
Metathetical Redox Reaction of (Diacetoxyiodo)arenes and Iodoarenes

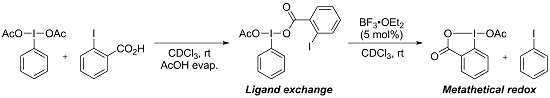
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
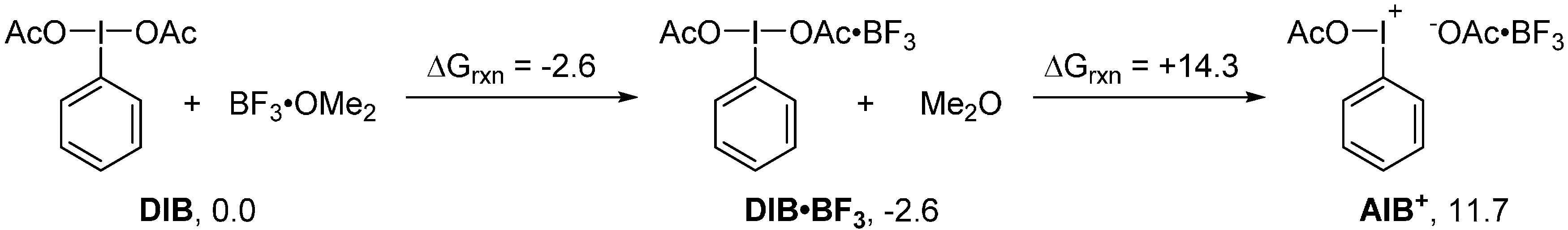{kind=link}
{kind=link}
{kind=link}
{kind=link}
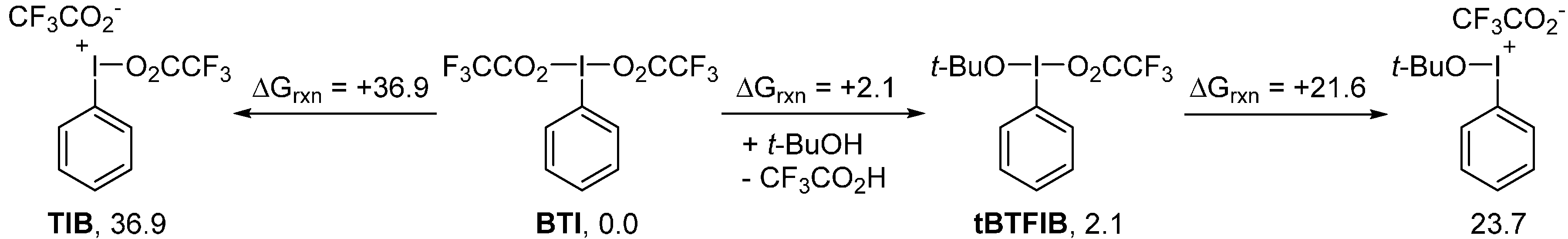{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
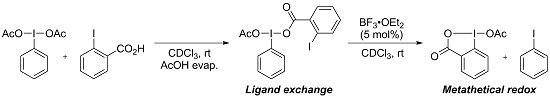
1. Introduction
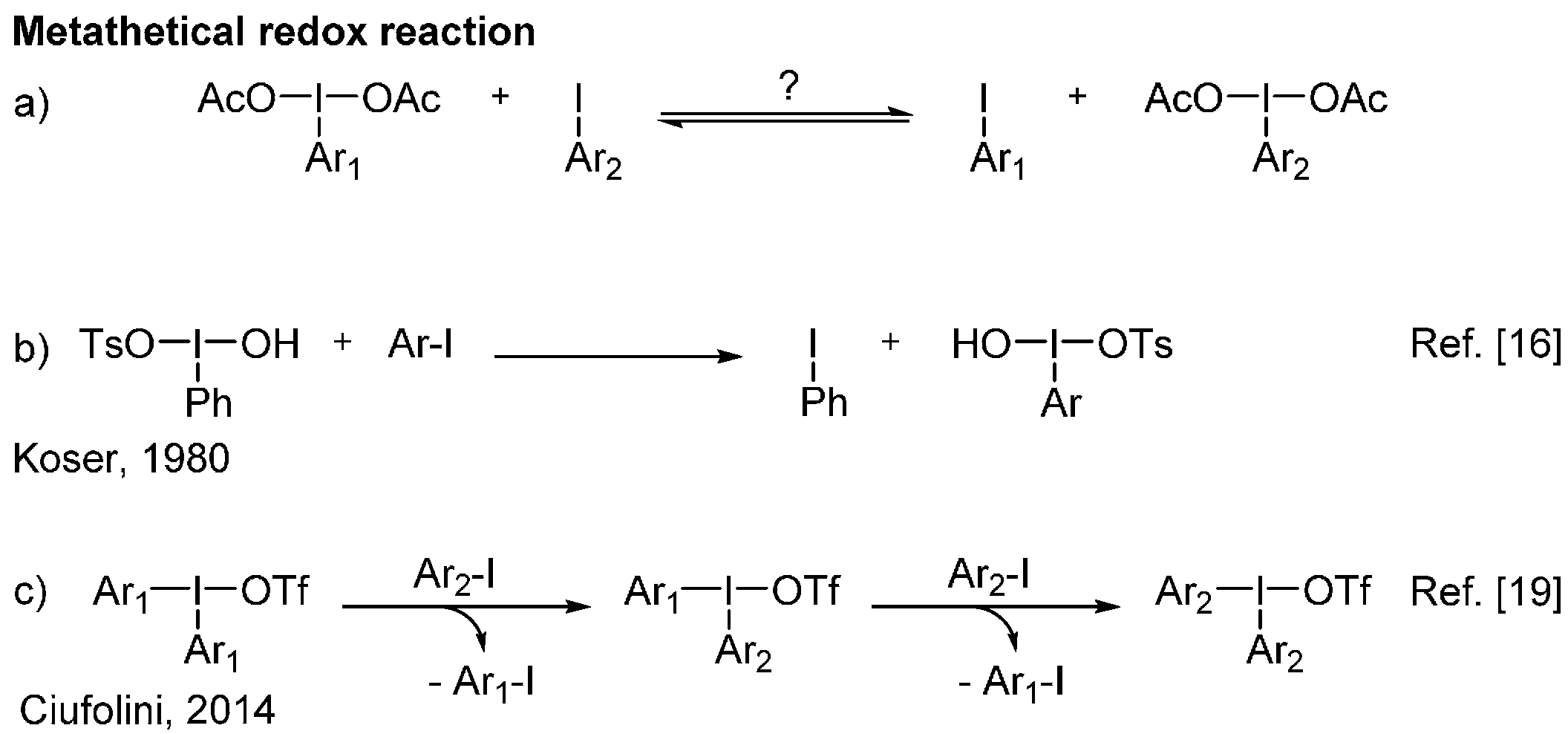
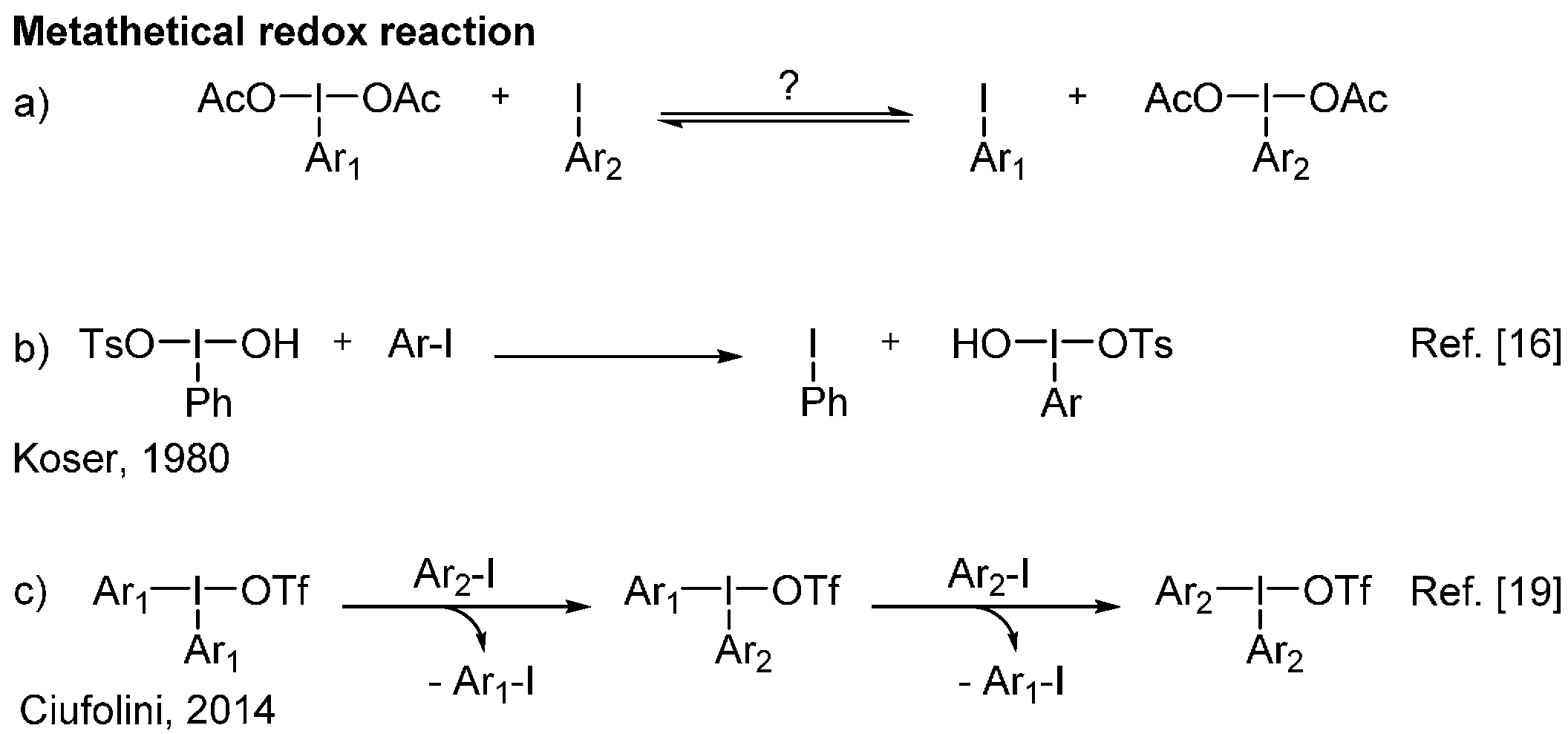
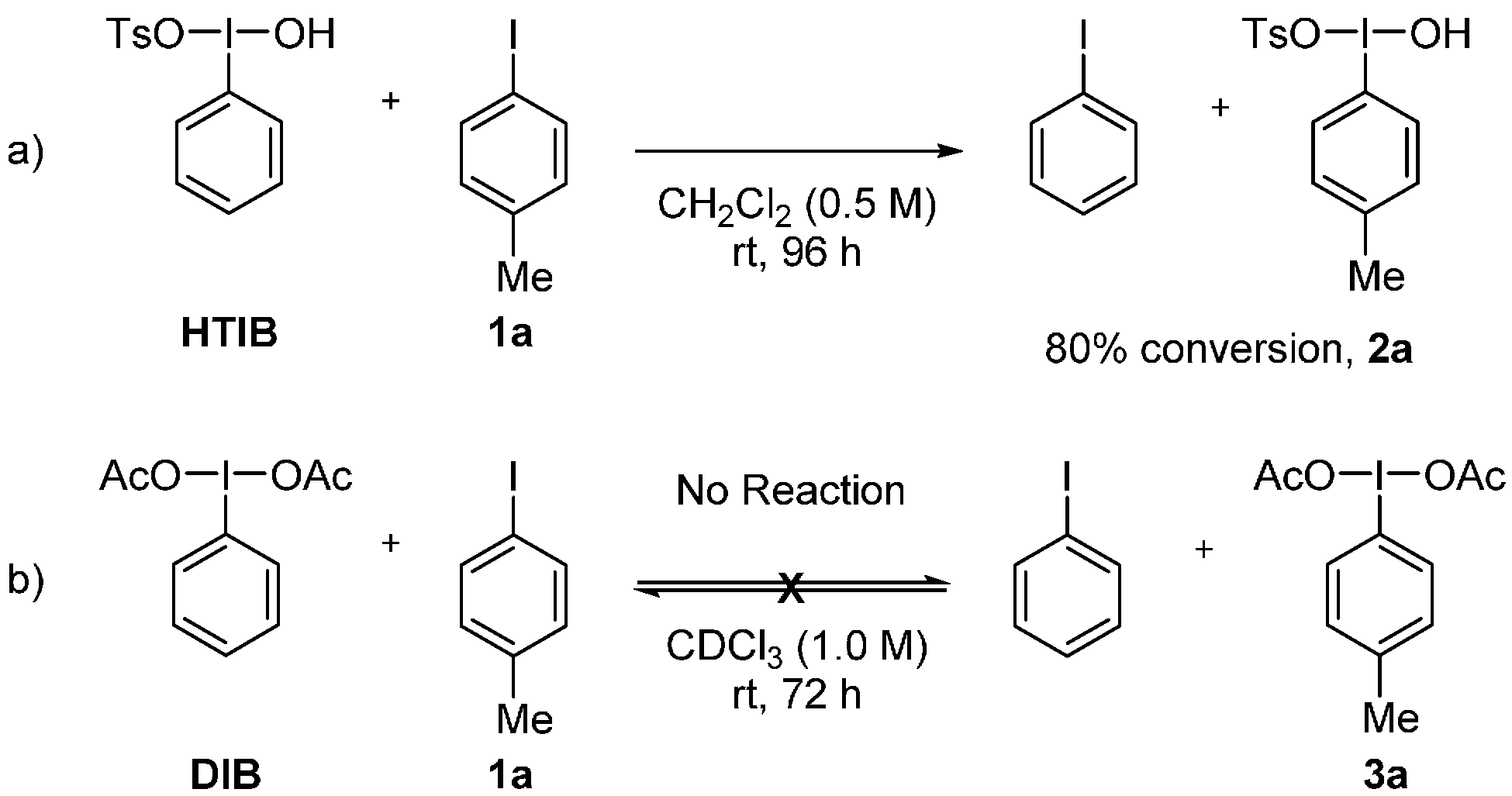
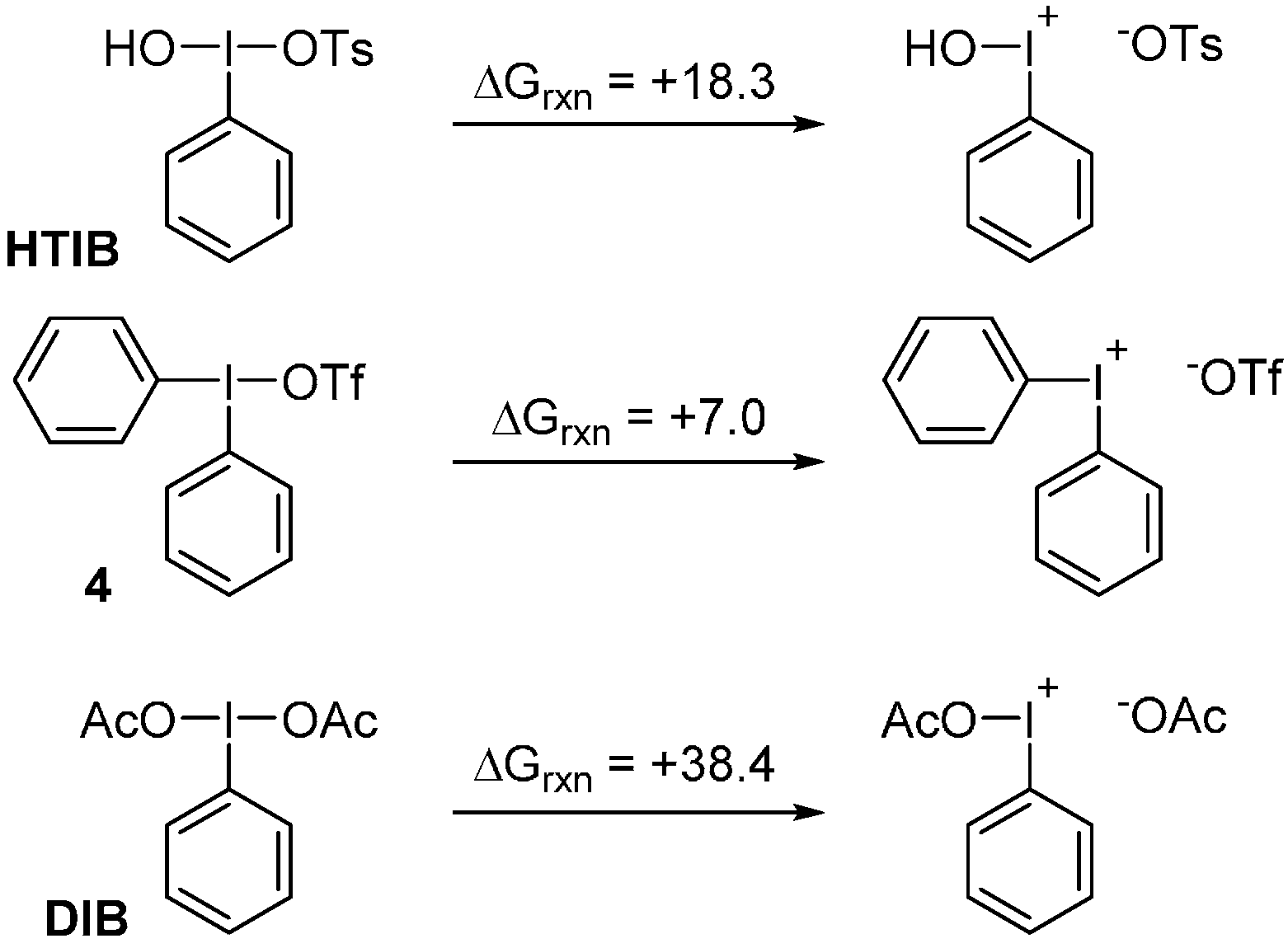
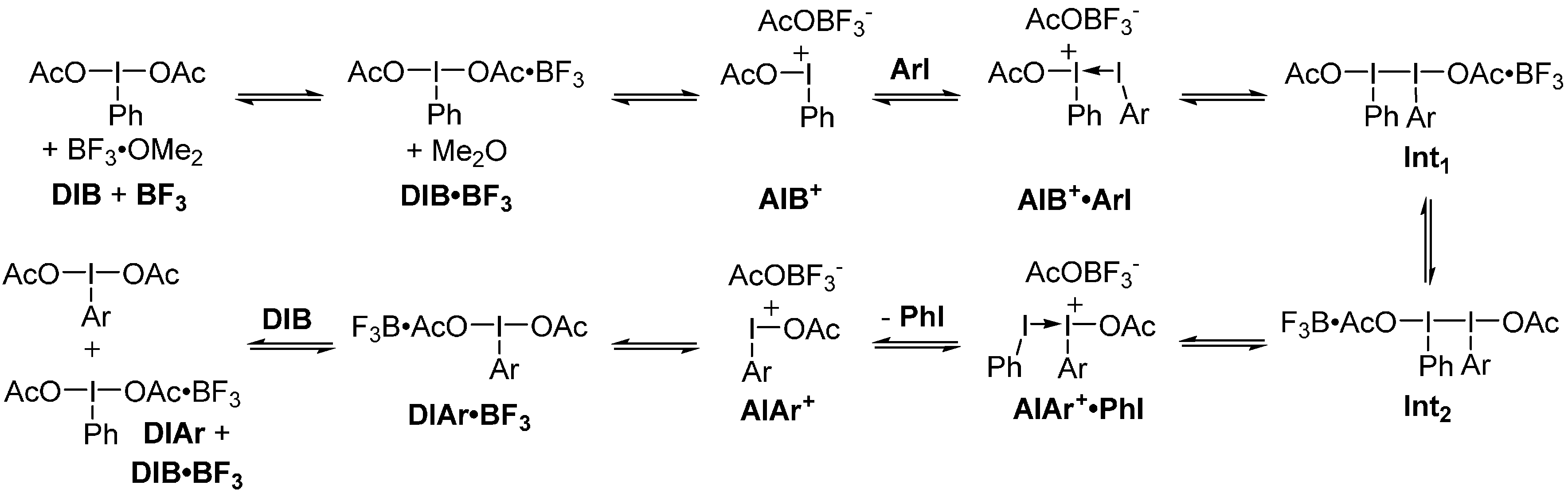
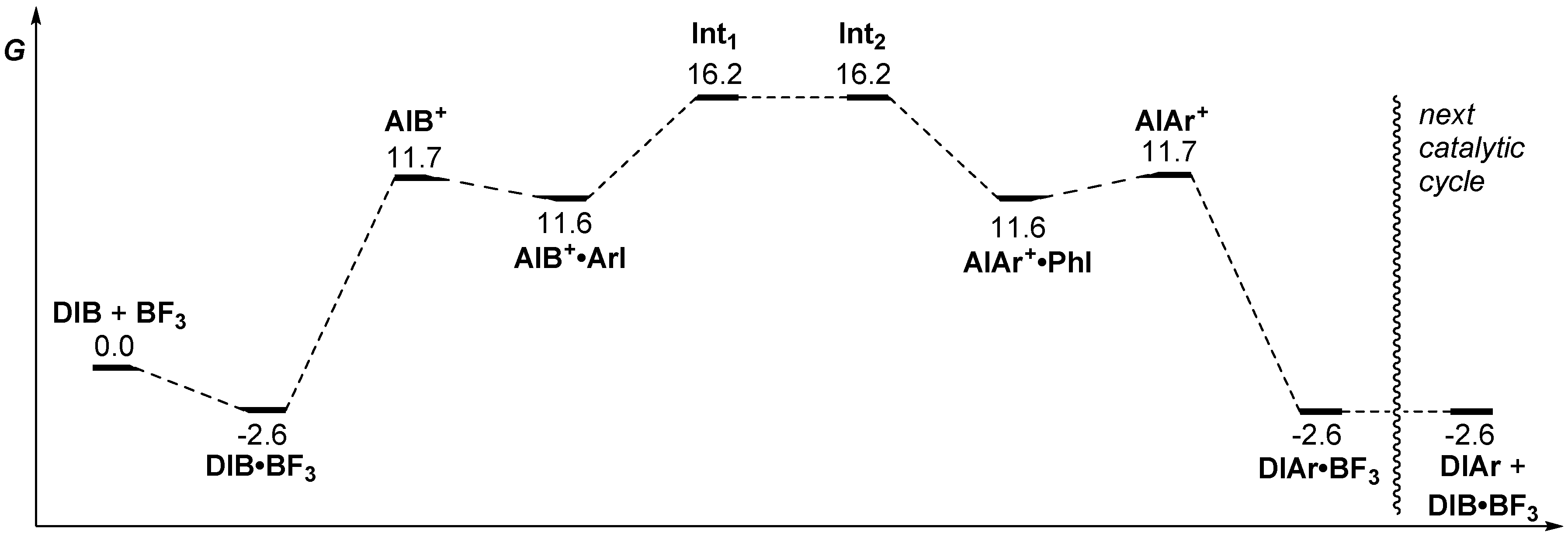
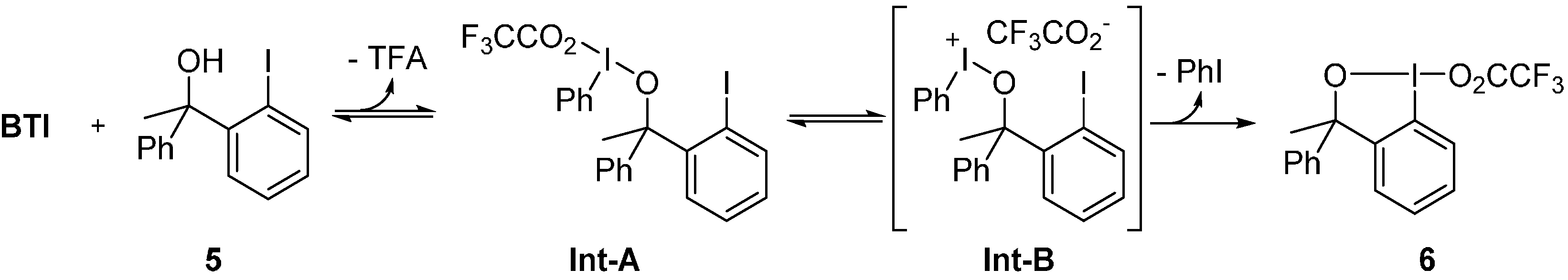
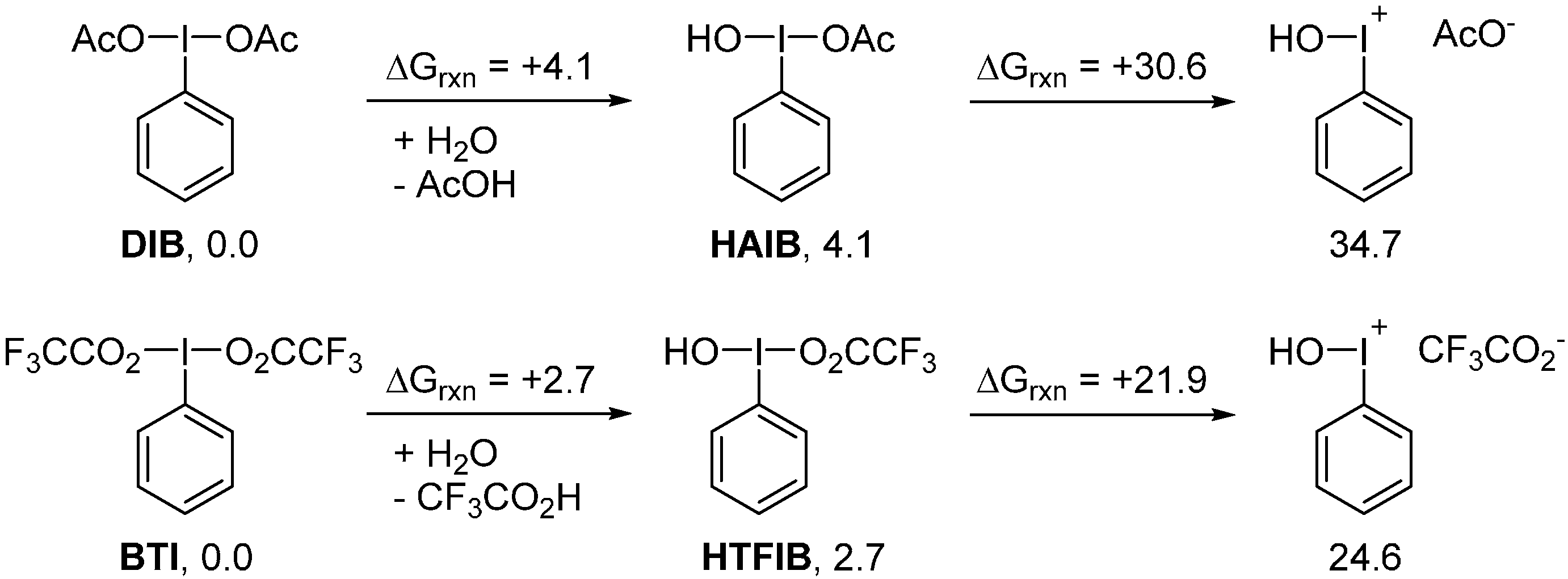
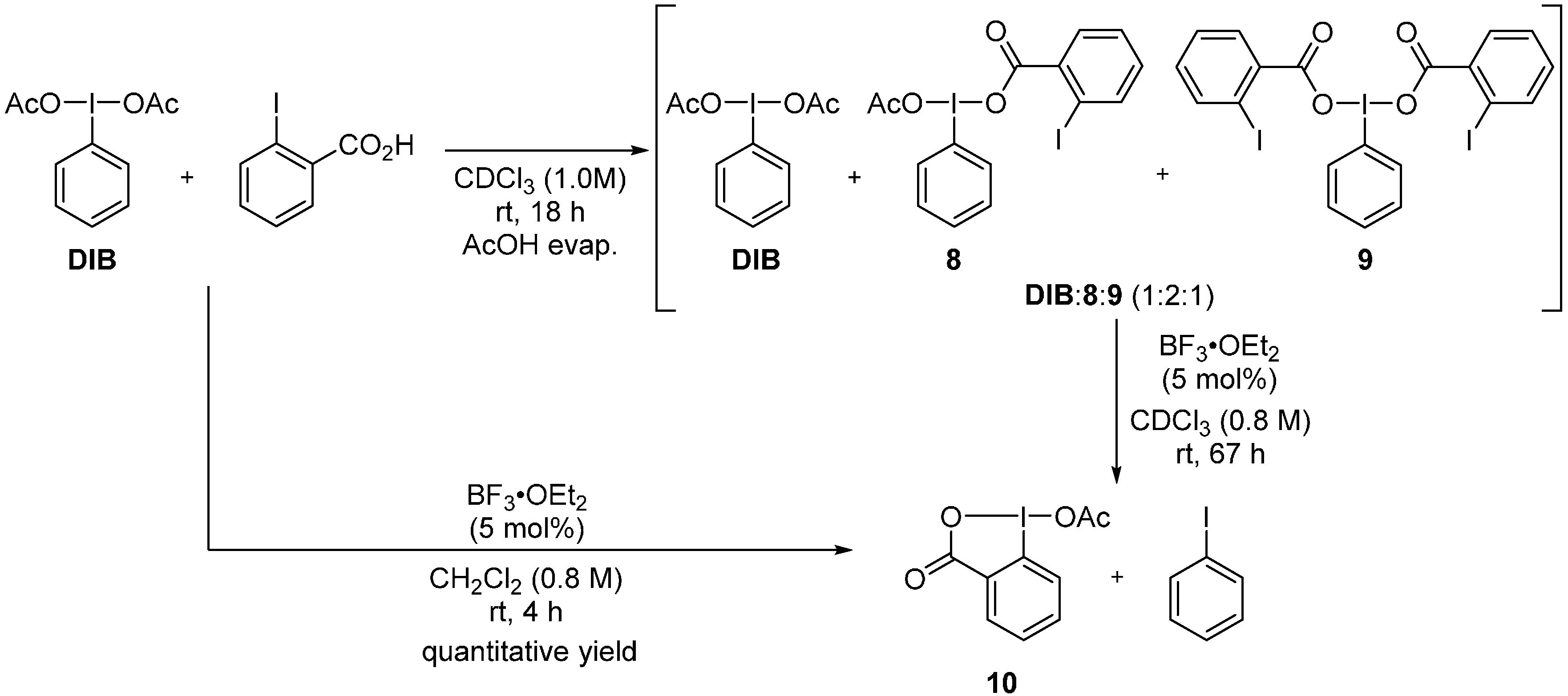
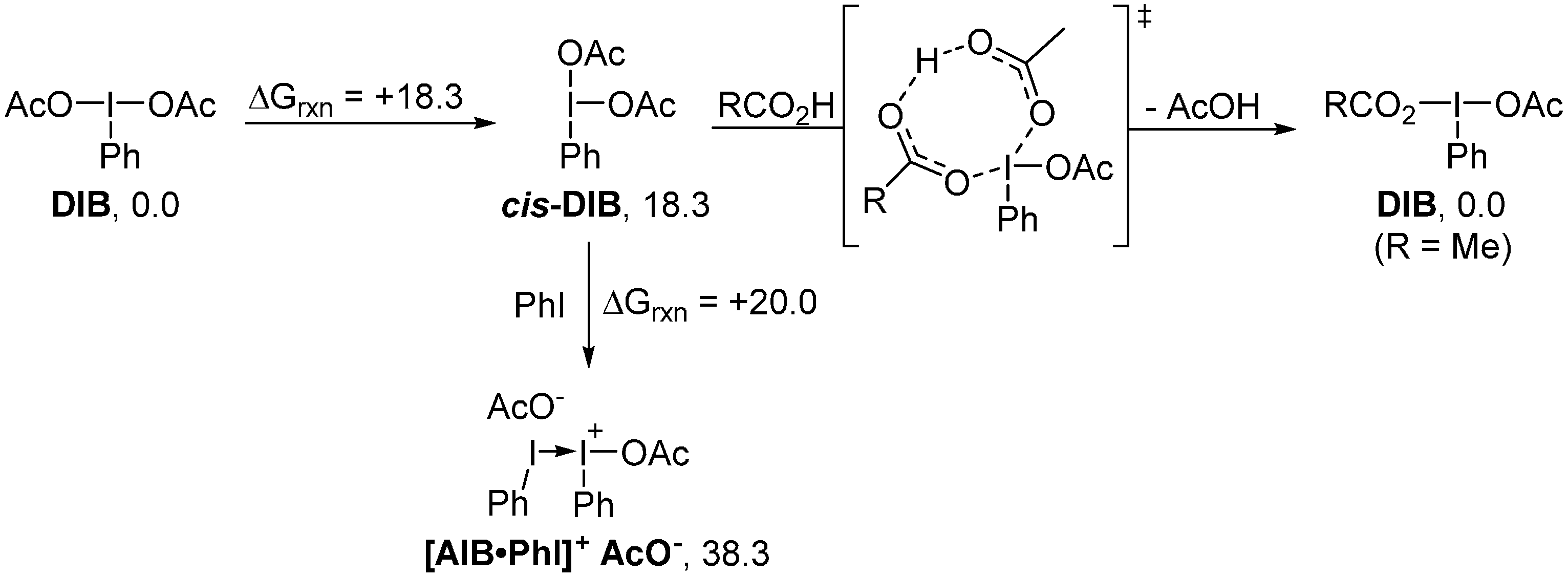
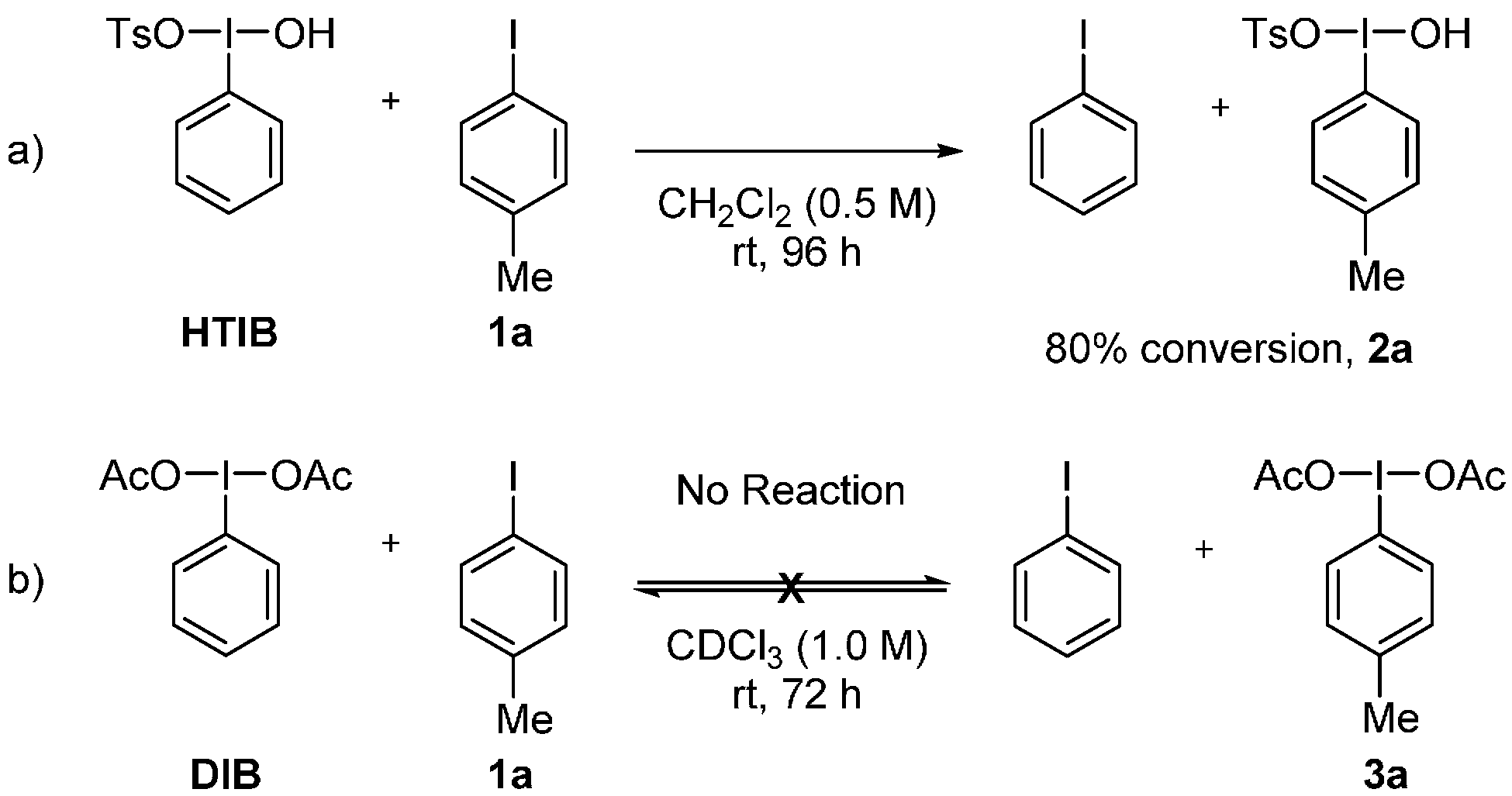
2. Results and Discussion
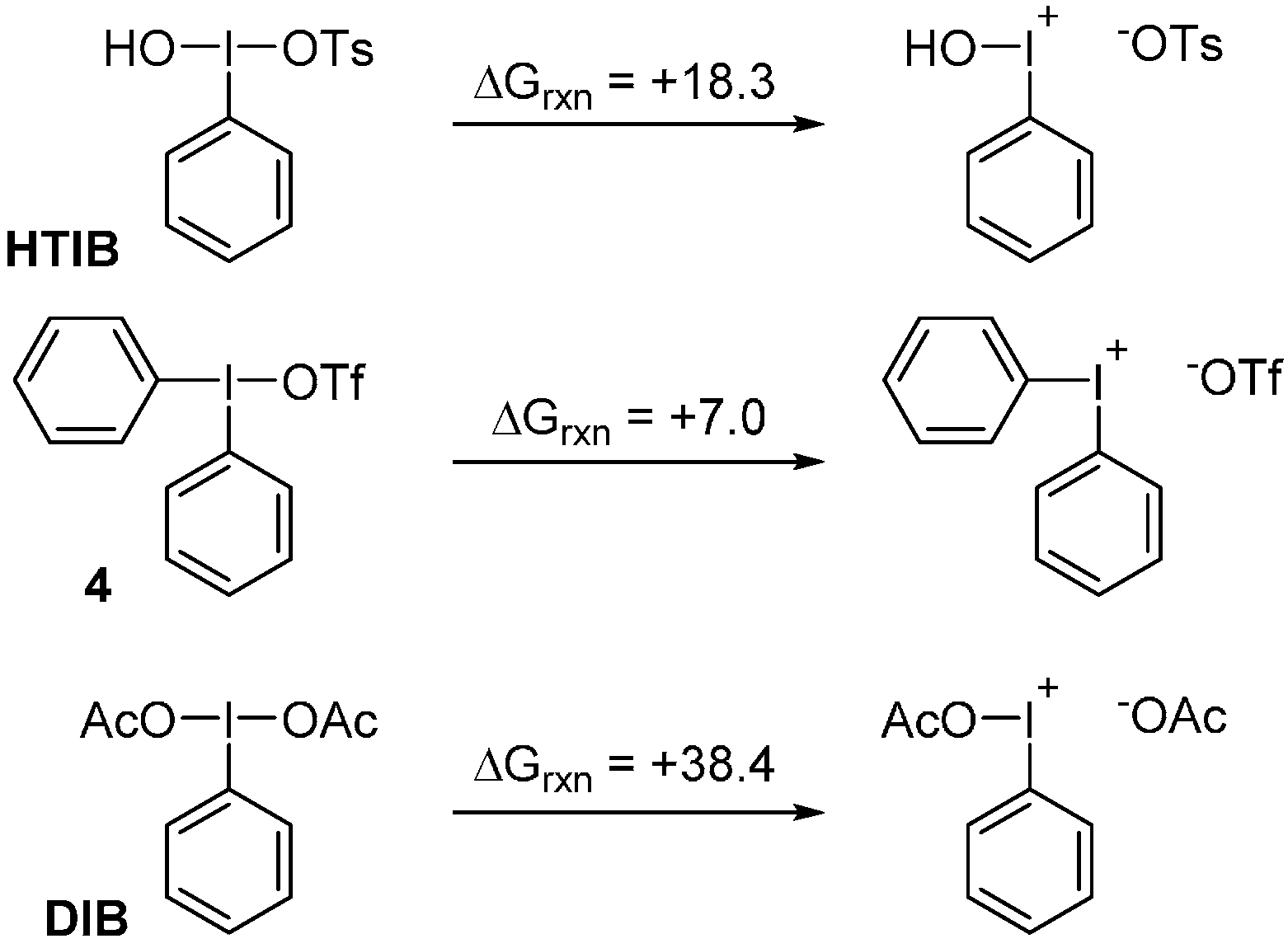
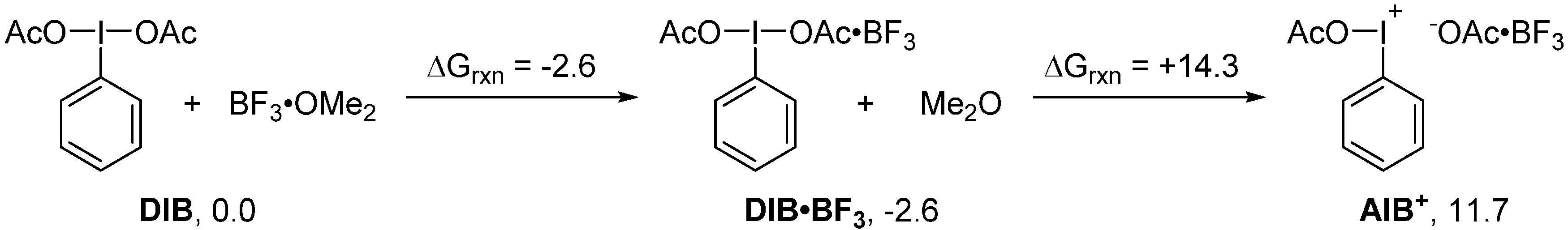
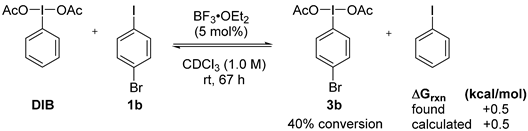
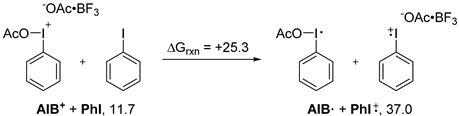
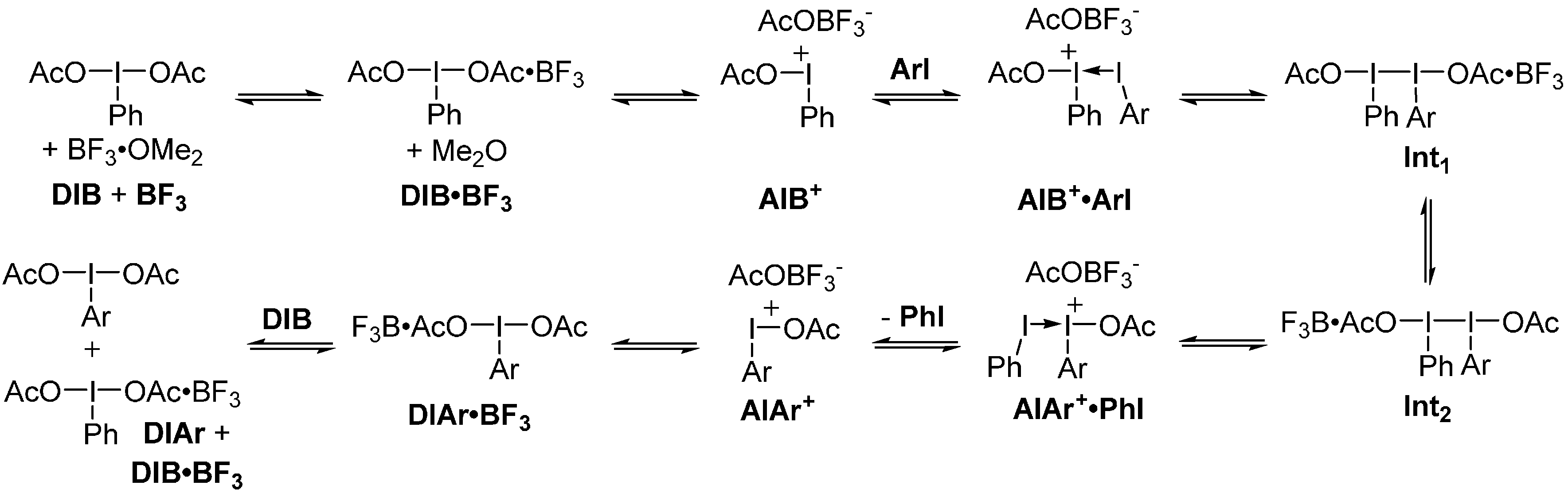

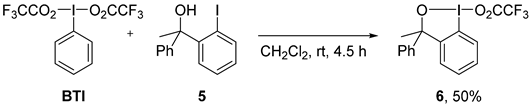
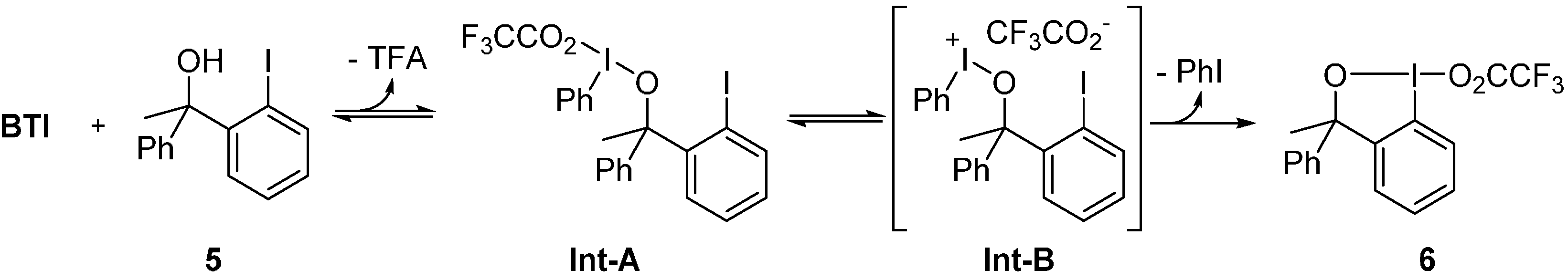
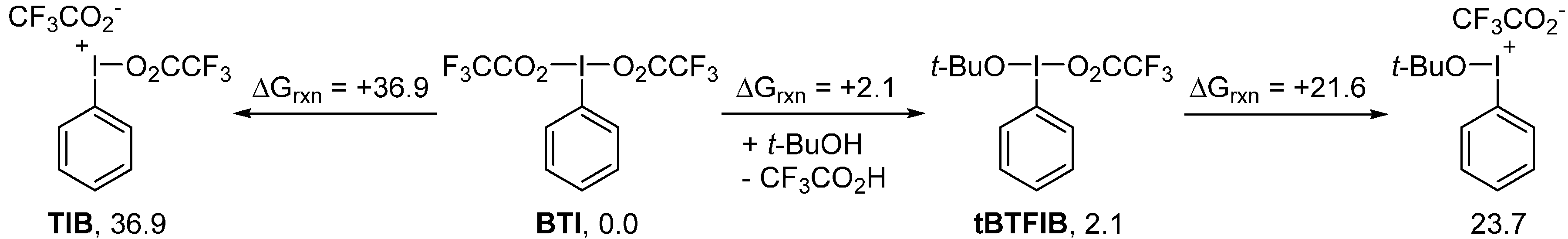
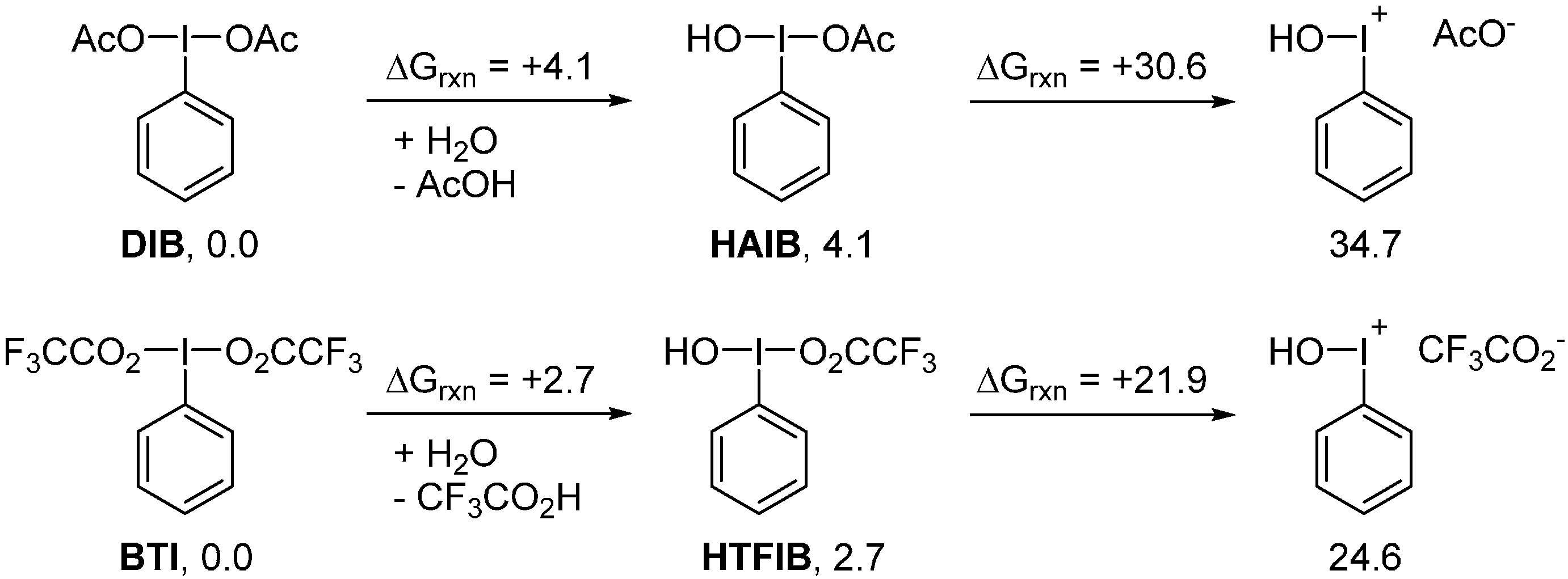
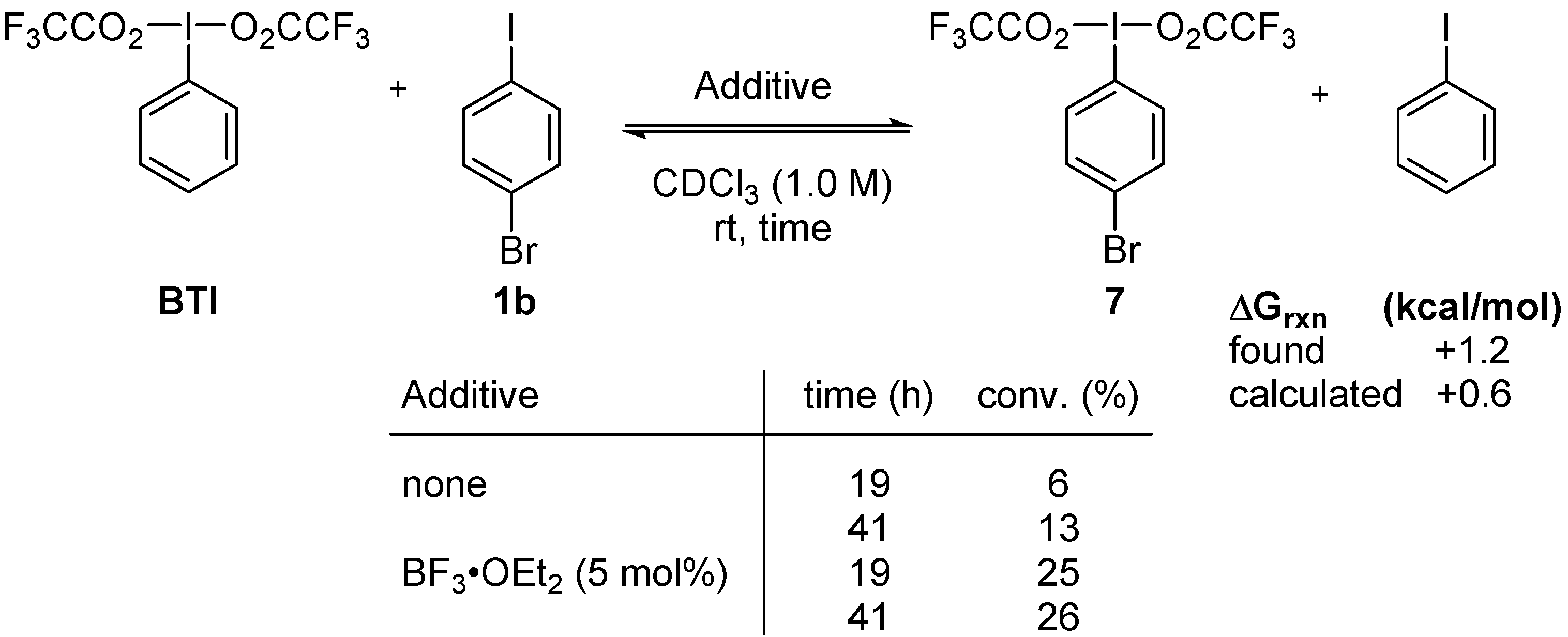
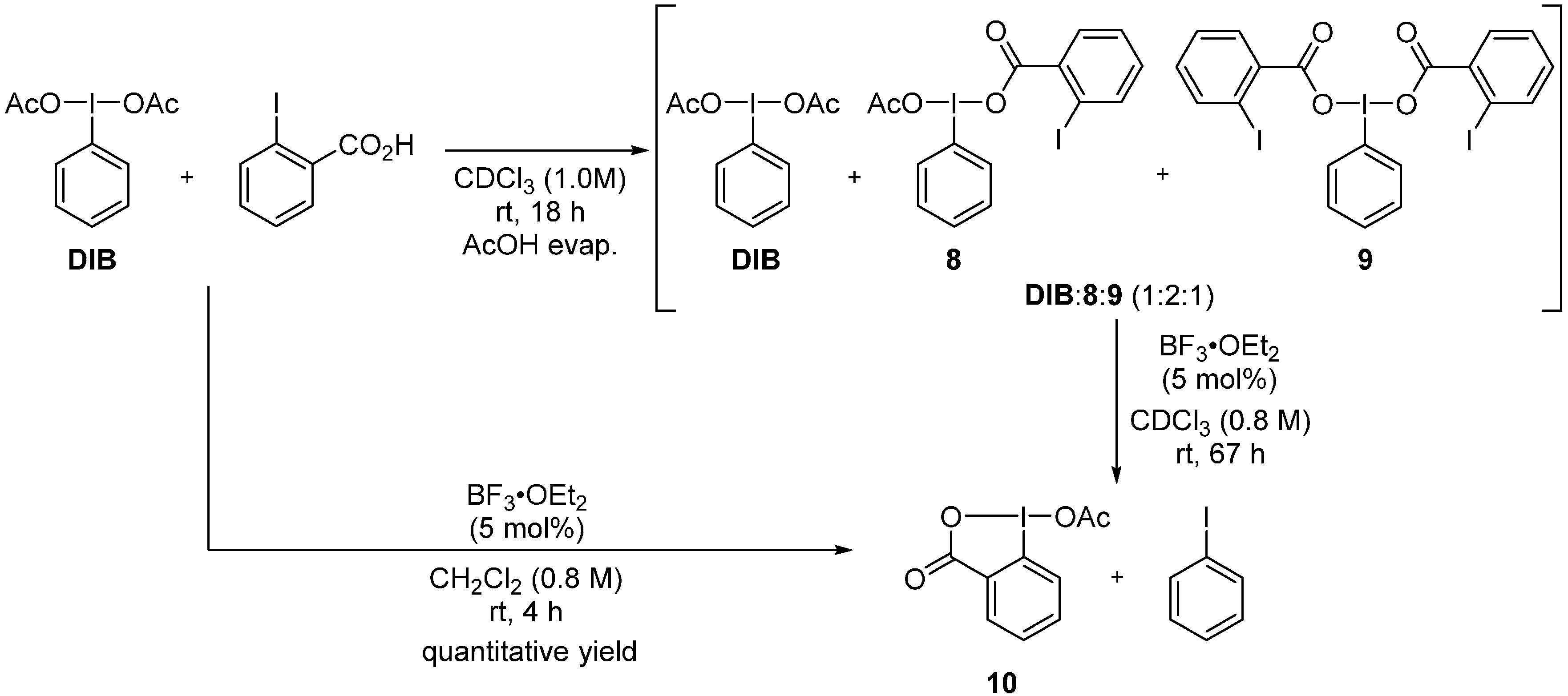
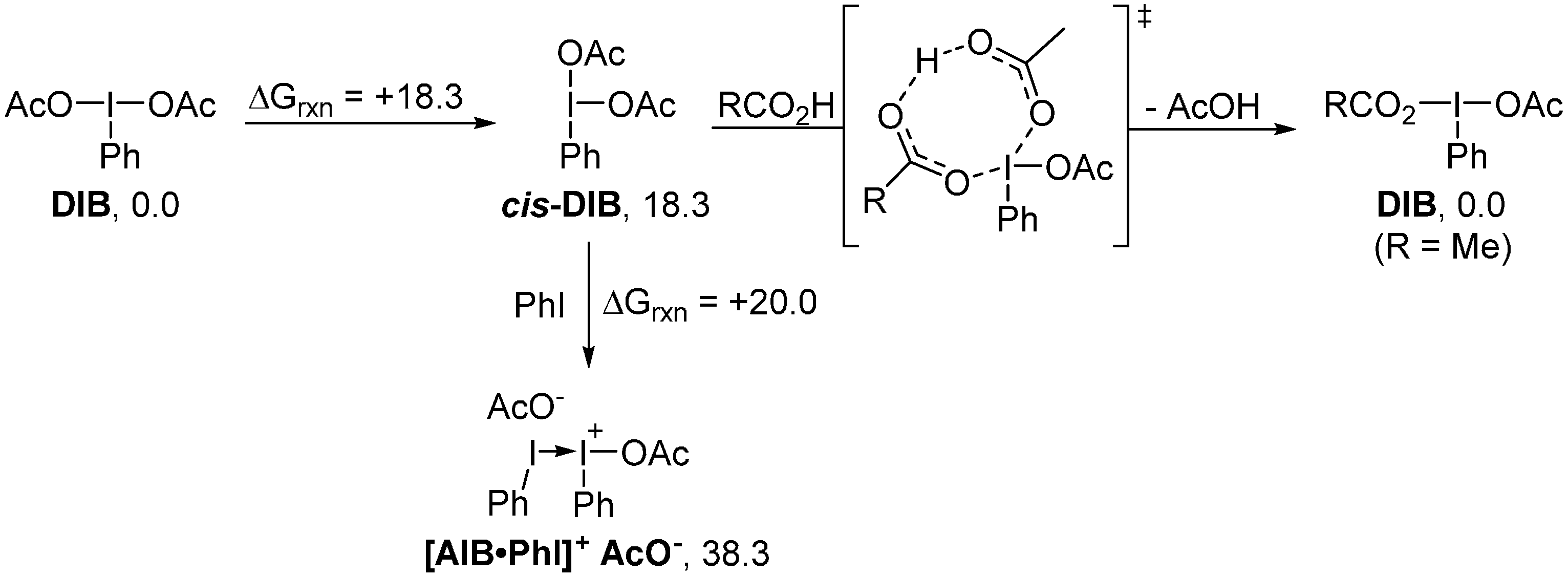
3. Experimental Section
3.1. General Information
3.2. Metathetical Redox Reaction of HTIB and p-Methyliodobenzene
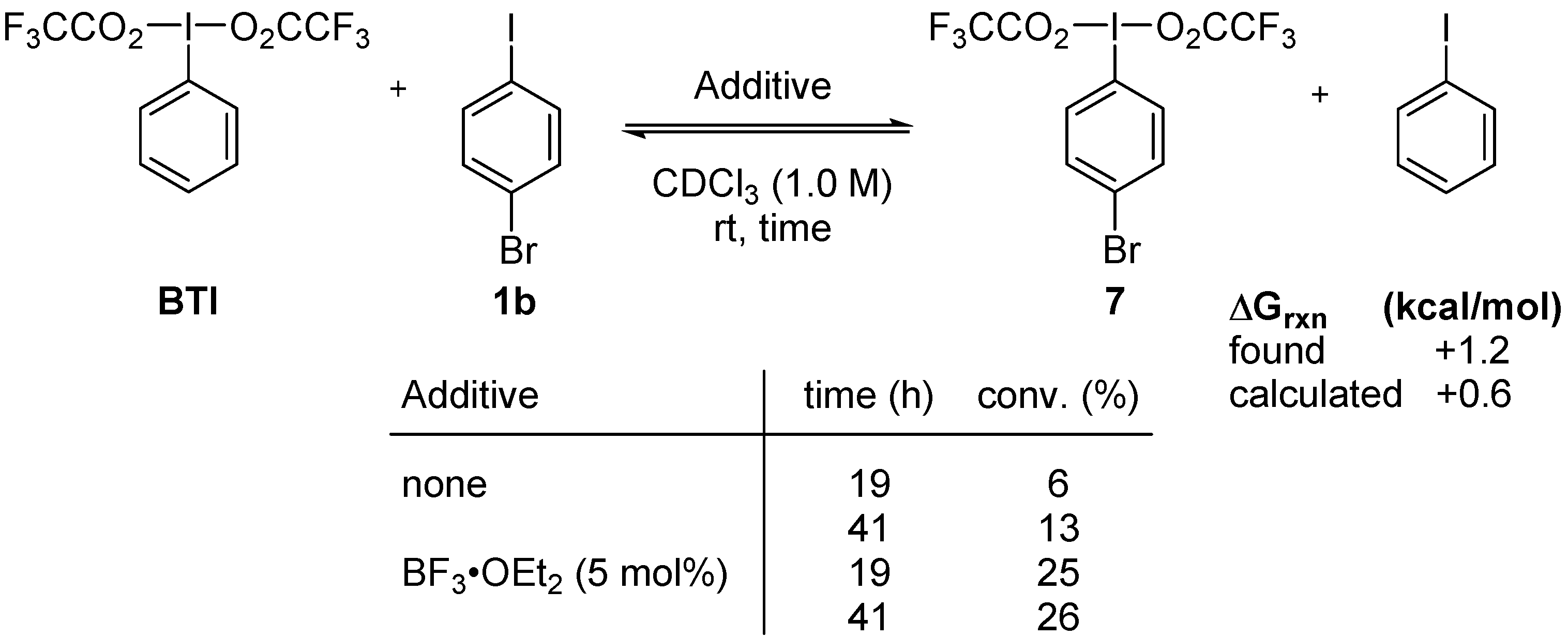
3.3. Lewis-Acid-Catalyzed Metathetical Redox Reaction of DIB and p-Bromoiodobenzene
3.4. One-Pot Metathetical Redox Reaction of DIB and o-Iodobenzoic Acid
4. Computational Details
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhdankin, V.V. Hypervalent. Iodine Chemistry; John Wiley & Sons: Chichester, UK, 2014. [Google Scholar]
- Wirth, T. Hypervalent. Iodine Chemistry; Springer: Berlin, Germany, 2003. [Google Scholar]
- Moriarty, R.M.; Prakash, O. Oxidation of phenolic compounds with organohypervalent iodine reagents. Org. React. 2001, 57, 327–415. [Google Scholar]
- Varvoglis, A. Hypervalent. Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Varvoglis, A. The Organic Chemistry of Polycoordinated Iodine; VCH: New York, NY, USA, 1992. [Google Scholar]
- Uyanik, M.; Ishihara, K. Hypervalent iodine-mediated oxidation of alcohols. Chem. Commun. 2009, 16, 2086–2099. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Stang, P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, R.M. Organohypervalent iodine: Development, applications, and future directions. J. Org. Chem. 2005, 70, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Stang, P.J. Recent developments in the chemistry of polyvalent Iodine compounds. Chem. Rev. 2002, 102, 2523–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V. Hypervalent iodine(III) reagents in organic synthesis. ARKIVOC 2009, 2009, 1–62. [Google Scholar]
- Varvoglis, A. Aryliodine(III) dicarboxylates. Chem. Soc. Rev. 1981, 10, 377–407. [Google Scholar] [CrossRef]
- Yusubov, M.S.; Wirth, T. Solvent-free reactions with hypervalent iodine reagents. Org. Lett. 2005, 7, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Togo, H. Facile One-pot preparation of [hydroxy(sulfonyloxy)iodo]arenes from iodoarenes with MCPBA in the Presence of Sulfonic Acids. Synlett 2005, 2486–2488. [Google Scholar] [CrossRef]
- Sharefkin, J.G.; Saltzman, H. Iodosobenzene diacetate. Org. Synth. 1963, 43, 62. [Google Scholar]
- Koser, G.F.; Wettach, R.H. [Hydroxy(tosyloxy)iodo]benzene, a versatile reagent for the mild oxidation of aryl iodides at the iodine atom by ligand transfer. J. Org. Chem. 1980, 45, 1542–1543. [Google Scholar] [CrossRef]
- Rabah, G.A.; Koser, G.F. Facile synthetic entry into the 1,3-dihydro-3-methyl-3-phenyl-1,2-benziodoxole family of λ3-iodanes. Tetrahedron Lett. 1996, 37, 6453–6456. [Google Scholar] [CrossRef]
- Geary, G.C.; Hope, E.G.; Singh, K.; Stuart, A.M. Electrophilic fluorination using a hypervalent iodine reagent derived from fluoride. Chem. Commun. 2013, 49, 9263–9265. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Jang, Y.J.; Racicot, L.; Panagopoulos, D.; Liang, S.H.; Ciufolini, M.A. Iodonium metathesis reactions. Angew. Chem. Int. Ed. 2014, 53, 9637–9639. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, S.; Legault, C.Y. Mechanistic insights on the iodine(III)-mediated α-oxidation of ketones. Chem. Eur. J. 2015, 21, 11206–11211. [Google Scholar] [CrossRef] [PubMed]
- Koser, G.F. [Hydroxy(tosyloxy)iodo]benzene and closely related iodanes: The second stage of development. Aldrichimica Acta 2001, 34, 89–102. [Google Scholar] [CrossRef]
- Richter, H.W.; Cherry, B.R.; Zook, T.D.; Koser, G.F. Characterization of species present in aqueous solutions of [hydroxy(mesyloxy)iodo]benzene and [hydroxy(tosyloxy)iodo]benzene. J. Am. Chem. Soc. 1997, 119, 9614–9623. [Google Scholar] [CrossRef]
- Ochiai, M.; Sueda, T.; Miyamoto, K.; Kiprof, P.; Zhdankin, V.V. Trans Influences on hypervalent bonding of aryl λ3-iodanes: Their stabilities and isodesmic reactions of benziodoxolones and benziodazolones. Angew. Chem. Int. Ed. 2006, 118, 8383–8386. [Google Scholar] [CrossRef]
- Zhong, W.; Yang, J.; Meng, X.; Li, Z. BF3·OEt2-Promoted diastereoselective diacetoxylation of alkenes by PhI(OAc)2. J. Org. Chem. 2011, 76, 9997–10004. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. Iodobenzene-catalyzed α-acetoxylation of ketones. In situ generation of hypervalent (diacyloxyiodo)benzenes using m-chloroperbenzoic acid. J. Am. Chem. Soc. 2005, 127, 12244–12245. [Google Scholar] [CrossRef] [PubMed]
- Waser, J.; Vita, M.V. Azidation of β-keto esters and silyl enol ethers with a benziodoxole reagent. Org. Lett. 2013, 15, 3246–3249. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—consistent molecular orbital methods. XII. Further extensions of gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for P-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jobin-Des Lauriers, A.; Legault, C.Y. Metathetical Redox Reaction of (Diacetoxyiodo)arenes and Iodoarenes. Molecules 2015, 20, 22635-22644. https://doi.org/10.3390/molecules201219874
Jobin-Des Lauriers A, Legault CY. Metathetical Redox Reaction of (Diacetoxyiodo)arenes and Iodoarenes. Molecules. 2015; 20(12):22635-22644. https://doi.org/10.3390/molecules201219874
Chicago/Turabian StyleJobin-Des Lauriers, Antoine, and Claude Y. Legault. 2015. "Metathetical Redox Reaction of (Diacetoxyiodo)arenes and Iodoarenes" Molecules 20, no. 12: 22635-22644. https://doi.org/10.3390/molecules201219874
APA StyleJobin-Des Lauriers, A., & Legault, C. Y. (2015). Metathetical Redox Reaction of (Diacetoxyiodo)arenes and Iodoarenes. Molecules, 20(12), 22635-22644. https://doi.org/10.3390/molecules201219874