
Synthesis and Chemistry of Organic Geminal Di- and Triazides

Abstract
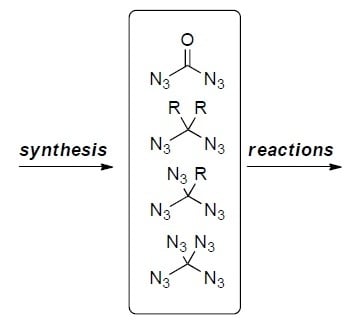
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
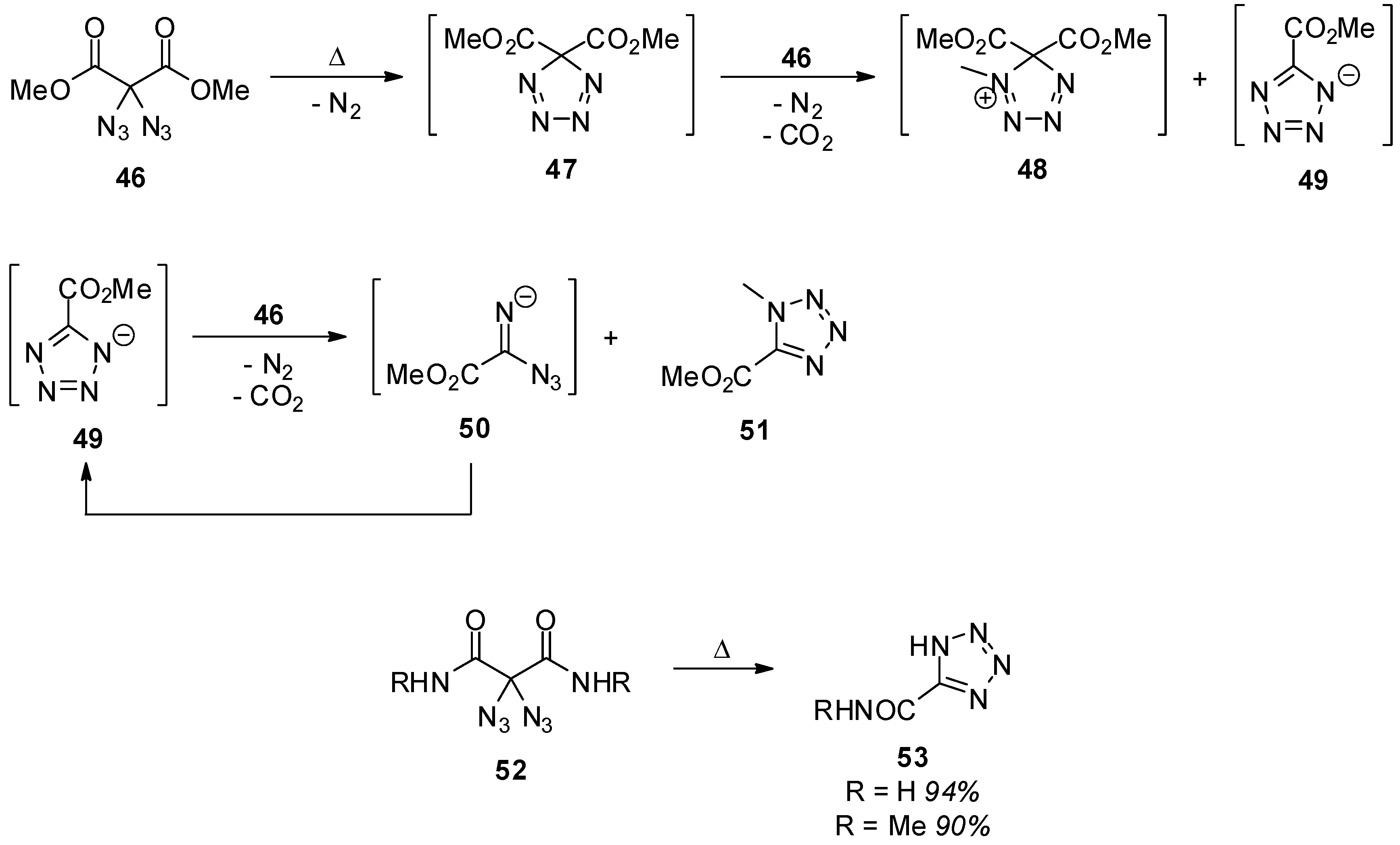{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
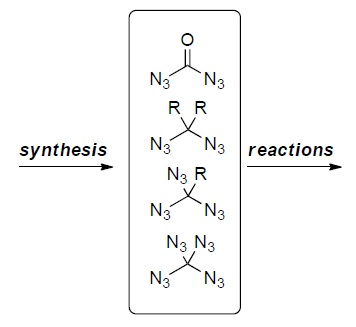
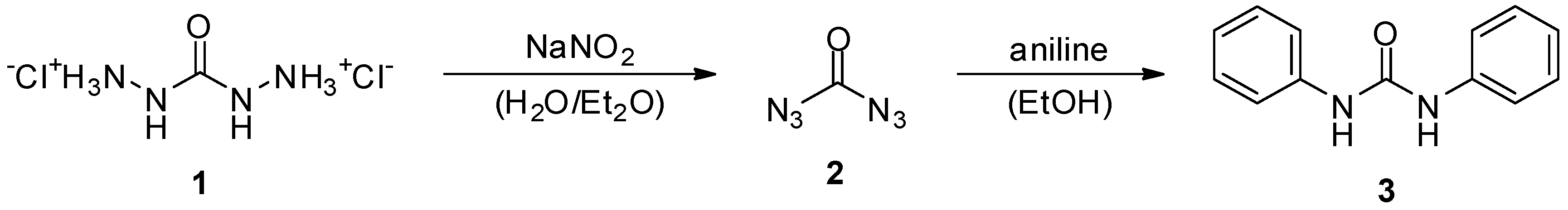
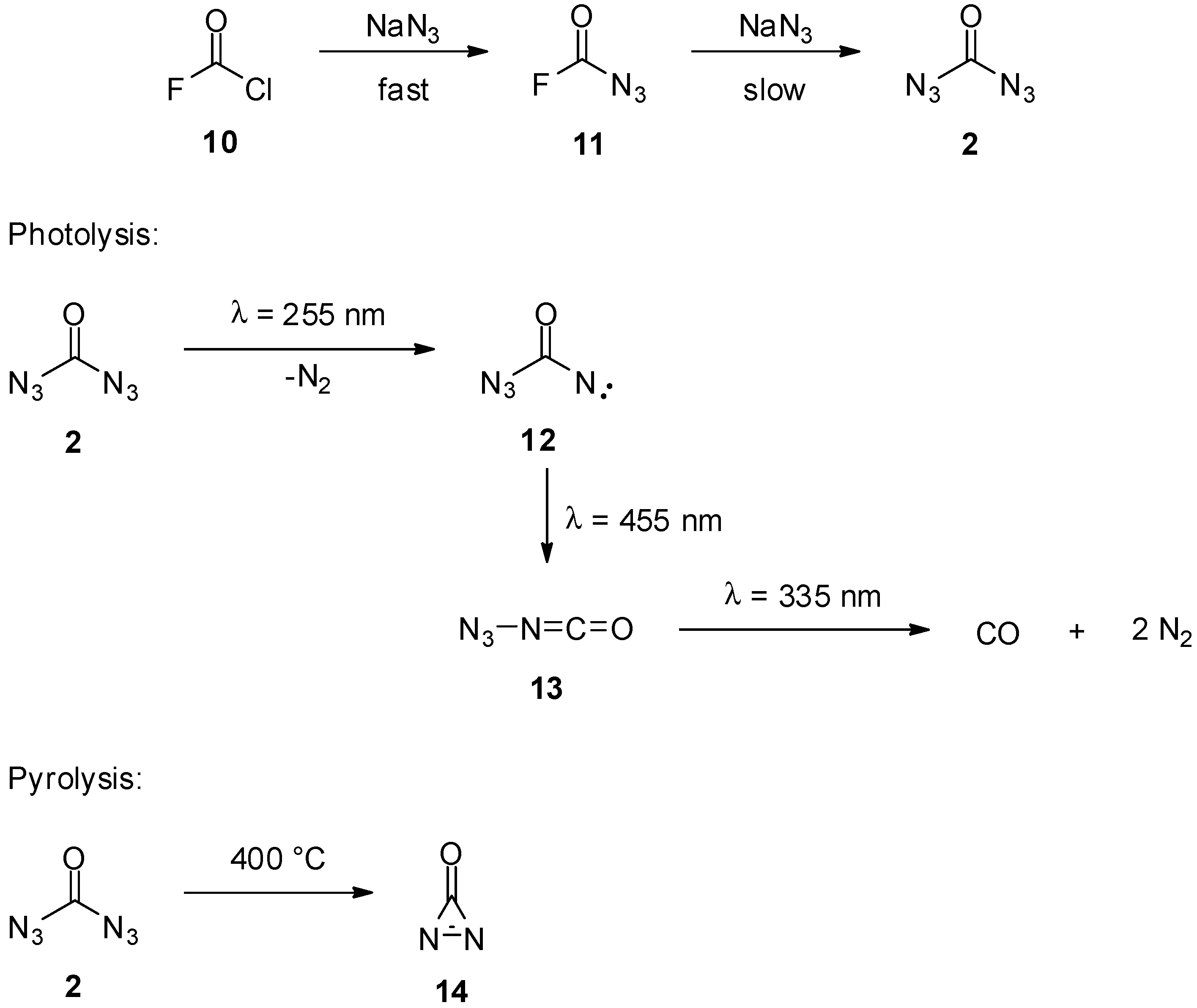
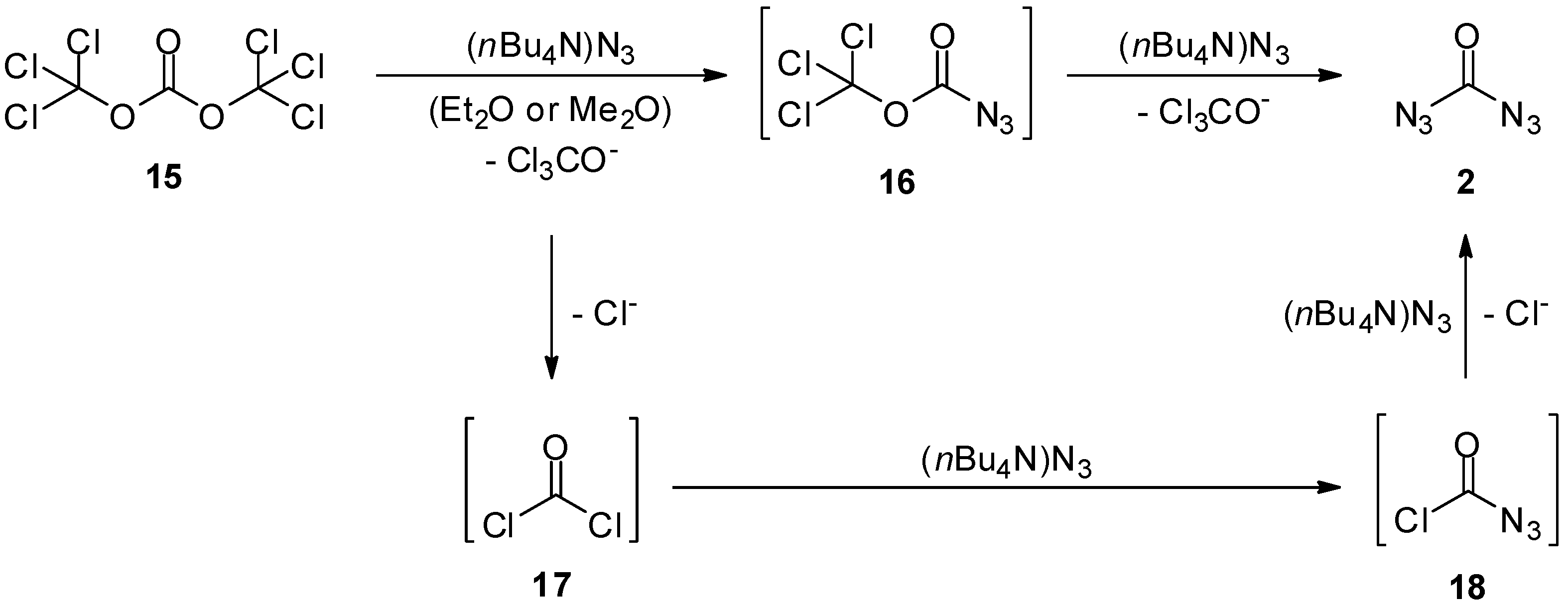
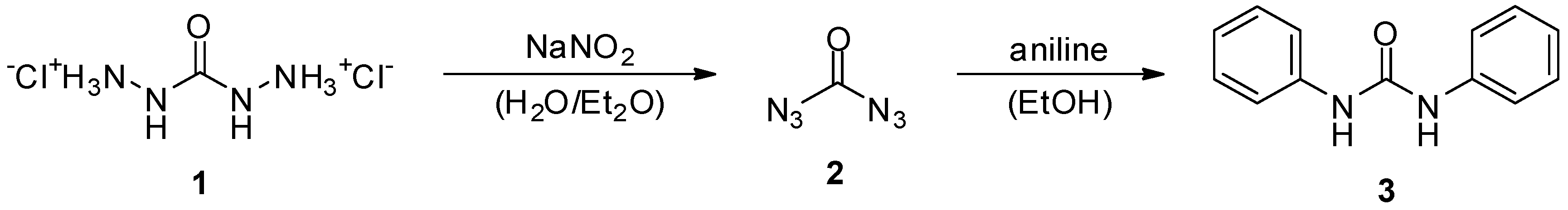
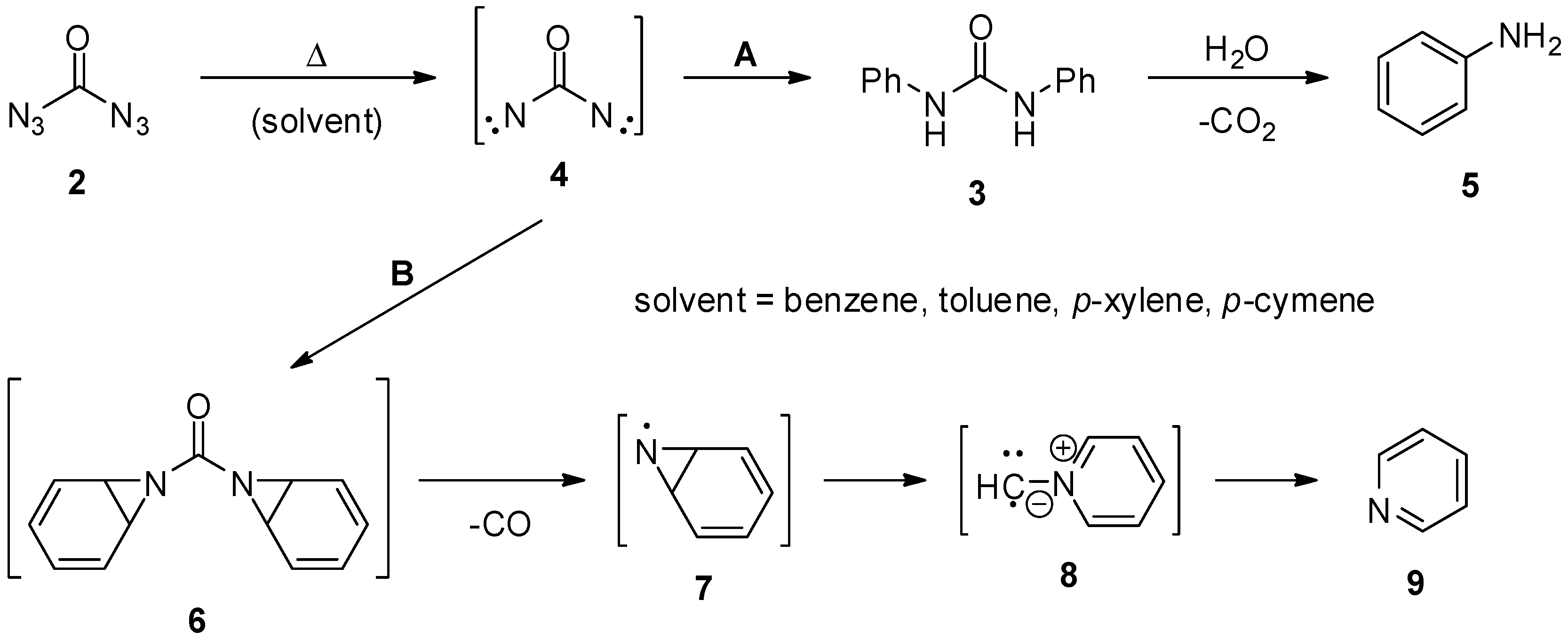
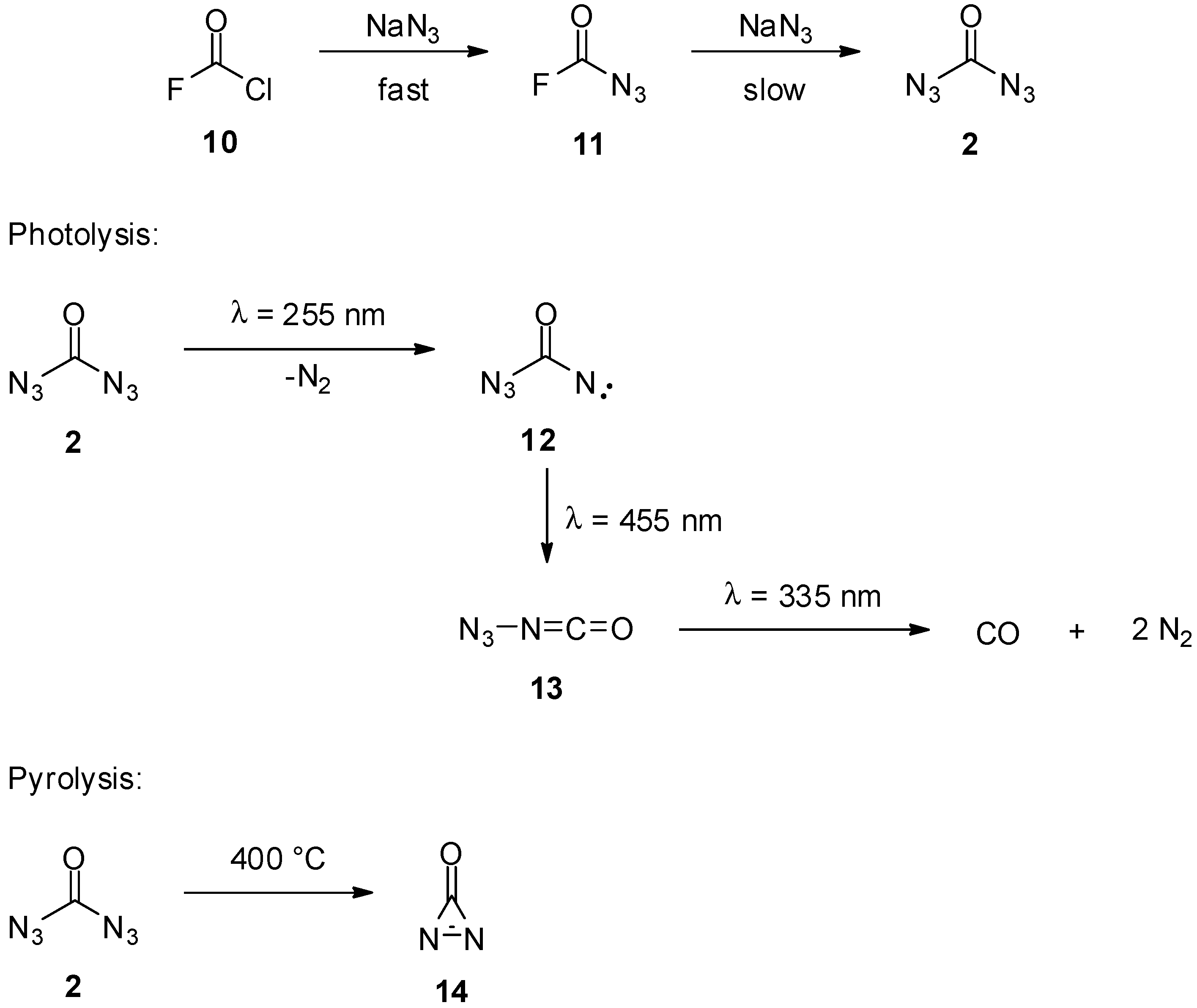
2. Carbonyl Diazide

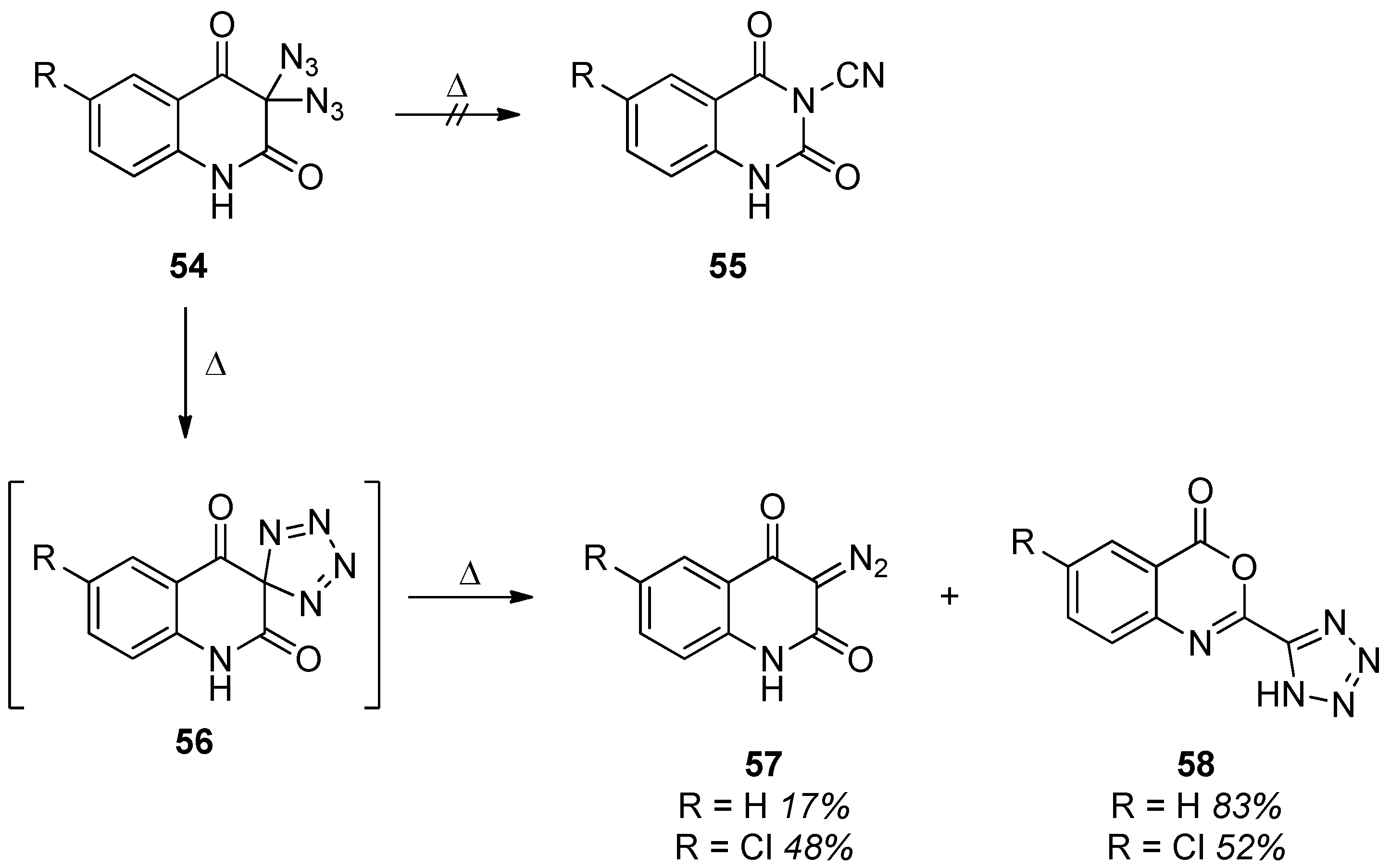
3. Geminal Diazides
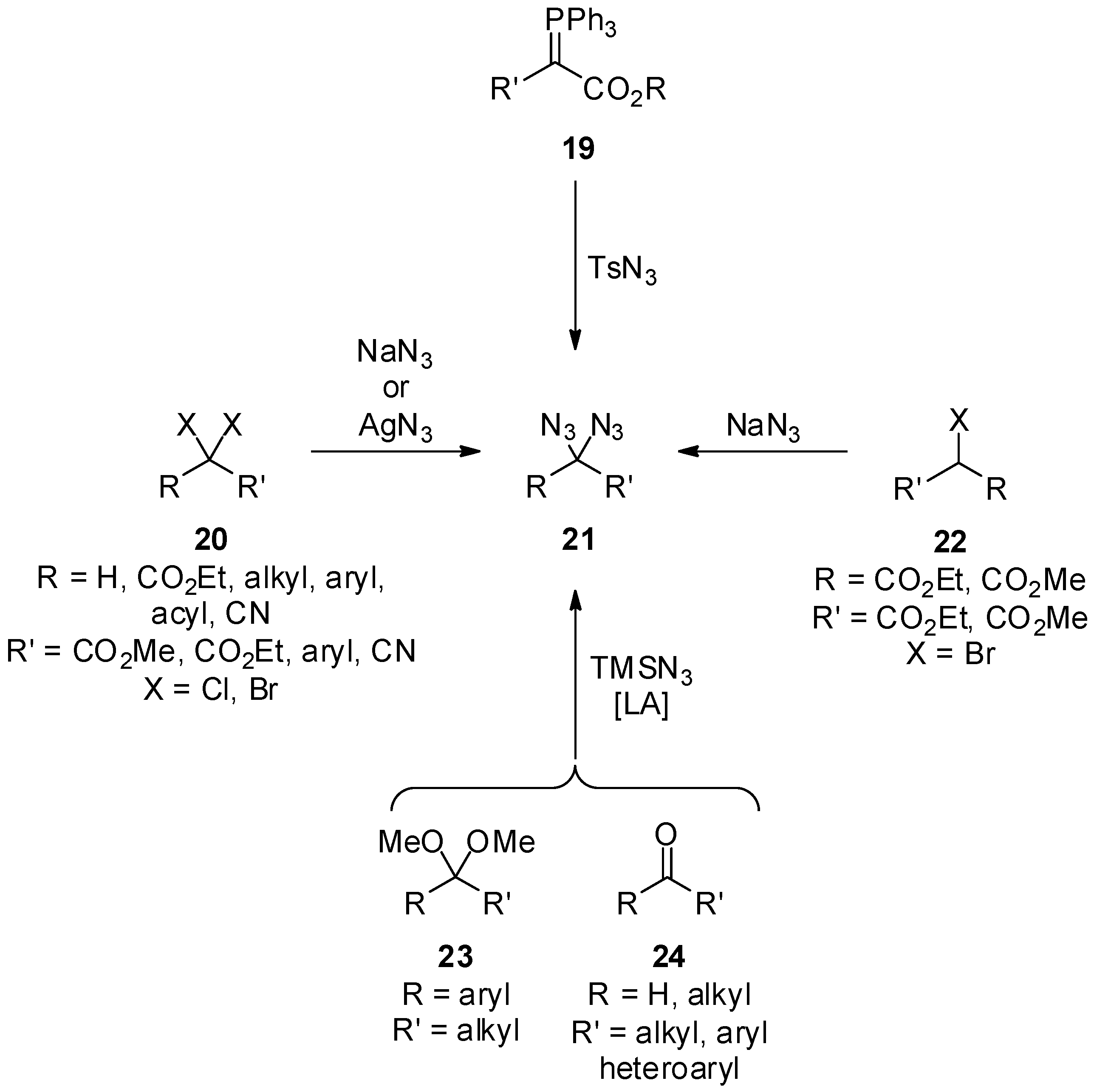
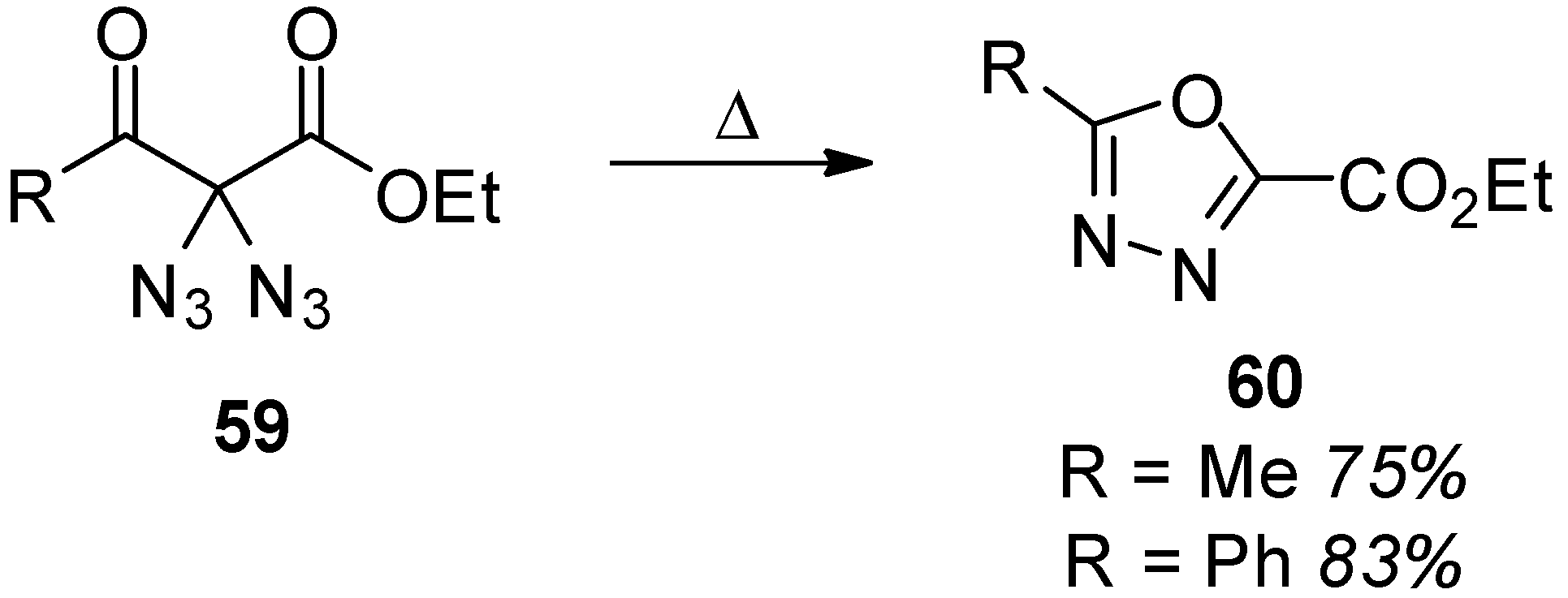
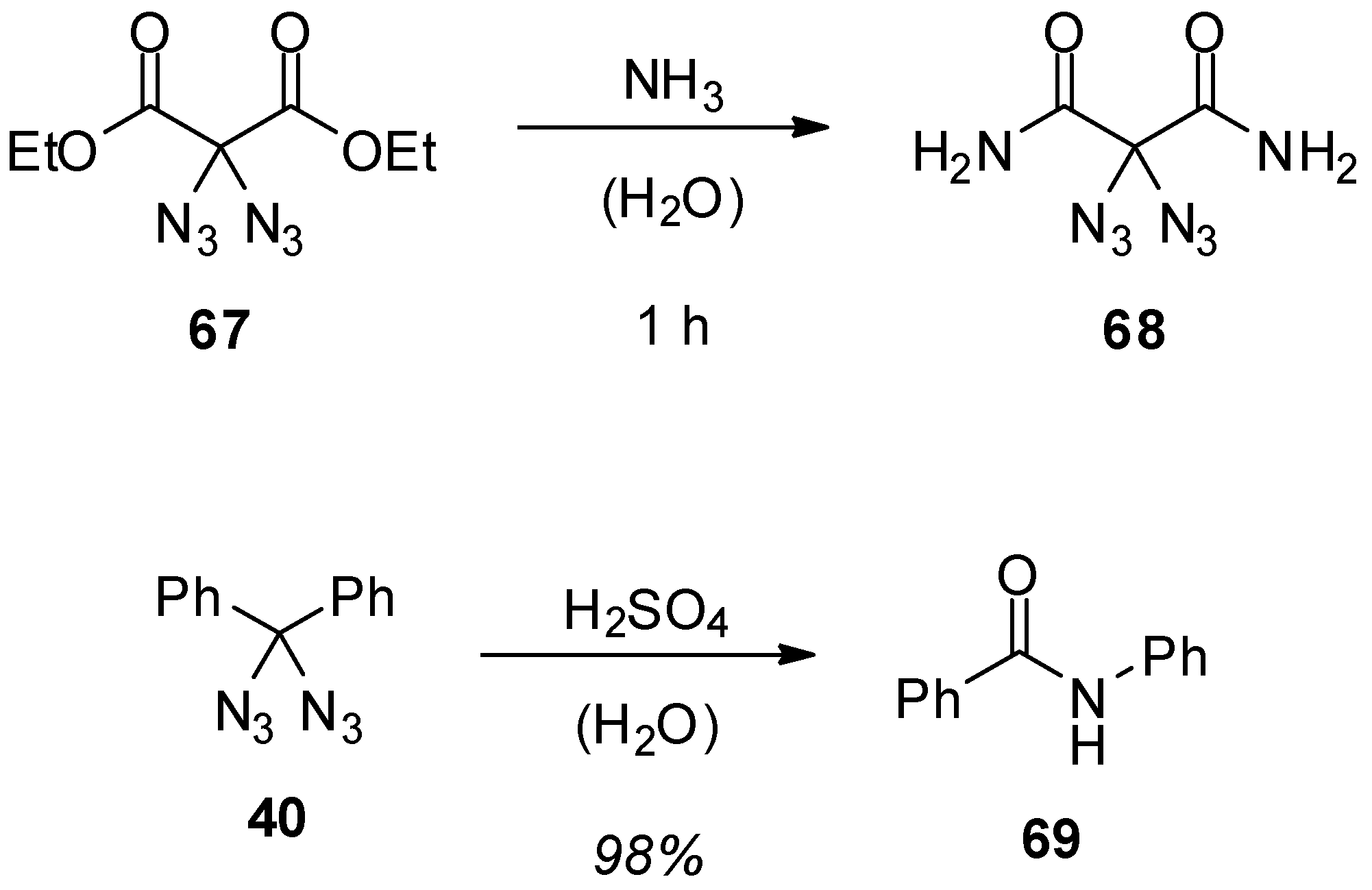
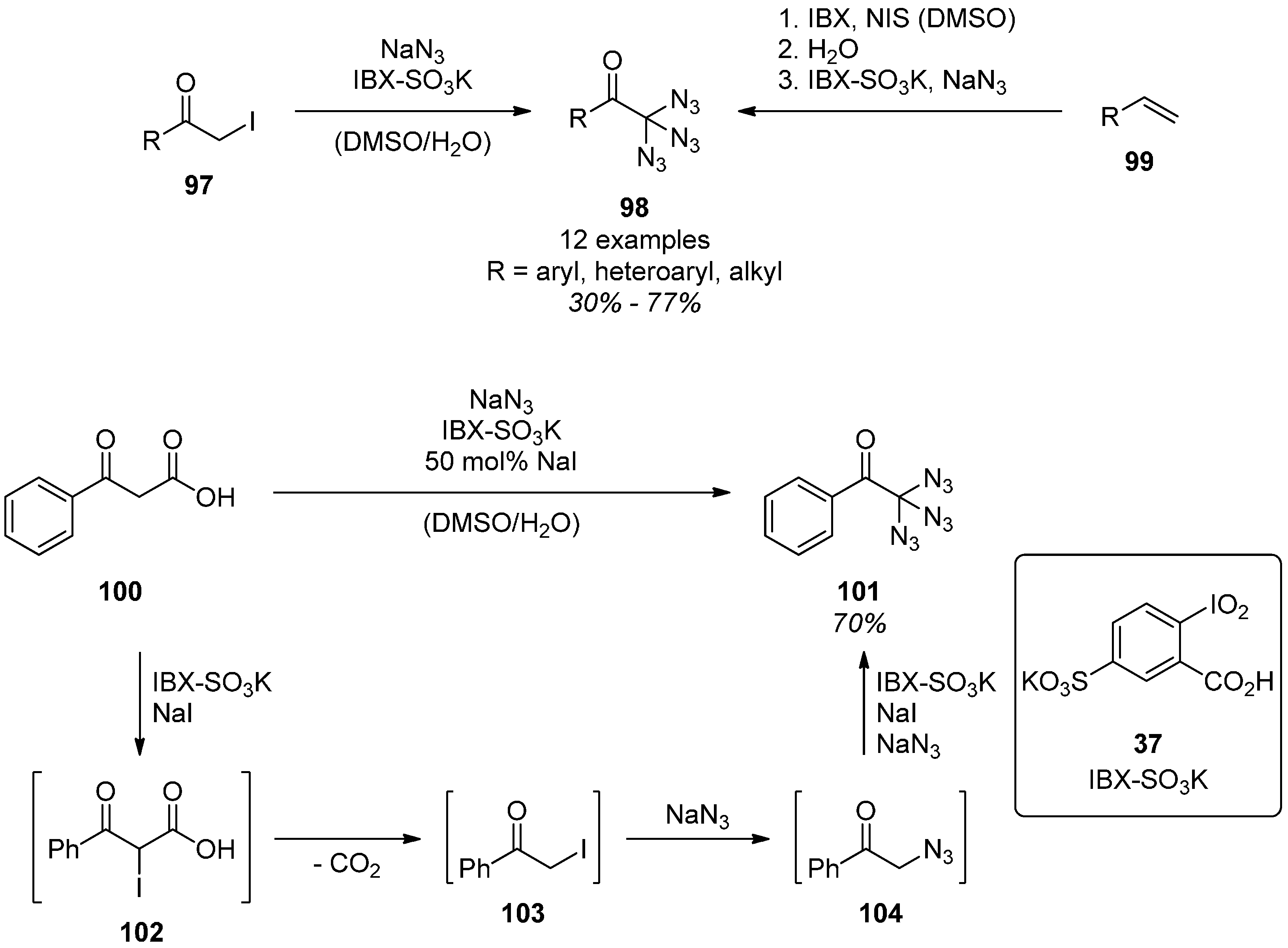
3.1. Geminal Aliphatic Diazides
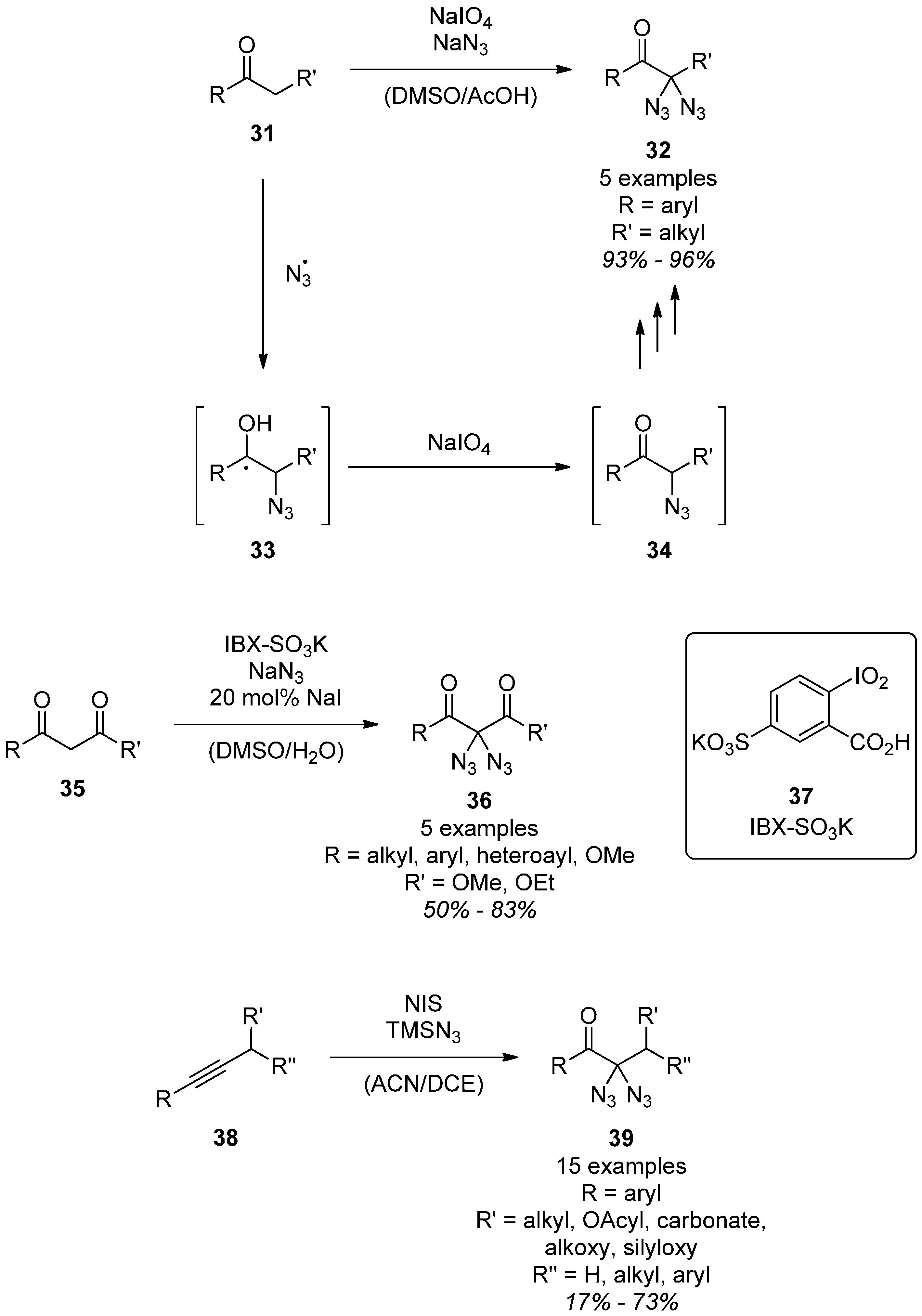
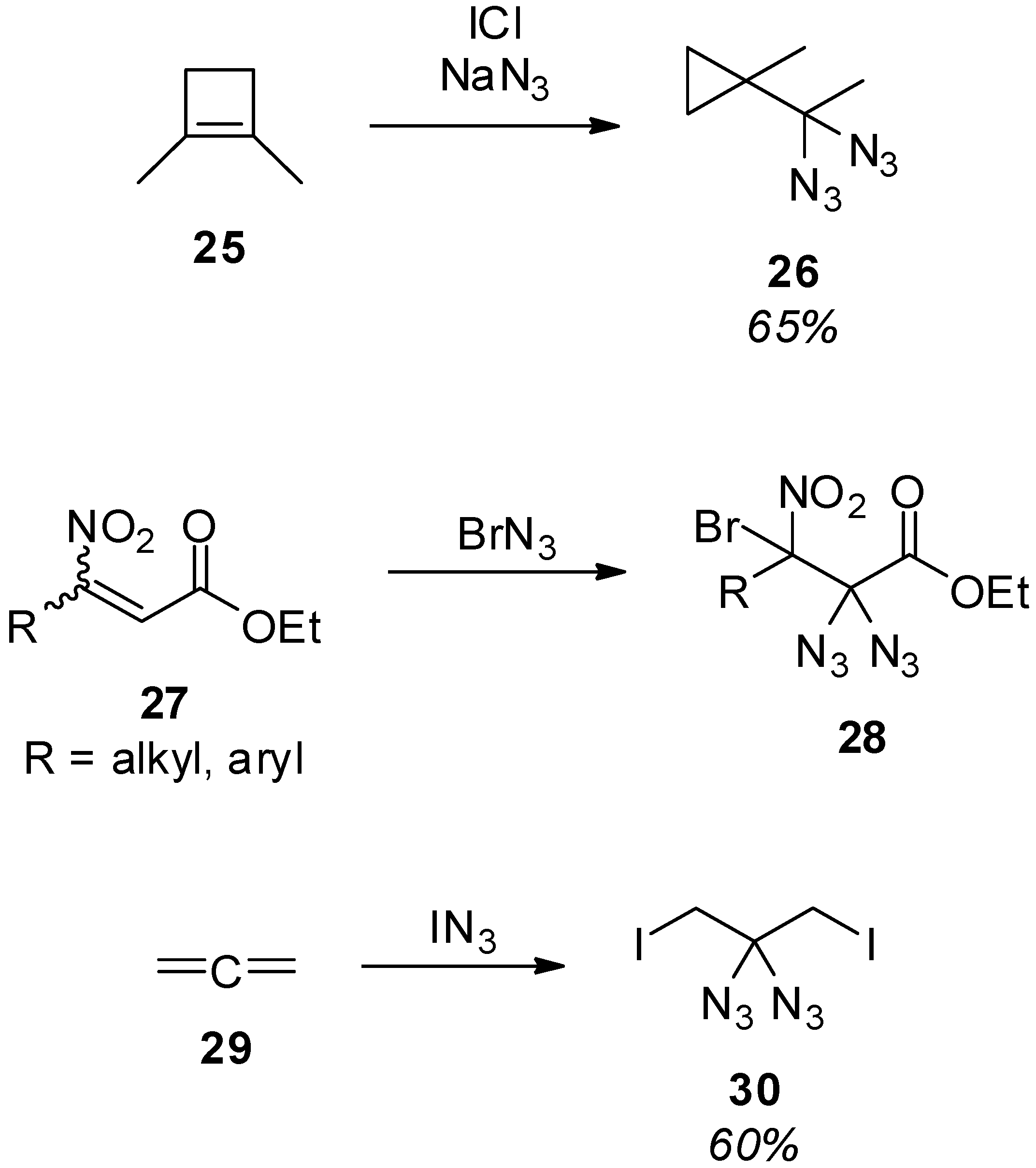
3.1.1. Synthesis

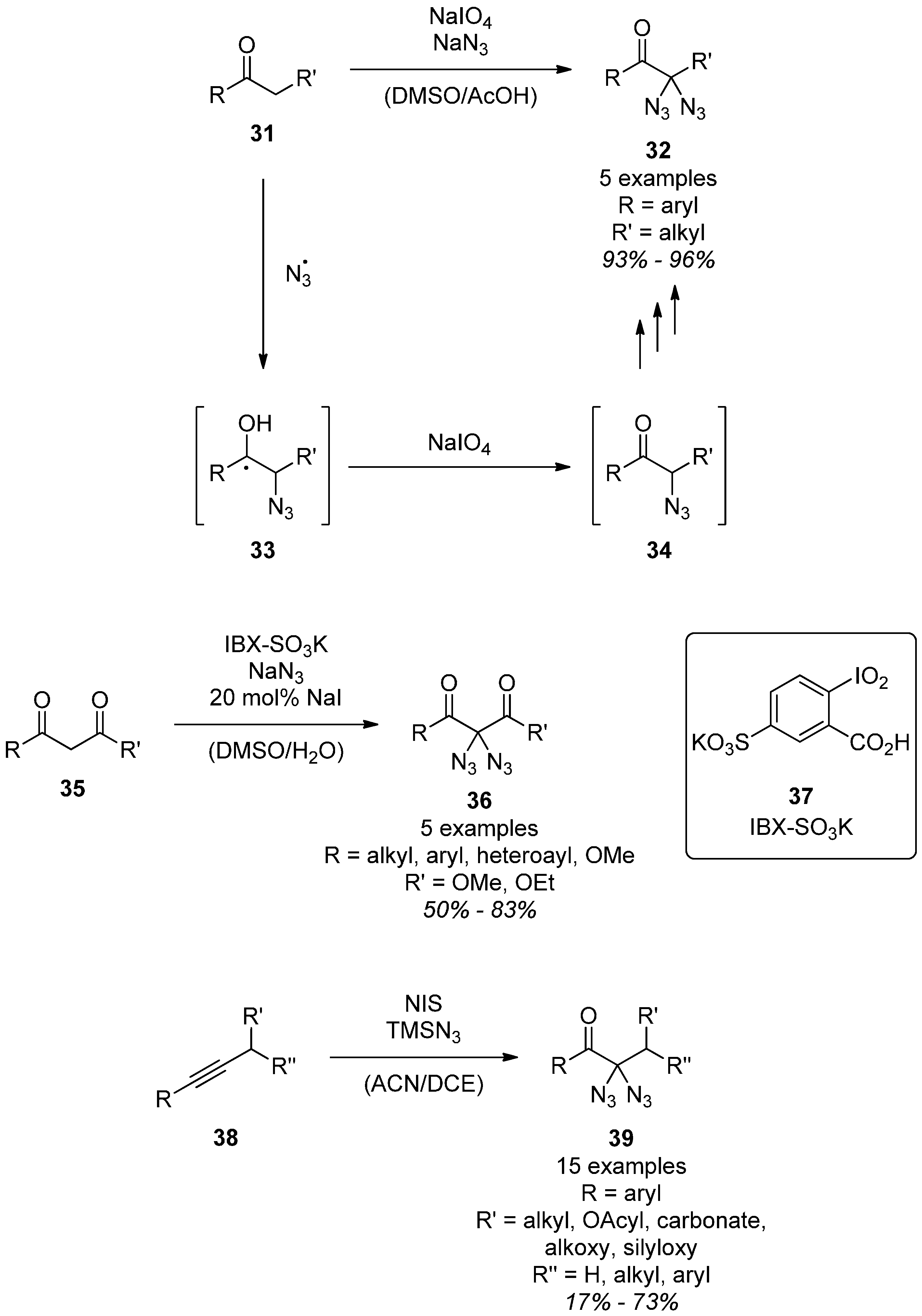
3.1.2. Reactivity
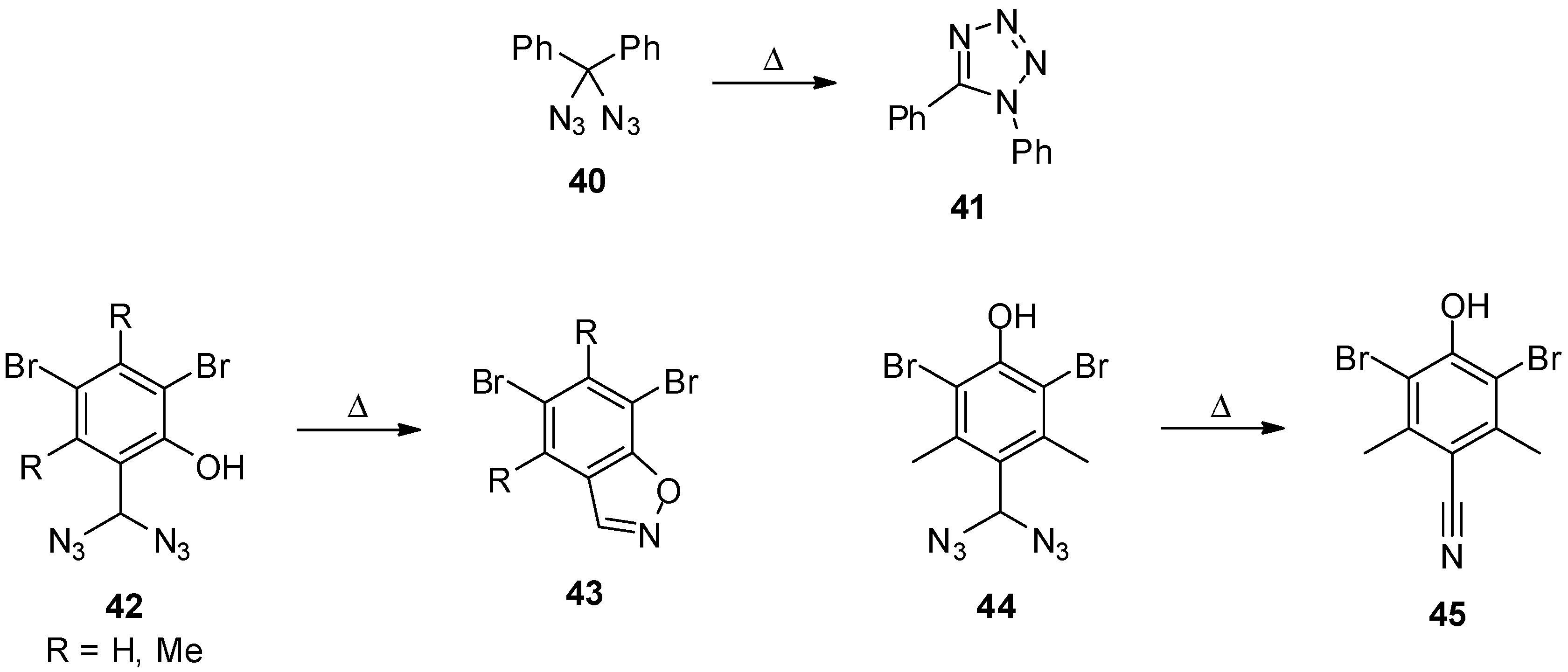
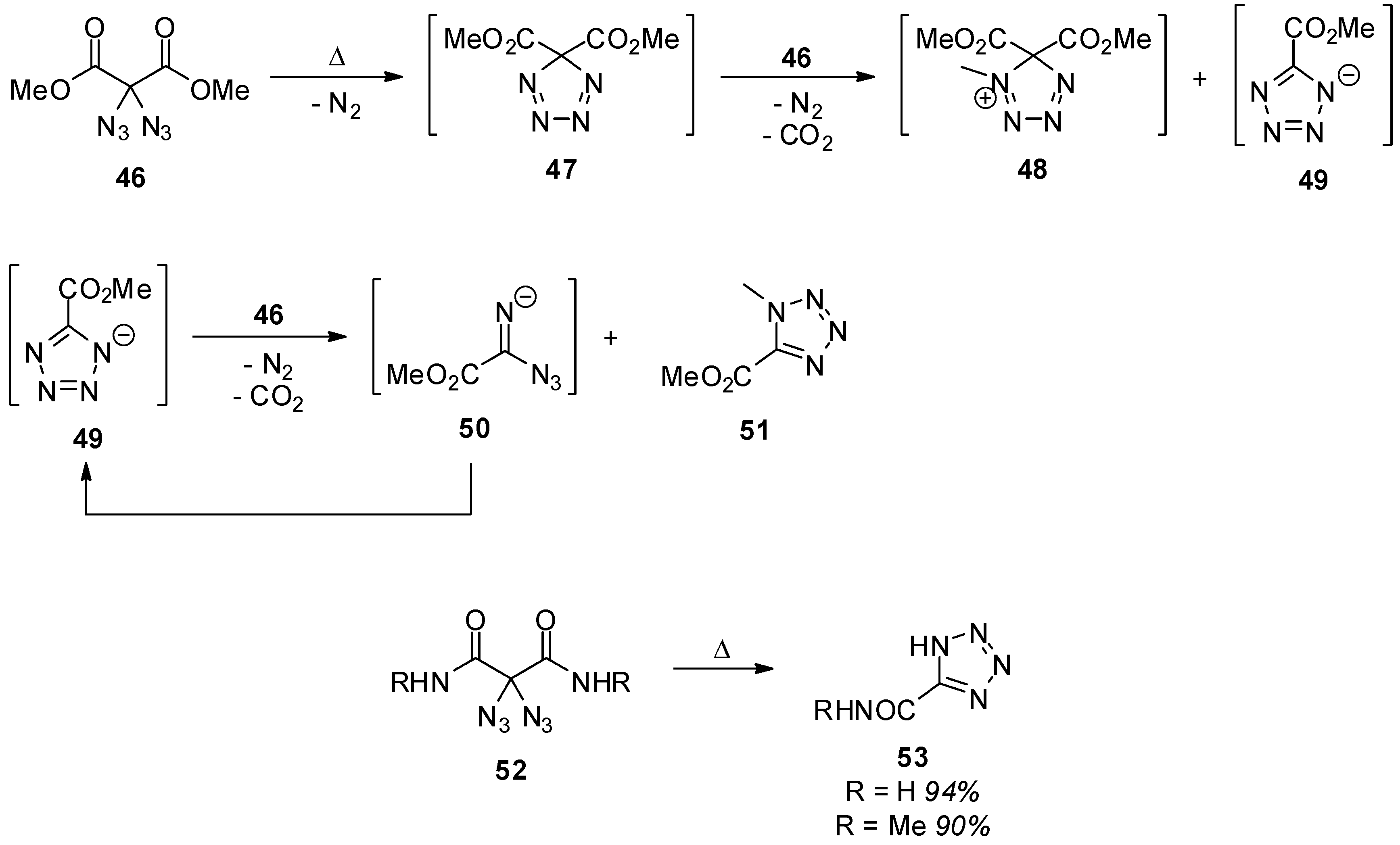
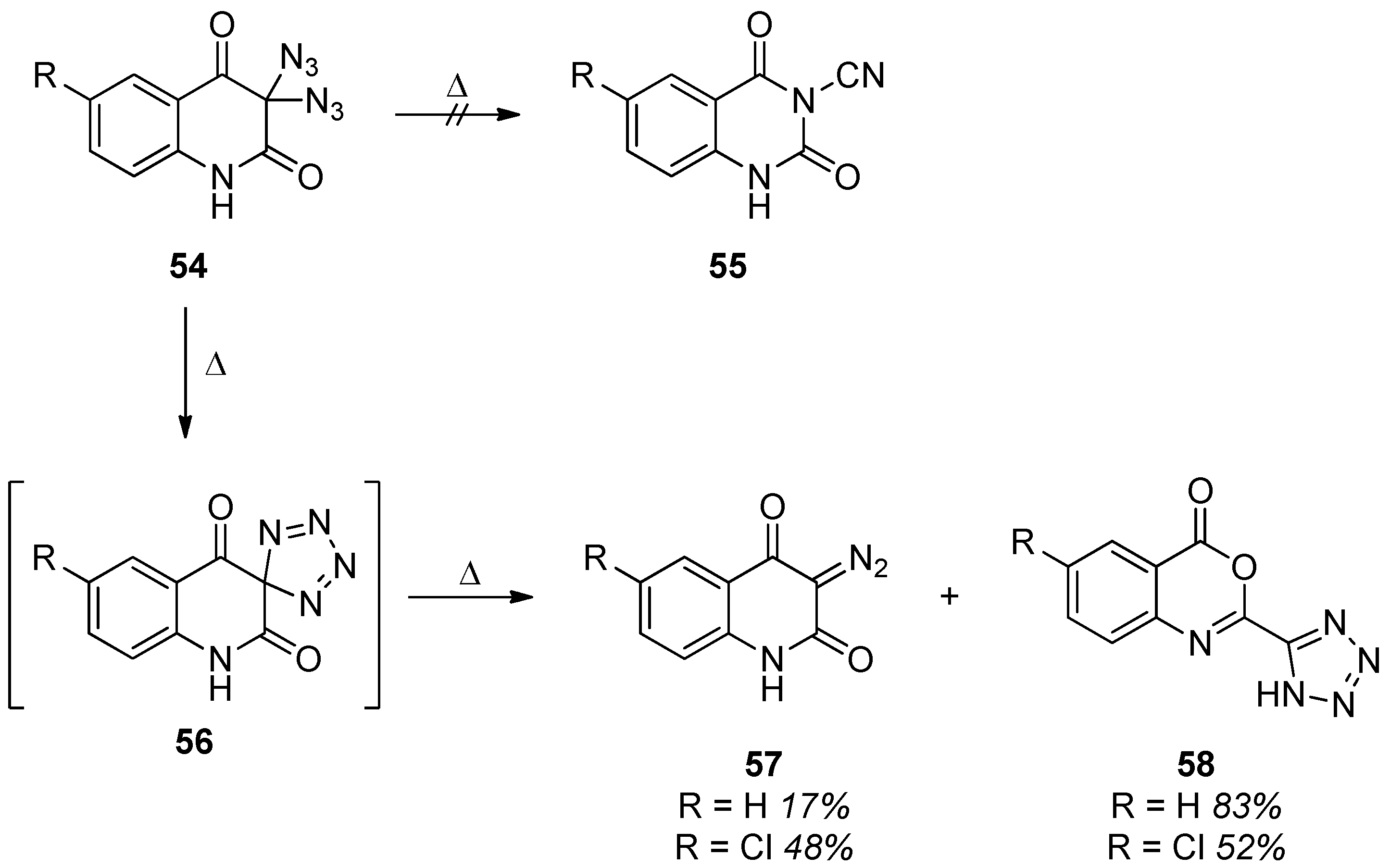

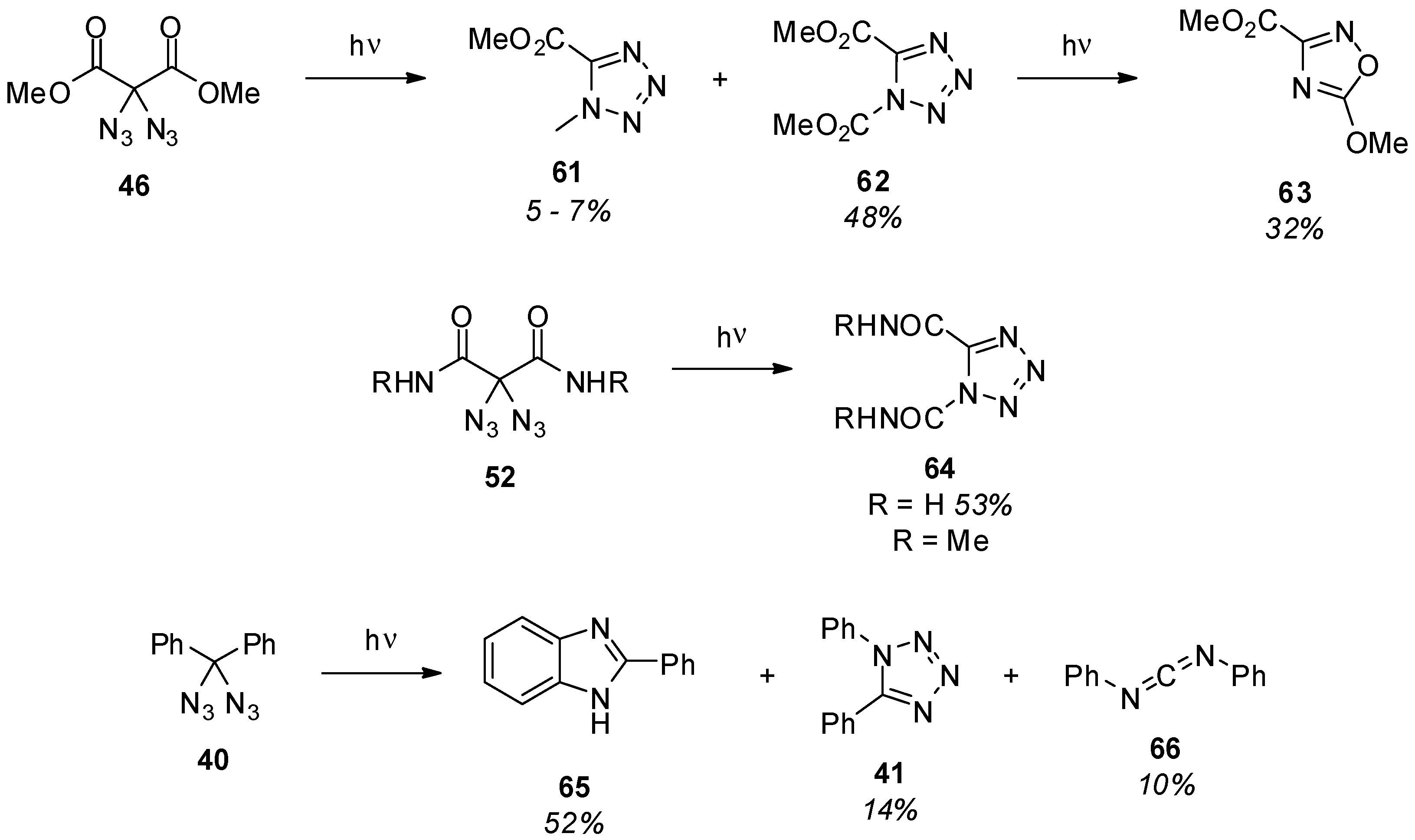
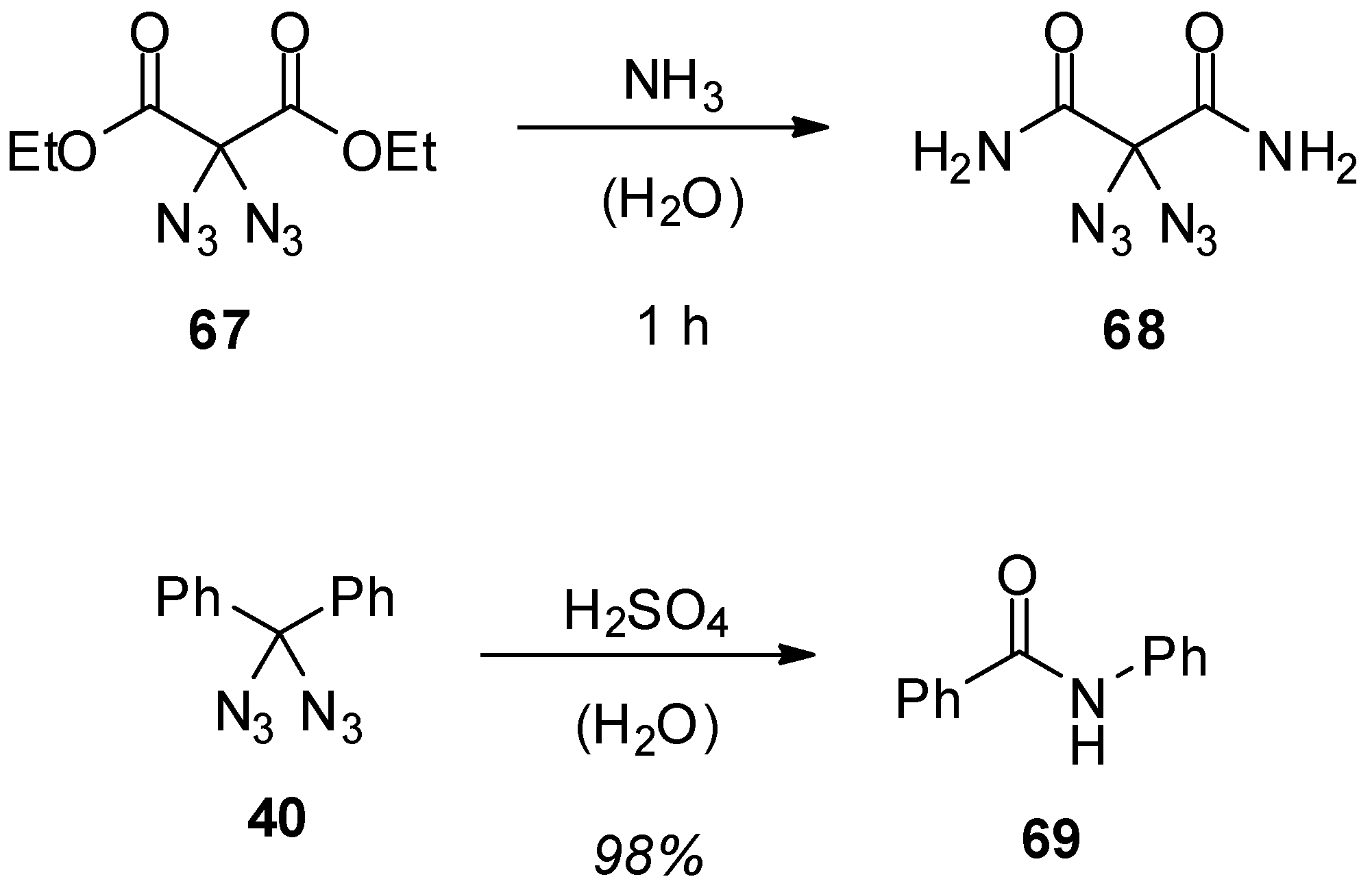
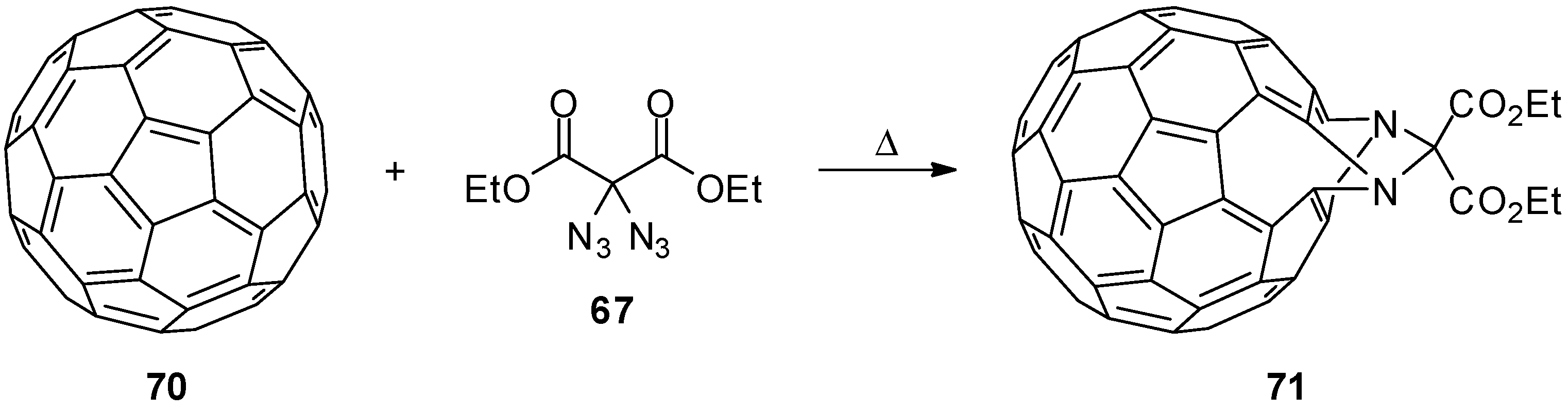
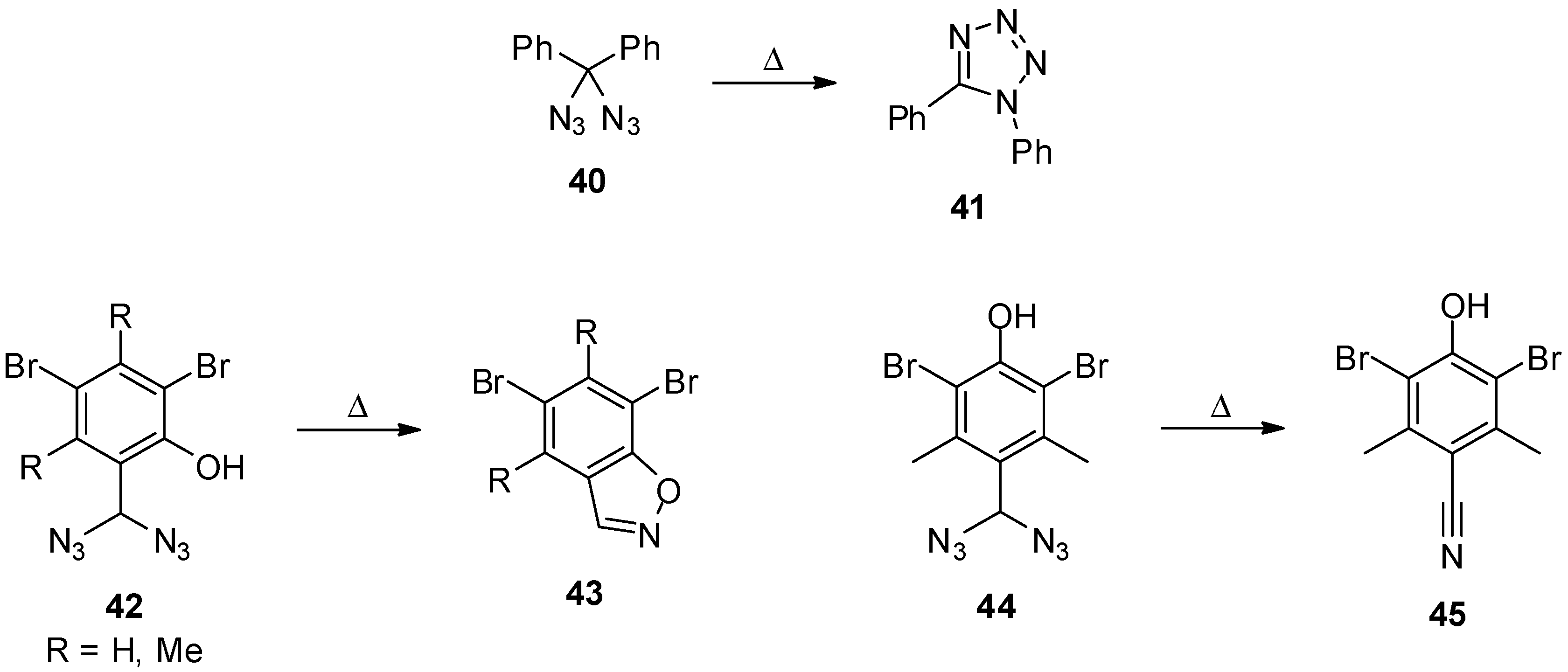
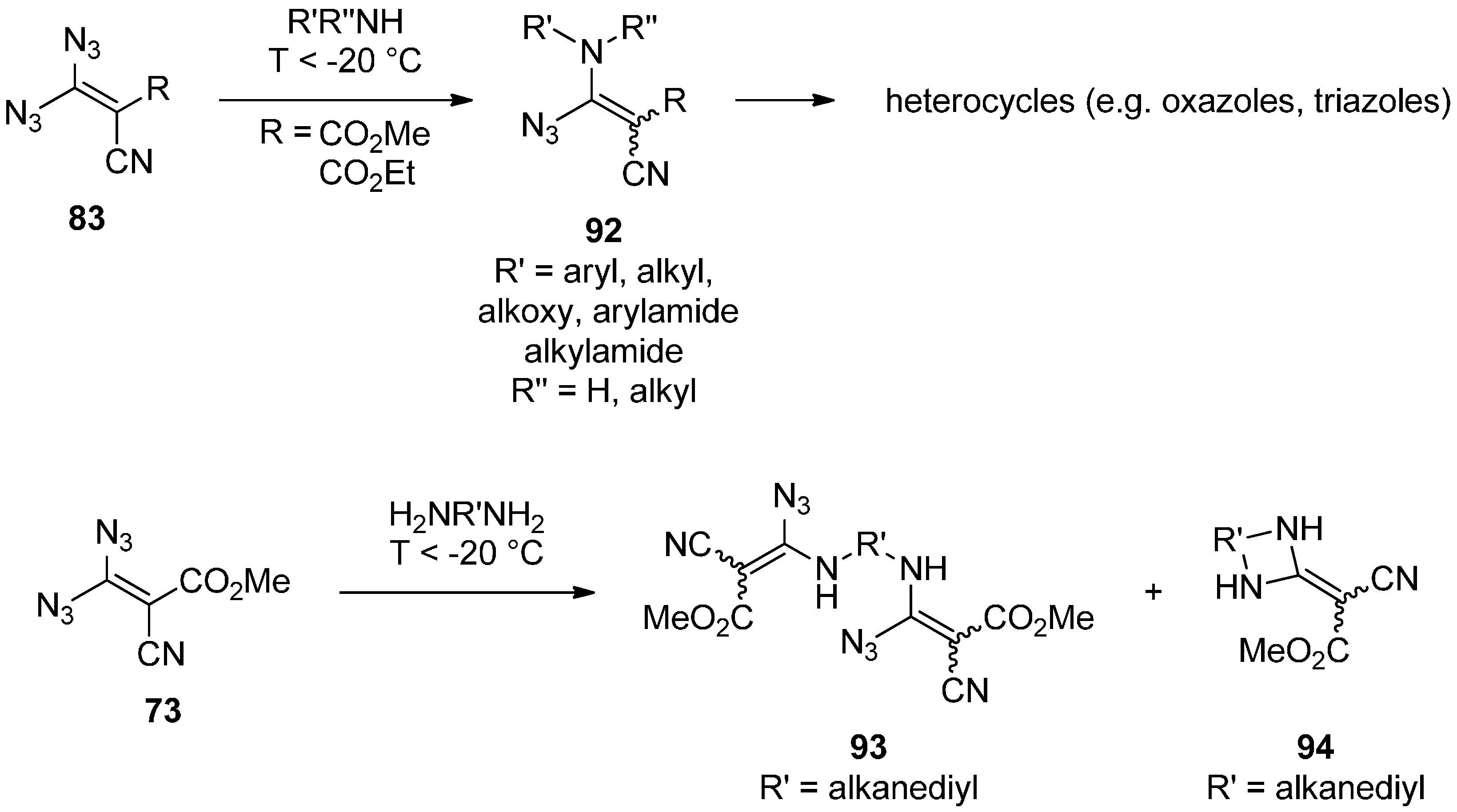
3.2. Geminal Vinyl Diazides


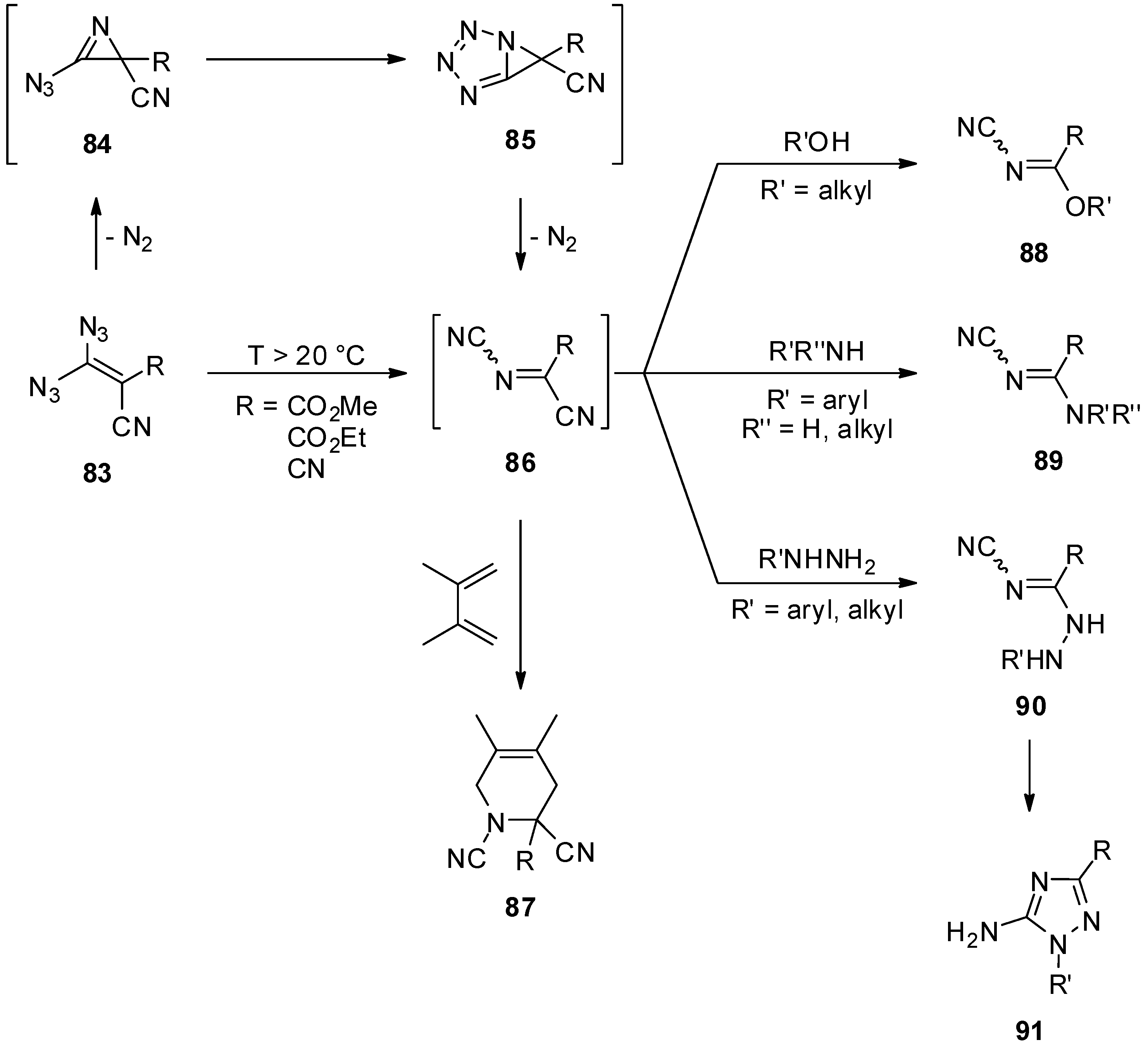

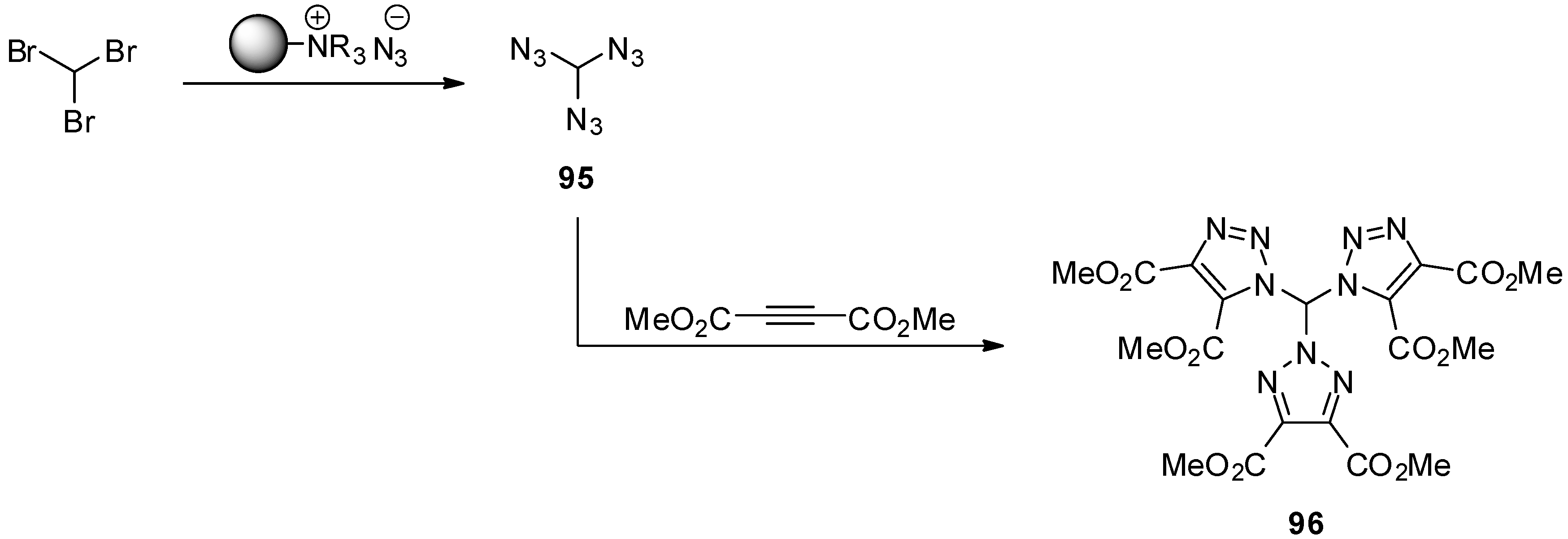
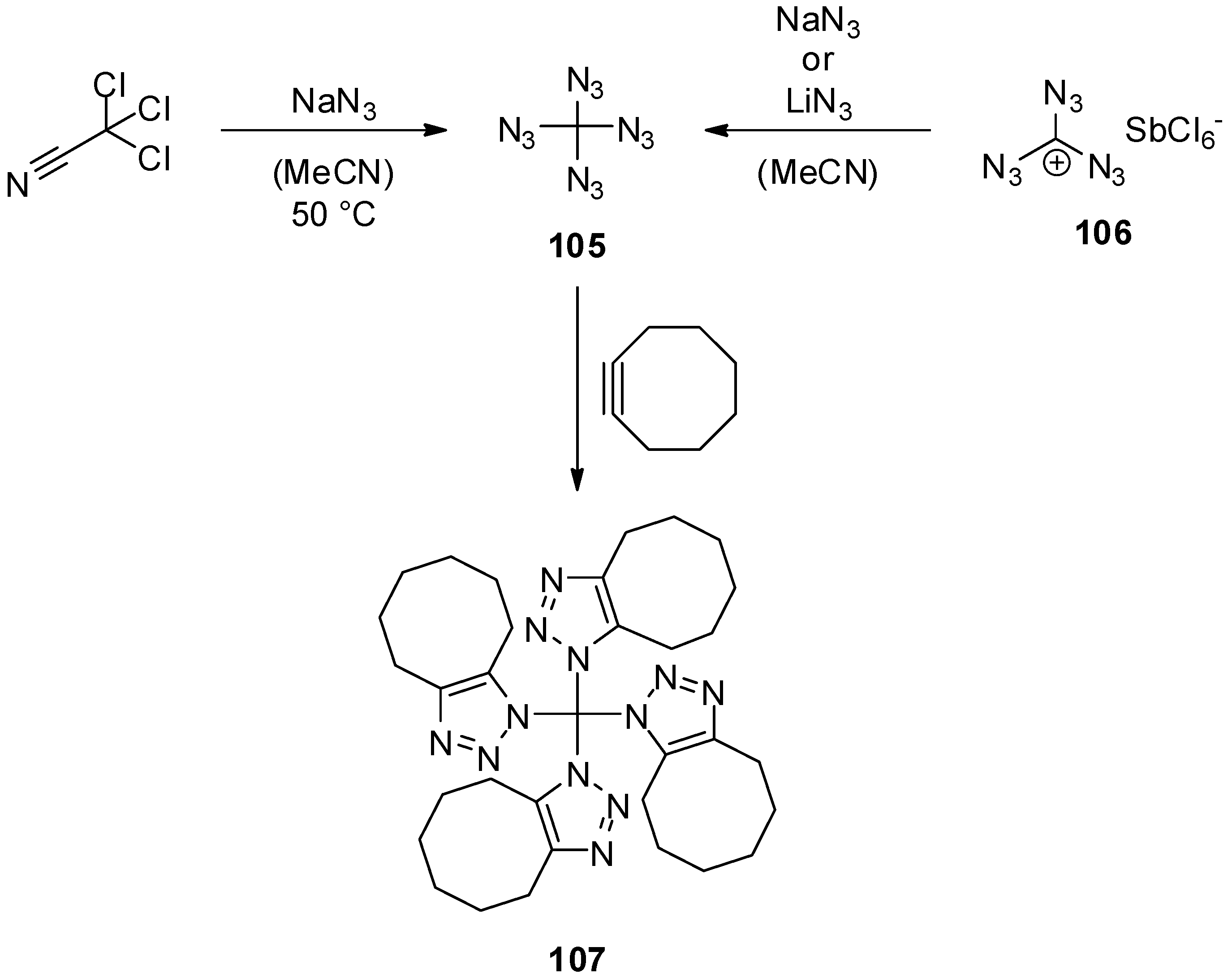
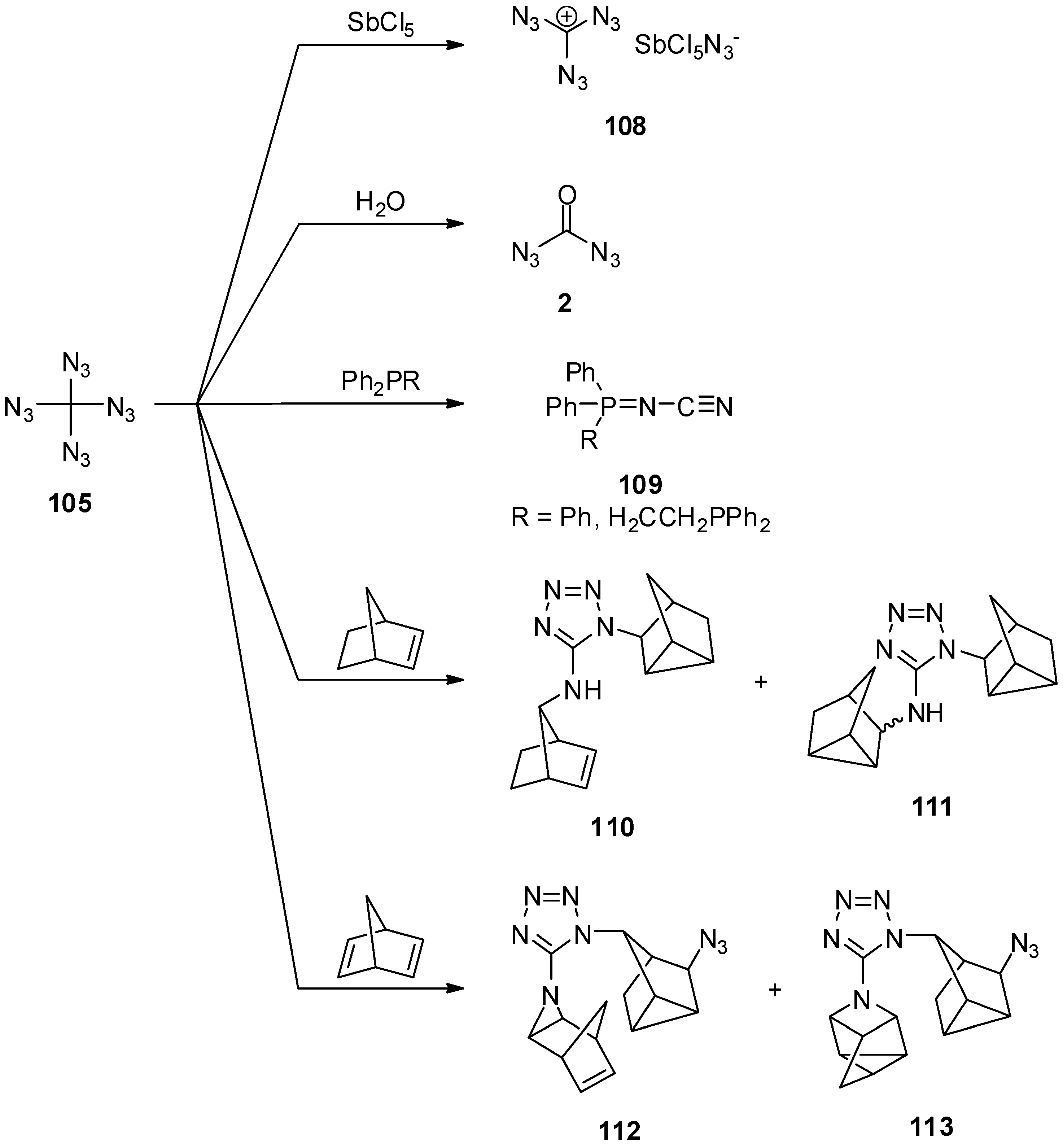
4. Geminal Triazides

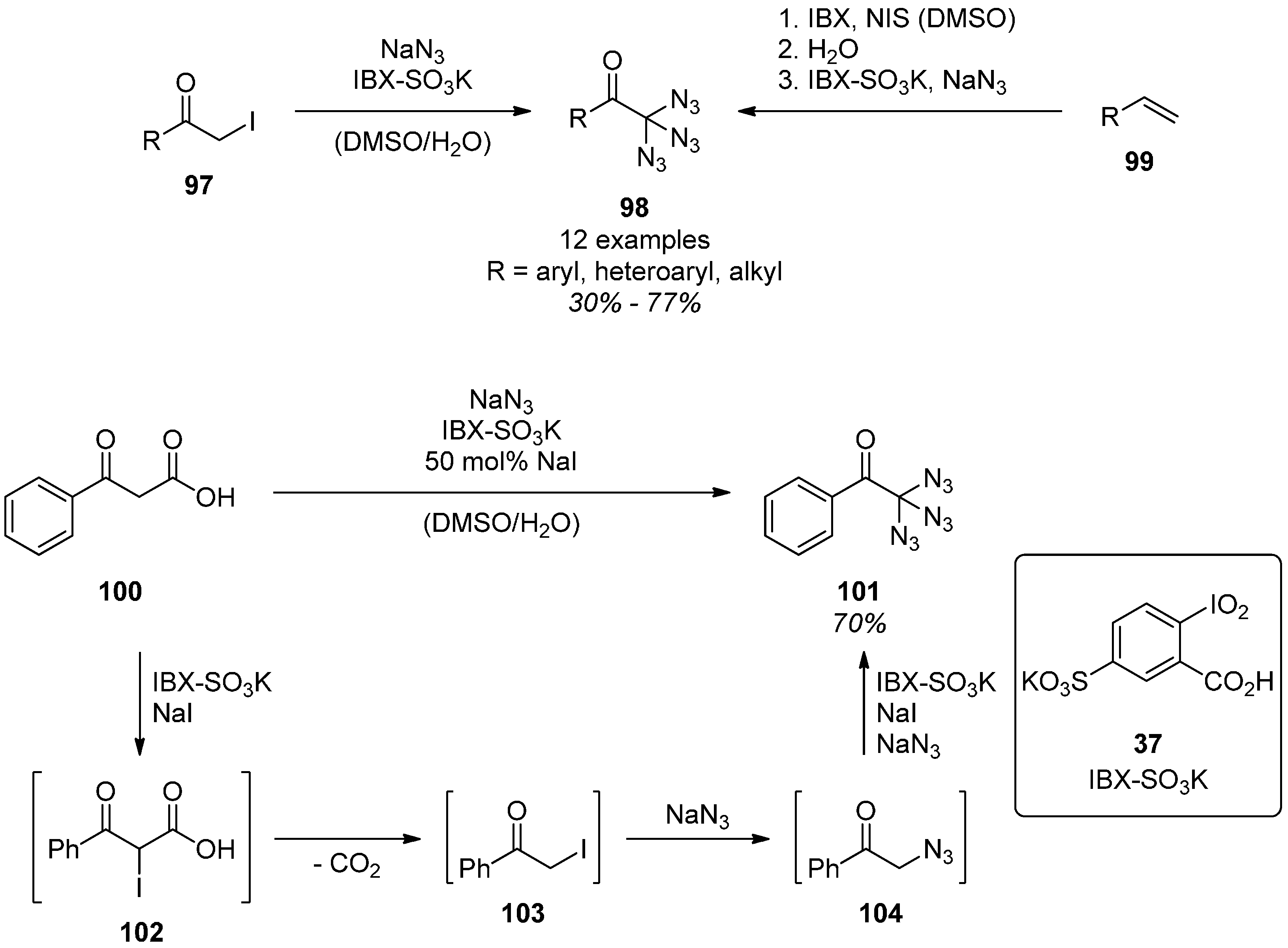
5. Tetraazidomethane
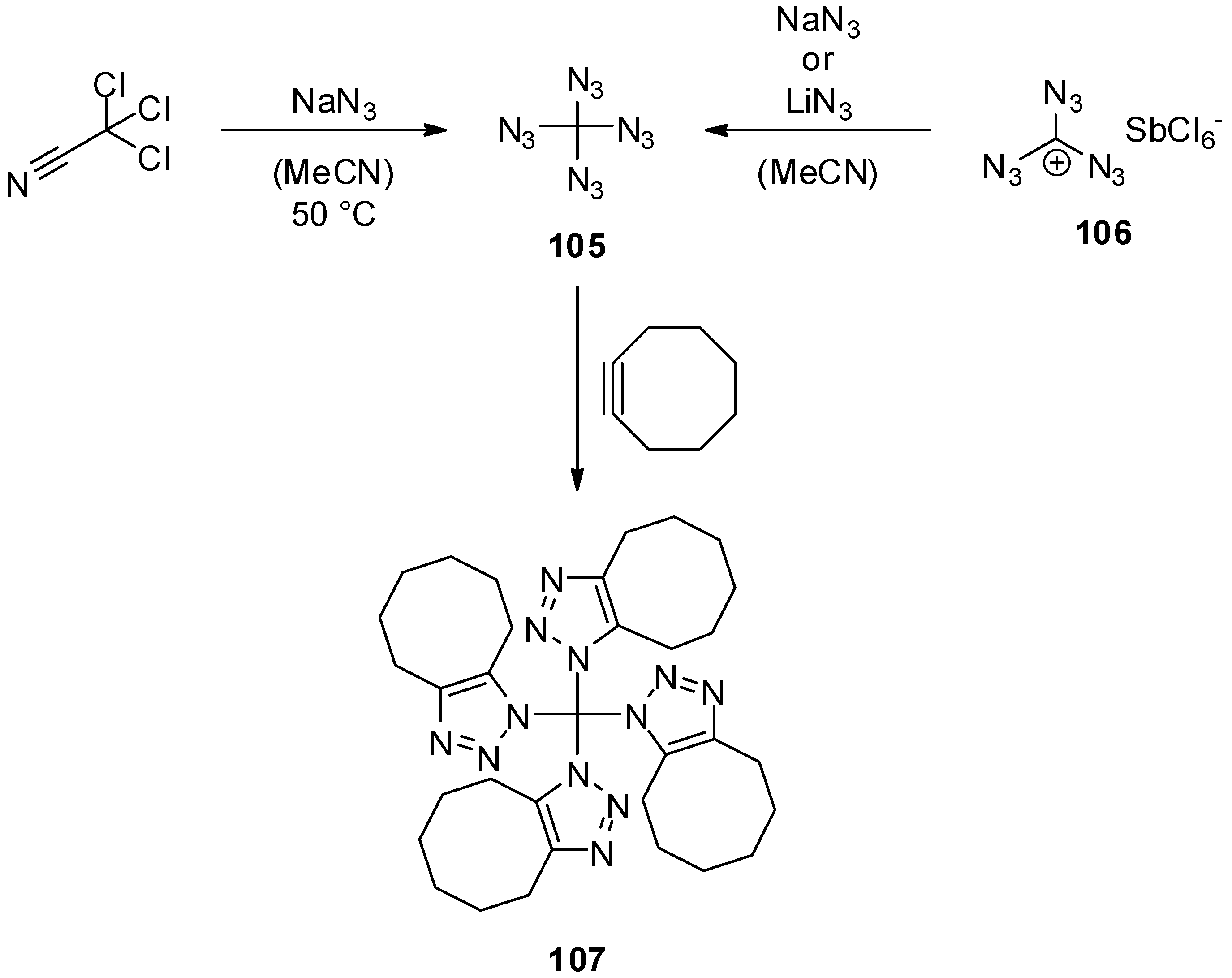
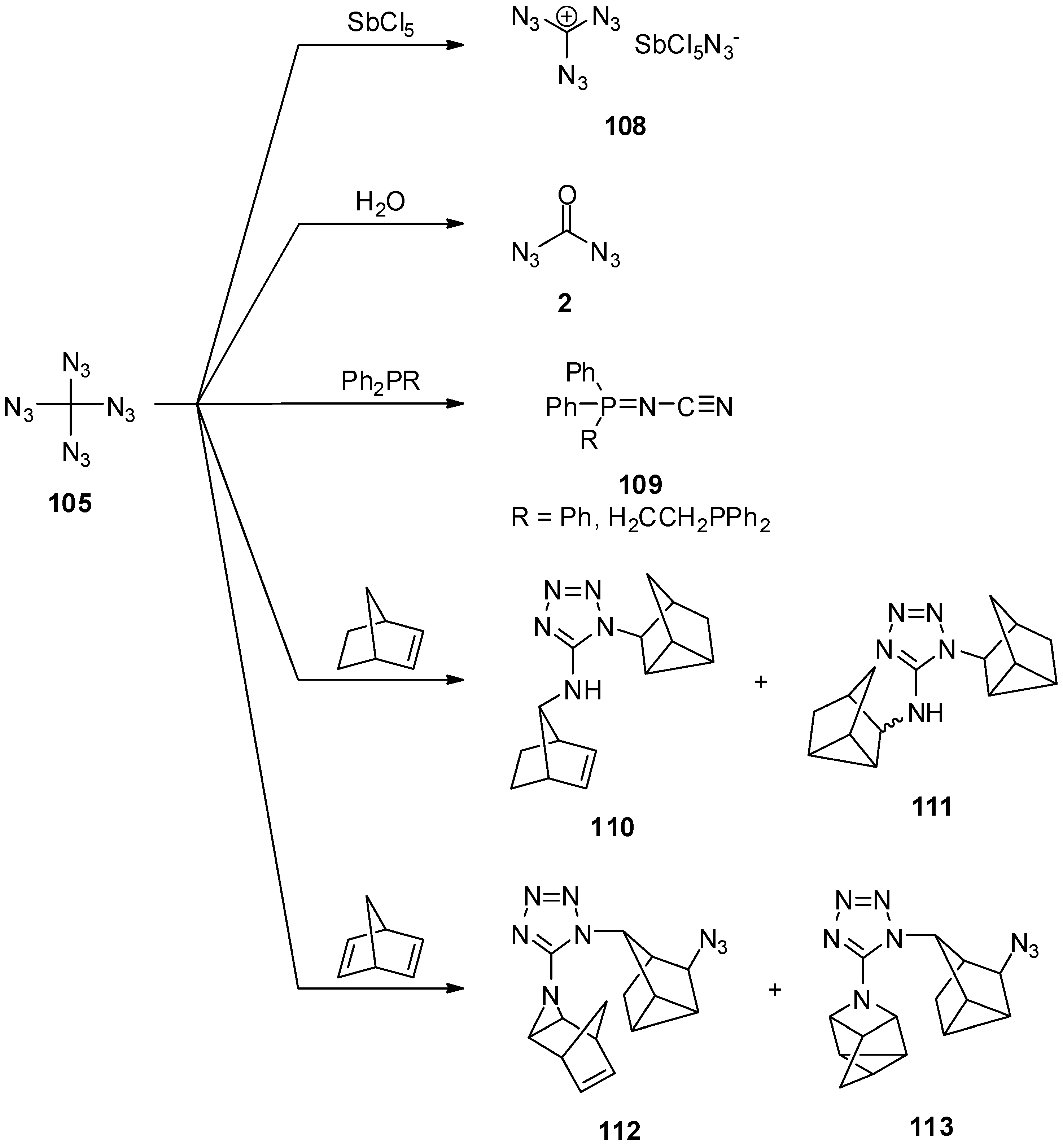
6. Outlook
Acknowledgments
Conflicts of Interest
References
- Bräse, S.; Banert, K. Organic azides: Syntheses and Applications; John Wiley: Chichester, West Sussex, UK, 2010. [Google Scholar]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Schilling, C.I.; Jung, N.; Biskup, M.; Schepers, U.; Bräse, S. Bioconjugation via azide–Staudinger ligation: An overview. Chem. Soc. Rev. 2011, 40, 4840–4871. [Google Scholar] [CrossRef] [PubMed]
- Mamidyala, S.K.; Finn, M.G. In situ click chemistry: Probing the binding landscapes of biological molecules. Chem. Soc. Rev. 2010, 39, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Hackenberger, C.P.R.; Schwarzer, D. Chemoselective Ligation and Modification Strategies for Peptides and Proteins. Angew. Chem. Int. Ed. 2008, 47, 10030–10074. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Spiteri, C.; Moses, J.E. Copper-Catalyzed Azide-Alkyne Cycloaddition: Regioselective Synthesis of 1,4,5-Trisubstituted 1,2,3-Triazoles. Angew. Chem. Int. Ed. 2010, 49, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Finn, M.G.; Koberstein, J.T.; Turro, N.J. Construction of Linear Polymers, Dendrimers, Networks, and Other Polymeric Architectures by Copper-Catalyzed Azide-Alkyne Cycloaddition “Click” Chemistry. Macromol. Rapid Commun. 2008, 29, 1052–1072. [Google Scholar] [CrossRef]
- Franc, G.; Kakkar, A.K. “Click” methodologies: Efficient, simple and greener routes to design dendrimers. Chem. Soc. Rev. 2010, 39, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Iha, R.K.; Wooley, K.L.; Nyström, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of orthogonal “click” chemistries in the synthesis of functional soft materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef] [PubMed]
- Curtius, T.; Heidenreich, K. Stickstoffkohlenoxyd und Diharnstoff. Ber. Dtsch. Chem. Ges. 1894, 27, 2684–2685. [Google Scholar] [CrossRef]
- Curtius, T.; Heidenreich, K. Die Hydrazide und Azide der Kohlensäure. J. Prakt. Chem. 1895, 52, 454–489. [Google Scholar] [CrossRef]
- Kesting, W. Über das Hydrazidicarbonazid und seine Entstehung als Nebenprodukt bei der Diazotierung von Carbohydrazid. Ber. Dtsch. Chem. Ges. A/B 1924, 57, 1321–1324. [Google Scholar] [CrossRef]
- Curtius, T.; Bertho, A. Die Einwirkung von Carbonylazid, CON6, auf aromatische Kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. A/B 1926, 59, 565–589. [Google Scholar] [CrossRef]
- Zeng, X.; Gerken, M.; Beckers, H.; Willner, H. Synthesis and Characterization of Carbonyl Diazide, OC(N3)2. Inorg. Chem. 2010, 49, 9694–9699. [Google Scholar] [CrossRef] [PubMed]
- Ball, D.W. Carbonyl diazide, OC(N3)2: Calculated thermodynamic properties. Comput. Theor. Chem. 2011, 965, 176–179. [Google Scholar] [CrossRef]
- Nolan, A.M.; Amberger, B.K.; Esselman, B.J.; Thimmakondu, V.S.; Stanton, J.F.; Woods, R.C.; McMahon, R.J. Carbonyl Diazide, OC(N3)2: Synthesis, Purification, and IR Spectrum. Inorg. Chem. 2012, 51, 9846–9851. [Google Scholar] [CrossRef] [PubMed]
- Napolion, B.; Watts, J.D.; Huang, M.-J.; McFarland, F.M.; McClendon, E.E.; Walters, W.L.; Williams, Q.L. Accurate theoretical predictions for carbonyl diazide molecules: A coupled-cluster study of the potential energy surface and thermochemical properties. Chem. Phys. Lett. 2013, 559, 18–25. [Google Scholar] [CrossRef]
- Li, H.; Li, D.; Zeng, X.; Liu, K.; Beckers, H.; Schaefer, H.F.; Esselman, B.J.; McMahon, R.J. Toward Understanding the Decomposition of Carbonyl Diazide (N3)2C–O and Formation of Diazirinone cycl-N2CO: Experiment and Computations. J. Phys. Chem. A 2015, 119, 8903–8911. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Beckers, H.; Willner, H. Matrix Isolation of Two Isomers of N4CO. Angew. Chem. Int. Ed. 2011, 50, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Beckers, H.; Willner, H.; Stanton, J.F. Elusive Diazirinone, N2CO. Angew. Chem. Int. Ed. 2011, 50, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Beckers, H.; Willner, H.; Stanton, J.F. Fascinating Diazirinone: A Violet Gas. Eur. J. Inorg. Chem. 2012, 2012, 3403–3409. [Google Scholar] [CrossRef]
- Banert, K.; Joo, Y.-H.; Rüffer, T.; Walfort, B.; Lang, H. The Exciting Chemistry of Tetraazidomethane. Angew. Chem. Int. Ed. 2007, 46, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.O.; Fierz, H.E.; Joshua, W.P. Bistriazoderivatives of Ethane and Acetic Ester. J. Chem. Soc. Trans. 1908, 93, 1070–1074. [Google Scholar] [CrossRef]
- Gilbert, E.E. A novel energetic polyazide: 1,2,4,5-Tetrakis (diazidomethyl) benzene. J. Energ. Mater. 1987, 5, 77–81. [Google Scholar] [CrossRef]
- Conrow, R.E.; Dean, W.D. Diazidomethane Explosion. Org. Process Res. Dev. 2008, 12, 1285–1286. [Google Scholar] [CrossRef]
- Sohn, M.B.; Jones, M., Jr.; Hendrick, M.E.; Rando, R.R.; Doerin, W.V.E. On Syntheses of Alkyl Diazopropionates. Tetrahedron Lett. 1972, 13, 53–56. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Takagi, K.; Yamaba, T.; Nabetani, M.; Koizumi, T. Synthetic studies for novel structure of α-nitrogenously functionalized α-fluorocarboxylic acids. Part III. Some reactions of α-bromo-α-fluorocarboxylic acids and their ethyl esters with sodium azide. J. Fluorine Chem. 1994, 68, 149–154. [Google Scholar] [CrossRef]
- Forster, M.O.; Newman, S.H. Azoimides of the Acetoacetic Series. J. Chem. Soc., Trans. 1910, 97, 1360–1368. [Google Scholar] [CrossRef]
- Ogilvie, W.; Rank, W. Thermolysis of geminal diazides: A novel route to 1,3,4-oxadiazoles. Can. J. Chem. 1987, 65, 166–169. [Google Scholar] [CrossRef]
- Zapol’skii, V.A.; Namyslo, J.C.; Gjikaj, M.; Kaufmann, D.E. Chemistry of Polyhalogenated Nitrobutadienes, Part 5: Synthesis and Reactions of Dichloromethyl Nitrovinylidene Ketones of Heterocycles. Synlett 2007, 1507–1512. [Google Scholar] [CrossRef]
- Landen, G.; Moore, H.W. Chemistry of Geminal Diazides. Rearrangements to N-Cyano Compounds. Tetrahedron Lett. 1976, 17, 2513–2516. [Google Scholar] [CrossRef]
- Forster, M.O.; Müller, R. Substituted Triazomalonic and Phenyltriazoacetic Acids. J. Chem. Soc., Trans. 1910, 97, 126–142. [Google Scholar] [CrossRef]
- Badawey, E.-S.A.M.; Rida, S.M.; Soliman, F.S.G.; Kappe, T. Benzimidazole Condensed Ring Systems, VI: Organic Azides in Heterocyclic Synthesis, X: Synthesis of Some Substituted Pyrimido[1,6-a]-benzimidazoles as Potential Antimicrobial Agents. Monatsh. Chem. 1989, 120, 1159–1164. [Google Scholar] [CrossRef]
- Malle, E.; Stadlbauer, W.; Ostermann, G.; Hofmann, B.; Leis, H.J.; Kostner, G.M. Synthesis of new 2-, 3-, and 4-substituted azidoquinolines: Inhibitors of human blood platelet aggregation in vitro. Eur. J. Med. Chem. 1990, 25, 137–142. [Google Scholar] [CrossRef]
- Kappe, C.O. Unexpected Formation of Nitriles from Reactions of 6-(Dibromomethyl)-1,2,3,4-tetrahydro-2-oxo-5-pyrimidinecarboxylates with Sodium Azide. Liebigs Ann. Chem. 1990, 5, 505–507. [Google Scholar] [CrossRef]
- Schroeter, G. Über die Hofmann-Curtiussche, die Beckmannsche und die Benzilsäure-Umlagerung. Ber. Dtsch. Chem. Ges. 1909, 42, 2336–2349. [Google Scholar] [CrossRef]
- Lindemann, H.; Mühlhaus, A. Oxy-benzal-azide und Indoxazene. Justus Liebigs Ann. Chem. 1926, 446, 1–13. [Google Scholar] [CrossRef]
- Götzky, S. Über das Benzophenon-diazid. Ber. Dtsch. Chem. Ges. A/B 1931, 64, 1555–1560. [Google Scholar] [CrossRef]
- Barash, L.; Wasserman, E.; Yager, W.A. Generation of Methylenes from Germinal Diazides via Excited Nitrenes. J. Am. Chem. Soc. 1967, 89, 3931–3932. [Google Scholar] [CrossRef]
- Wieland, M.; Su, K.; Wagner, G.; Brinker, U.H.; Arion, V.B. 1,2,4,5-Tetrakis(diazidomethyl)benzene. Acta Crystallogr. Sect. C Struct. Chem. 2009, 65, o240–o242. [Google Scholar] [CrossRef] [PubMed]
- Hassner, A.; Stern, M.; Gottlieb, H.E.; Frolow, F. Utility of a Polymeric Azide Reagent in the Formation of Di- and Triazidomethane. Their NMR Spectra and the X-ray Structure of Derived Triazoles. J. Org. Chem. 1990, 55, 2304–2306. [Google Scholar] [CrossRef]
- Ranaweera, R.A.A.U.; Sankaranarayanan, J.; Casey, L.; Ault, B.S.; Gudmundsdottir, A.D. Triplet-Sensitized Photoreactivity of a Geminal Diazidoalkane. J. Org. Chem. 2011, 76, 8177–8188. [Google Scholar] [CrossRef] [PubMed]
- Bretschneider, H.; Karpitschka, N. Bildung von Diazidomalonester aus Monobrommalonester mit Natriumazid, eine neue Disproportionierungsreaktion. Monatsh. Chem. 1953, 84, 1091–1096. [Google Scholar] [CrossRef]
- Moriarty, K.M.; Kliegman, J.M.; Shovlin, C. The Photochemical Decomposition of Geminal Diazides. I. Dimethyl Diazidomalonate. J. Am. Chem. Soc. 1967, 89, 5958–5959. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Bailey, B.R., III; Prakash, I.; Miller, R.S. Thermal Decomposition of Geminal Diazidomalonic Acid Derivatives. An Intermolecular Process. J. Org. Chem. 1985, 50, 3710–3713. [Google Scholar] [CrossRef]
- Evans, D.A.; Britton, T.C.; Ellman, J.A.; Dorow, R.L. The Asymmetric Synthesis of α-amino acids. Electrophilic Azidation of Chiral Imide Enolates, a Practical Approach to the Synthesis of (R)- and (S)-α-Azido Carboxylic Acids. J. Am. Chem. Soc. 1990, 112, 4011–4030. [Google Scholar] [CrossRef]
- Kirchmeyer, S.; Mertens, A.; Olah, G.A. Synthetic Methods and Reactions; 115. Conversion of Acetals and Ketone Acetals into Azido Compounds using Trimethylsilyl Azide. Synthesis 1983, 1983, 500–502. [Google Scholar] [CrossRef]
- Nishiyama, K.; Watanabe, A. Addition Reaction of Trimethylsilyl Azide towards Ketones and Facile Formation of Tetrazole Derivatives. Chem. Lett. 1984, 13, 455–458. [Google Scholar] [CrossRef]
- Nishiyama, K.; Yamaguchi, T. Selective Formation of Alkyl Azides Using Trimethylsilyl Azide and Carbonyl Compounds. Synthesis 1988, 1988, 106–108. [Google Scholar] [CrossRef]
- Nishiyama, K.; Oba, M.; Watanabe, A. Reactions of Trimethylsilyl Azide with Aldehydes: Facile and Convenient Syntheses of Diazides, Tetrazoles, and Nitriles. Tetrahedron 1987, 43, 693–700. [Google Scholar] [CrossRef]
- Ye, C.; Gao, H.; Boatz, J.A.; Drake, G.W.; Twamley, B.; Shreeve, J.M. Polyazidopyrimidines: High-Energy Compounds and Precursors to Carbon Nanotubes. Angew. Chem. Int. Ed. 2006, 45, 7262–7265. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Nakaya, C. A Convenient One-Step Method of Converting Electron-Rich Aromatic Aldehydes into Nitriles. Synthesis 1992, 1992, 641–642. [Google Scholar] [CrossRef]
- Suzuki, H.; Hwang, Y.S.; Nakaya, C.; Matano, Y. Improved Schmidt Synthesis of 1,5-Disubstituted 1H-Tetrazoles from Ketones. Synthesis 1993, 1993, 1218–1220. [Google Scholar] [CrossRef]
- O’Hare, M.J.; Swern, D. Novel Ring Contractions induced by LAH Reduction of Addition Products of Iodine Azide to Alkyl substituted Cyclobutenes. Tetrahedron Lett. 1973, 14, 1607–1610. [Google Scholar] [CrossRef]
- Shin, C.-G.; Yonezawa, Y.; Suzuki, K.; Yoshimura, J. α,β-Unsaturated Carboxylic Acid Derivatives. XV. The Reaction of Ethyl 3-Nitro-2-alkenoate with Bromine Azide or Bromine, and Transformations of the Products. Bull. Chem. Soc. Jpn. 1978, 51, 2614–2617. [Google Scholar] [CrossRef]
- Hassner, A.; Keogh, J. Regiochemistry of Halogen Azide Addition to Allenes. J. Org. Chem. 1986, 51, 2767–2770. [Google Scholar] [CrossRef]
- Kamble, D.A.; Karabal, P.U.; Chouthaiwale, P.V.; Sudalai, A. NaIO4–NaN3-mediated diazidation of styrenes, alkenes, benzylic alcohols, and aryl ketones. Tetrahedron Lett. 2012, 53, 4195–4198. [Google Scholar] [CrossRef]
- Klahn, P.; Erhardt, H.; Kotthaus, A.; Kirsch, S.F. The Synthesis of α-Azidoesters and Geminal Triazides. Angew. Chem. Int. Ed. 2014, 53, 7913–7917. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Sueda, T.; Minami, H.; Miwa, Y.; Yanada, R. Regioselective Iodoazidation of Alkynes: Synthesis of α,α-Diazidoketones. Org. Lett. 2015, 17, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Harschneck, T.; Hummel, S.; Kirsch, S.F.; Klahn, P. Practical Azidation of 1,3-Dicarbonyls. Chem. Eur. J. 2012, 18, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Bredenkamp, A.; Mohr, F.; Kirsch, S.F. Synthesis of Isatins through Direct Oxidation of Indols with IBX-SO3K/NaI. Synthesis 2015, 47, 1937–1943. [Google Scholar] [CrossRef]
- McGirk, R.H.; Cyr, C.R.; Ellis, W.D.; White, E.H. Application of the Nitrosoamide Reaction to Hydrazones. J. Org. Chem. 1974, 39, 3851–3855. [Google Scholar] [CrossRef]
- Temple, C., Jr.; Kussner, C.L.; Montgomery, J.A. Pyrimido[5,4-e]-as-triazines. VIII. Synthesis of 7-Azaaminopterin. J. Org. Chem. 1975, 40, 2205–2208. [Google Scholar] [CrossRef] [PubMed]
- Szeimies, G.; Mannhardt, K.; Mickler, W. Synthese und thermisches Verhalten einiger 3-Aryl-3-azidoaziridin-2-carbonsäure-ethylester. Chem. Ber. 1977, 110, 2922–2938. [Google Scholar] [CrossRef]
- Al-Khalil, S.I.; Bowman, W. Radical-Nucleophilic Substitution (SRN1) Reactions: Preparation and Reactions of Aliphatic α-Nitro-Azides. Tetrahedron Lett. 1982, 23, 4513–4516. [Google Scholar] [CrossRef]
- Al-Khalil, S.I.; Bowman, W.R.; Symons, M.C.R. Radical-nucleophilic (SRN1) reactions. Part 4. Preparation, Reactions, and Electron Spin Resonance studies of α-Nitro Azides. J. Chem. Soc. Perkin Trans. 1 1986, 555–565. [Google Scholar] [CrossRef]
- Arimoto, M.; Yamaguchi, H.; Fujita, E.; Ochiai, M.; Nagao, Y. Iodosylbenzene-Trimethylsilyl Azide-Boron Trifluoride Etherate: A Highly Efficient System for Direct Synthesis of Allyl Azides from Allylsilanes. Tetrahedron Lett. 1987, 28, 6289–6292. [Google Scholar] [CrossRef]
- Hassner, A.; Fibiger, R.; Amarasekara, A.S. TiCl4-Catalyzed Addition of HN3 to Aldehydes and Ketones. Thermolysis and Photolysis of α-Azido Ethers. J. Org. Chem. 1988, 53, 22–27. [Google Scholar] [CrossRef]
- Grayson, E.J.; Whitham, G.H. Addition of Iodine-Based Electrophilic Reagents to Some Vinylsilanes. Tetrahedron 1988, 44, 4087–4094. [Google Scholar] [CrossRef]
- Maag, H.; Rydzewski, R.M. An Allylic Azide Route to 4′-Azido Carbocyclic Nucleosides. Synthesis of (+−)-(1′α,2′α,3′β)- and (+−)-(1′α,2′β,3′β)-1-[1-Azido-2-hydroxy-1-(hydroxymethyl)-3-cyclopentyl]thymine. J. Org. Chem. 1992, 57, 5823–5831. [Google Scholar] [CrossRef]
- Nagy, J.; Nyitrai, J.; Kajtár-Peredy, M. Monobactams Acylated with Isoxazoleacetic and Isoxazolecarboxylic Acids. Liebigs Ann. Chem. 1993, 8, 815–821. [Google Scholar] [CrossRef]
- Kapeller, H.; Griengl, H. Synthesis of Methyl 5-azido-5-deoxy-2,3-O-isopropylidenecarba-α-d-allo-hexafuranuronate, the Sugar Part of Carbapolyoxins and Carbanikkomycins. Tetrahedron 1997, 53, 14635–14644. [Google Scholar] [CrossRef]
- Hein, J.E.; Tripp, J.C.; Krasnova, L.B.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Cycloaddition of Organic Azides and 1-Iodoalkynes. Angew. Chem. Int. Ed. 2009, 48, 8018–8021. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Hutait, S.; Biswas, S.; Batra, S. Versatility of Substituted 1-Formyl-9H-β-carbolines for the Synthesis of New Fused β-Carbolines via Intramolecular 1,3-Dipolar Cycloaddition. Eur. J. Org. Chem. 2010, 2010, 531–539. [Google Scholar] [CrossRef]
- Kuang, G.-C.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Zhu, L. Chelation-Assisted, Copper(II)-Acetate-Accelerated Azide-Alkyne Cycloaddition. J. Org. Chem. 2010, 75, 6540–6548. [Google Scholar] [CrossRef] [PubMed]
- Banert, K.; Joo, Y.-H.; Rüffer, T.; Walfort, B.; Lang, H. Synthesis of azidochloromethane and azidobromomethane. Tetrahedron Lett. 2010, 51, 2880–2882. [Google Scholar] [CrossRef]
- Pramanik, S.; Ghorai, P. Chemoselective three-component synthesis of homoallylic azides using an FeCl3 catalyst. RSC Adv. 2013, 3, 23157–23165. [Google Scholar] [CrossRef]
- Pramanik, S.; Ghorai, P. Trapping of Azidocarbenium Ion: A Unique Route for Azide Synthesis. Org. Lett. 2014, 16, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Amyes, T.L.; Richard, J.P. Kinetic and Thermodynamic Stability of α-Azidobenzyl Carbocations: Putative Intermediates in the Schmidt Reaction. J. Am. Chem. Soc. 1991, 113, 1867–1869. [Google Scholar] [CrossRef]
- Amyes, T.L.; Stevens, I.W.; Richard, J.P. The Effects of α-Substituents on the Kinetic and Thermodynamic stability of 4-Methoxybenzyl Carbocations: Carbocation Lifetimes That Are Independent of Their Thermodynamic Stability. J. Org. Chem. 1993, 58, 6057–6066. [Google Scholar] [CrossRef]
- Richard, J.P.; Amyes, T.L.; Lee, Y.-G.; Jagannadham, V. Demonstration of the Chemical Competence of an Iminodiazonium Ion to Serve as the Reactive Intermediate of a Schmidt Reaction. J. Am. Chem. Soc. 1994, 116, 10833–10834. [Google Scholar] [CrossRef]
- Richard, J.P.; Amyes, T.L.; Jagannadham, V.; Lee, Y.-G.; Rice, D.J. Spontaneous Cleavage of gem-Diazides: A Comparison of the Effects of α-Azido and Other Electron-Donating Groups on the Kinetic and Thermodynamic Stability of Benzyl and Alkyl Carbocations in Aqueous Solution. J. Am. Chem. Soc. 1995, 117, 5198–5205. [Google Scholar] [CrossRef]
- Liang, Y.; Li, N. Chain or Ring: Which One is Favorable in Nitrogen-Rich Molecules N6XHm, N8XHm, and N10XHm (X = B, Al, Ga, m = 1 and X = C, Si, Ge, m = 2)? J. Phys. Chem. A 2014, 118, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, L.; Jiao, N. Direct Transformation of Methyl Arenes to Aryl Nitriles at Room Temperature. Angew. Chem. Int. Ed. 2009, 48, 7094–7097. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Sydnes, L.K. The Chemistry of Acylals. Part II. Formation of Nitriles by Treatment of Acylals with Trimethylsilyl Azide in the Presence of a Lewis Acid. Tetrahedron Lett. 1998, 39, 6361–6364. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Serridge, P. Thermal Decomposition of Geminal Diazides. J. Am. Chem. Soc. 1971, 93, 1534–1535. [Google Scholar] [CrossRef]
- Yokoyama, M.; Hirano, S.; Matsushita, M.; Hachiya, T.; Kobayashi, N.; Kubo, M.; Togo, H.; Seki, H. Synthesis of tetrazoles bearing a sugar moiety (sugar tetrazoles). X-Ray molecular structure of “(7R,8R,9S,10R)-8,9,10-tribenzyloxy-7-benzyloxymethyl-6-oxa-1,5-pentamethylenetetrazole”. J. Chem. Soc. Perkin Trans. 1 1995, 13, 1747–1753. [Google Scholar] [CrossRef]
- Moore, H.W.; Pearce, D.S. Pyrolysis of 2-Azido-1,3-indanediones. Azanaphthoquinone Synthesis. Tetrahedron Lett. 1971, 12, 1621–1624. [Google Scholar] [CrossRef]
- Pearce, D.S.; Locke, M.J.; Moore, H.W. Rearrangements of Azidoquinones. XV. Thermal Rearrangement of 2,3-Diazido-1,4-quinones to 2-Aza-3-cyano-1,4-quinones. J. Am. Chem. Soc. 1975, 97, 6181–6186. [Google Scholar] [CrossRef]
- Kappe, T.; Lang, G.; Pongratz, E. Thermolysis of Geminal Diazido Compounds. J. Chem. Soc. Chem. Commun. 1984, 338–339. [Google Scholar] [CrossRef]
- Kappe, C.O.; Färber, G. Thermolysis and Photolysis of 6-Diazidomethyl-1,2,3,4-tetrahydro-2-oxopyrimidine-5-carboxylates. J. Chem. Soc. Perkin Trans. 1 1991, 5, 1342–1344. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Kliegman, J.M. The Photochemical Decomposition of Geminal Diazides. II. The Solution Photolysis of Benzophenone Diazide. J. Am. Chem. Soc. 1967, 89, 5959–5960. [Google Scholar] [CrossRef]
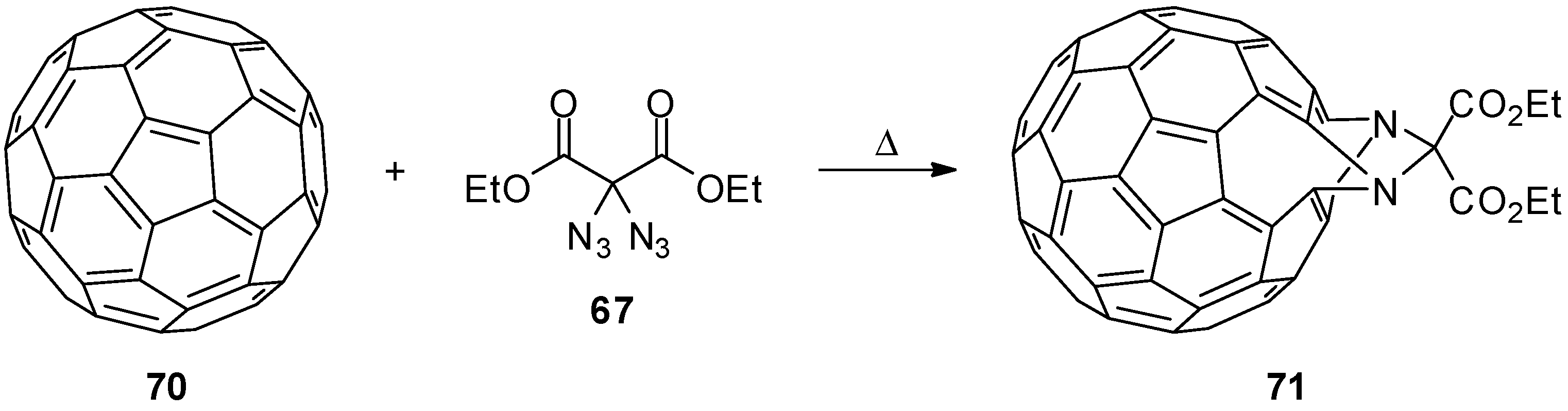
- Dong, G.-X.; Li, J.-S.; Chan, T.-H. Reaction of [60]Fullerene with Diethyl Diazidomalonate: A Doubly Bridged Fulleroid. J. Chem. Soc. Chem. Commun. 1995, 1725–1726. [Google Scholar] [CrossRef]
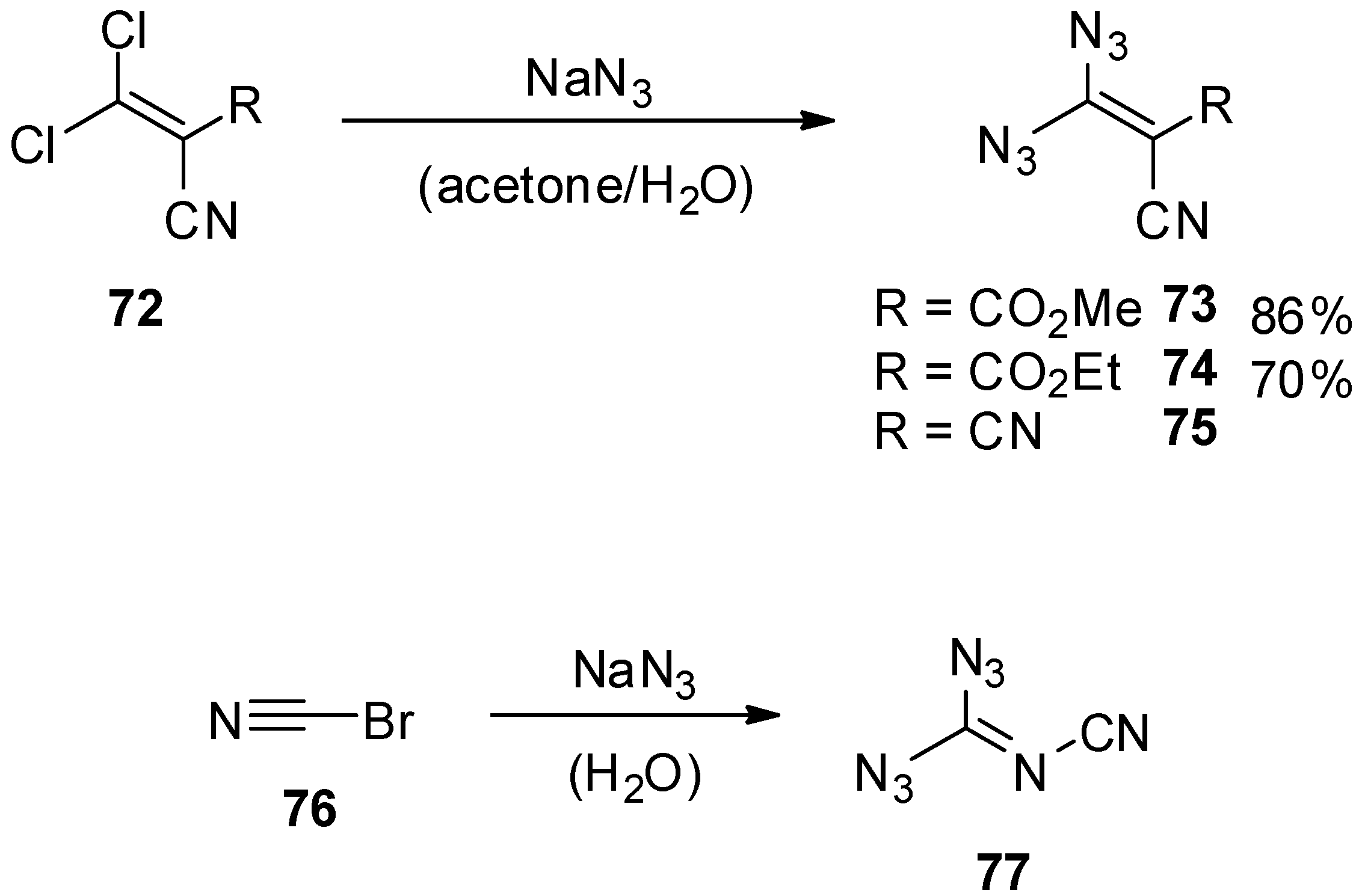
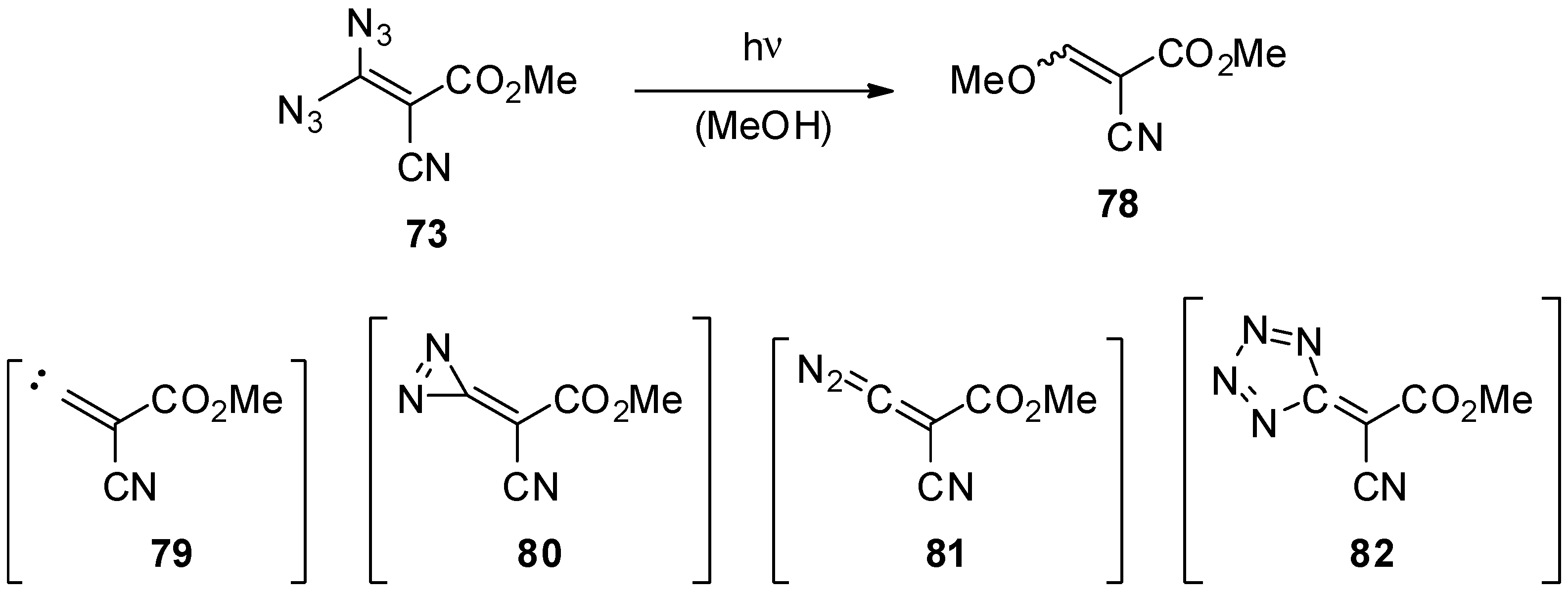
- Carrié, R.; Danion, D.; Ackermann, E.; Saalfrank, R.W. Geminal Vinyl Diazides: Potential Precursors of Functionalized Alkylidenecarbenes; Synthesis and Reactions of 3,3-Diazido-2-cyanoacrylic Acid Methyl Ester. Angew. Chem. Int. Ed. 1982, 21, 287. [Google Scholar] [CrossRef]
- Carrié, R.; Danion, D.; Ackermann, E.; Saalfrank, R.W. Geminale Vinyldiazide: Potentielle Vorstufen funktionalisierter Alkylidencarbene Synthese und Umsetzungen von 3,3-Diazido-2-cyan-acrylsäuremethylester. Angew. Chem. Suppl. 1982, 21, 660–667. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Ramezanian, M.; Saeva, F.D. A New Azacyanocarbon, C4N4: Tricyanomethanimine. Tetrahedron Lett. 1988, 29, 1235–1238. [Google Scholar] [CrossRef]
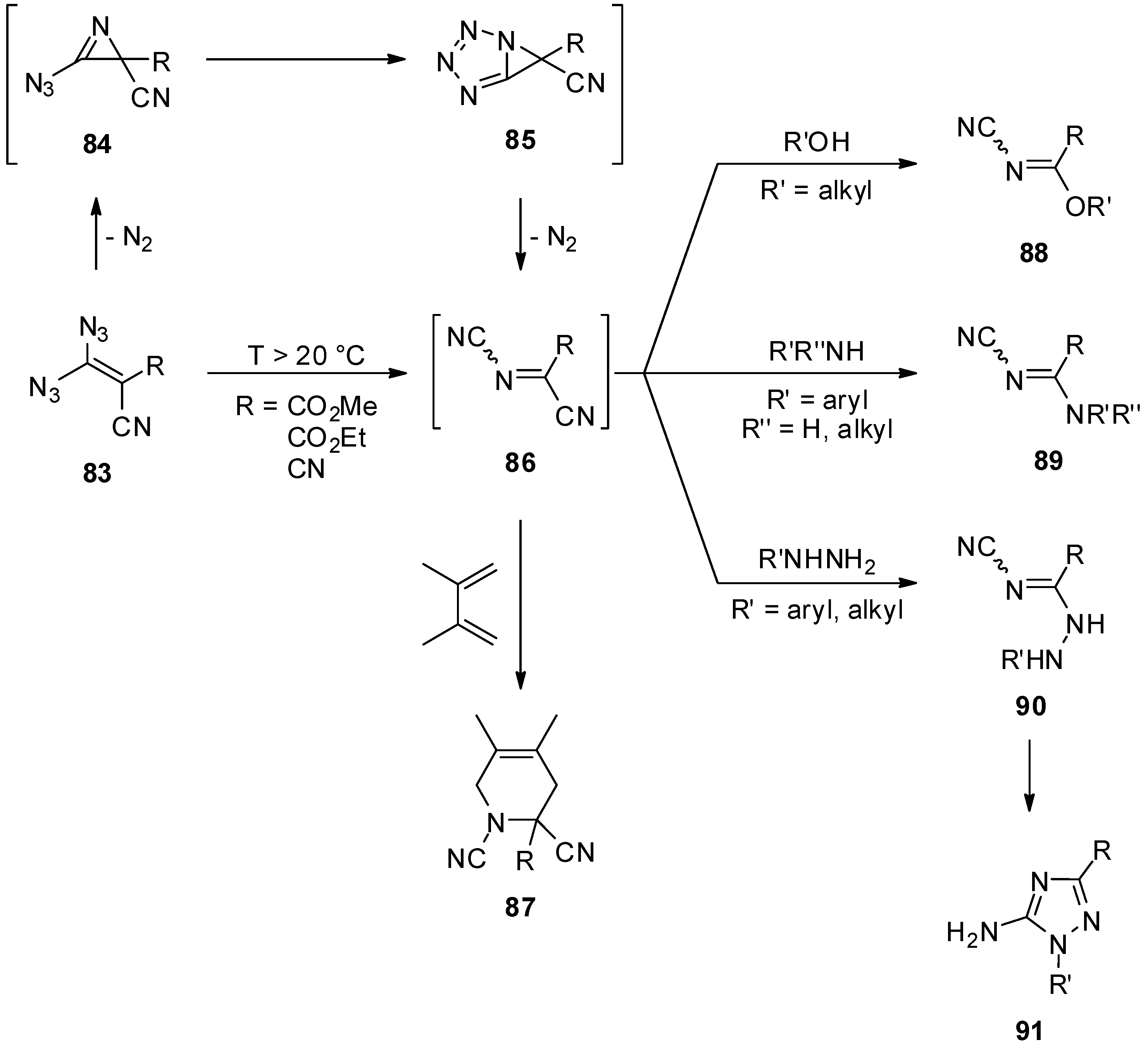
- Saalfrank, R.W.; Wirth, U. 4,5-Dihydro-1H-tetrazol-5-ylidene aus 3,3-Diazido-2-cyanacrylsäureestern und Hydrazinen, Hydraziden sowie O-substituierten Hydroxylaminen. Chem. Ber. 1989, 122, 519–522. [Google Scholar] [CrossRef]
- Darzens, M.G. Sur un perazoture de carbone. C. R. Hebd. Séances Acad. Sci. 1912, 154, 1232–1234. [Google Scholar]
- Hart, C.V. Carbonic Acid Azides. J. Am. Chem. Soc. 1928, 50, 1922–1930. [Google Scholar] [CrossRef]
- Ott, E.; Weißenburger, H. Über das Dibrom-malonitril und seine Umwandlung in Natrium-azidomalonitril und in ein dimolekulares Cyan-azid C2N8. Ber. Dtsch. Chem. Ges. A/B 1937, 70, 1829–1834. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Ackermann, E.; Fischer, M.; Wirth, U. Substituentenabhängige Konkurrenz zwischen 1,5- und 3,5-Cyclisierung bei Vinylaziden; 1,2,3-Triazole und 2H-Azirine aus 3,3-Diazido-2-cyanacrylsäure-methylester und Aminen. Chem. Ber. 1987, 120, 2003–2006. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Maid, H. Roots: From carbenes to allenes and coordination polymers. Ever present never twice the same. Chem. Commun. 2005, 5953–5967. [Google Scholar] [CrossRef] [PubMed]
- Carrié, R.; Danion, D.; Ackermann, E.; Saalfrank, R.W. Substituent-Dependent Competition between 1,5- and 3,5-Cyclization in Vinly Azides; 4H-Triazoles from 3,3-Diazido-2-cyanoacrylic Acid Methyl Ester and Primary Amines. Angew. Chem. Int. Ed. 1982, 21, 288. [Google Scholar] [CrossRef]
- Carrié, R.; Danion, D.; Ackermann, E.; Saalfrank, R.W. Substituentenabhängige Konkurrenz zwischen 1,5-und 3,5-Cyclisierung bei Vinylaziden 4H-Triazole aus 3,3-Diazido-2-cyan-acrylsäuremethylester und primären Aminen. Angew. Chem. Suppl. 1982, 21, 668–674. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Wirth, U. Bis(oxazole) und Tetrazolylammonium-Betaine aus Bis(vinylaziden). Chem. Ber. 1989, 122, 969–973. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Ackermann, E.; Fischer, M.; Wirth, U.; Zimmermann, H. Oxazole aus 3,3-Diazido-2-cyanacrylsäure-methylester und Aminen. Chem. Ber. 1990, 123, 115–120. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Lurz, C.-J.; Hassa, J.; Danion, D.; Toupet, L. 3,3-Diazido-2-cyanacrylsäure-methylester: Synthese von Vinylaziden, 4,5-Dihydro-1H-tetrazol-5-ylidenen, Oxazolen und N-Cyaniminen. Chem. Ber. 1991, 124, 595–608. [Google Scholar] [CrossRef]
- Ramezanian, M.; Padias, A.B.; Saeva, F.D.; Hall, H.K., Jr. Synthesis and Reactions of Highly Electrophilic Imines Containing the N-Cyano group. J. Org. Chem. 1990, 55, 1768–1771. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Fischer, M.; Wirth, U.; Zimmermann, H. Methyl (E)-2-(1-Aryl-4,5-dihydro-1H-tetrazol-5-ylidene)-2-cyanoacetate from Methyl 3,3-Diazido-2-cyanoacrylate and Primary Aromatic Amines. Angew. Chem. Int. Ed. 1987, 26, 1160–1161. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Wirth, U.; Lurz, C.-J. Substituent-Dependent Competition between 1,5- and 1,5′-Cyclization of Vinyl Azides. 1,2,3-Triazoles and 4,5-Dihydro-1H-tetrazol-5-ylidenes from Methyl 3,3-Diazido-2-cyanoacrylate with Amines. J. Org. Chem. 1989, 54, 4356–4359. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Häring, A.P.; Kirsch, S.F. Synthesis and Chemistry of Organic Geminal Di- and Triazides. Molecules 2015, 20, 20042-20062. https://doi.org/10.3390/molecules201119675
Häring AP, Kirsch SF. Synthesis and Chemistry of Organic Geminal Di- and Triazides. Molecules. 2015; 20(11):20042-20062. https://doi.org/10.3390/molecules201119675
Chicago/Turabian StyleHäring, Andreas P., and Stefan F. Kirsch. 2015. "Synthesis and Chemistry of Organic Geminal Di- and Triazides" Molecules 20, no. 11: 20042-20062. https://doi.org/10.3390/molecules201119675
APA StyleHäring, A. P., & Kirsch, S. F. (2015). Synthesis and Chemistry of Organic Geminal Di- and Triazides. Molecules, 20(11), 20042-20062. https://doi.org/10.3390/molecules201119675