Palladium-Catalyzed Direct Addition of 2-Aminobenzonitriles to Sodium Arylsulfinates: Synthesis of o-Aminobenzophenones


Abstract
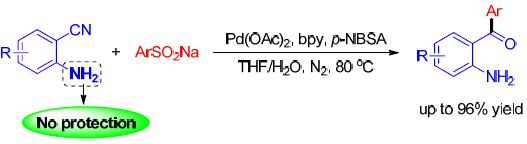
:
1. Introduction
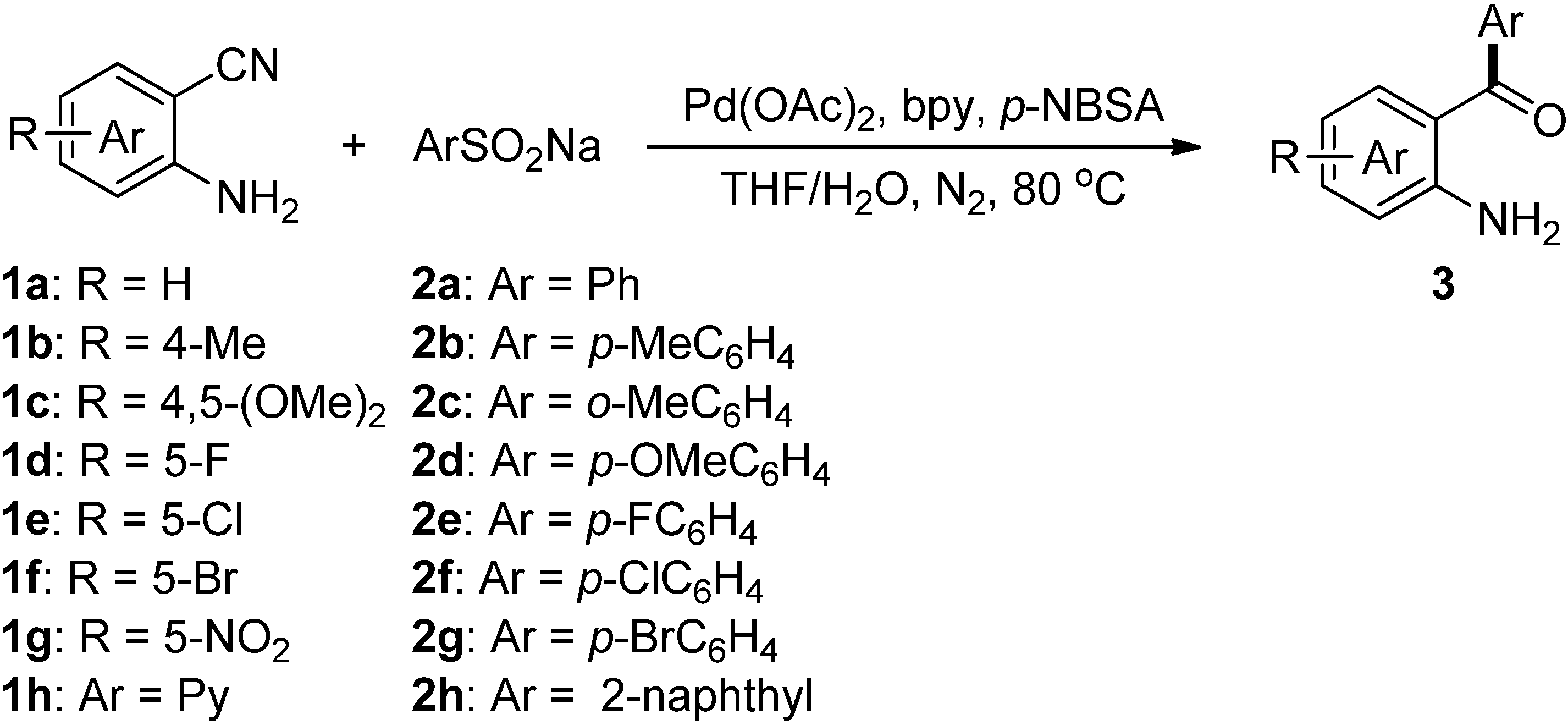
2. Results and Discussion


| Entry | Pd source | Additive | Solvent | Yield (%) b |
|---|---|---|---|---|
| 1 | Pd(CF3CO2)2 | CF3CO2H | THF | 18 |
| 2 | PdCl2 | CF3CO2H | THF | 11 |
| 3 | Pd(OAc)2 | CF3CO2H | THF | 32 |
| 4 | Pd(acac)2 | CF3CO2H | THF | 13 |
| 5 | Pd(PPh3)4 | CF3CO2H | THF | trace |
| 6 | PdCl2(dppe) | CF3CO2H | THF | 0 |
| 7 | Pd(OAc)2 | CH3CO2H | THF | 10 |
| 8 | Pd(OAc)2 | CH3SO3H | THF | 44 |
| 9 | Pd(OAc)2 | PhSO3H | THF | 59 |
| 10 | Pd(OAc)2 | p-TSA c | THF | 61 |
| 11 | Pd(OAc)2 | p-NBSA d | THF | 73 |
| 12 | Pd(OAc)2 | p-NBSA | toluene | 42 |
| 13 | Pd(OAc)2 | p-NBSA | 2-MeTHF | 69 |
| 14 | Pd(OAc)2 | p-NBSA | dioxane | 53 |
| 15 | Pd(OAc)2 | p-NBSA | DMF | trace |
| 16 | Pd(OAc)2 | p-NBSA | THF | 91 e |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | ArSO2Na (2) | Product (3) | Yield (%) b |
|---|---|---|---|
| 1 |  | 3a | 91 |
| 2 |  | 3b | 88 |
| 3 |  | 3c | 64 |
| 4 |  | 3d | 85 |
| 5 |  | 3e | 81 |
| 6 |  | 3f | 83 |
| 7 |  | 3g | 80 |
| 8 |  | 3h | 90 |
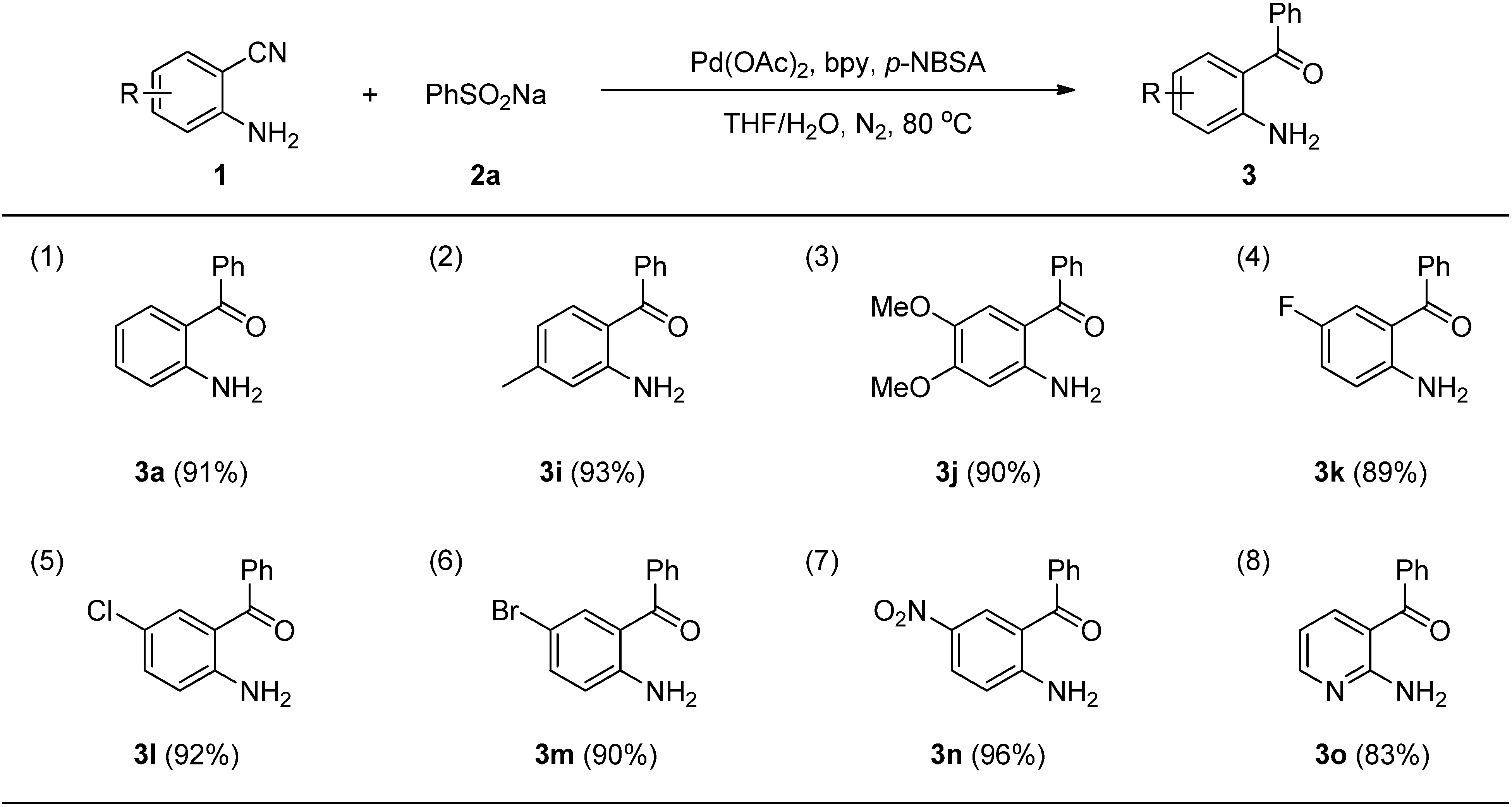
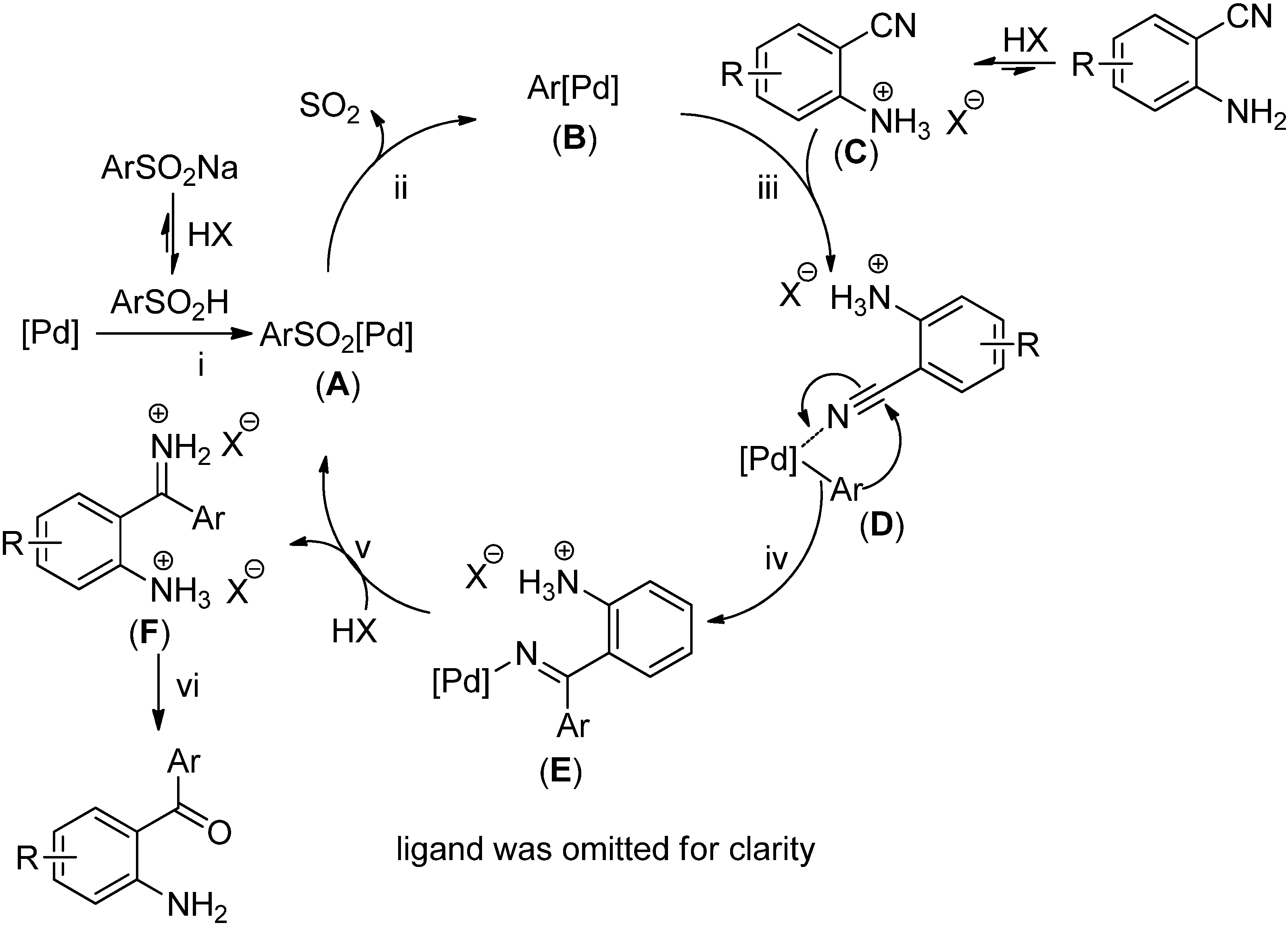
3. Experimental
General Information
General Procedure for the Synthesis of o-Aminobenzophenones
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, R.K.; Prasad, D.N.; Bhardwaj, T.R. Design, synthesis and evaluation of aminobenzophenone derivatives containing nitrogen mustard moiety as potential central nervous system antitumor agent. Med. Chem. Res. 2013, 22, 5901–5911. [Google Scholar] [CrossRef]
- Liou, J.-P.; Chang, C.-W.; Song, J.-S.; Yang, Y.-N.; Yeh, C.-F.; Tseng, H.-Y.; Lo, Y.-K.; Chang, Y.-L.; Chang, C.-M.; Hsieh, H.-P. Synthesis and structure activity relationship of 2-aminobenzophenone derivatives as antimitotic agents. J. Med. Chem. 2002, 45, 2556–2562. [Google Scholar] [CrossRef]
- Castellano, S.; Taliani, S.; Viviano, M.; Milite, C.; da Pozzo, E.; Costa, B.; Barresi, E.; Bruno, A.; Cosconati, S.; Marinelli, L.; et al. Structure–activity relationship refinement and further assessment of 4-phenylquinazoline-2-carboxamide translocator protein ligands as antiproliferative agents in human glioblastoma tumors. J. Med. Chem. 2014, 57, 2413–2428. [Google Scholar] [CrossRef]
- Kobayashi, K.; Fujita, S.; Fukamachi, S.; Konishi, H. One-pot synthesis of quinoline-2(1H)-thiones from 2-isocyanostyrenes via electrocyclic reaction of the corresponding 2-isothiocyanatestyrenes. Synthesis 2009, 2009, 3378–3382. [Google Scholar] [CrossRef]
- Cai, S.; Zeng, J.; Bai, Y.; Liu, X. Access to Quinolines through gold-catalyzed intermolecular cycloaddition of 2-aminoaryl carbonyls and internal alkynes. J. Org. Chem. 2012, 77, 801–807. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, Z. Metal-free intramolecular oxidative decarboxylative amination of primary a-amino acids with product selectivity. Chem. Commun. 2011, 47, 9513–9515. [Google Scholar] [CrossRef]
- Anand, N.; Reddy, K.H.P.; Satyanarayana, T.; Rao, K.S.R.; Burri, D.R. A magnetically recoverable γ-Fe2O3 nanocatalyst for the synthesis of 2-phenylquinazolines under solvent-free conditions. Catal. Sci. Technol. 2012, 2, 570–574. [Google Scholar] [CrossRef]
- Ren, L.; Lei, T.; Ye, J.; Gong, L. Step-economical synthesis of tetrahydroquinolines by asymmetric relay catalytic Friedläender condensation/transfer hydrogenation. Angew. Chem. Int. Ed. Engl. 2012, 51, 771–774. [Google Scholar] [CrossRef]
- Earmme, T.; Ahmed, E.; Jenekhe, S.A. Solution-processed highly efficient blue phosphorescent polymer light-emitting diodes enabled by a new electron transport material. Adv. Mater. 2010, 22, 4744–4748. [Google Scholar] [CrossRef]
- Walsh, D.A. The Synthesis of 2-aminobenzophenones. Synthesis 1980, 1980, 677–688. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, Y.; Huang, L.; Zhang, X.; Zhang, Y. Pd-Catalyzed arylation/oxidation of benzylic C-H bond. Org. Lett. 2012, 14, 1238–1241. [Google Scholar]
- Khedkar, M.V.; Tambade, P.J.; Qureshi, Z.S.; Bhanage, B.M. Pd/C: An efficient, heterogeneous and reusable catalyst for phosphane-free carbonylative suzuki coupling reactions of aryl and heteroaryl iodides. Eur. J. Org. Chem. 2010, 2010, 6981–6986. [Google Scholar]
- Cai, M.; Peng, J.; Hao, W.; Ding, G. A phosphine-free carbonylative cross-coupling reaction of aryl iodides with arylboronic acids catalyzed by immobilization of palladium in MCM-41. Green Chem. 2011, 13, 190–196. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamano, M. A new practical one-pot conversion of phenols to anilines. Org. Lett. 2005, 7, 3629–3631. [Google Scholar] [CrossRef]
- Thakur, K.G.; Ganapathy, D.; Sekar, G. d-Glucosamine as a green ligand for copper catalyzed synthesis of primary aryl amines from aryl halides and ammonia. Chem. Commun. 2011, 47, 5076–5078. [Google Scholar] [CrossRef]
- Mateos, C.; Rincón, J.A.; Villanueva, J. Efficient and scalable synthesis of ketones via nucleophilic Grignard addition to nitriles using continuous flow chemistry. Tetrahedron Lett. 2013, 45, 2226–2230. [Google Scholar]
- Zhou, X.; Luo, J.; Liu, J.; Peng, S.; Deng, D. Pd-catalyzed desulfitative heck coupling with dioxygen as the terminal oxidant. Org. Lett. 2011, 13, 1432–1436. [Google Scholar]
- Rao, H.; Yang, L.; Shuai, Q.; Li, C. Rhodium-catalyzed aerobic coupling between aldehydes and arenesulfinic acid salts: A novel synthesis of aryl ketones. Adv. Synth. Catal. 2011, 353, 1701–1706. [Google Scholar] [CrossRef]
- Chen, J.; Sun, Y.; Liu, B.; Liu, D.; Cheng, J. The palladium-catalyzed desulfitative cyanation of arenesulfonyl chlorides and sodium sulfinates. Chem. Commun. 2012, 48, 449–451. [Google Scholar]
- Liu, B.; Guo, Q.; Cheng, Y.; Lan, J.; You, J. Palladium-catalyzed desulfitative C-H arylation of heteroarenes with sodium sulfinates. Chem. Eur. J. 2011, 17, 13415–13419. [Google Scholar] [CrossRef]
- Zhao, F.; Tan, Q.; Xiao, F.; Zhang, S.; Deng, G. Palladium-catalyzed desulfitative cross-coupling reaction of sodium sulfinates with benzyl chlorides. Org. Lett. 2013, 15, 1520–1523. [Google Scholar] [CrossRef]
- Fleming, F.F.; Wang, Q. Unsaturated nitriles: Conjugate additions of carbon nucleophiles to a recalcitrant class of acceptors. Chem. Rev. 2003, 103, 2035–2078. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Additions to metal-activated organonitriles. Chem. Rev. 2002, 102, 1771–1802. [Google Scholar] [CrossRef]
- Rach, S.F.; Kuhn, F.E. Nitrile ligated transition metal complexes with weakly coordinating counteranions and their catalytic applications. Chem. Rev. 2009, 109, 2061–2080. [Google Scholar] [CrossRef]
- Larock, R.C.; Tian, Q.; Pletnv, A.A. Carbocycle synthesis via carbopalladation of nitriles. J. Am. Chem. Soc. 1999, 121, 3238–3239. [Google Scholar] [CrossRef]
- Ueura, K.; Satoh, T.; Miura, M. Rhodium-catalyzed arylation using arylboron compounds: Efficient coupling with aryl halides and unexpected multiple arylation of benzonitrile. Org. Lett. 2005, 7, 2229–2231. [Google Scholar] [CrossRef]
- Wong, Y.-C.; Parthasarathy, K.; Cheng, C.-H. Direct synthesis of arylketones by nickel-catalyzed addition of arylboronic acids to nitriles. Org. Lett. 2010, 12, 1736–1739. [Google Scholar] [CrossRef]
- Tsui, G.C.; Glenadel, Q.; Lau, C.; Lautens, M. Rhodium(I)-catalyzed addition of arylboronic acids to (benzyl-/arylsulfonyl)acetonitriles: Efficient synthesis of (Z)-β-sulfonylvinylamines and β-keto sulfones. Org. Lett. 2011, 13, 208–211. [Google Scholar] [CrossRef]
- Lindh, J.; Sjöberg, P.J.R.; Larhed, M. Synthesis of aryl ketones by palladium(II)-catalyzed decarboxylative addition of benzoic acids to nitriles. Angew. Chem. Int. Ed. Engl. 2010, 49, 7733–7737. [Google Scholar]
- Hsieh, J.-C.; Chen, Y.-C.; Cheng, A.; Tseng, H.-C. Nickel-catalyzed intermolecular insertion of aryl iodides to nitriles: A novel method to synthesize arylketones. Org. Lett. 2012, 14, 1282–1285. [Google Scholar]
- Wang, X.; Liu, M.; Xu, L.; Wang, Q.; Chen, J.; Ding, J.; Wu, H. Palladium-catalyzed addition of potassium aryltrifluoroborates to aliphatic nitriles: Synthesis of alkyl aryl ketones, diketone compounds, and 2-arylbenzo[b]furans. J. Org. Chem. 2013, 78, 5273–5281. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Liu, M.; Ding, J.; Chen, J.; Wu, H. Palladium-catalyzed reaction of arylboronic acids with aliphatic nitriles: Synthesis of alkyl aryl ketones and 2-aryl benzofurans. Synthesis 2013, 45, 2241–2244. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, Q.; Chen, J.; Liu, M.; Cheng, S.; Ding, J.; Wu, H.; Su, W. Copper(II) acetate-catalyzed addition of arylboronic acids to aromatic aldehydes. J. Org. Chem. 2009, 74, 943–945. [Google Scholar]
- Qin, C.; Wu, H.; Chen, J.; Liu, M.; Cheng, J.; Su, W.; Ding, J. Palladium-catalyzed aromatic esterification of aldehydes with organoboronic acids and molecular oxygen. Org. Lett. 2008, 10, 1537–1540. [Google Scholar]
- Qin, C.; Wu, H.; Cheng, J.; Chen, X.; Liu, M.; Zhang, W.; Su, W.; Ding, J. The palladium-catalyzed addition of aryl- and heteroarylboronic acids to aldehydes. J. Org. Chem. 2007, 72, 4102–4107. [Google Scholar]
- Zheng, H.; Ding, J.; Chen, J.; Liu, M.; Gao, W.; Wu, H. Copper-catalyzed arylation of arylboronic acids with aldehydes. Synlett 2011, 2011, 1626–1630. [Google Scholar] [CrossRef]
- Qin, C.; Chen, J.; Wu, H.; Cheng, J.; Zhang, Q.; Zuo, B.; Su, W.; Ding, J. One-pot synthesis of diaryl ketones from aldehydes via palladium-catalyzed reaction with aryl boronic acids. Tetrahedron Lett. 2008, 49, 1884–1888. [Google Scholar] [CrossRef]
- Lothrop, W.C.; Goodwin, P.A. New modification of the Ullmann synthesis of fluorene derivatives. J. Am. Chem. Soc. 1943, 65, 363–367. [Google Scholar] [CrossRef]
- Martinez-Viturro, C.M.; Dominguez, D. Synthesis of aza analogues of the anticancer agent batracylin. Tetrahedron Lett. 2007, 48, 4707–4710. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, J.; Li, J.; Su, W. Palladium-Catalyzed Direct Addition of 2-Aminobenzonitriles to Sodium Arylsulfinates: Synthesis of o-Aminobenzophenones. Molecules 2014, 19, 6439-6449. https://doi.org/10.3390/molecules19056439
Chen J, Li J, Su W. Palladium-Catalyzed Direct Addition of 2-Aminobenzonitriles to Sodium Arylsulfinates: Synthesis of o-Aminobenzophenones. Molecules. 2014; 19(5):6439-6449. https://doi.org/10.3390/molecules19056439
Chicago/Turabian StyleChen, Jiuxi, Jianjun Li, and Weike Su. 2014. "Palladium-Catalyzed Direct Addition of 2-Aminobenzonitriles to Sodium Arylsulfinates: Synthesis of o-Aminobenzophenones" Molecules 19, no. 5: 6439-6449. https://doi.org/10.3390/molecules19056439
APA StyleChen, J., Li, J., & Su, W. (2014). Palladium-Catalyzed Direct Addition of 2-Aminobenzonitriles to Sodium Arylsulfinates: Synthesis of o-Aminobenzophenones. Molecules, 19(5), 6439-6449. https://doi.org/10.3390/molecules19056439