zhang, X.; Hayashi, T. Mizoroki-heck-type reaction mediated by potassium tert-butoxide. Angew. Chem. Int. Ed. 2011, 50, 4671–4674. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Ueda, K.; Taniguchi, T.; Itami, K. Potassium t-butoxide alone can promote the biaryl coupling of electron-deficient nitrogen heterocycles and haloarenes. Org. Lett. 2008, 10, 4673–4676. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-L.; Li, H.; Yu, D.-G.; Yu, M.; Zhou, X.; Lu, X.-Y.; Huang, K.; Zheng, S.-F.; Li, B.-J.; Shi, Z.-J. An efficient organocatalytic method for constructing biaryls through aromatic C-H activation. Nat. Chem. 2010, 2, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cao, H.; Zhang, H.; Zhang, H.; Chung, K.H.; He, C.; Wang, H.; Kwong, F.Y.; Lei, A. Organocatalysis in cross-coupling: DMEDA-catalyzed direct C-H arylation of unactivated benzene. J. Am. Chem. Soc. 2010, 132, 16737–16740. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, E.; Itoh, K.-I.; Higashino, T.; Hayashi, T. tert-Butoxide-mediated arylation of benzene with aryl halides in the presence of a catalytic 1,10-phenanthroline derivative. J. Am. Chem. Soc. 2010, 132, 15537–15539. [Google Scholar] [CrossRef] [PubMed]
- Studer, A.; Curran, D.P. Organocatalysis and C-H activation meet radical- and electron-transfer reactions. Angew. Chem. Int. Ed. 2011, 50, 5018–5022. [Google Scholar] [CrossRef]
- Shirakawa, E.; Hayashi, T. Transition-metal-free coupling reactions of aryl halides. Chem. Lett. 2012, 41, 130–134. [Google Scholar] [CrossRef]
- Sun, C.-L.; Gu, Y.-F.; Wang, B.; Shi, J.-S. Direct arylation of alkenes with aryl iodides/bromides through an organocatalytic radical process. Chem. Eur. J. 2011, 17, 10844–10847. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Leiendecker, M.; Das, A.; Poisson, T.; Bui, L. Potassium tert-butoxide mediated Heck-type cyclization/isomerization-benzofurans from organocatalytic radical cross-coupling reactions. Chem. Commun. 2011, 47, 10629–10631. [Google Scholar] [CrossRef]
- Zhou, S.; Anderson, G.M.; Mondal, B.; Doni, E.; Ironmonger, V.; Kranz, M.; Tuttle, T.; Murphy, J.A. Organic super-electron-donors: Initiators in transition metal-free haloarene-arene coupling. Chem. Sci. 2014, 5, 476–482. [Google Scholar] [CrossRef]
- Cuthbertson, J.; Gray, V.J.; Wilden, J.D. Observations on transition metal free biaryl coupling: Potassium tert-butoxide alone promotes the reaction without diamine or phenanthroline catalysts. Chem. Commun. 2014, 50, 2575–2578. [Google Scholar]
- Yi, H.; Jutand, A.; Lei, A. Evidence for the interaction between tBuOK and 1,10-phenanthroline to form the 1,10-phenanthroline radical anion: a key step for the activation of aryl bromides by electron transfer. Chem. Commun. 2015, 51, 545–548. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, L.; Wang, L.; Gong, M.; Wang, W.; Yuan, R. Visible light photoredox catalyzed biaryl synthesis using nitrogen heterocycles as promoter. ACS Catal. 2015, 5, 45–50. [Google Scholar] [CrossRef]
- Zhou, S.; Doni, E.; Anderson, G.M.; Kane, R.G.; MacDougall, S.W.; Ironmonger, V.; Tuttle, T.; Murphy, J.A. Identifying the roles of amino acids, alcohols and 1,2-diamines as mediators in coupling of haloarenes to arenes. J. Am. Chem. Soc. 2014, 136, 17818–17826. [Google Scholar] [CrossRef] [PubMed]
- Tanimoro, K.; Ueno, M.; Takeda, K.; Kirihata, M.; Tanimori, S. Proline catalyzes direct C-H arylations of unactivated arenes. J. Org. Chem. 2012, 77, 7844–7849. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tian, F.; Wang, X.; Yu, H.; Bi, Y. Simple alcohols promoted direct C-H arylation of unactivated arenes with aryl halides. Chem. Commun. 2013, 49, 2983–2985. [Google Scholar] [CrossRef]
- Bradshaw, J.S.; Hales, R.H. Reaction of bromonaphthalene with potassium tert-butoxide and tert-butyl alcohol in dimethyl sulfoxide. J. Org. Chem. 1971, 36, 318–322. [Google Scholar] [CrossRef]
- Brocks, J.J.; Welle, F.M.; Beckhaus, H.-D.; R üchardt, C. Are α-centered peptide radicals stabilized by a capto-dative effect? Tetrahedron Lett. 1997, 38, 7721–7724. [Google Scholar] [CrossRef]
- Jaimes, M.C.B.; Weingand, V.; Rominger, F.; Hashmi, A.S.K. From ynamides to highly substituted benzo[b]furans: Gold(I)-catalyzed 5-endo-dig-cyclization/rearrangement of alkylic oxonium intermediates. Chem. Eur. J. 2013, 19, 12504–12511. [Google Scholar] [CrossRef] [PubMed]
- Garland, P.B.; Serafinowski, P.J. High yield detritylation of surface-attached nucleosides with photoacid generated in an overlying solid film: Roles of translational diffusion and scavenging. Org. Biomol. Chem. 2009, 7, 451–459. [Google Scholar] [CrossRef]
- Yang, F.-L.; Ma, X.-T.; Tian, S.-K. Oxidative Mizoroki-heck-type reaction of arylsulfonyl hydrazides for a highly regio- and stereoselective synthesis of polysubstituted alkenes. Chem. Eur. J. 2012, 18, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- King, F.D.; Caddick, S. Triflic acid-mediated phenylation of N-acylaminoalkyl diethylacetals and N-acyl-2-phenyl cyclic amides. Org. Biomol. Chem. 2011, 9, 4361–4366. [Google Scholar] [CrossRef] [PubMed]
- Donck, S.; Baroudi, A.; Fensterbank, L.; Goddard, J.-P.; Ollivier, C. Visible-light photocatalytic reduction of sulfonium salts as a source of aryl radicals. Adv. Synth. Catal. 2013, 355, 1477–1482. [Google Scholar] [CrossRef]
- Weidert, P.J.; Geyer, E.; Horner, L. Phosphoorganische verbindungen 120.1 Versuche zum nachweis von “phosphenen” aus geeigneten phosphinsaurederivaten durch eliminierung. Phosphorus Sulfur Silicon Relat. Elem. 1989, 45, 55–59. [Google Scholar] [CrossRef]
- Xiao, Q.; Ma, J.; Yang, Y.; Zhang, Y.; Wang, J. Pd-catalyzed C═C double-bond formation by coupling of N-tosylhydrazones with benzyl halides. Org. Lett. 2009, 11, 4732–4735. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bosco, M.; Locatelli, M.; Marcantoni, E.; Melchiorre, P.; Sambri, L. Unusual and unexpected reactivity of t-butyl dicarbonate (boc2o) with alcohols in the presence of magnesium perchlorate. A new and general route to t-butyl ethers. Org. Lett. 2005, 7, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Kanagaraj, K.; Pitchumani, K. The Aminocyclodextrin/Pd(OAc)2 complex as an efficient catalyst for the mizoroki–heck cross-coupling reaction. Chem. Eur. J. 2013, 19, 14425–14431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-G.; Liu, X.-L.; He, Z.-Y.; Li, X.-M.; Kang, H.-J.; Tian, S.-K. Palladium/copper-catalyzed oxidative arylation of terminal alkenes with aroyl hydrazides. Chem. Eur. J. 2014, 20, 2765–2769. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Shibata, I.; Baba, A. Inter- and intramolecular radical couplings of ene-ynes or halo-alkenes promoted by an InCl3/MeONa/Ph2SiH2 system. Org. Lett. 2005, 7, 3093–3096. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doni, E.; Zhou, S.; Murphy, J.A. Electron Transfer-Induced Coupling of Haloarenes to Styrenes and 1,1-Diphenylethenes Triggered by Diketopiperazines and Potassium tert-Butoxide. Molecules 2015, 20, 1755-1774. https://doi.org/10.3390/molecules20021755
Doni E, Zhou S, Murphy JA. Electron Transfer-Induced Coupling of Haloarenes to Styrenes and 1,1-Diphenylethenes Triggered by Diketopiperazines and Potassium tert-Butoxide. Molecules. 2015; 20(2):1755-1774. https://doi.org/10.3390/molecules20021755
Chicago/Turabian StyleDoni, Eswararao, Shengze Zhou, and John A. Murphy. 2015. "Electron Transfer-Induced Coupling of Haloarenes to Styrenes and 1,1-Diphenylethenes Triggered by Diketopiperazines and Potassium tert-Butoxide" Molecules 20, no. 2: 1755-1774. https://doi.org/10.3390/molecules20021755
APA StyleDoni, E., Zhou, S., & Murphy, J. A. (2015). Electron Transfer-Induced Coupling of Haloarenes to Styrenes and 1,1-Diphenylethenes Triggered by Diketopiperazines and Potassium tert-Butoxide. Molecules, 20(2), 1755-1774. https://doi.org/10.3390/molecules20021755