2.1. TiO2/ZSM-5 Adsorb and Shuttle
The chosen adsorbent is the high silica zeolite ZSM-5 in a configuration similar to the system of Vaisman
et al. [
5] designed as an adsorbent for photocatalytic regeneration. ZSM-5, with a highly hydrophobic surface, presents an adsorption profile resembling activated carbon, but weaker for some highly hydrophobic molecules and stronger for more polar organics. It is used here in composite with Degussa P25 TiO
2 (PZS). The percentages of three different model substrates adsorbed on the surface of PZS and P25 after 30 min of stirring in the dark are shown in
Table 1. It can be seen that there is an unequivocal increase in the amount of the 2,4,6-trichlorophenol (2,4,6-TCP) substrate adsorbed on the surface of PZS with a drastic increase observed in the case of sulfamethoxazole (SMX), while for atrazine, the adsorption was the least. The overall order of increase in percent adsorption is SMX > 2,4,6-TCP > atrazine.
Table 1.
Adsorption percentage of the different model compounds at initial concentrations of 50.0 mg/L on the surface of PZS and P25 after the dark period. SMX, sulfamethoxazole; 2,4,6-TCP, 2,4,6-trichlorophenol.
Table 1.
Adsorption percentage of the different model compounds at initial concentrations of 50.0 mg/L on the surface of PZS and P25 after the dark period. SMX, sulfamethoxazole; 2,4,6-TCP, 2,4,6-trichlorophenol.
| Compound | Photocatalyst |
|---|
| Degussa P25% | PZS% |
|---|
| SMX | 2.60 | 18.00 |
| 2,4,6-TCP | 29.20 | 41.90 |
| Atrazine | 2.40 | 5.64 |
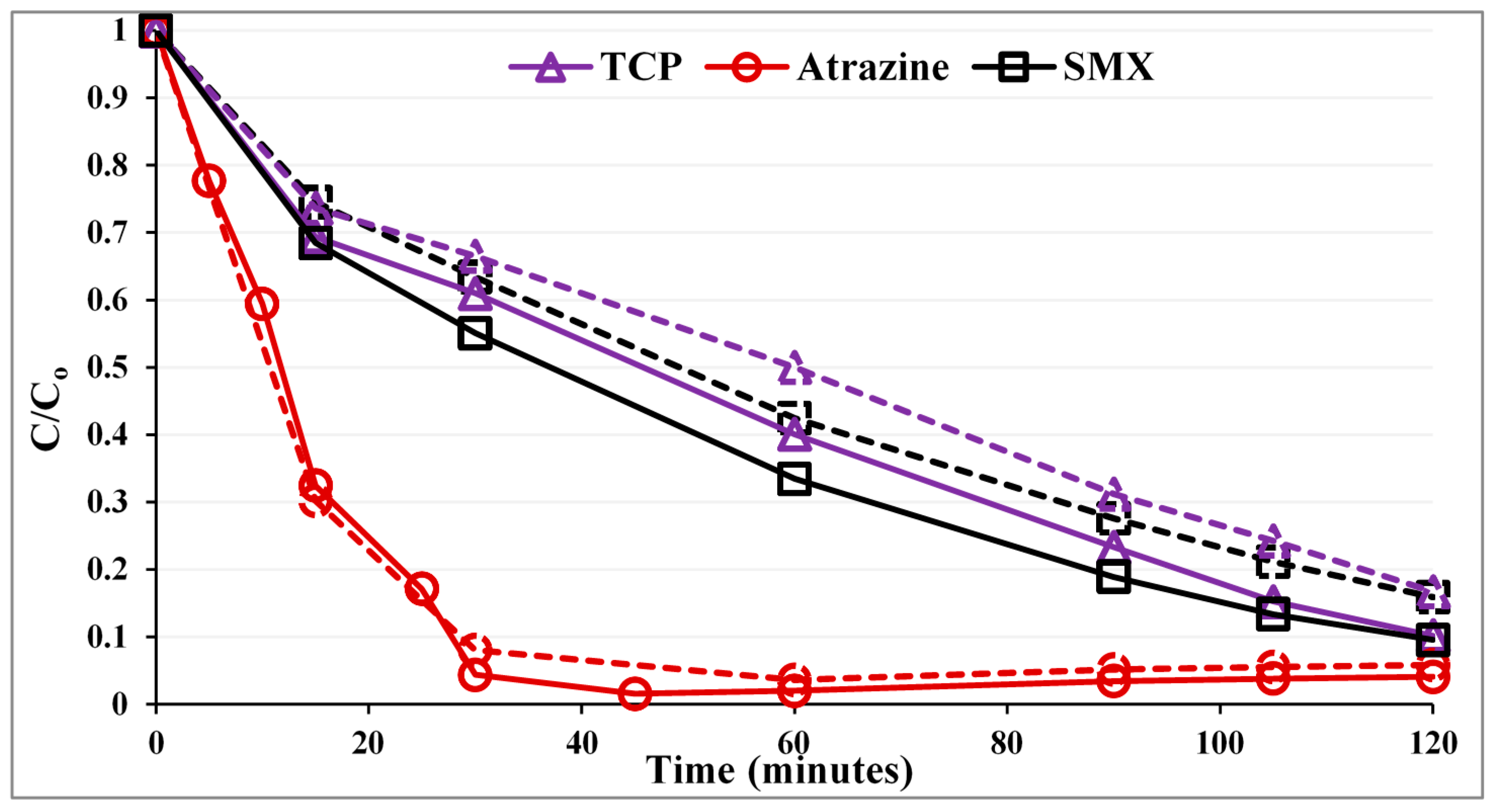
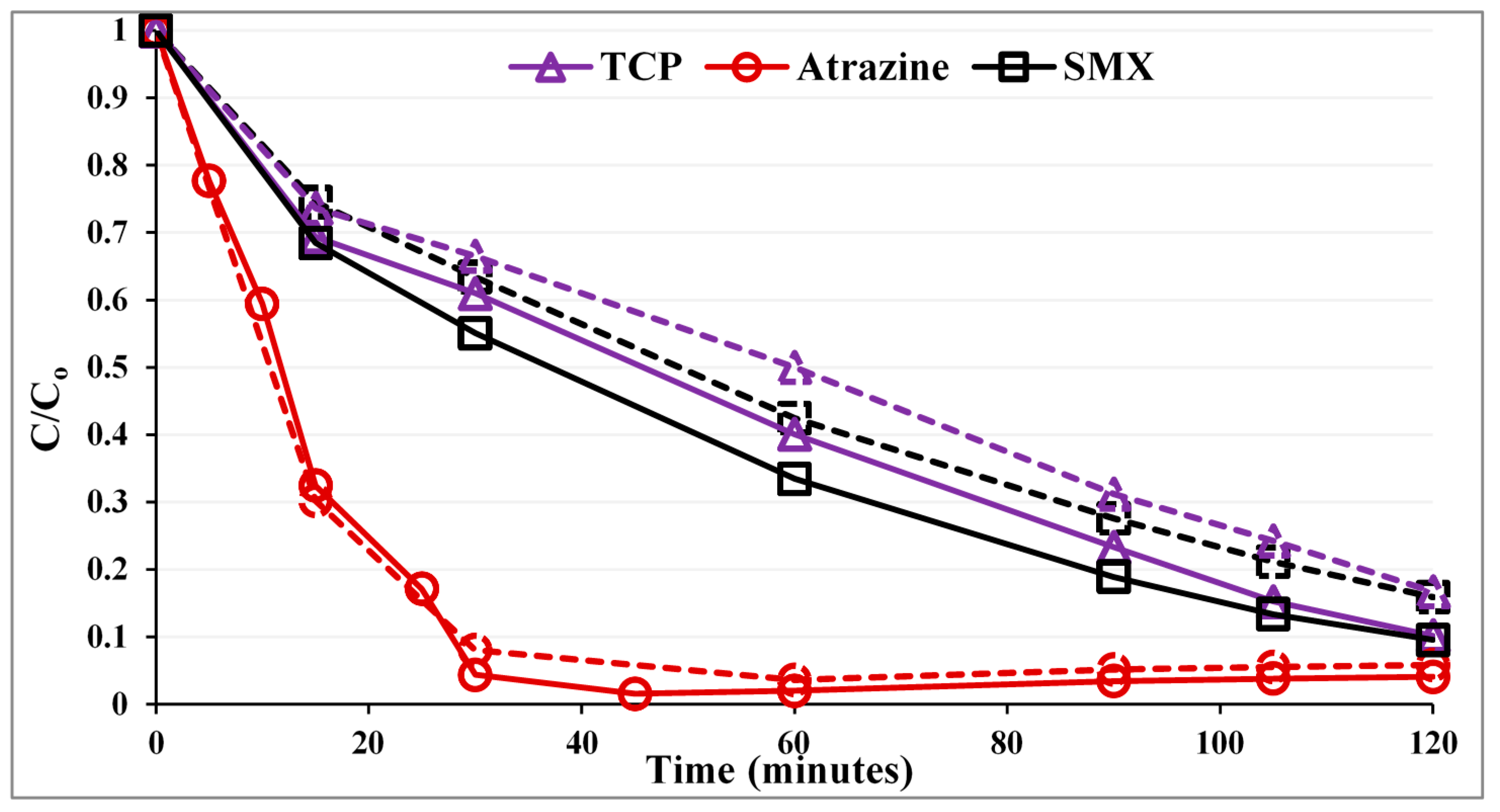
Figure 1 compares the photocatalytic performance of the PZS to that of commercial Degussa P25 as a reference. The first point corresponds to the normalized concentration after the dark adsorption period. Pseudo first-order rate constants (k) along with correlation coefficients (R
2) are listed in
Table 2 (because of the rapid reaction, it was not a certain that the atrazine data do fit first order kinetics).
Figure 1.
Photocatalytic degradation of the substrates with a UVA LED photoreactor. Solid lines, PZS (P25:ZSM-5:silica gel = 0.3:0.5:0.5 g/L); dashed lines, Degussa P25 (0.3 g/L).
Figure 1.
Photocatalytic degradation of the substrates with a UVA LED photoreactor. Solid lines, PZS (P25:ZSM-5:silica gel = 0.3:0.5:0.5 g/L); dashed lines, Degussa P25 (0.3 g/L).
Table 2.
Pseudo first order rate constants (min−1) and R2 fit measure for the substrates.
Table 2.
Pseudo first order rate constants (min−1) and R2 fit measure for the substrates.
| Compound | Degussa P25 min−1 | PZS min−1 |
|---|
| SMX | k = 0.0147 | R2 = 0.996 | k = 0.0189 | R2 = 0.9965 |
| 2,4,6-TCP | k = 0.0137 | R2 = 0.977 | k = 0.0179 | R2 = 0.982 |
| Atrazine | k = 0.055 | R2 = 0.923 | k = 0.0976 | R2 = 0.966 |
As can be seen in
Figure 1, for SMX and 2,4,6-TCP, the PZS shows noticeably higher photocatalytic activity than the Degussa P25. The enhancement in efficiency is not in proportion, as the adsorption strength of these compounds on the zeolite surface is different. While for atrazine, the photocatalytic activity was within the error, the same for both Degussa P25 and PZS. The order of photocatalytic activity improvement is SMX ~ 2,4,6-TCP > atrazine, which is consistent with the order of improved adsorption. This suggests that adsorb and shuttle may have been effective.
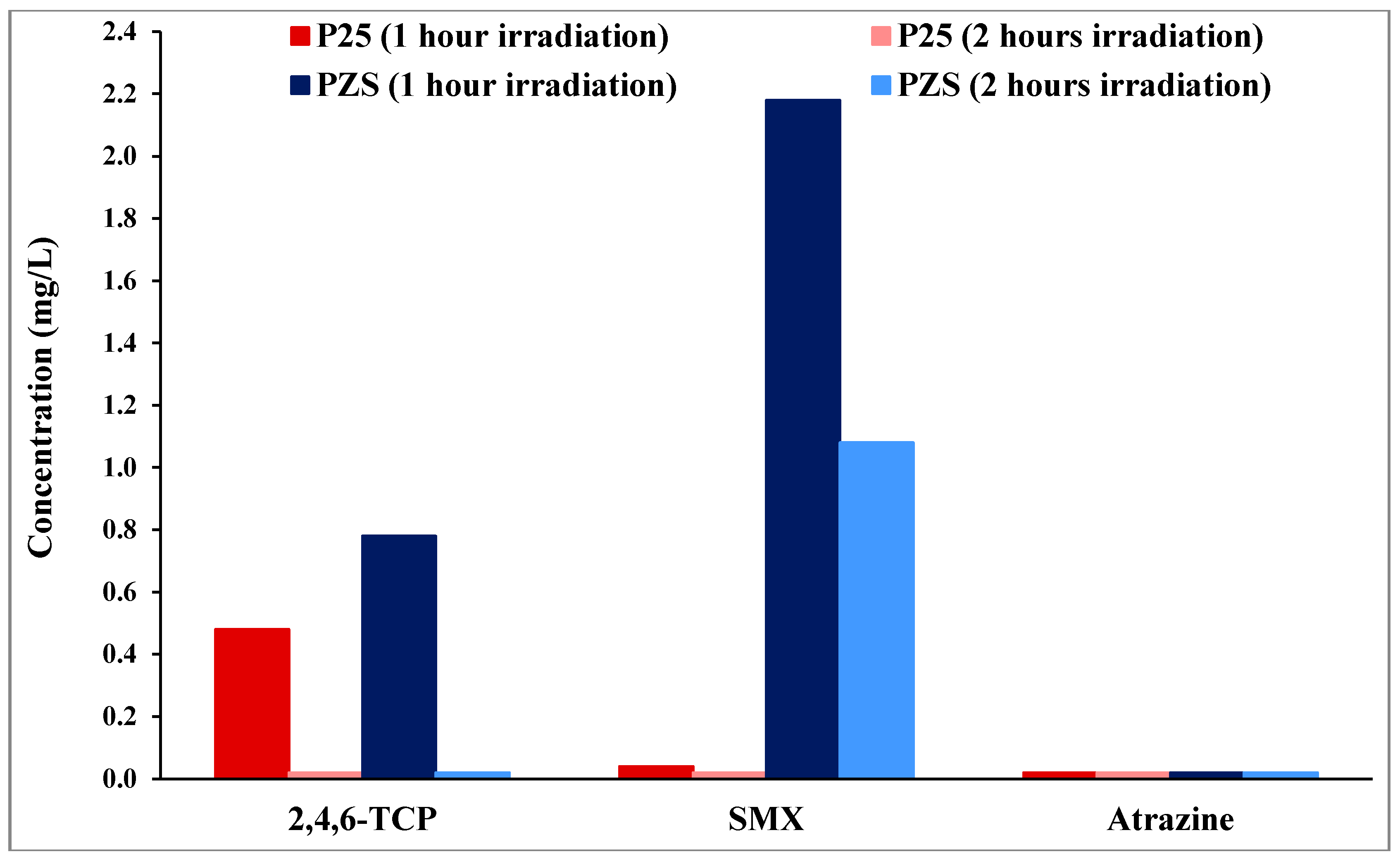
Substrates were extracted to track the extent of the retention of the initial substrate on the adsorbent surface during reaction. The amounts are reported in units of the solution concentration equivalent.
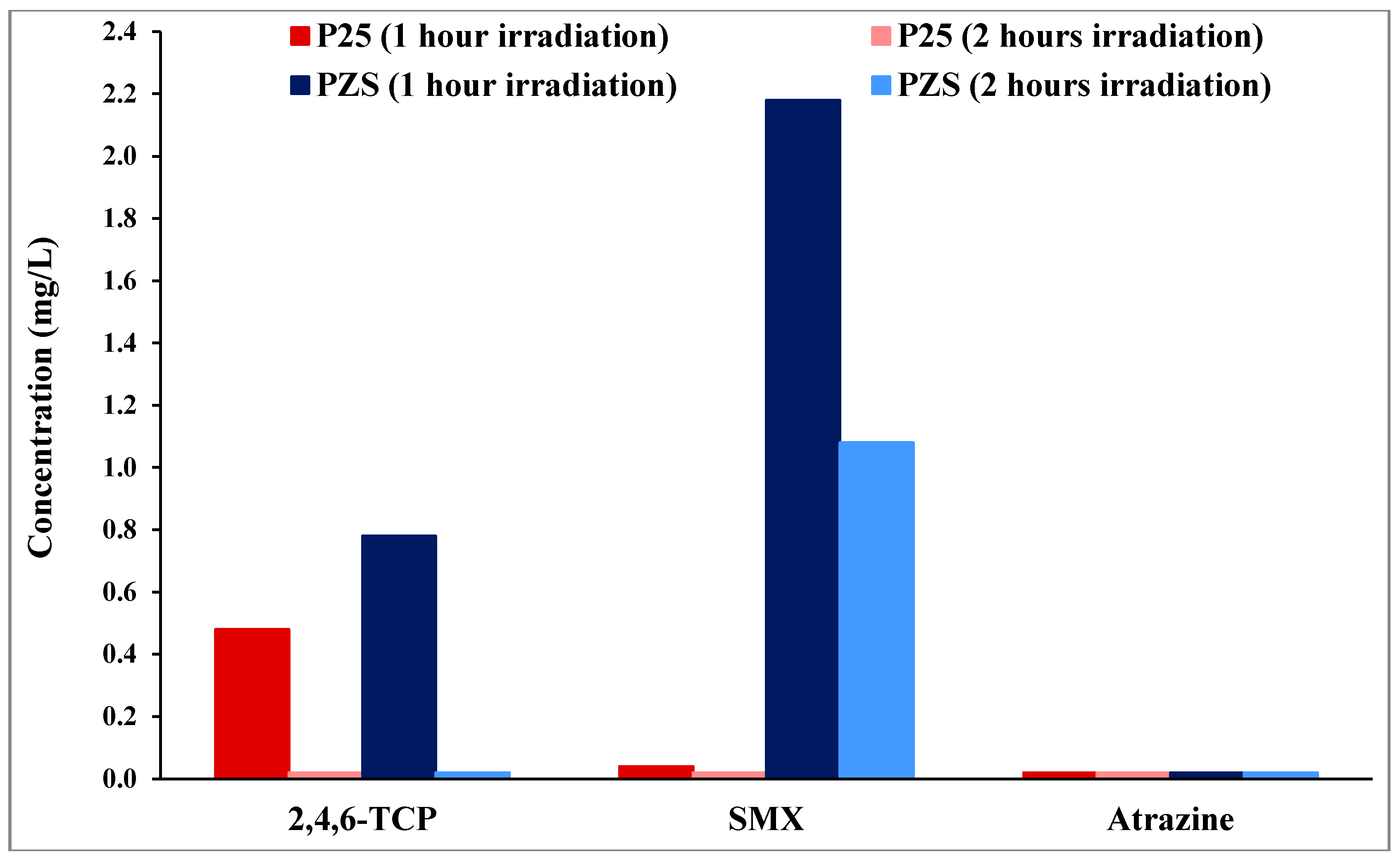
Figure 2 shows data for 60 and 120 min.
Figure 2.
Residue results of the substrates on the surface of PZS and Degussa P25 after one and two hour irradiation. The left bar shows 1-h data and the (clearly visible only for SMX) right bar 2-h data.
Figure 2.
Residue results of the substrates on the surface of PZS and Degussa P25 after one and two hour irradiation. The left bar shows 1-h data and the (clearly visible only for SMX) right bar 2-h data.
2,4,6-TCP residue on the surface of both photocatalysts after 1 h of irradiation was small (less than 1 mg/L solution equivalent compared to an initial total of 50 mg/L). After 2 h of irradiation, no 2,4,6-TCP residues were detected on the surface of either photocatalyst. Furthermore, it can be seen from
Figure 2 that, when the initial extent of dark adsorption is small, no residues were detected on the surface of either photocatalyst, as seen for atrazine on both photocatalysts and even for SMX on P25. The residue of SMX on the surface of PZS after 1 h of irradiation was 2.2 mg/L, which reduced to 1 mg/L after 2 h of irradiation. The comparison of the initial extent of dark adsorption results (
Table 1) with these results suggests that 2,4,6-TCP surface diffusion [
4] is faster than SMX.
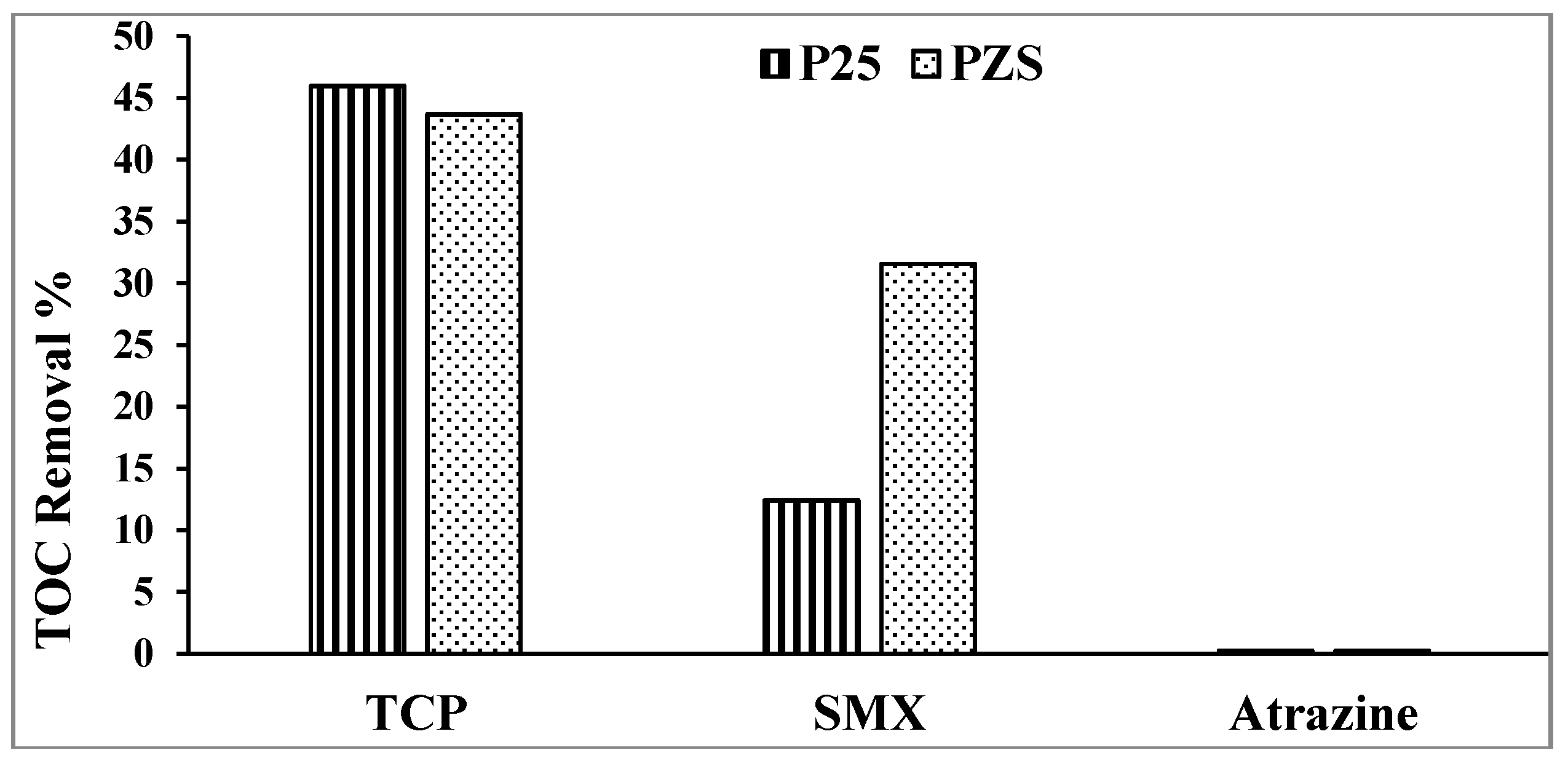
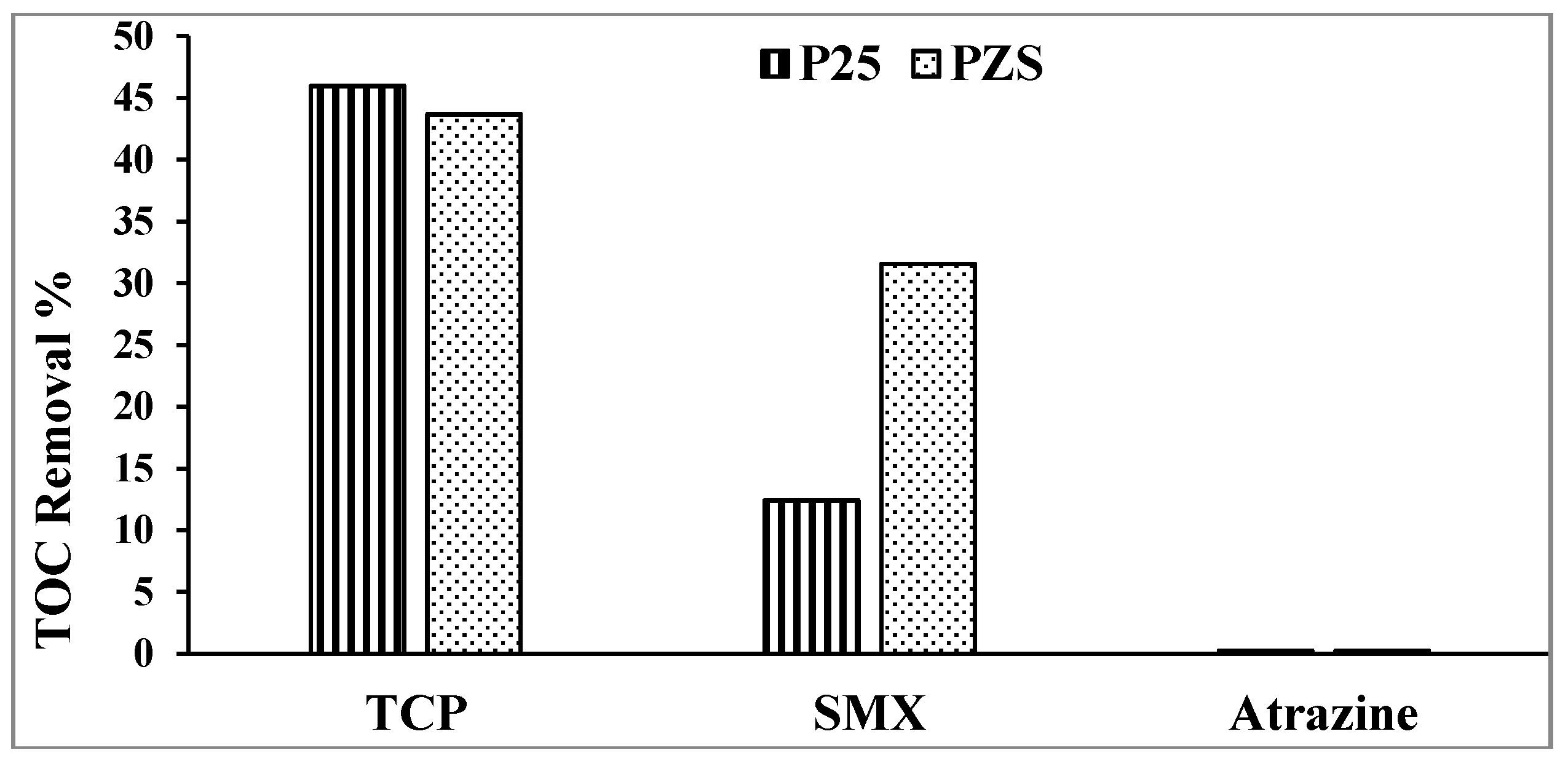
The key advantage of A/S is the promotion of mineralization [
1]. The percentage of TOC removal of the substrates using PZS and Degussa P25 after 1 h of irradiation is shown in
Figure 3. Using PZS improves the mineralization of SMX and shows that 2,4,6-TCP is ~45% mineralized after ~50% loss in the substrate. No mineralization of atrazine is seen, in agreement with McMurray
et al. [
10].
The results are a reminder that atrazine is one of the rare compounds not mineralized on TiO
2. The reaction stops at cyanuric acid [
10].
Figure 3.
Percentage TOC removal after 1 h of irradiation.
Figure 3.
Percentage TOC removal after 1 h of irradiation.
2.2. Interphase Charge Transfer: TiO2/WO3
One of the most striking demonstrations of the reduction of recombination rates arises from the persistence of reactivity after irradiation is terminated [
11]. In this experiment, 4-chlorophenol (4-CP) was chosen as a substrate to avoid any A/S contribution.
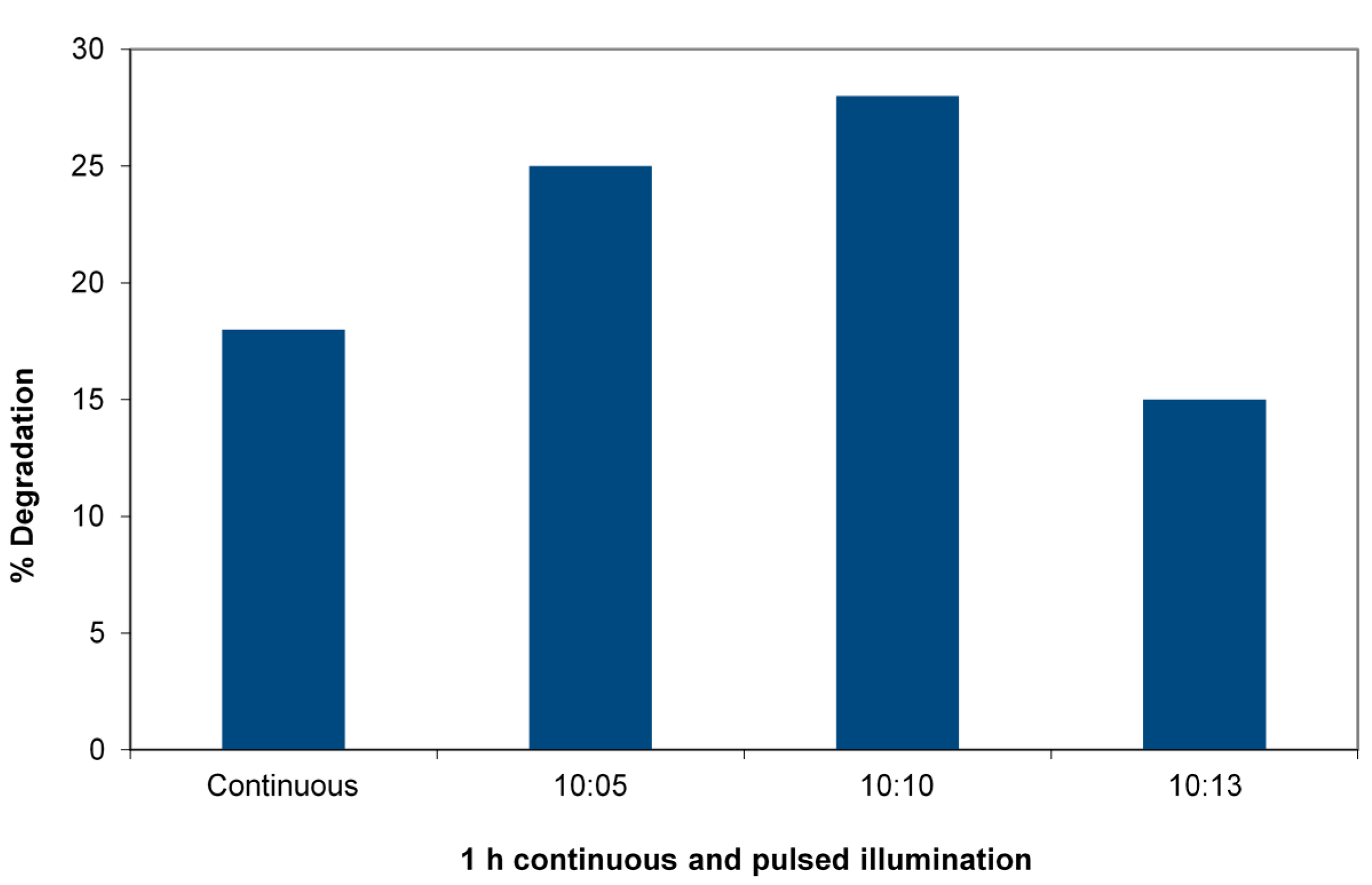
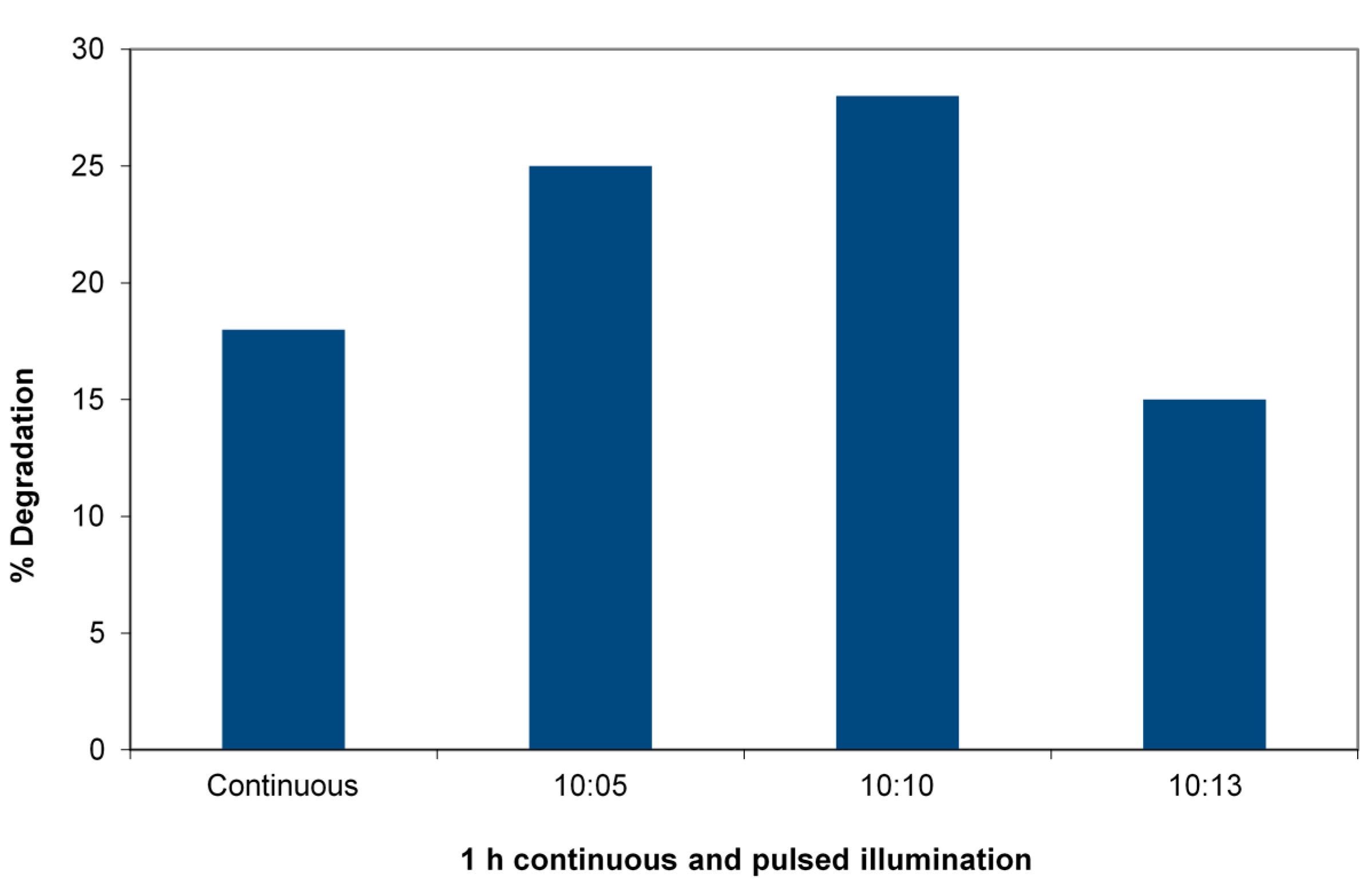
Figure 4 shows the percent degradation of 4-CP after one hour of irradiation in a slurry of TiO
2/WO
3 in a 33-ppm solution of the 4-CP using the 365-nm LED reactor to allow precise pulsing. The duty cycle is varied. The figure shows the striking case of the efficiency increase in degradation reactions of 4-chlorophenol. A 1:1 duty cycle at a constant total dose with a pulse time of 10 minutes (10
4 ms) increases the efficiency of energy utilization by almost a factor of two. Titration of available electrons after termination of illumination by Fe(III) reduction is shown in
Figure 5.
Figure 4.
Pulsed vs. continuous irradiation in a 365-nm LED reactor degrading 4-chlorophenol (4-CP). Charge is 0.05 g WO3/TiO2 and 5 mL of 33 mg/L 4-CP receiving (4.3 ± 0.2) × 1016 photons/s. Total irradiation: 60 min in each case, duty cycle (ratio minute light:minute dark) varied.
Figure 4.
Pulsed vs. continuous irradiation in a 365-nm LED reactor degrading 4-chlorophenol (4-CP). Charge is 0.05 g WO3/TiO2 and 5 mL of 33 mg/L 4-CP receiving (4.3 ± 0.2) × 1016 photons/s. Total irradiation: 60 min in each case, duty cycle (ratio minute light:minute dark) varied.
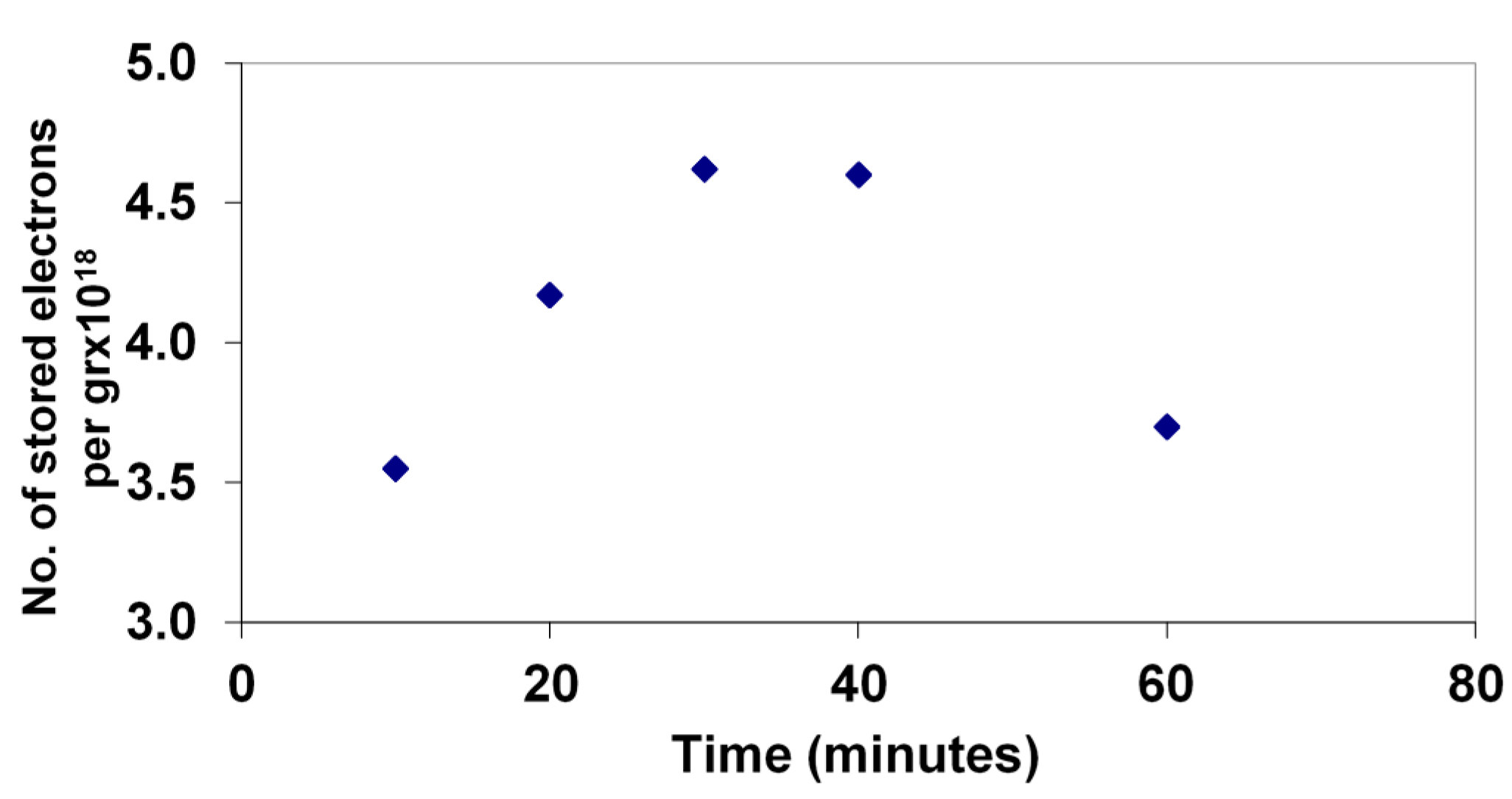
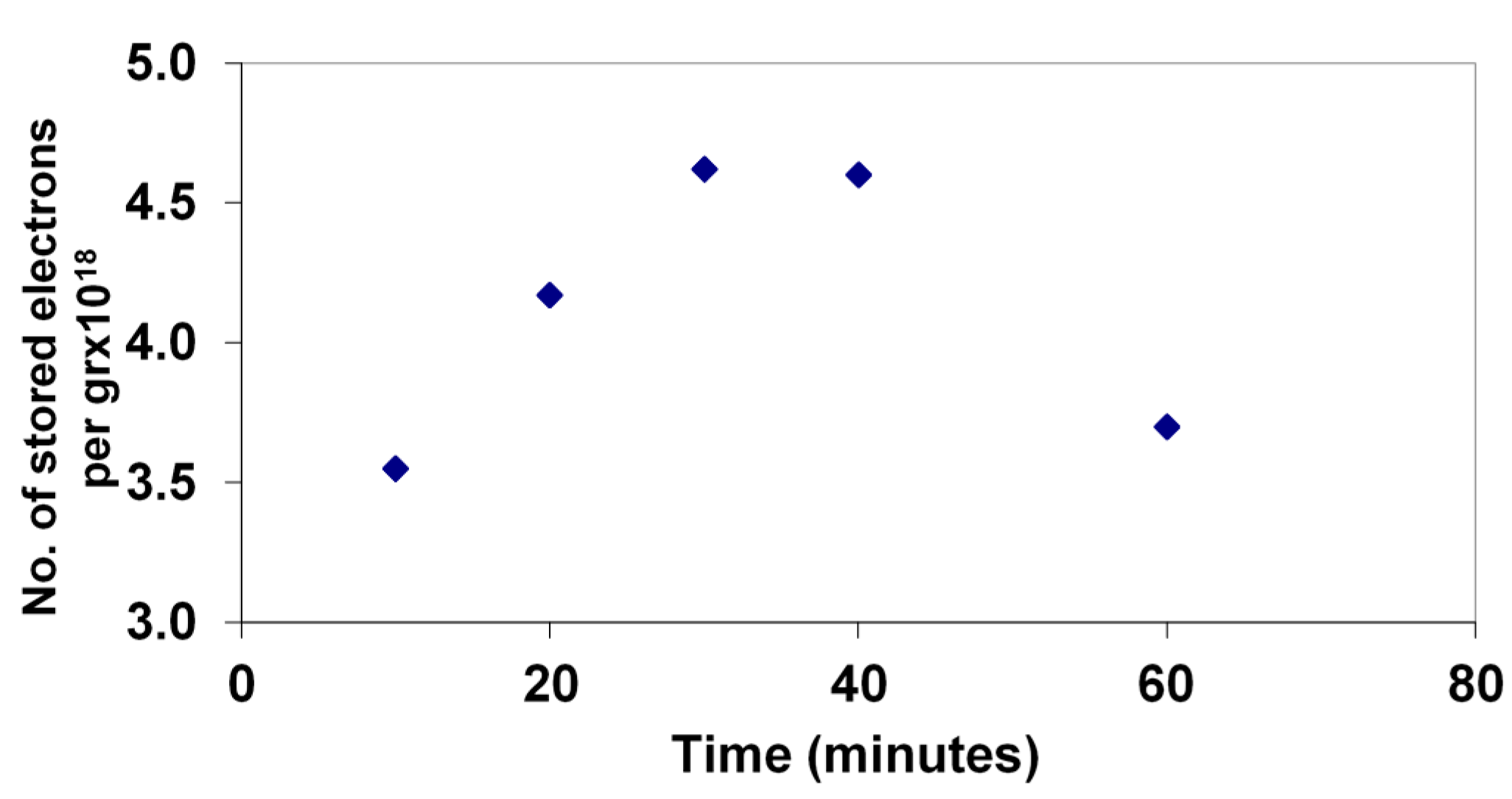
The extent of electron storage during the photodegradation of 4-CP was monitored by rapid titration with Fe(III) of a sample of the solution after successive times of illumination.
Figure 5 shows the levels during the degradation of the 33 mg/L sample, as described above.
Figure 5.
Stored electron levels during the photocatalytic degradation of 4-CP measured by Fe(III) titration with Fe(II) detection by phenanthroline complexation.
Figure 5.
Stored electron levels during the photocatalytic degradation of 4-CP measured by Fe(III) titration with Fe(II) detection by phenanthroline complexation.
It has been reported [
7] that charge storage with WO
3 allows inhibition of
E. coli that are exposed in the dark to TiO
2/WO
3. This is confirmed by data in
Table 3, where illumination was UVA (365 nm), and the sample was outflow water from a secondary wastewater treatment plant. This water contains a mix of other organic molecules that can compete for reaction with excited TiO
2/WO
3. A 50-mL waste water sample with 10-ppm fulvic acid (FA) solution (as a supplemental hole scavenger) + 0.33 g WO
3-TiO
2 was used. In a first experiment, 50 mL of contaminated water was added to the TiO
2 slurry, and the mixture was incubated for 5 h. In the subsequent experiment, after 30 min in the dark, the slurry was irradiated for 2 h in the 365-nm LED reactor. Post irradiation, the 50-mL contaminated water sample was added and incubated for 5 h. The samples were measured for coliforms by the Colilert tray method, widely used for water monitoring. Note that the initial sample counts are not reproducible due to the biological activity in the stock.
Table 3.
Coliform counts after exposure to TiO2/WO3 without and with pre-charging for 2 h. (MPN = most probable number of colonies).
Table 3.
Coliform counts after exposure to TiO2/WO3 without and with pre-charging for 2 h. (MPN = most probable number of colonies).
| Sample | Total Coliform (MPN) | E coli (MPN) | Decrease in E. coli |
|---|
| Stock solution | 1,921.2 | 246.8 |
| WO3/TiO2 (dark blank) | 872.0 | 161.0 | 35% |
| Stock solution | 1,732.9 | 488.4 |
| WO3/TiO2 charged | 108.4 | 26.6 | 95% |
2.3. Carbon
Carbon is a good adsorbent and can act as an electron acceptor [
12]. Consequently, both mechanisms may arise. An interesting potential application is the removal of emerging contaminants from water. For this reason, composites were made using the well-established TiO
2, Degussa P25 reference point.
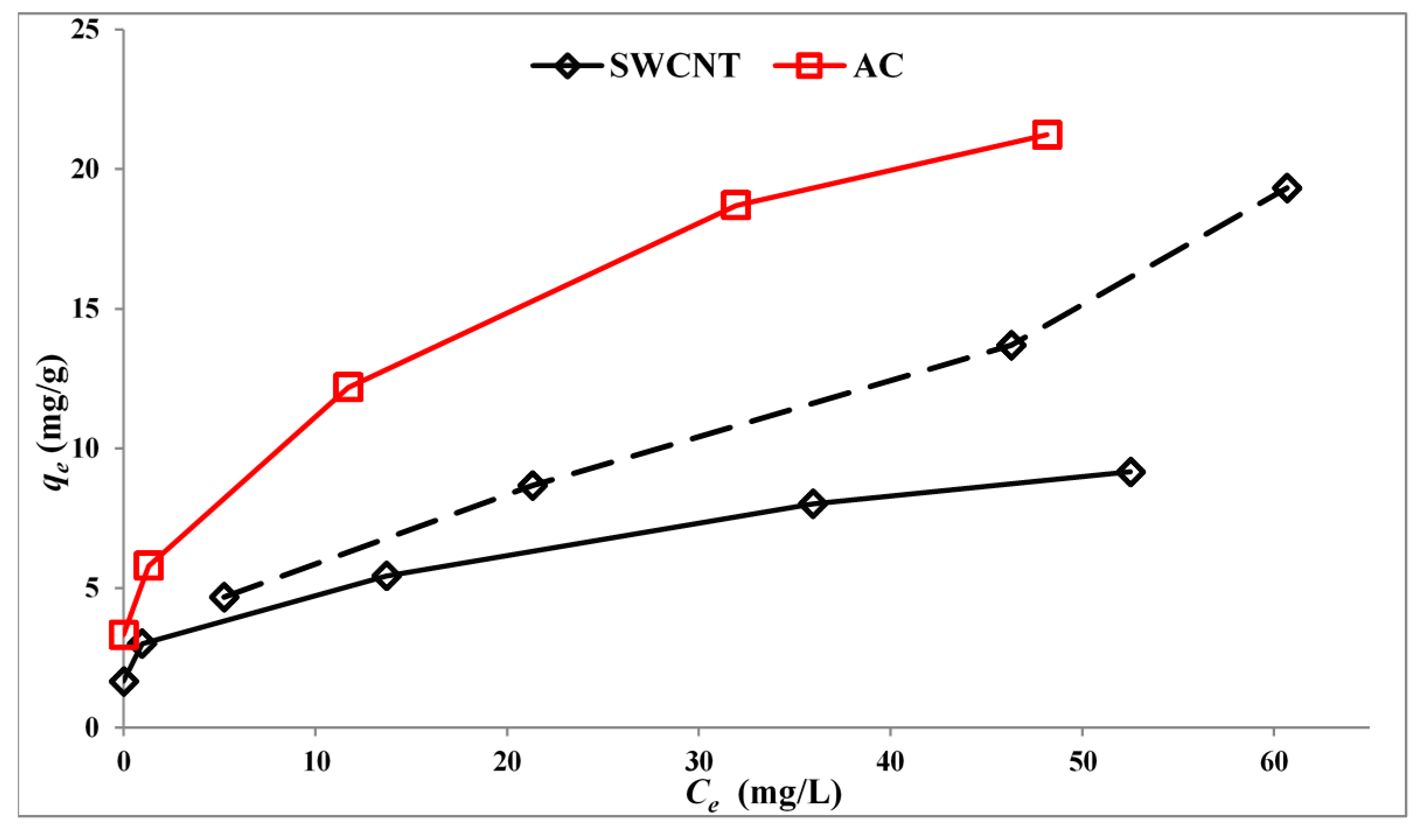
Figure 6 shows the adsorption isotherms for SMX on P25 loaded with 5% by weight of either AC or SWCNTs or 0.25% by weight of SWCNTs. These values are chosen, because preliminary results show good rates for photocatalysts with 0.25% SWCNTs and 5% activated carbon (AC).
The comparative amounts adsorbed as a function of the loading of carbon on TiO2 is interesting and offers a possible explanation for the preliminary observation that 5.0% AC and 0.25% SWCNTs were both favourable loadings for the study of the photocatalytic reaction. The amounts of SMX adsorbed are similar.
Figure 6.
Adsorption isotherms at 22 °C for SMX on activated carbon (AC) or single-walled carbon nanotubes (SWCNT). SWCNT: solid line, 5.0% by weight; dashed line, 0.25% by weight. Ce = solution concentration; qe = adsorbed weight per unit mass (mg/g).
Figure 6.
Adsorption isotherms at 22 °C for SMX on activated carbon (AC) or single-walled carbon nanotubes (SWCNT). SWCNT: solid line, 5.0% by weight; dashed line, 0.25% by weight. Ce = solution concentration; qe = adsorbed weight per unit mass (mg/g).
A series of representative results for SMX degradation on the three photocatalysts are collected below for a photocatalyst loading of 1.0 g/L and initial solution SMX concentrations of 2.4 × 10
−4 M. After the dark adsorption equilibration period, solution concentrations were 1.1 × 10
−4 M for P25/AC, 2.3 × 10
−4 M for P25 and 1.4 × 10
−4 M for P25/SWCNT.
Table 4 collects the rate constants. The rate constants for the total loss of substrate are compared in the table to apparent “rate constants” calculated from solution concentrations alone. Where adsorption is small (P25), the solution value is in agreement. However, differences are quite significant when carbon is added. The data shown here are a reminder that many rate constants in the literature have been reported on solution data where adsorption may have been significant. This can be quite misleading. It is especially true for studies involving dye molecules [
9].
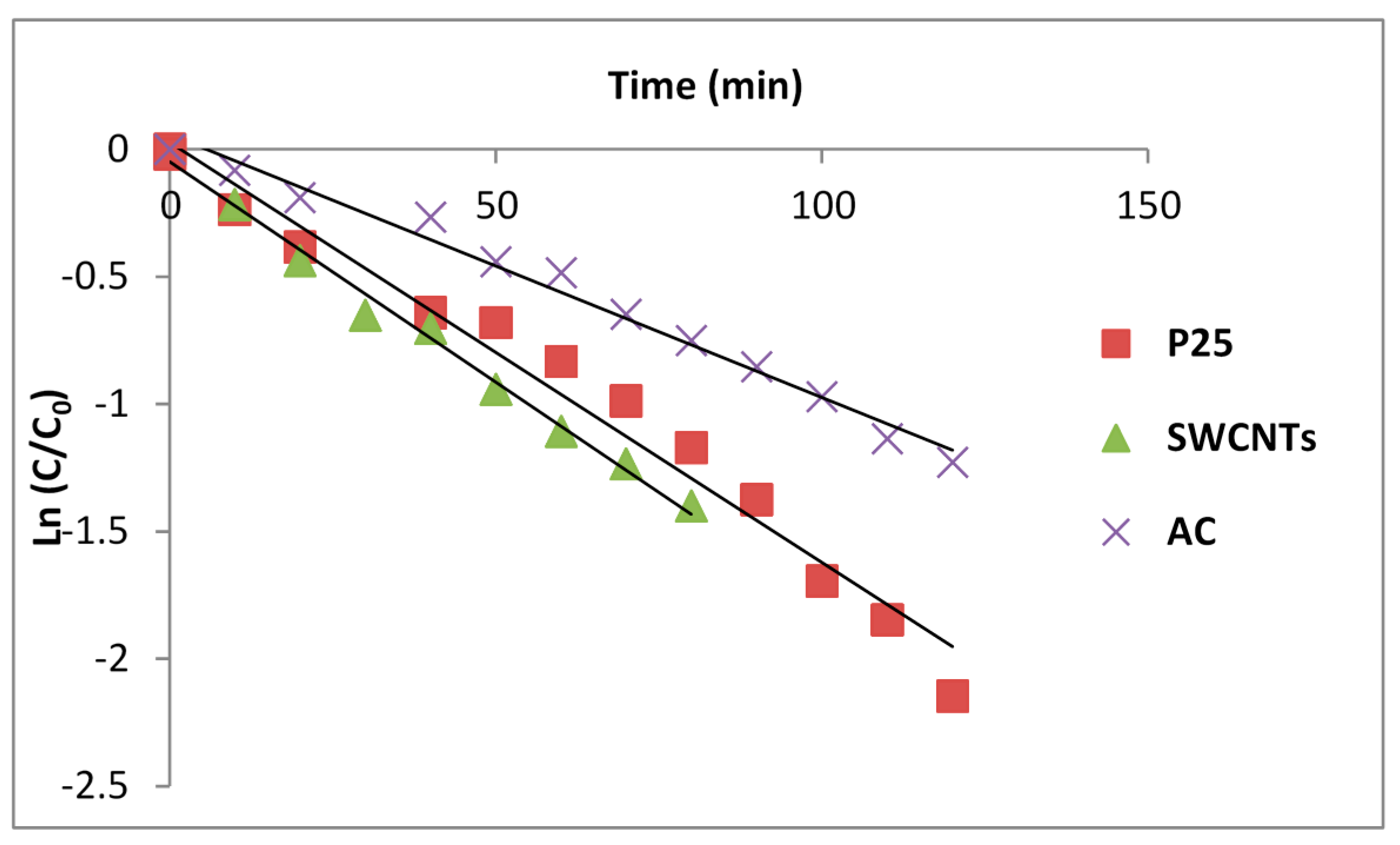
Figure 7 shows the kinetics of total SMX loss with P25, P25/AC and P25/SWCNT as photocatalysts plotted as first order reactions. Powdered activated carbon appears to act as an inhibitor in these circumstances. SWCNTs may be marginally accelerating. This is consistent with reports that surface diffusion is faster on SWCNTs than powdered activated carbon [
8].
Table 4.
First order rate constants for the total loss of SMX (4.2 × 10−4 M) with three photocatalysts.
Table 4.
First order rate constants for the total loss of SMX (4.2 × 10−4 M) with three photocatalysts.
| Photocatalyst | Rate Constant (min−1) | R2 Fitting Parameter | Apparent Solution “Rate Const.” (min−1) |
|---|
| P25/AC | 0.0103 | 0.988 | 0.0154 |
| P25 | 0.0165 | 0.980 | 0.0166 |
| P25/SWCNT | 0. 0173 | 0. 990 | 0.0227 |
In an effort to detect possible electron storage from charge transfer in TiO
2/SWCNTs composites, reactions were carried out in the pulse irradiation mode with pulse cycle times down to 100 ms, with duty cycles of 50%, 30% and 10%. (with the relaxation time of TiO
2 alone near 70 ms. [
13]l a shorter pulse time would not distinguish carbon effects). In no case was a significant difference between pulse irradiation and continuous irradiation observed for a constant energy dose. This could suggest that the SWCNTs function only in the A/S mode, but the alternative explanation, given the direct evidence for electron transfer, is that carbon mediates rapid electron transfer to O
2. There is evidence that nanocarbon can function as an efficient conductor, delivering the electrons [
8] to an acceptor. Yao
et al. [
12] reported results for a physical mixture of carbon nanotubes with TiO
2 nanotubes. The solution conversion rates were superior to TiO
2 alone, but the physical mixture rate was only about half of the composite rate. The positive effect in a physical mixture was taken to be evidence of electron transfer. It does seem unlikely that the time of an encounter would allow efficient shuttle action, unless encounters are “sticky”. Yao
et al. used a dispersant to minimize this. An experiment with a physical mixture of P25 with SWCNTs (ratio as above) started from 95 mg/L SMX, which led to a surface concentration equivalent to 24 mg/L after dark adsorption (clearly carbon loading). Four hours of irradiation reduced the solution concentration and the surface residual to values comparable to those for the composite, tending to support a role for electron transfer.
Figure 7.
Kinetics of the total loss of SMX on irradiation of slurries in a 365-nm batch reactor.
Figure 7.
Kinetics of the total loss of SMX on irradiation of slurries in a 365-nm batch reactor.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}