Activity of Antifungal Organobismuth(III) Compounds Derived from Alkyl Aryl Ketones against S. cerevisiae: Comparison with a Heterocyclic Bismuth Scaffold Consisting of a Diphenyl Sulfone

Abstract
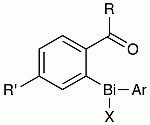
:
1. Introduction
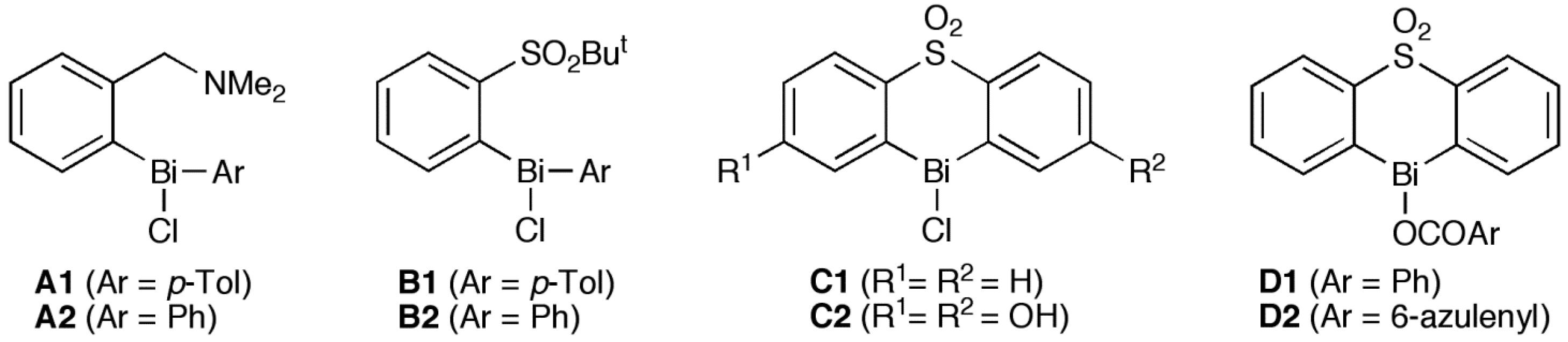

{kind=link}
{kind=link}
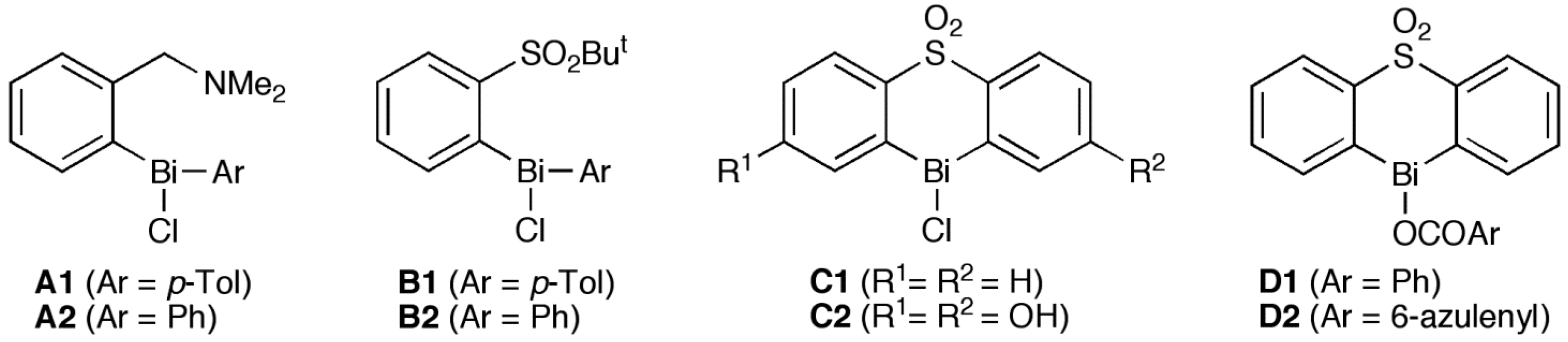{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
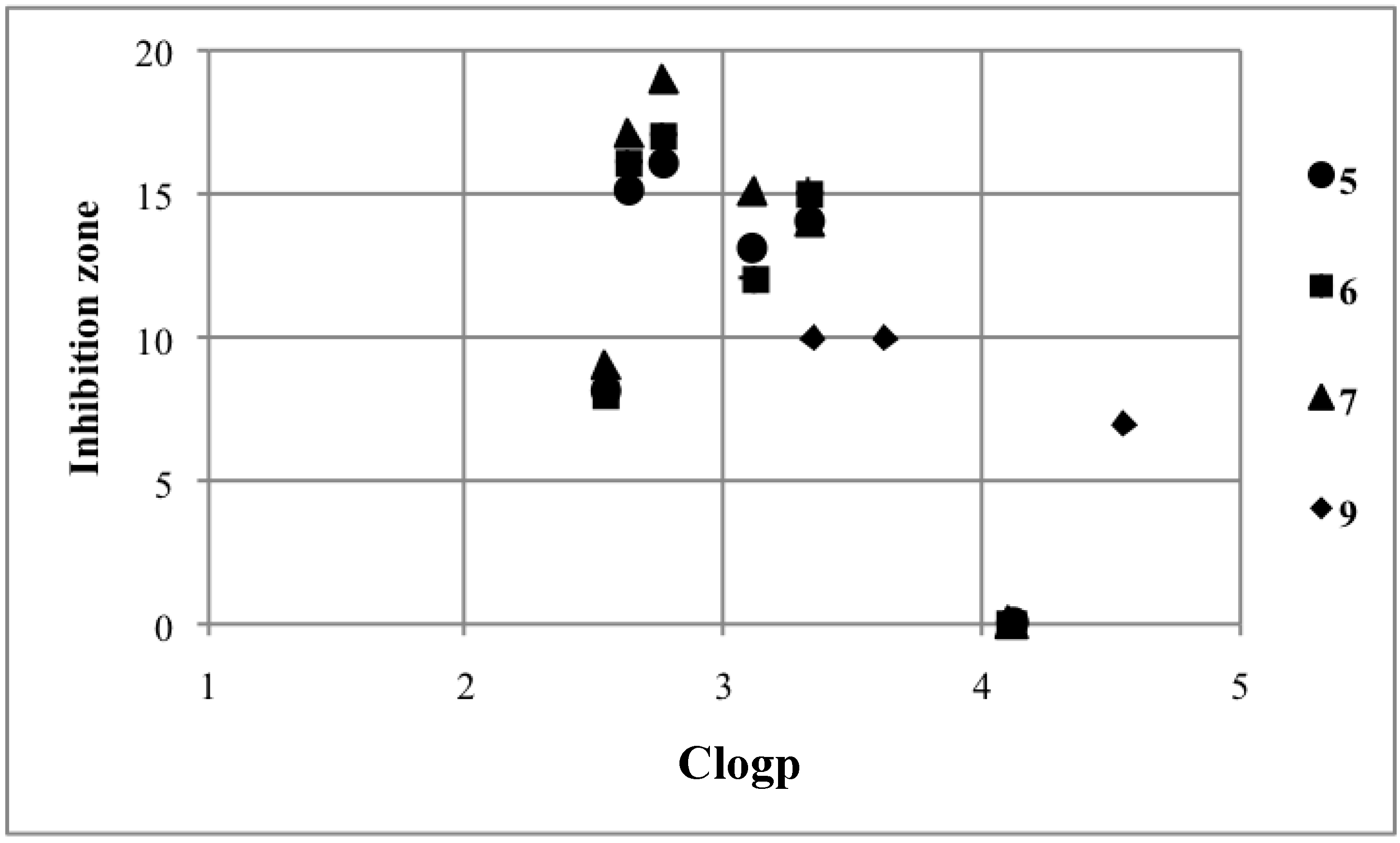
| Compound | Inhibition Zone (mm) | ClogP | Compound | Inhibition Zone (mm) | ClogP |
|---|---|---|---|---|---|
| A1 | 13 | 3.52 | 6a | 8 | 2.54 |
| A2 | 12 | 3.02 | 6b | 12 | 3.12 |
| B1 | 8 | 3.28 | 6c | 16 | 2.63 |
| B2 | 12 | 2.78 | 6d | 17 | 2.77 |
| C1 | 18 | 1.18 | 6e | 15 | 3.34 |
| C2 | 8 | 0.81 | 6f | 0 | 4.12 |
| D1 | 17 | 3.49 | 7a | 9 | 2.54 |
| D2 | 12 | 4.66 | 7b | 15 | 3.12 |
| 7c | 17 | 2.63 | |||
| 4b | 0 | 5.81 | 7d | 19 | 2.77 |
| 4c | 0 | 4.82 | 7e | 14 | 3.34 |
| 4d | 0 | 5.10 | 7f | 0 | 4.12 |
| 4e | 0 | 6.24 | 9a | 10 | 3.62 |
| 5a | 8 | 2.54 | 9b | 7 | 4.55 |
| 5b | 13 | 3.12 | 9c | 10 | 3.34 |
| 5c | 15 | 2.63 | 11a | 9 | 4.18 |
| 5d | 16 | 2.77 | 11b | 8 | 4.71 |
| 5e | 14 | 3.34 | 11c | 10 | 3.96 |
| 5f | 0 | 4.12 | Nystatin | 30 |
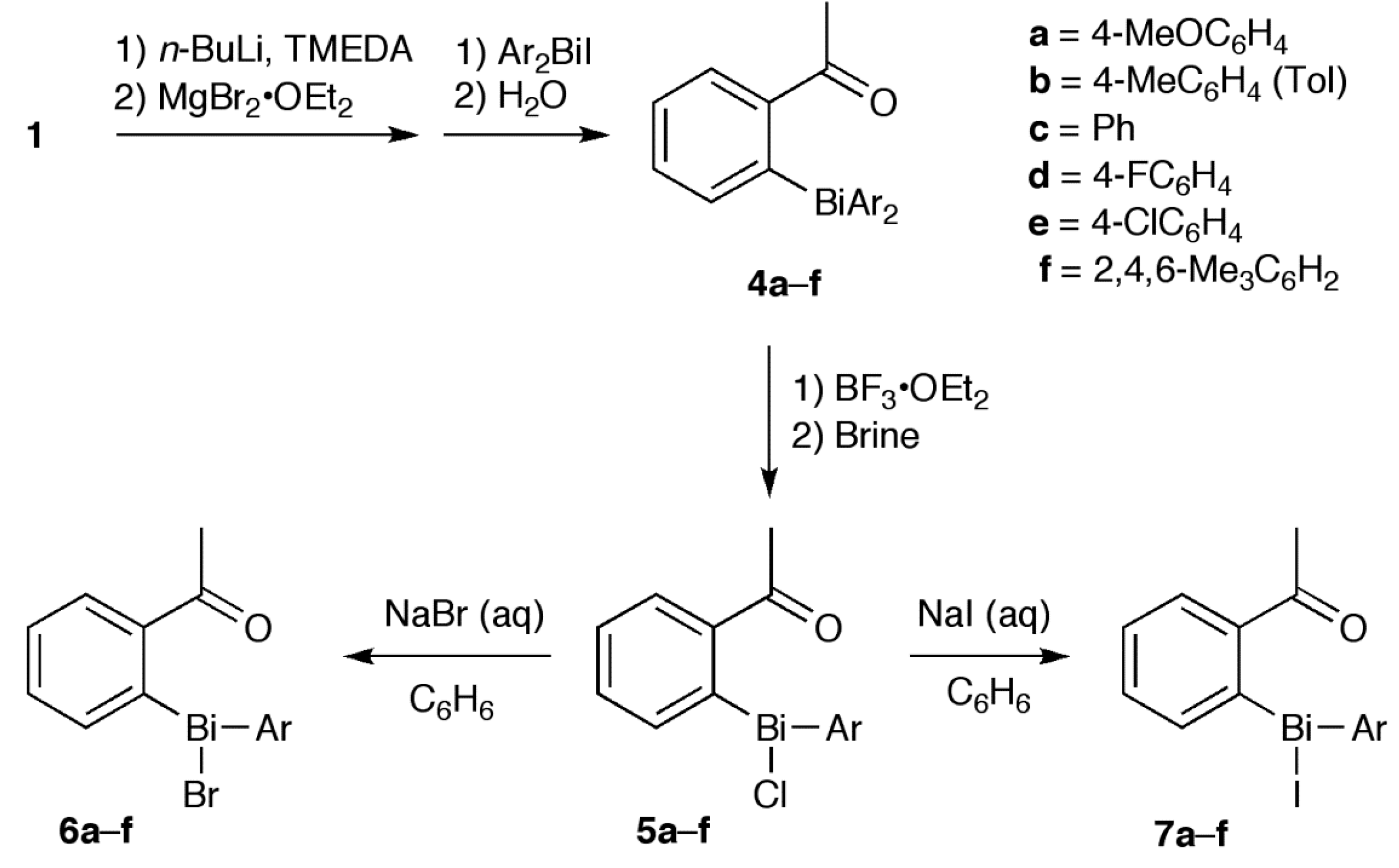
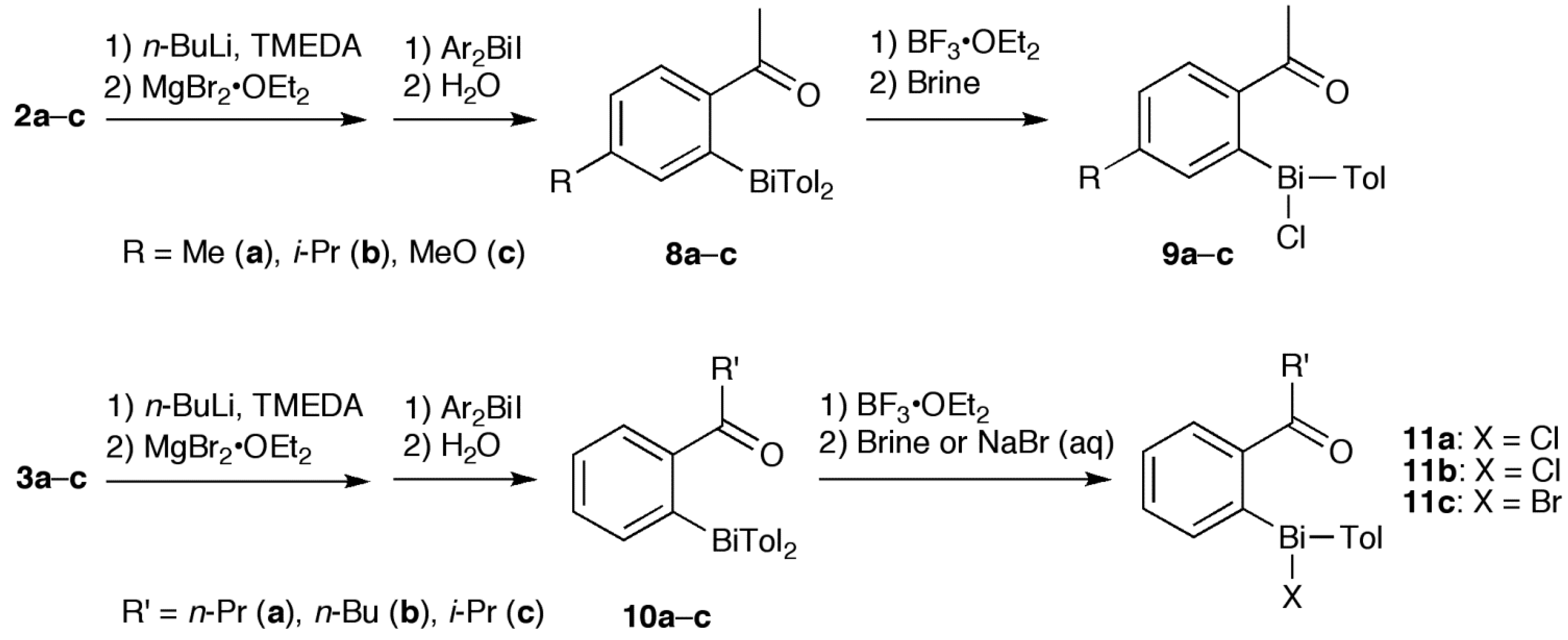
2. Results and Discussion
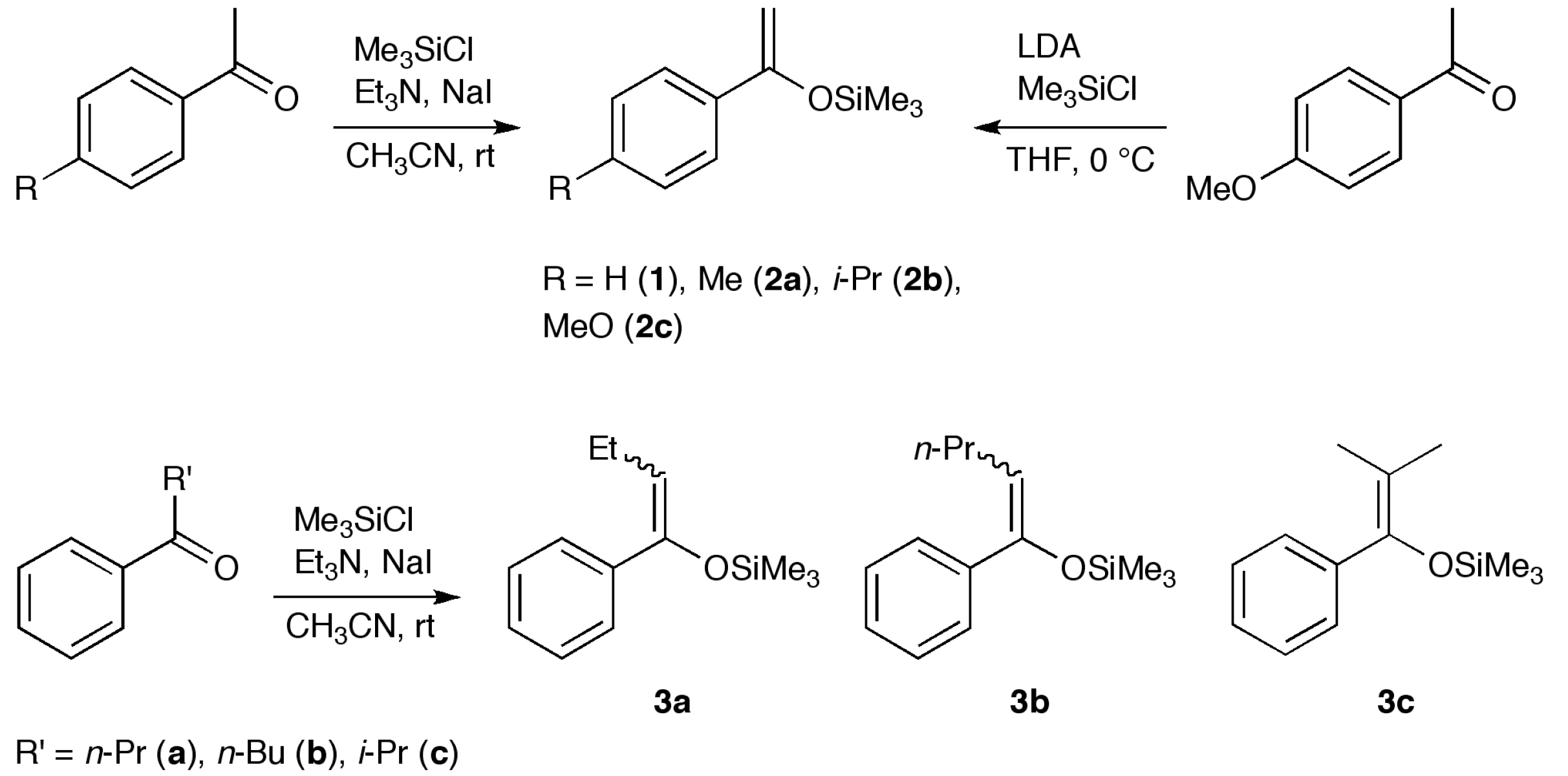
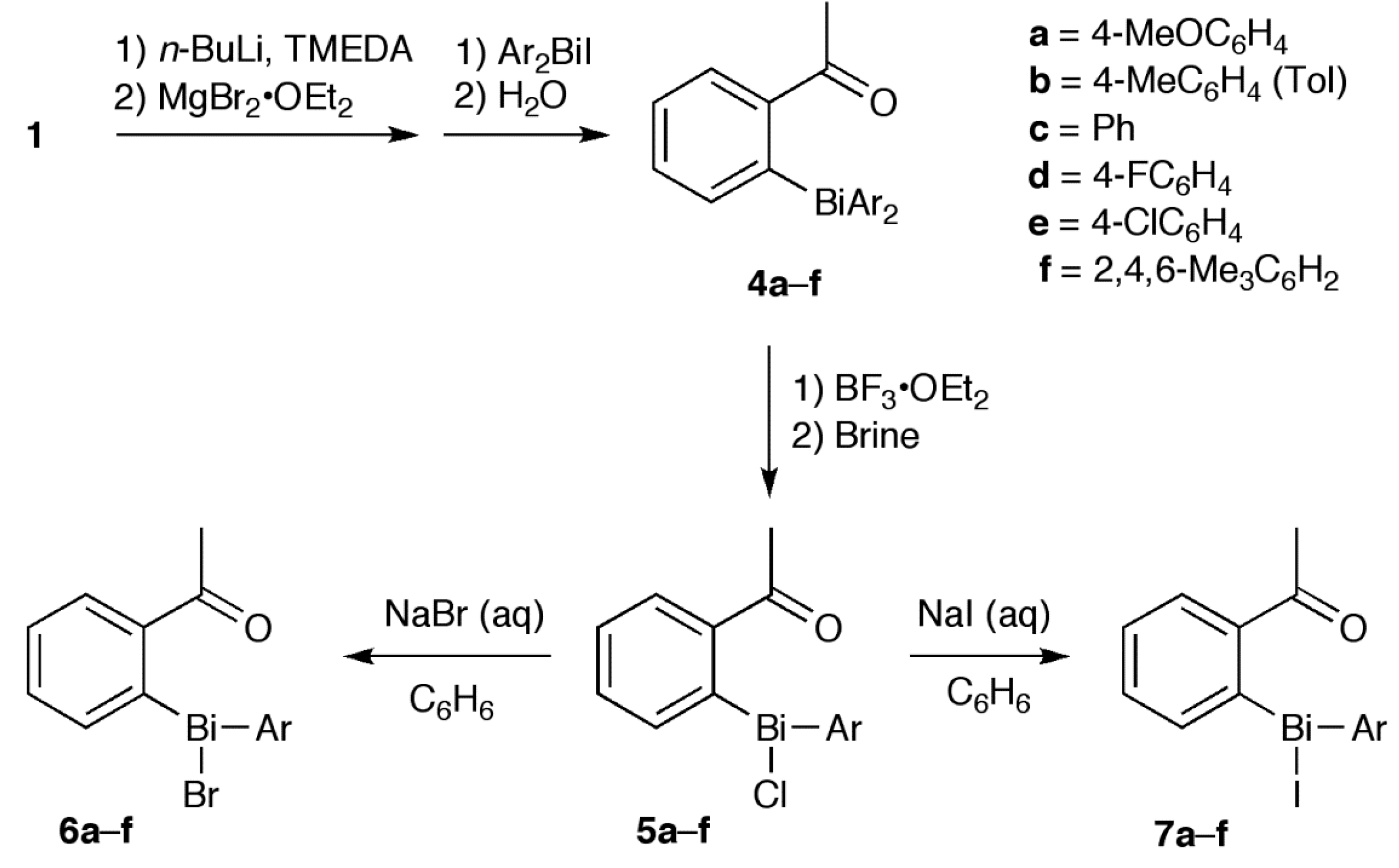
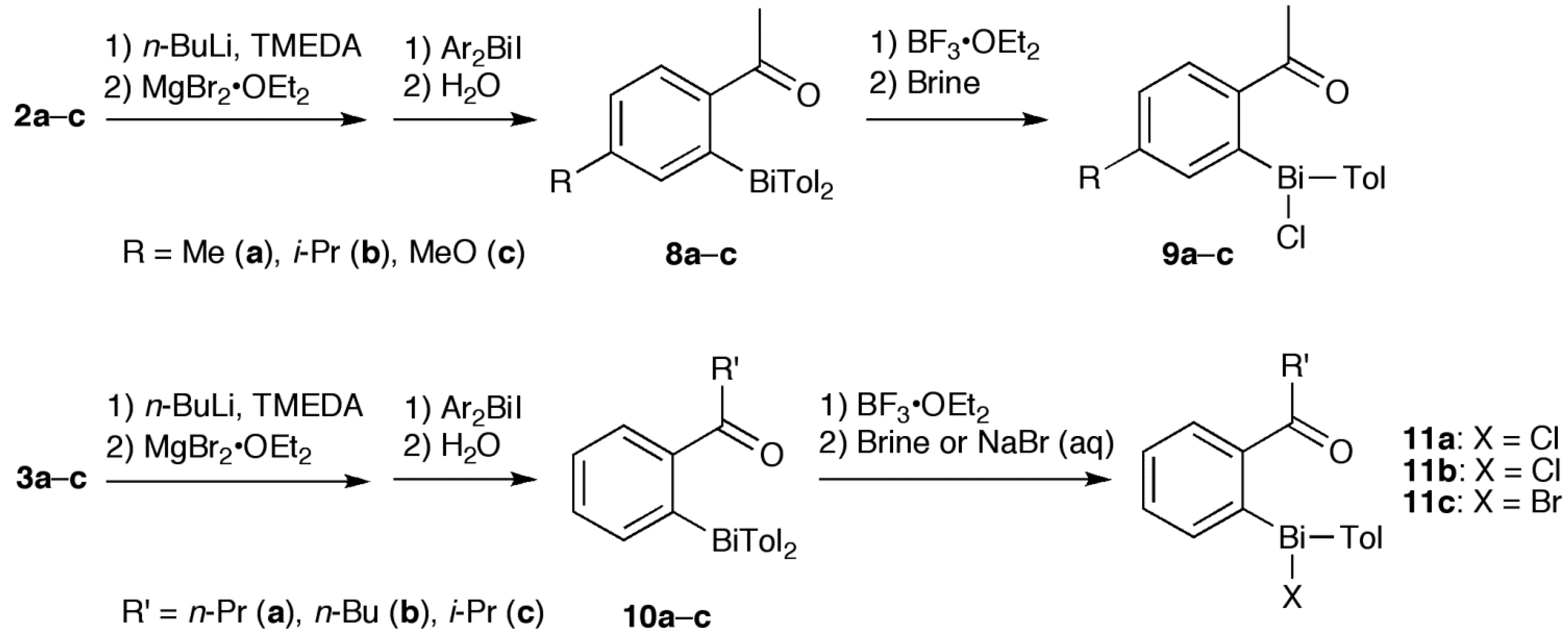
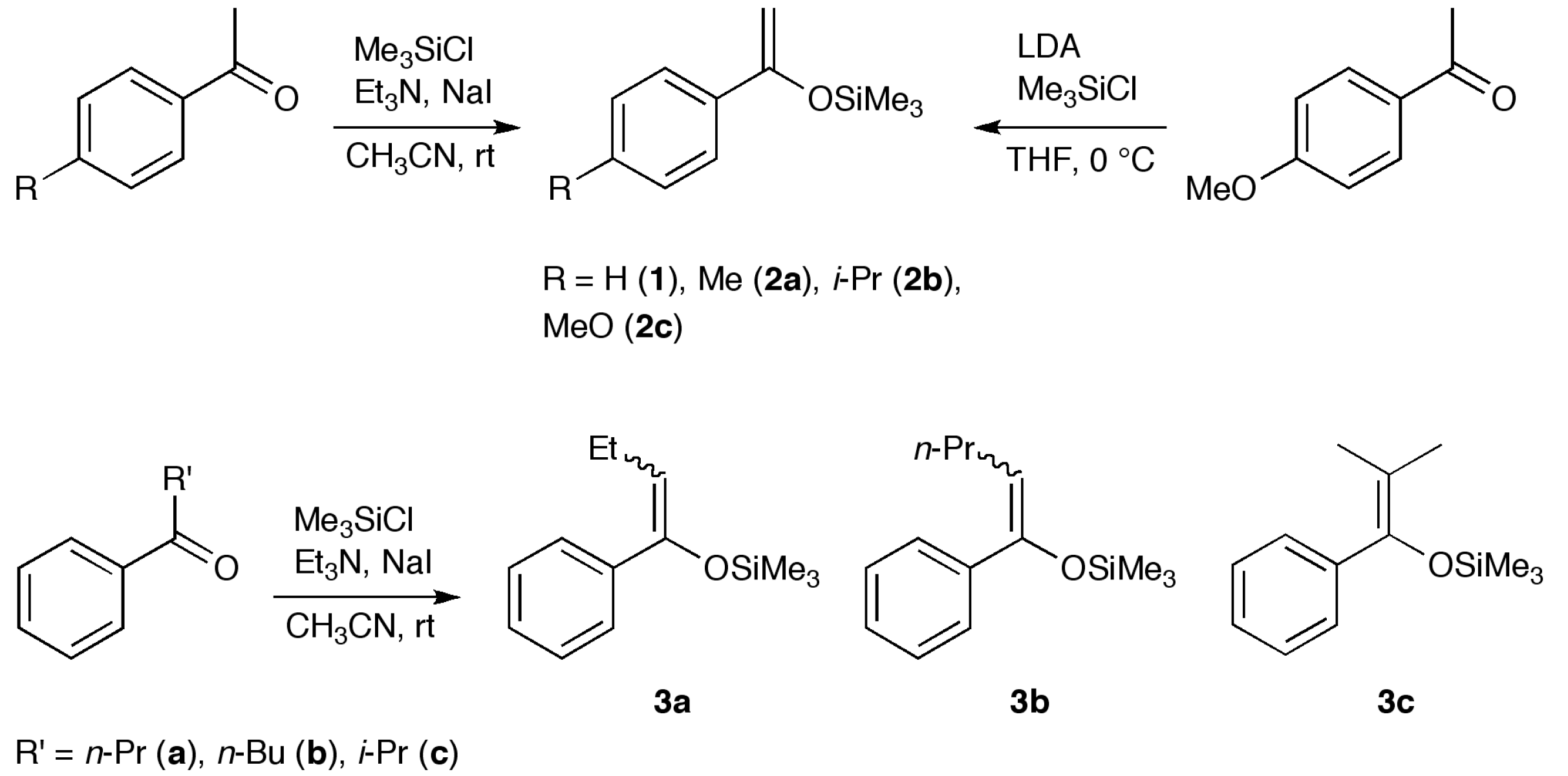
2.1. Synthesis
2.2. Growth Inhibition Tests against S. cerevisiae
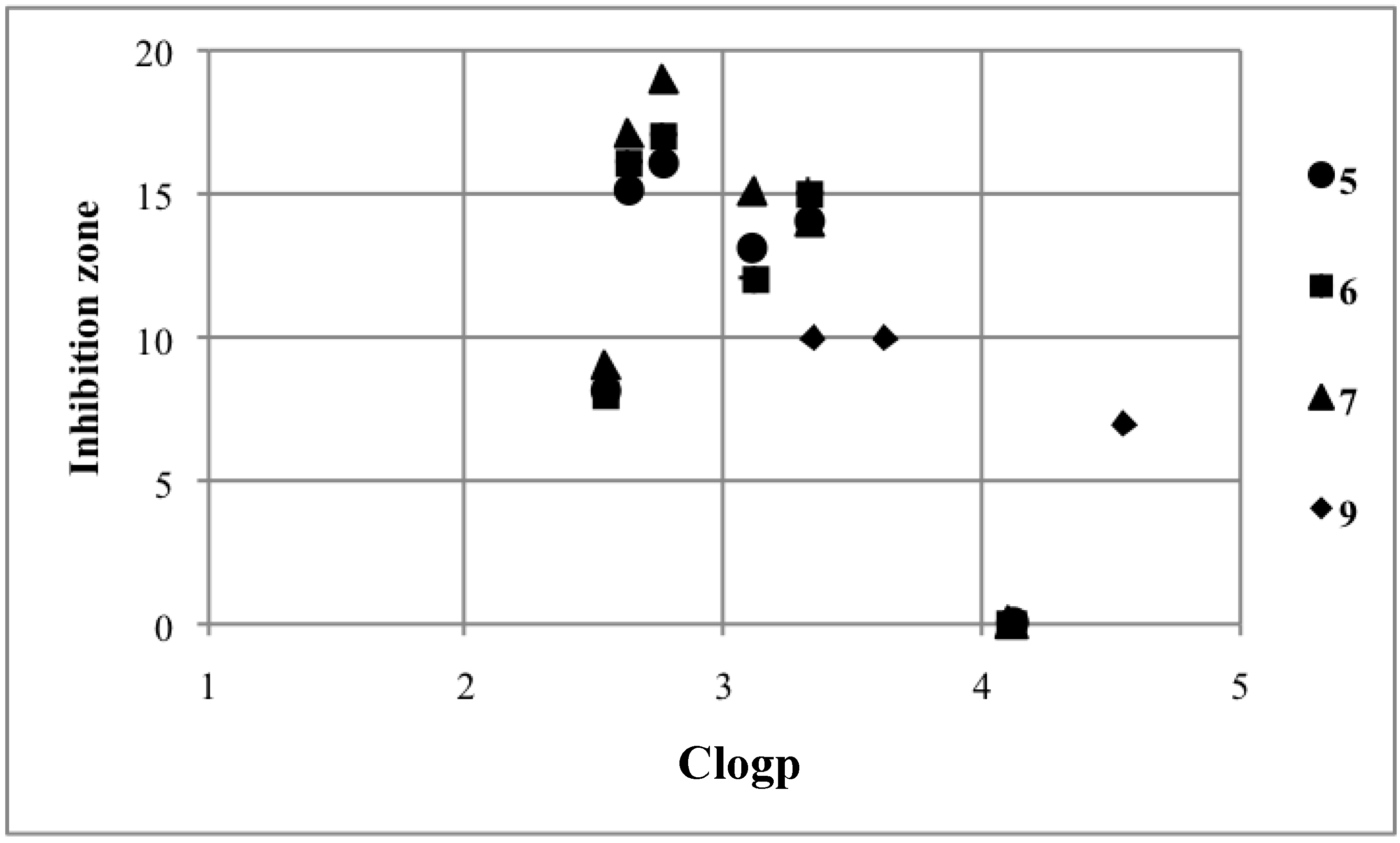
2.3. Structure-Activity Relationship
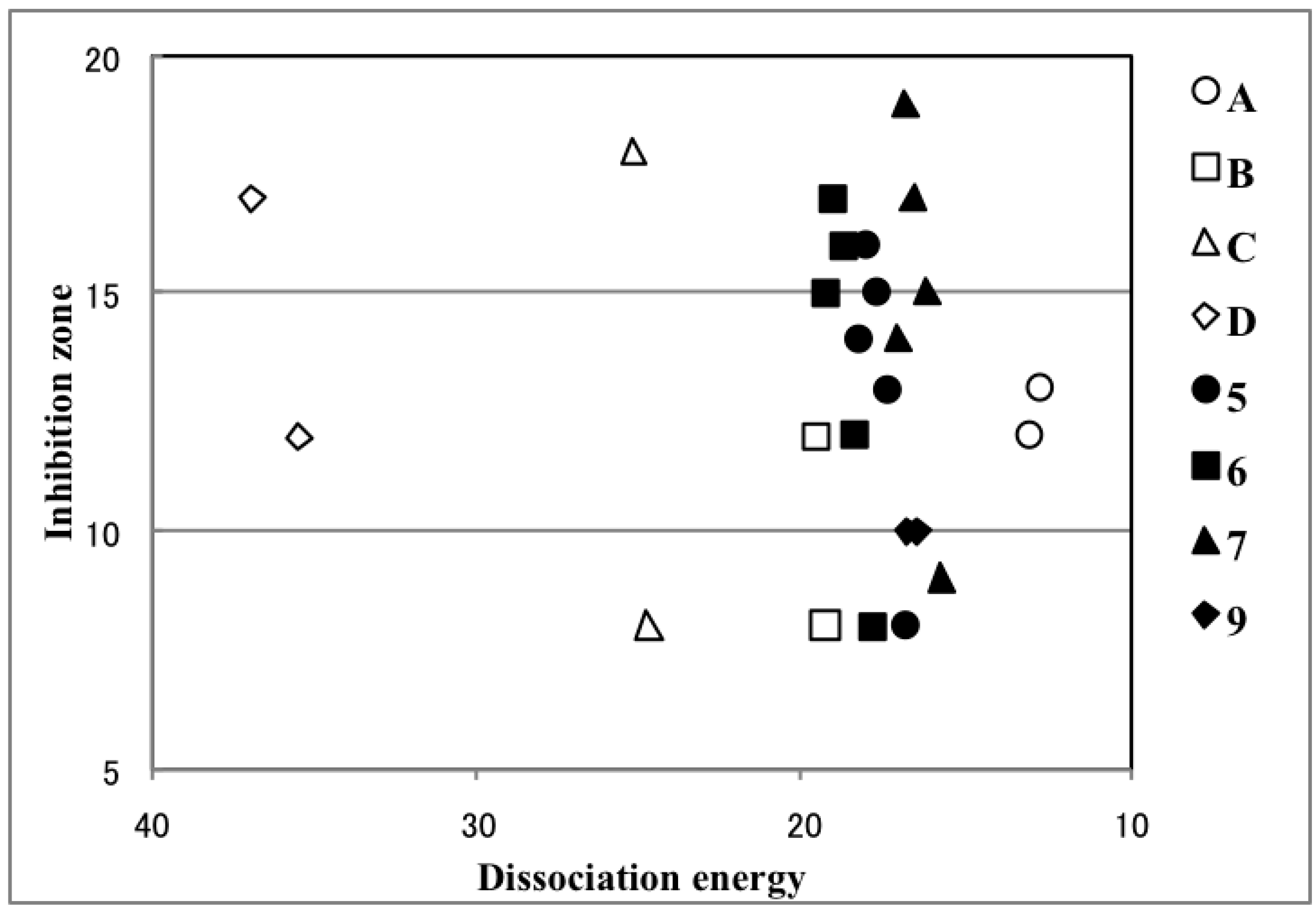
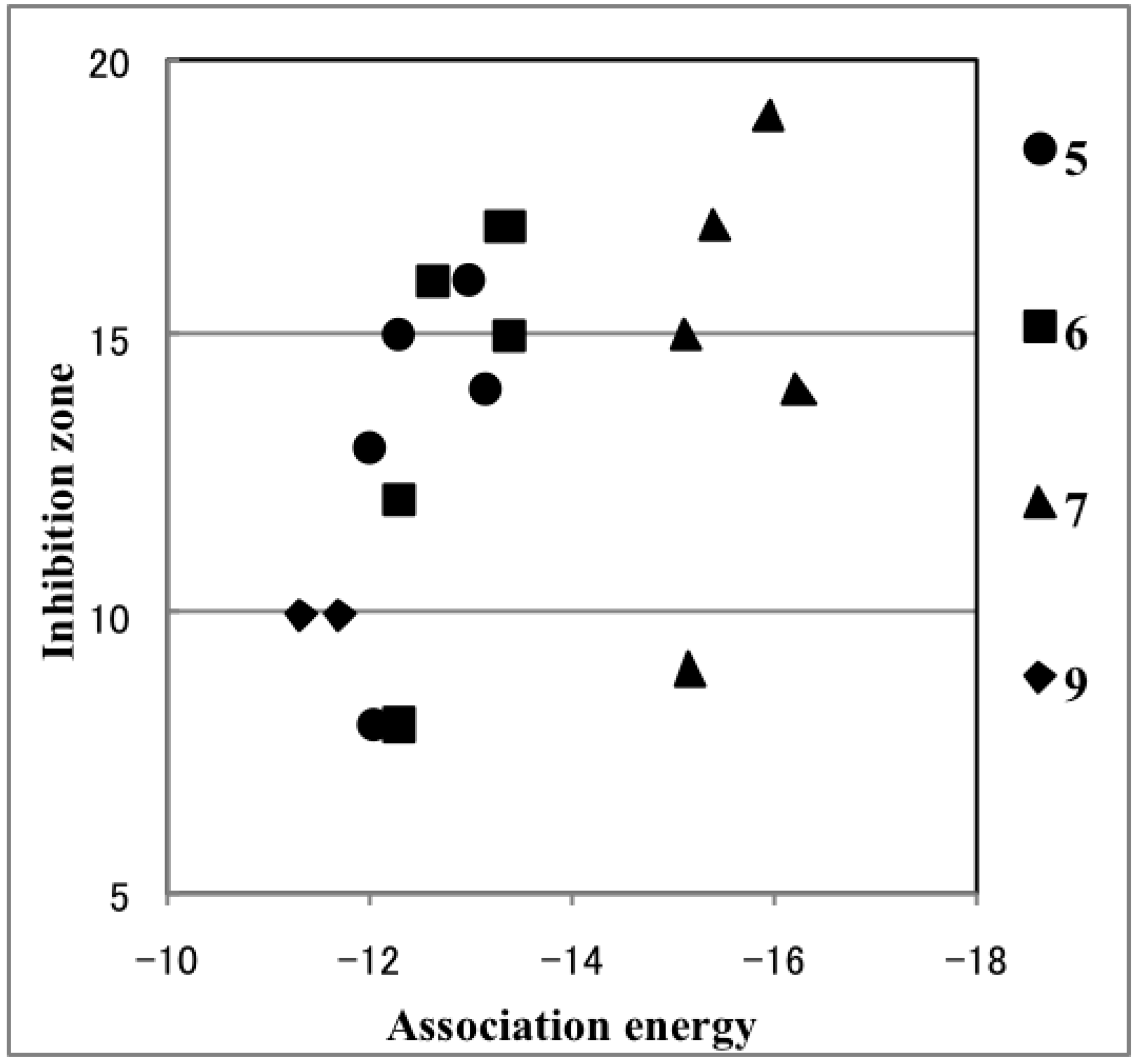
2.4. Plausible Mechanism of Action for Hypervalent Bismuthanes
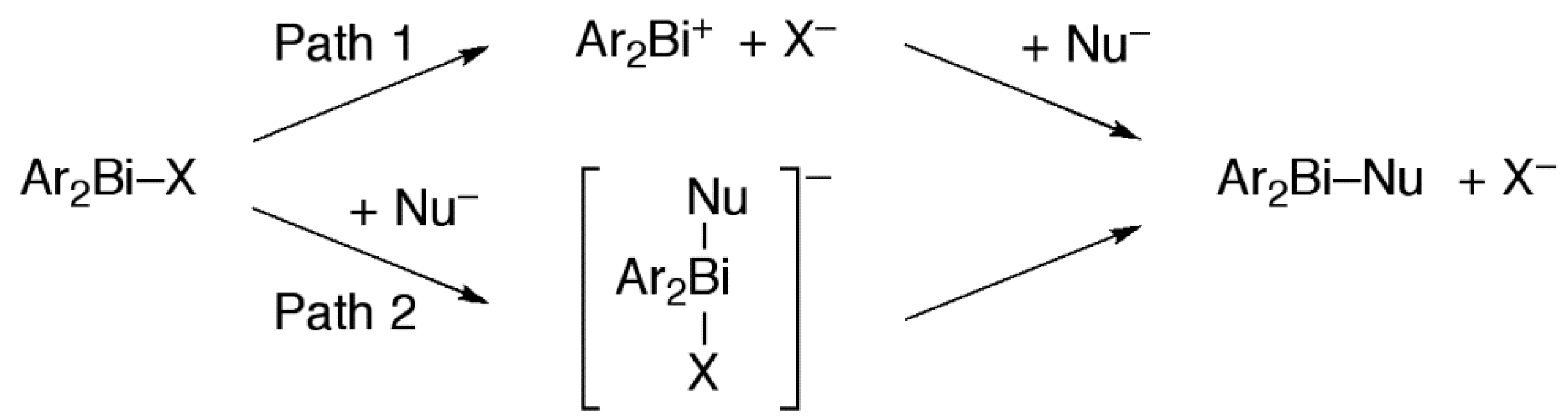
| Ar2BiX | Inhibition Zone (mm) | Path 1 (kcal/mol) | Path 2 (kcal/mol) | Ar2BiX | Inhibition Zone (mm) | Path 1 (kcal/mol) | Path 2 (kcal/mol) |
|---|---|---|---|---|---|---|---|
| A1 | 13 | 12.82 | –8.18 | 6a | 8 | 17.93 | –12.29 |
| A2 | 12 | 13.12 | –8.46 | 6b | 12 | 18.38 | –12.33 |
| B1 | 8 | 19.33 | –13.51 | 6c | 16 | 18.73 | –12.65 |
| B2 | 12 | 19.66 | –13.85 | 6d | 17 | 19.06 | –13.29 |
| C1 | 18 | 25.22 | –17.22 | 6e | 15 | 19.37 | –13.38 |
| C2 | 8 | 24.75 | –17.15 | 6f | 0 | 18.15 | – |
| D1 | 17 | 36.93 | –10.09 | 7a | 9 | 15.81 | –15.18 |
| D2 | 12 | 35.49 | –10.97 | 7b | 15 | 16.22 | –15.13 |
| 5a | 8 | 16.93 | –12.08 | 7c | 17 | 16.59 | –15.43 |
| 5b | 13 | 17.37 | –12.00 | 7d | 19 | 16.89 | –16.00 |
| 5c | 15 | 17.75 | –12.33 | 7e | 14 | 17.14 | –16.26 |
| 5d | 16 | 18.07 | –13.02 | 7f | 0 | 18.15 | - |
| 5e | 14 | 18.36 | –13.14 | 9a | 10 | 16.83 | –11.71 |
| 5f | 0 | 18.15 | - | 9c | 10 | 16.54 | –11.32 |
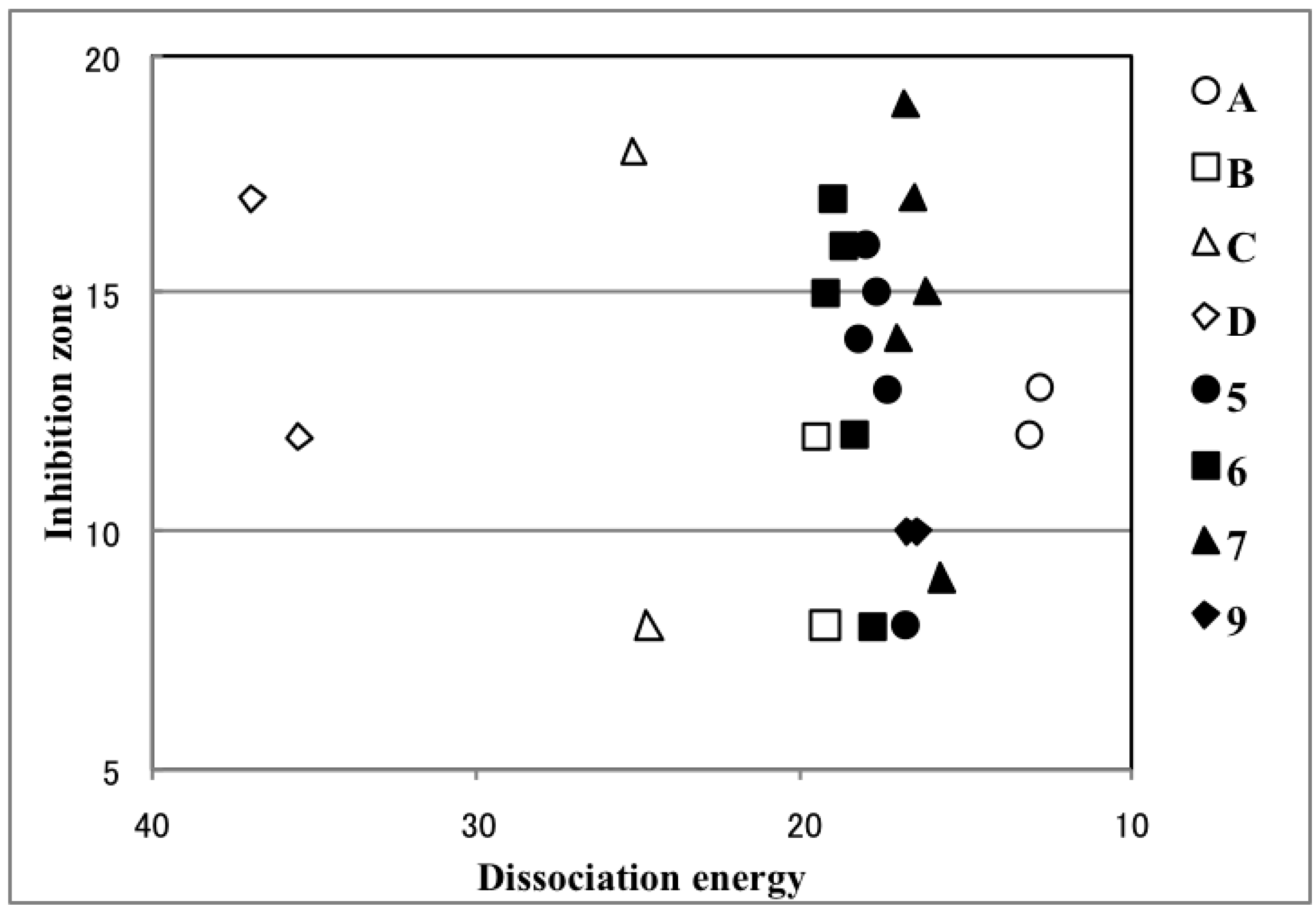
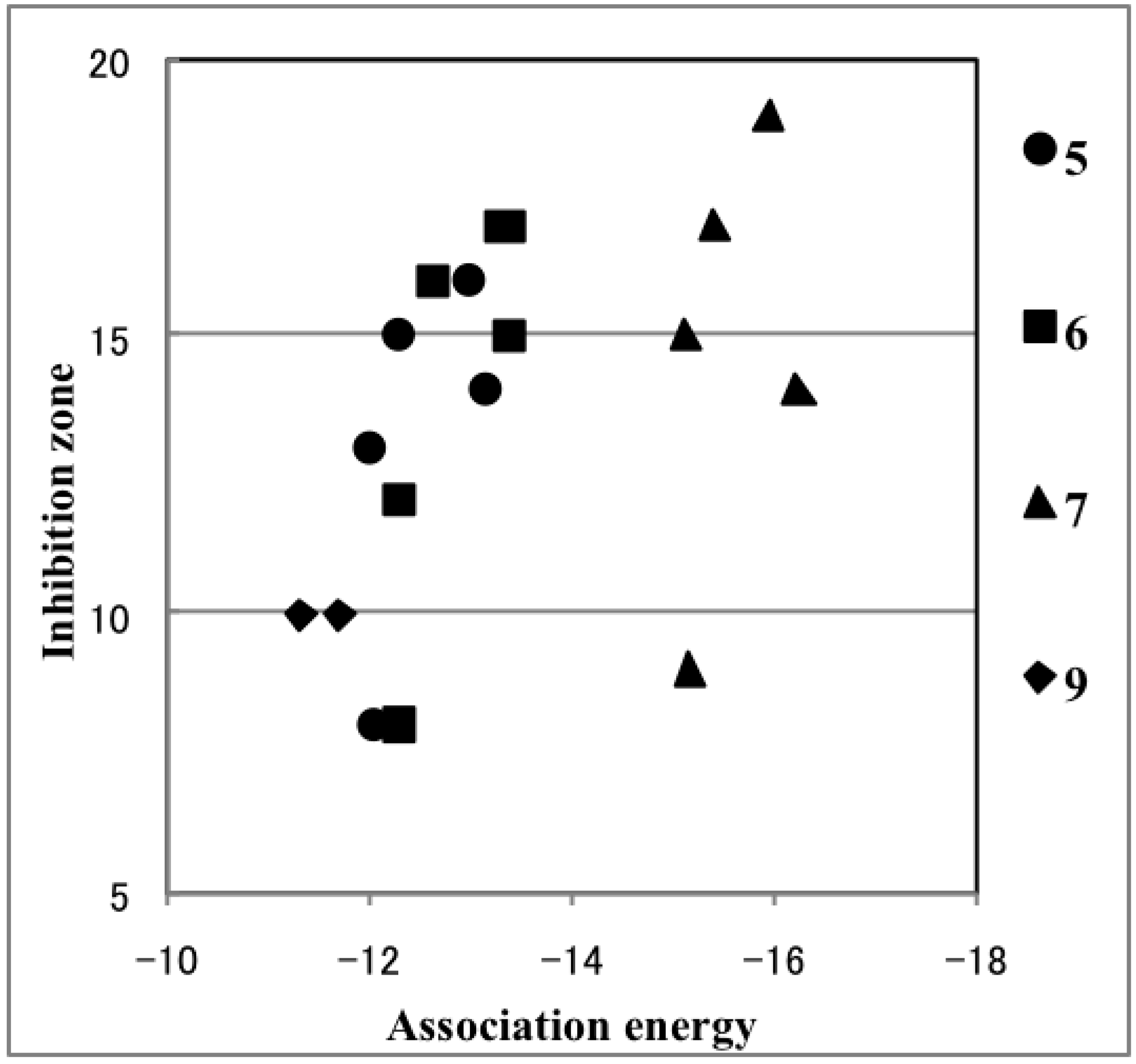
3. Experimental Section
3.1. General Information
3.2. Synthesis of 4, 8 and 10
3.3. Synthesis of 5a, 5f and 11
3.4. Synthesis of 5b–e and 9
3.5. Synthesis of 6 and 7
3.6. Qualitative Antifungal Assay
3.7. Lipophilicity
3.8. DFT Calculation of the Dissociation and Association Energies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Onishi, K.; Douke, M.; Nakamura, T.; Ochiai, Y.; Kakusawa, N.; Yasuike, S.; Kurita, J.; Yamamoto, C.; Kawahata, M.; Yamaguchi, K.; et al. A novel organobismuth compound, 1-[(2-di-p-tolylbismuthanophenyl)diazenyl]pyrrolidine, induces apoptosis in the human acute promyelocytic leukemia cell line NB4 via reactive oxygen species. J. Inorg. Biochem. 2012, 117, 77–84. [Google Scholar] [CrossRef]
- Andrews, P.C.; Frank, R.; Junk, P.C.; Kedzierski, L.; Kumar, I.; MacLellan, J.G. Anti-Leishmanial activity of homo- and heteroleptic bismuth(III) carboxylates. J. Inorg. Biochem. 2011, 105, 454–461. [Google Scholar] [CrossRef]
- Iuchi, K.; Hatano, Y.; Yagura, T. Heterocyclic organobismuth(III) induces apoptosis of human promyelocytic leukemic cells through activation of caspases and mitochondrial perturbation. Biochem. Pharmacol. 2008, 76, 974–986. [Google Scholar] [CrossRef]
- Li, H.; Lai, C.S.; Wu, J.; Ho, P.C.; de Vos, D.; Tiekink, E.R.T. Cytotoxicity, qualitative structure-activity relationship (QSAR), and anti-tumor activity of bismuth dithiocarbamate complexes. J. Inorg. Biochem. 2007, 101, 809–816. [Google Scholar] [CrossRef]
- Islam, A.; da Silva, J.G.; Berbet, F.M.; da Silva, S.M.; Rodrigues, B.L.; Beraldo, H.; Melo, M.N.; Frézard, F.; Demicheli, C. Novel triphenylantimony(V) and triphenylbismuth(V) complexes with benzoic acid derivatives: Structural characterization, in vitro antileishmanial and antibacterial activities and cytotoxicity against macrophages. Molecules 2014, 19, 6009–6030. [Google Scholar] [CrossRef]
- Briand, G.G.; Burford, N. Bismuth compounds and preparations with biological or medicinal relevance. Chem. Rev. 1999, 99, 2601–2657. [Google Scholar] [CrossRef]
- Condon, S.; Pichon, C.; Davi, M. Preparation and synthetic applications of trivalent arylbismuth compounds as arylating reagents. A review. Org. Prep. Proced. Int. 2014, 46, 89–131. [Google Scholar] [CrossRef]
- Shimada, S.; Rao, M.L.N. Transition-metal catalyzed C-C bond formation using organobismuth compounds. Top. Curr. Chem. 2012, 311, 199–228. [Google Scholar] [CrossRef]
- Braunschweig, H.; Cogswell, P.; Schwab, K. Synthesis, structure and reactivity of complexes containing a transition metal-bismuth bond. Coord. Chem. Rev. 2011, 255, 101–117. [Google Scholar] [CrossRef]
- Murafuji, T.; Miyoshi, Y.; Ishibashi, M.; Rahman, A.F.M.M.; Sugihara, Y.; Miyakawa, I.; Uno, H. Antifungal activity of organobismuth compounds against the yeast Saccharomyces cerevisiae: Structure-activity relationship. J. Inorg. Biochem. 2004, 98, 547–552. [Google Scholar] [CrossRef]
- Murafuji, T.; Fujiwara, Y.; Yoshimatsu, D.; Miyakawa, I.; Migita, K.; Mikata, Y. Bismuth heterocycles based on a diphenyl sulfone scaffold: Synthesis and substituent effect on the antifungal activity against Saccharomyces cerevisiae. Eur. J. Med. Chem. 2011, 46, 519–525. [Google Scholar] [CrossRef]
- Murafuji, T.; Kitagawa, K.; Yoshimatsu, D.; Kondo, K.; Ishiguro, K.; Tsunashima, R.; Miyakawa, I.; Mikata, Y. Heterocyclic bismuth carboxylates based on a diphenyl sulfone scaffold: Synthesis and antifungal activity against Saccharomyses cerevisiae. Eur. J. Med. Chem. 2013, 63, 531–535. [Google Scholar] [CrossRef]
- Chen, X.; Yamamoto, Y.; Akiba, K.-Y. Hypervalent tetracoordinate organobismuth compounds (10-Bi-4). Heteroatom Chem. 1995, 6, 293–303. [Google Scholar] [CrossRef]
- Ohkata, K.; Takemoto, S.; Ohnishi, M.; Akiba, K.-Y. Synthesis and chemical behaviors of 12-substituted dibenz[c,f][1,5]azastibocine and dibenz[c,f][1,5]azabismocine derivatives: Evidence of 10-Pn-4 type hypervalent interaction. Tetrahedron Lett. 1989, 30, 4841–4844. [Google Scholar]
- Qiu, R.; Yin, S.; Zhang, X.; Xia, J.; Xu, X.; Luo, S. Synthesis and structure of an air-stable cationic organobismuth complex and its use as a highly efficient catalyst for the direct diastereoselective Mannich reaction in water. Chem. Comm. 2009, 4759–4761. [Google Scholar]
- Bao, M.; Hayashi, T.; Shimada, S. Cationic organobismuth complex with 5,6,7,12-tetrahydrodibenz[c,f][1,5]azabismocine framework and its coordination complexes with neutral molecules. Organometallics 2007, 26, 1816–1822. [Google Scholar] [CrossRef]
- Rajabi, L.; Courreges, C.; Montoya, J.; Aguilera, R.J.; Primm, T.P. Acetophenones with selective antimycobacterial activity. Lett. Appl. Microbiol. 2005, 40, 212–217. [Google Scholar] [CrossRef]
- Sivakumar, P.M.; Sheshayan, G.; Doble, M. Experimental and QSAR of acetophenones as antibacterial agents. Chem. Biol. Drug. Des. 2008, 72, 303–313. [Google Scholar] [CrossRef]
- Li, Q.; Ning, P.; Zheng, L.; Huang, J.; Li, G.; Hsiang, T. Fumigant activity of volatiles of Streptomyces globisporus JK-1 against Penicillium italicum on Citrus microcarpa. Postharv. Biol. Technol. 2010, 58, 157–165. [Google Scholar] [CrossRef]
- Cazeau, P.; Duboudin, F.; Moulines, F.; Babot, O.; Dunogues, J. A new practical synthesis of silyl enol ethers: Part. 1. From simple aldehydes and ketones. Tetrahedron 1987, 43, 2075–2088. [Google Scholar] [CrossRef]
- Prantz, K.; Mulzer, J. Synthesis of (Z)-trisubstituted olefins by decarboxylative Grob-type fragmentations: Epothilone D, discodermolide, and peloruside A. Chem. Eur. J. 2010, 16, 485–506. [Google Scholar] [CrossRef]
- Murafuji, T.; Mutoh, T.; Satoh, K.; Tsunenari, K.; Azuma, N.; Suzuki, H. Hypervalent bond formation in halogeno(2-acylphenyl)bismuthanes. Organometallics 1995, 14, 3848–3854. [Google Scholar] [CrossRef]
- Klein, J.; Medlik-Balan, A. Metalation reactions. 18. Polymetalation substituted acetophenones. J. Org. Chem. 1976, 41, 3307–3312. [Google Scholar] [CrossRef]
- Suzuki, H.; Murafuji, T.; Azuma, N. Synthesis and first X-ray structure analysis of a stabilized chiral chlorobismuthine: Fixation of molecular geometry induced by the intramolecular coordination of a sulfonyl group. J. Chem. Soc. Perkin Trans. 1 1993, 1169–1175. [Google Scholar] [CrossRef]
- Compounds 5a, 6a, and 7a were partially deposited on the surface of the agar medium.
- Fujiwara, Y.; Mitani, M.; Yasuike, S.; Kurita, J.; Kaji, T. An organobismuth compound that exhibits selective cytotoxicity to vascular endothelial cells in vitro. J. Health Sci. 2005, 51, 333–340. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest. Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Murphy, C.D.; Clark, B.R.; Amadio, J. Metabolism of fluoroorganic compounds in microorganisms: Impacts for the environment and the production of fine chemicals. Appl. Microbiol. Biotechnol. 2009, 84, 617–629. [Google Scholar] [CrossRef]
- Timoshkin, A.L.; Bodensteiner, M.; Sevastianova, T.N.; Lisovenko, A.S.; Davydova, E.I.; Scheer, M.; Graßl, C.; Butlak, A.V. Do solid-state structures reflect Lewis acidity trends of heavier group 13 trihalides? Experimental and theoretical case study. Inorg. Chem. 2012, 51, 11602–11611. [Google Scholar] [CrossRef]
- Dakternieks, D.; Jurkschat, K.; Tozer, R.; Hook, J.; Tiekink, E.R.T. Novel coordination isomerization in organotin(IV) compounds. Synthesis, molecular structures, and NMR studies of LSnPhX2 (X = Ph, Cl, Br, I, SPh), LCH2SnPhX2 (X = Ph, Cl, Br, I), and LSiPh3, where LH is (2-MeO-3-tBu-5-Me-C6H2)2CH2. Organometallics 1997, 16, 3696–3706. [Google Scholar] [CrossRef]
- Suzuki, H.; Murafuji, T.; Matano, Y.; Azuma, N. Chiral chlorobismuthines stabilized by the intramolecular coordination of an N,N-dimethylamino group: X-ray structure analysis, asymmetric induction at the bismuth centre, and dynamic behaviour in solution. J. Chem. Soc. Perkin Trans. 1 1993, 2969–2973. [Google Scholar]
- Beleaga, A.; Bojan, V.R.; Pöllnitz, A.; Raț, C.I.; Silvestru, C. Organomercury(II) and tellurium(II) compounds with the “pincer” ligand 2,6-[O(CH2CH2)2NCH2]2C6H3 - stabilization of an unusual organotellurium(II) cationic species. Dalton Trans. 2011, 40, 8830–8838. [Google Scholar] [CrossRef]
- Denmark, S.E.; Beutner, G.L. Lewis base catalysis in organic synthesis. Angew. Chem. Int. Ed. 2008, 47, 1560–1638. [Google Scholar] [CrossRef]
- Holmes, R.R. Comparison of phosphorus and silicon: Hypervalency, stereochemistry, and reactivity. Chem. Rev. 1996, 96, 927–950. [Google Scholar] [CrossRef]
- Chuit, C.; Corriu, R.J.P.; Reye, C.; Young, J.C. Reactivity of penta- and hexacoordinate silicon compounds and their role as reaction intermediates. Chem. Rev. 1993, 93, 1371–1448. [Google Scholar] [CrossRef]
- Trinquier, G.; Daudey, J.-P.; Caruana, G.; Madaule, Y. Theoretical data on the multicoordination of phosphorus and arsenic. J. Am. Chem. Soc. 1984, 106, 4794–4799. [Google Scholar] [CrossRef]
- Deiters, J.A.; Holmes, R.R. Enhanced reactivity of pentacoordinated silicon species. An ab initio approach. J. Am. Chem. Soc. 1990, 112, 7197–7202. [Google Scholar] [CrossRef]
- Burford, N.; Eelman, M.D.; Mahony, D.E.; Morash, M. Definitive identification of cysteine and glutathione complexes of bismuth by mass spectrometry: Assessing the biochemical fate of bismuth pharmaceutical agents. Chem. Comm. 2003, 146–147. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01 Gaussian,Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Kolb, U.; Dräger, M.; Jousseaume, B. Triorganotin compounds SnXMe2-1,4-cyclohexadiene-COOMe: Intra- and intermolecular unfolding of an inner tetrahedron to distorted trigonal bipyramids (X = Cl, Br, I) and a distorted octahedron (X = F). Organometallics 1991, 10, 2737–2742. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds A-D and 4-11 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Murafuji, T.; Tomura, M.; Ishiguro, K.; Miyakawa, I. Activity of Antifungal Organobismuth(III) Compounds Derived from Alkyl Aryl Ketones against S. cerevisiae: Comparison with a Heterocyclic Bismuth Scaffold Consisting of a Diphenyl Sulfone. Molecules 2014, 19, 11077-11095. https://doi.org/10.3390/molecules190811077
Murafuji T, Tomura M, Ishiguro K, Miyakawa I. Activity of Antifungal Organobismuth(III) Compounds Derived from Alkyl Aryl Ketones against S. cerevisiae: Comparison with a Heterocyclic Bismuth Scaffold Consisting of a Diphenyl Sulfone. Molecules. 2014; 19(8):11077-11095. https://doi.org/10.3390/molecules190811077
Chicago/Turabian StyleMurafuji, Toshihiro, Mai Tomura, Katsuya Ishiguro, and Isamu Miyakawa. 2014. "Activity of Antifungal Organobismuth(III) Compounds Derived from Alkyl Aryl Ketones against S. cerevisiae: Comparison with a Heterocyclic Bismuth Scaffold Consisting of a Diphenyl Sulfone" Molecules 19, no. 8: 11077-11095. https://doi.org/10.3390/molecules190811077
APA StyleMurafuji, T., Tomura, M., Ishiguro, K., & Miyakawa, I. (2014). Activity of Antifungal Organobismuth(III) Compounds Derived from Alkyl Aryl Ketones against S. cerevisiae: Comparison with a Heterocyclic Bismuth Scaffold Consisting of a Diphenyl Sulfone. Molecules, 19(8), 11077-11095. https://doi.org/10.3390/molecules190811077