Chemical Compositions, Chromatographic Fingerprints and Antioxidant Activities of Andrographis Herba

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of the Extraction Method
2.2. Optimization of Chromatographic Conditions
2.3. Validation of Quantitative Analytical Method
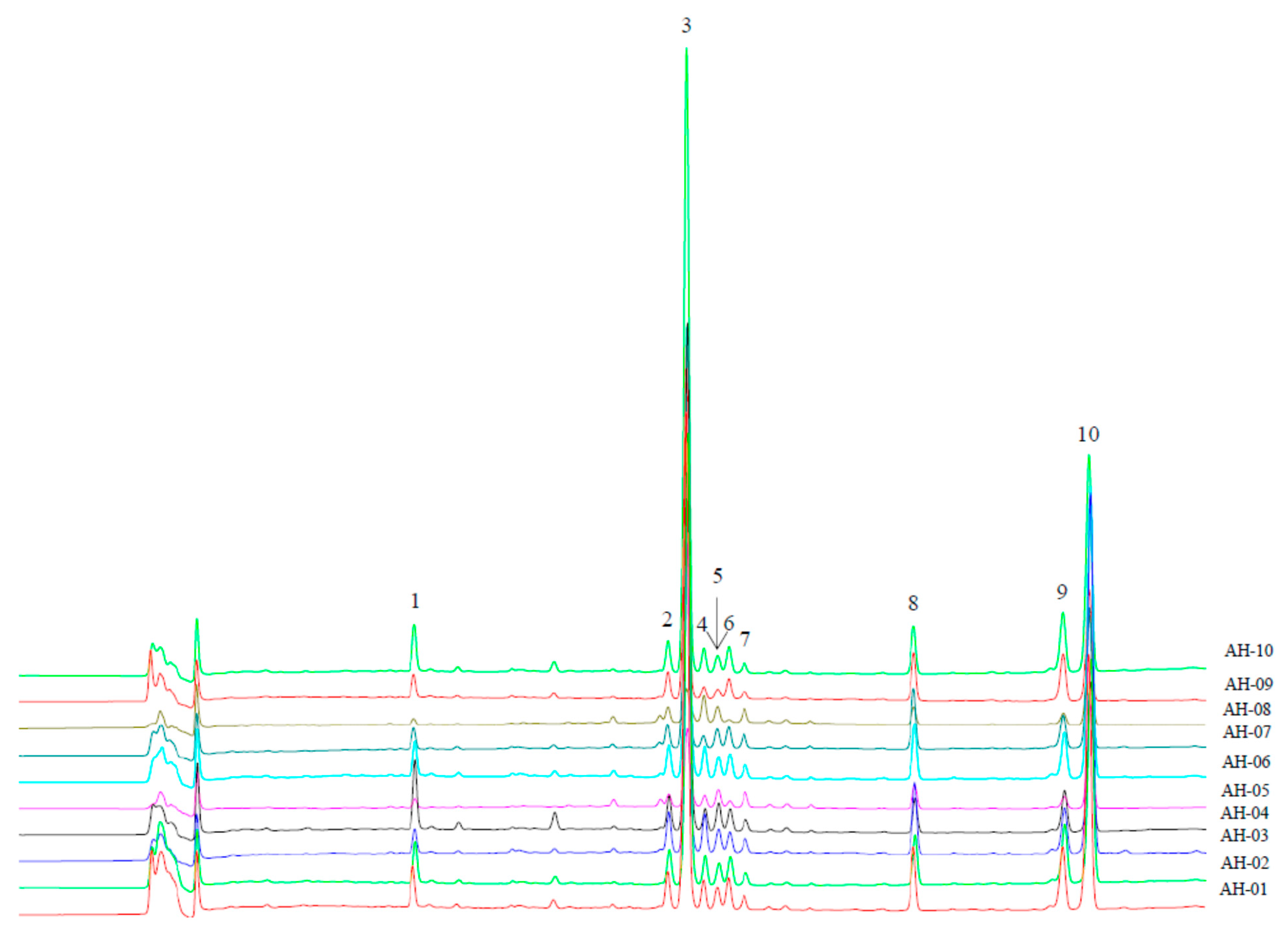
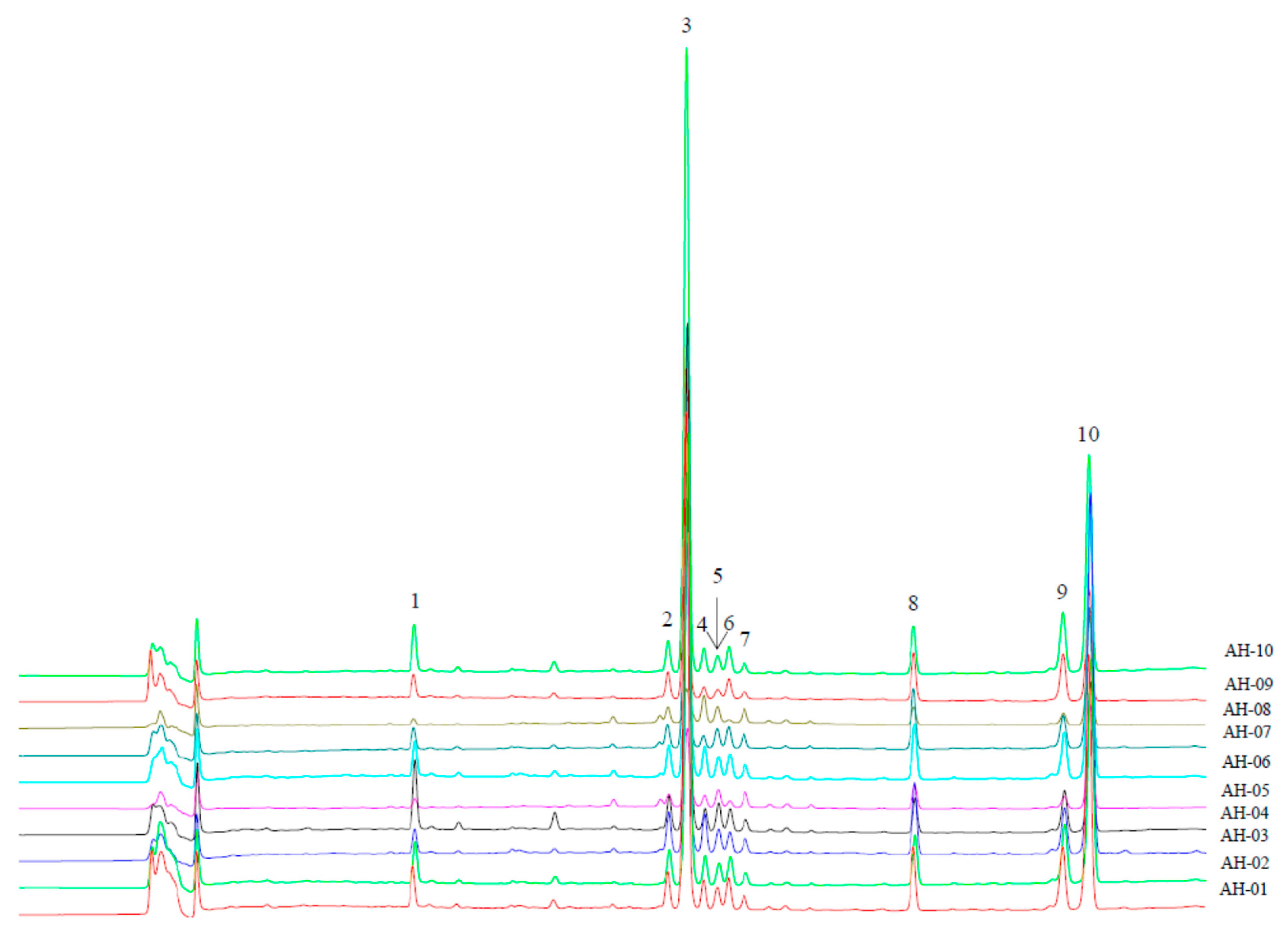
2.4. Validation of the Chromatographic Fingerprinting Method
2.5. Quantitative Determination of the Two Analytes in AH Samples
2.6. Assignments of the Characteristic Peaks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Regression Equation | Correlation Coefficient | Linearity Range (μg/mL) | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|---|---|---|
| Andrographolide | y = 20427x − 4906.5 | 0.9999 | 3.91–250 | 0.06 | 0.21 |
| Dehydroandrographolide | y = 15967x − 11707 | 0.9999 | 15.62–250 | 0.12 | 0.49 |
| Analyte | Precision (RSD, %) | Repeatability (RSD, %) | Stability (RSD, %) | |
|---|---|---|---|---|
| Intra-day (n = 5) | Inter-day (n = 5) | |||
| Andrographolide | 0.08 | 2.28 | 1.13 | 1.07 |
| Dehydroandrographolide | 0.07 | 2.69 | 1.33 | 1.02 |
| Analyte | Sample Weight (g) | Original (mg) | Spiked (mg) | Found (mg) | Recovery (%) | Mean Recovery (%) | RSD (%) |
|---|---|---|---|---|---|---|---|
| Andrographolide | 0.1002 | 1.1373 | 1.0000 | 2.2264 | 108.92 | 107.02 | 1.52 |
| 0.1015 | 1.1520 | 1.0000 | 2.2198 | 106.78 | |||
| 0.1019 | 1.1566 | 1.0000 | 2.2368 | 108.02 | |||
| 0.1023 | 1.1611 | 1.0000 | 2.2070 | 104.59 | |||
| 0.1034 | 1.1736 | 1.0000 | 2.2417 | 106.81 | |||
| Dehydroandrographolide | 0.1002 | 0.8427 | 0.8000 | 1.6436 | 100.11 | 98.86 | 1.46 |
| 0.1015 | 0.8536 | 0.8000 | 1.6423 | 98.58 | |||
| 0.1019 | 0.8570 | 0.8000 | 1.6616 | 100.57 | |||
| 0.1023 | 0.8603 | 0.8000 | 1.6409 | 97.57 | |||
| 0.1034 | 0.8696 | 0.8000 | 1.6491 | 97.44 |
| Sample No. | Content a (%) | Similarity | |||||
|---|---|---|---|---|---|---|---|
| Correlation Coefficient | Angle Cosin | ||||||
| Andrographolide | Dehydroandrographolide | Total | Mean Value b | Median Value c | Mean Value | Median Value | |
| AH-01 | 1.2504 | 0.9214 | 2.1718 | 0.9953 | 0.9963 | 0.9964 | 0.9972 |
| AH-02 | 1.1316 | 0.8053 | 1.9368 | 0.9937 | 0.9948 | 0.9952 | 0.9961 |
| AH-03 | 0.8043 | 1.3472 | 2.1515 | 0.9422 | 0.9382 | 0.9590 | 0.9558 |
| AH-04 | 1.2835 | 0.5493 | 1.8328 | 0.9601 | 0.9623 | 0.9688 | 0.9706 |
| AH-05 | 0.2235 | 0.3991 | 0.6226 | 0.9177 | 0.9127 | 0.9412 | 0.9371 |
| AH-06 | 1.1932 | 1.1167 | 2.3100 | 0.9998 | 0.9995 | 0.9998 | 0.9996 |
| AH-07 | 0.7361 | 0.5217 | 1.2578 | 0.9916 | 0.9930 | 0.9939 | 0.9950 |
| AH-08 | 0.1122 | 0.4195 | 0.5317 | 0.6526 | 0.6435 | 0.7605 | 0.7530 |
| AH-09 | 0.9147 | 0.4269 | 1.3415 | 0.9669 | 0.9702 | 0.9739 | 0.9765 |
| AH-10 | 1.5926 | 0.8157 | 2.4083 | 0.9759 | 0.9783 | 0.9794 | 0.9815 |
| No. | RT (min) | UV (nm) | MS in Neg. Mode | MS2 in Neg. Mode | MS in Pos. Mode | MS2 in Pos. Mode | Assignment | References |
|---|---|---|---|---|---|---|---|---|
| 1 | 7.4 | 225 | 511 [M−H]− | 331 | 551 [M+K]+ | |||
| 493 [M−H−H2O]− | ||||||||
| 557 [M−H+HCOOH]− | ||||||||
| 2 | 12.0 | 200 | 495 [M−H]− | 333 [M−H−glucosyl]− | 519 [M+Na]+ | Glucosyl-deoxyandrographolide | ||
| 541 [M−H+HCOOH]− | 535 [M+K]+ | |||||||
| 991 [2M−H]− | ||||||||
| 3 | 12.4 | 225 | 349 [M−H]− | 331 [M−H−H2O]− | 297 [M+H−3H2O]+ | Andrographolide | [16,17,18,19,20,25] | |
| 331 [M−H−H2O]− | 315 [M+H−2H2O]+ | |||||||
| 395 [M−H+HCOOH]− | 333 [M+H−H2O]+ | |||||||
| 4 | 12.7 | 251 | 495 [M−H]− | 333 [M−H−glucosyl]− | 519 [M+Na]+ | Glucosyl-deoxyandrographolide | ||
| 541 [M−H+HCOOH]− | 535 [M+K]+ | |||||||
| 5 | 12.9 | 287 | 495 [M−H]− | 333 [M−H−glucosyl]− | 519 [M+Na]+ | Glucosyl-deoxyandrographolide | ||
| 541 [M−H+HCOOH]− | 535 [M+K]+ | |||||||
| 6 | 13.1 | 227 | 349 [M−H]− | 331 [M−H−H2O]− | 315 [M+H−2H2O]+ | Isoandrographolide | [17,25] | |
| 331 [M−H−H2O]− | ||||||||
| 395 [M−H+HCOOH]− | ||||||||
| 7 | 13.4 | 264 | 347 [M−H]− | 349 [M+H]+ | 14-deoxy-11-oxoandrographolide | [16] | ||
| 313 [M+H−2H2O]+ | ||||||||
| 331 [M+H−H2O]+ | ||||||||
| 8 | 16.6 | 201 | 479 [M−H]− | 317 [M−H−glucosyl]− | 481 [M+H]+ | 319 [M+H−glucosyl]+ | Neoandrographolide | [16,17,18,19,25] |
| 525 [M−H+HCOOH]− | 301 [M+H−glucosyl−H2O]+ | |||||||
| 959 [2M−H]− | 319 [M+H−glucosyl]− | |||||||
| 503 [M+Na]+ | ||||||||
| 519 [M+K]+ | ||||||||
| 9 | 19.3 | 200 | 333 [M−H]− | 305 [M−H−CO]− | 335 [M+H]+ | 299 [M+H−2H2O]+ | Deoxyandrographolide | [16,18,19,25] |
| 299 [M+H−2H2O]+ | 317 [M+H−H2O]+ | |||||||
| 317 [M+H−H2O]+ | ||||||||
| 10 | 19.8 | 251 | 331 [M−H]− | 303 [M−H−CO]− | 333 [M+H]+ | Dehydroandrographolide | [16,19,20,25] | |
| 297 [M+H−2H2O]+ | ||||||||
| 315 [M+H−H2O]+ |
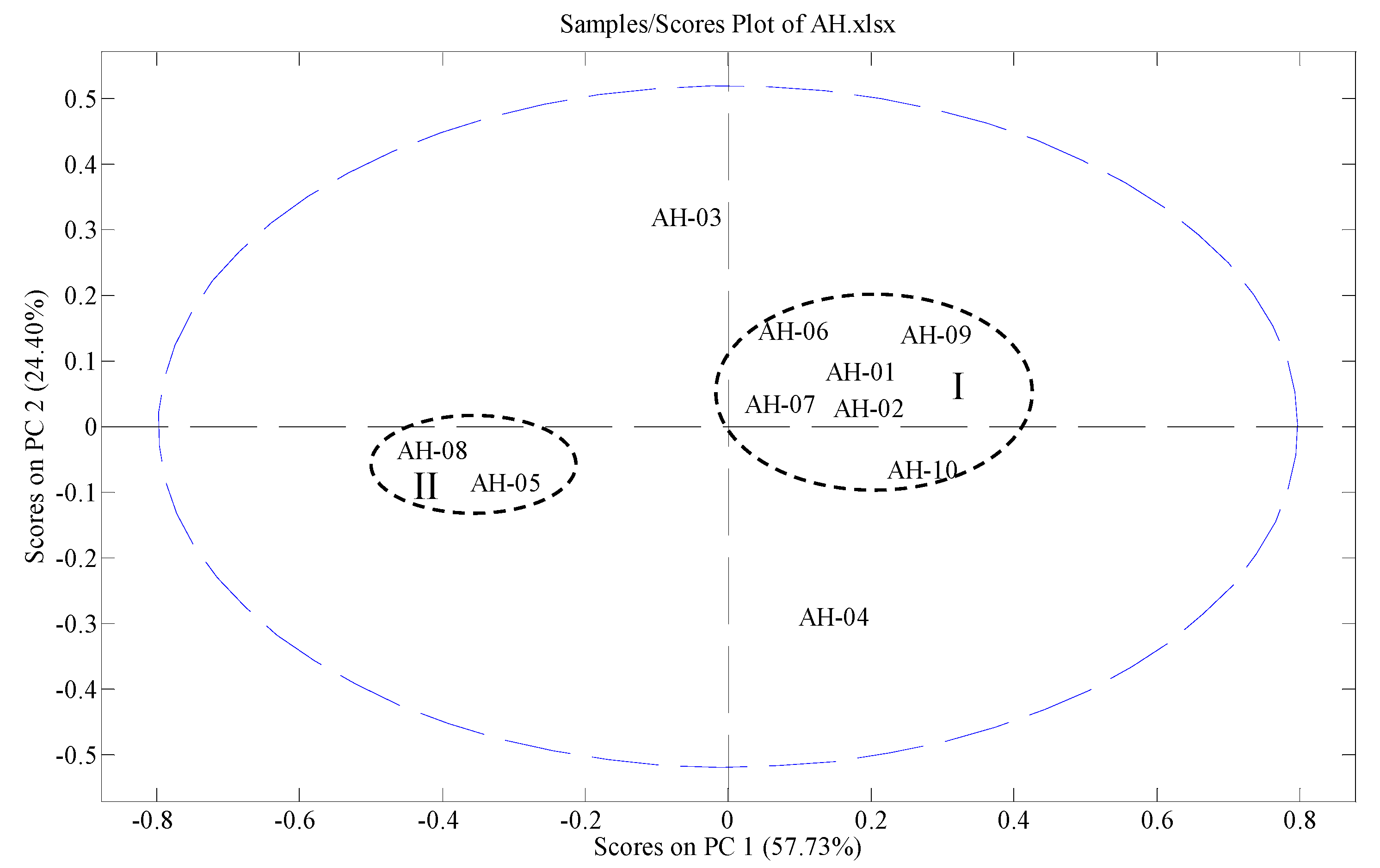
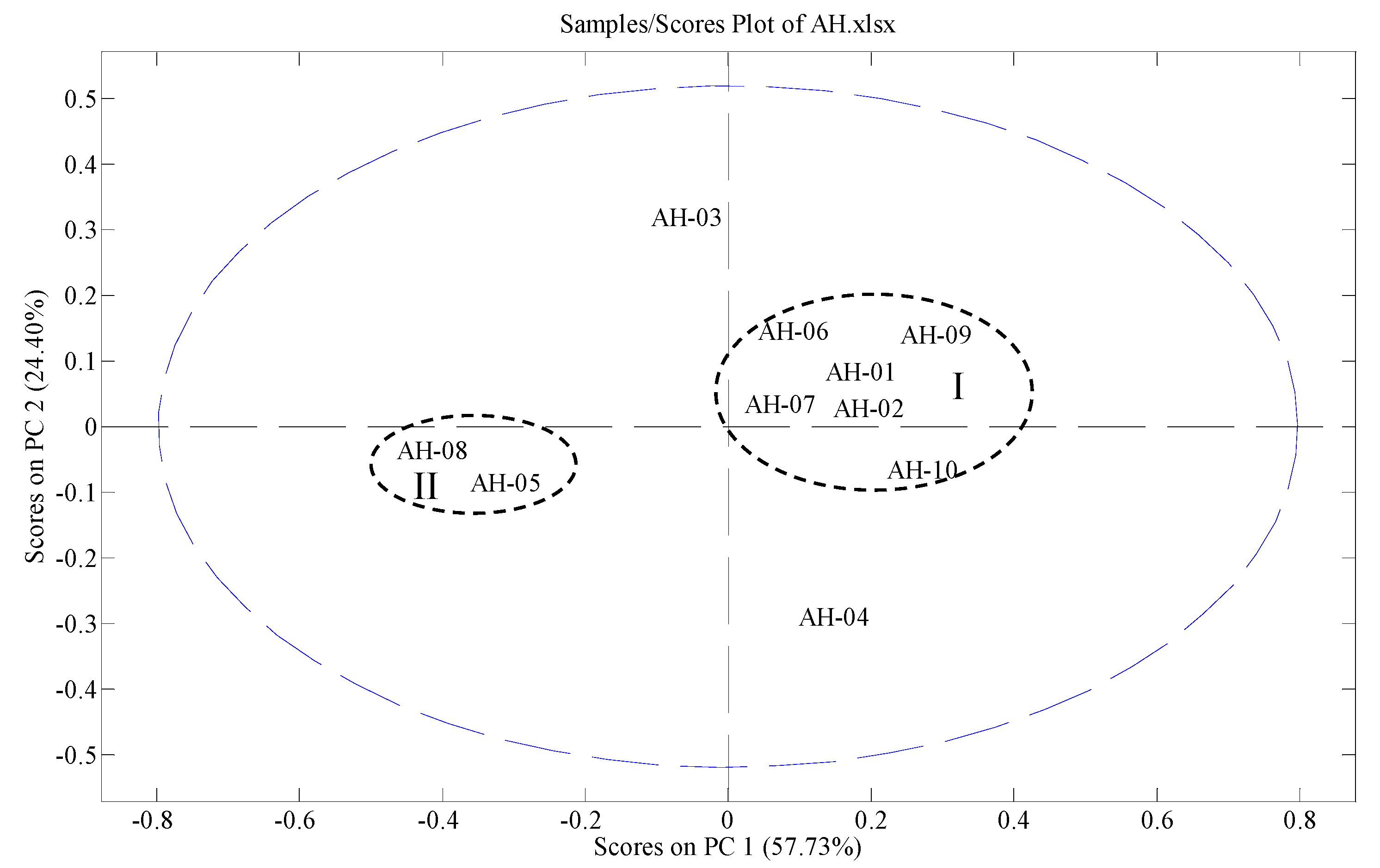
2.7. Fingerprinting and Chemometrics Analyses
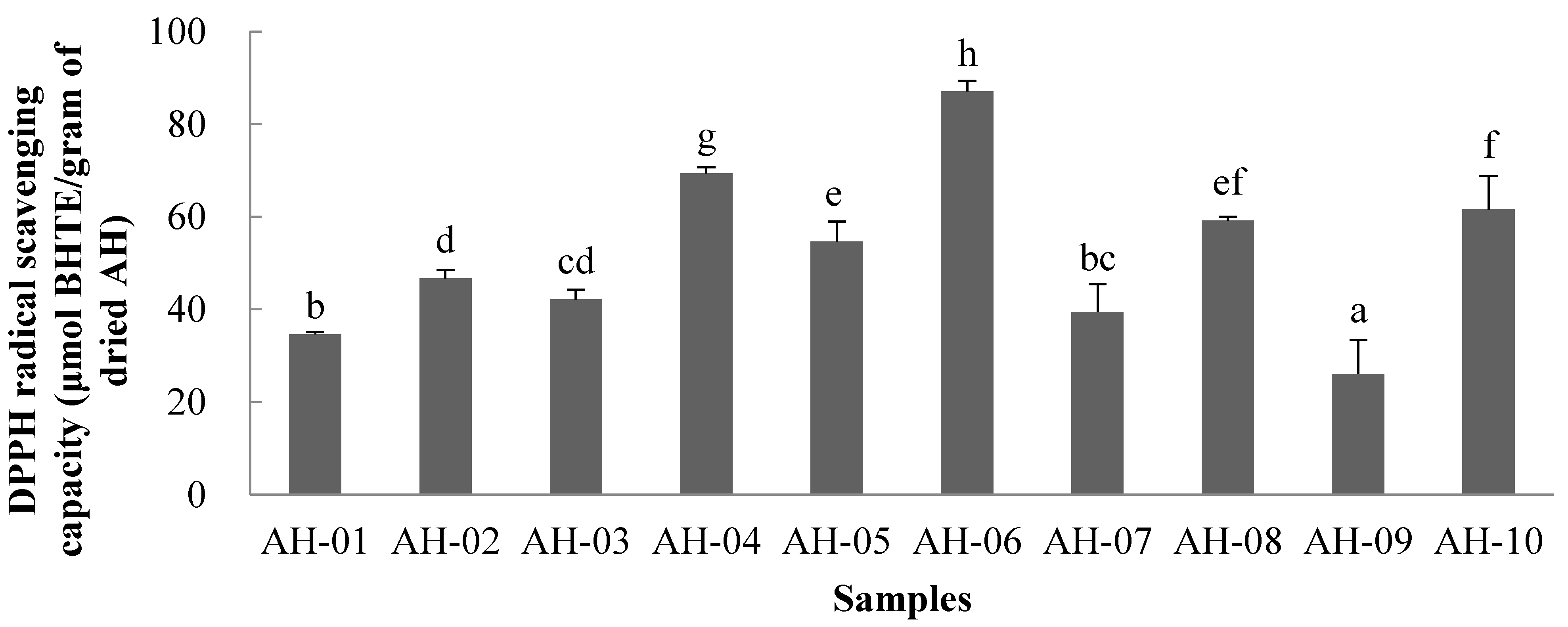
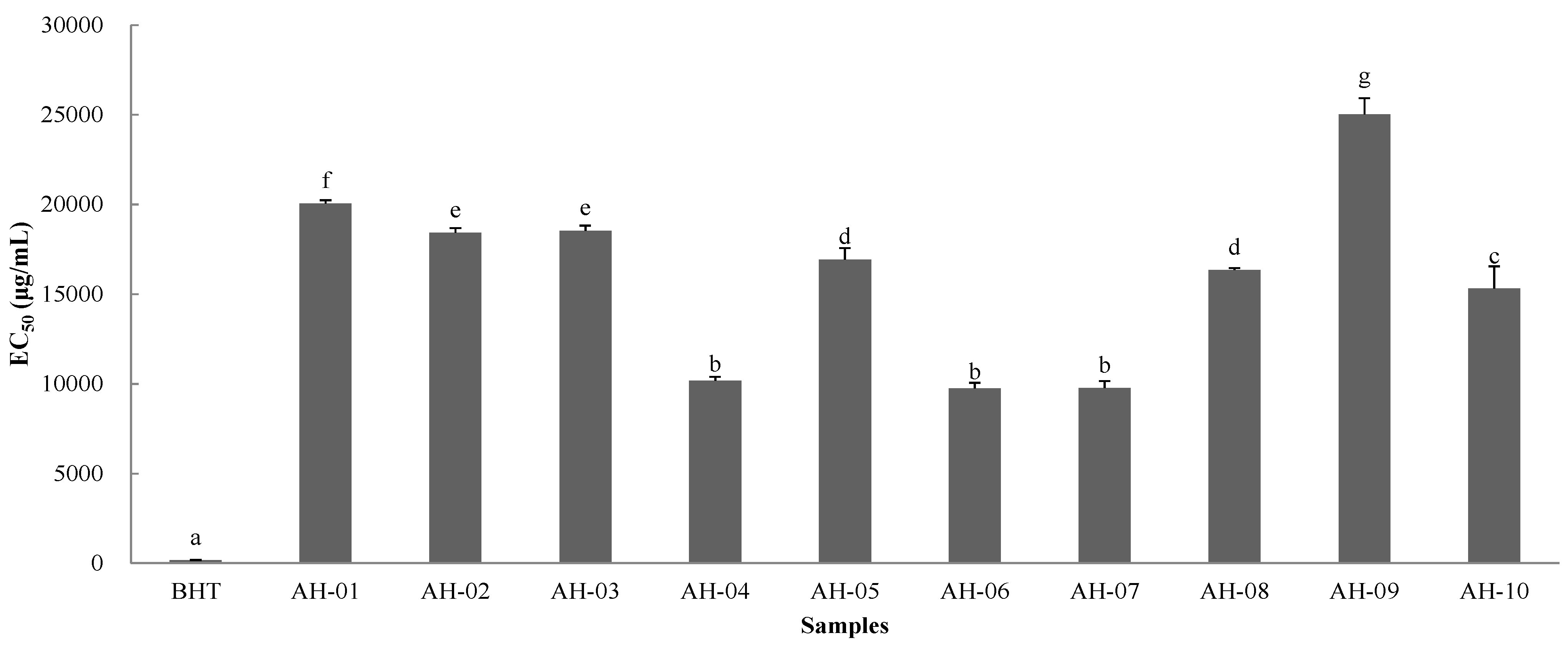
2.8. Antioxidant Activity of AH Samples
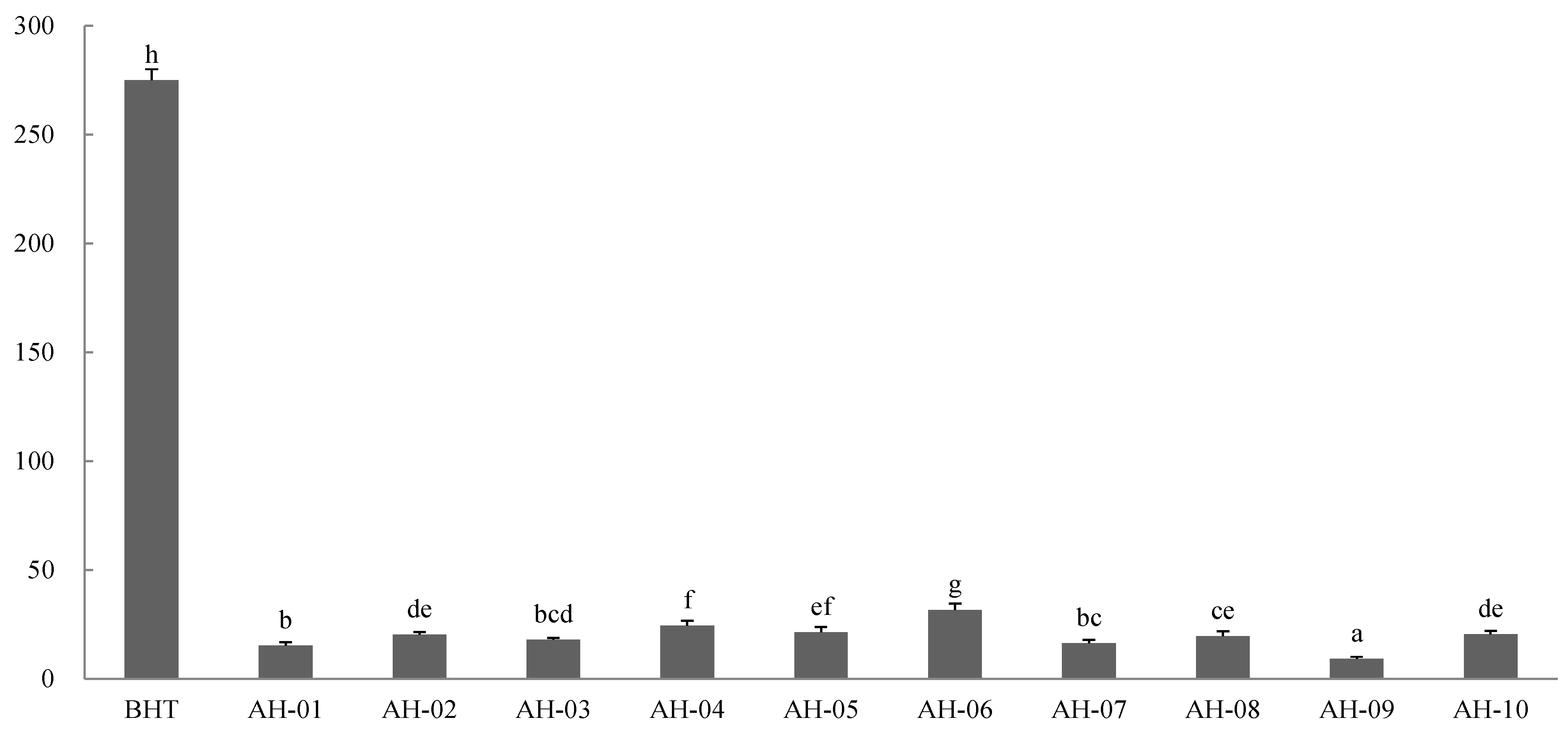
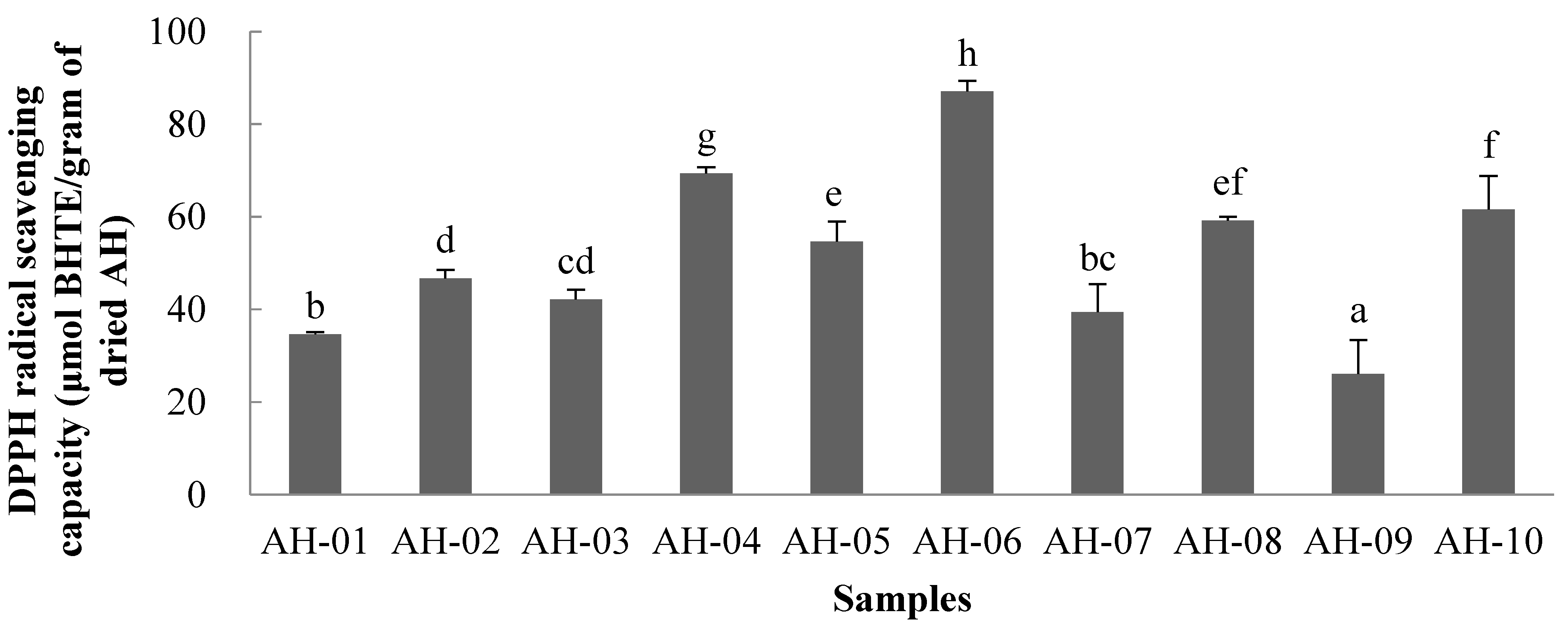
2.8.1. DPPH Assay
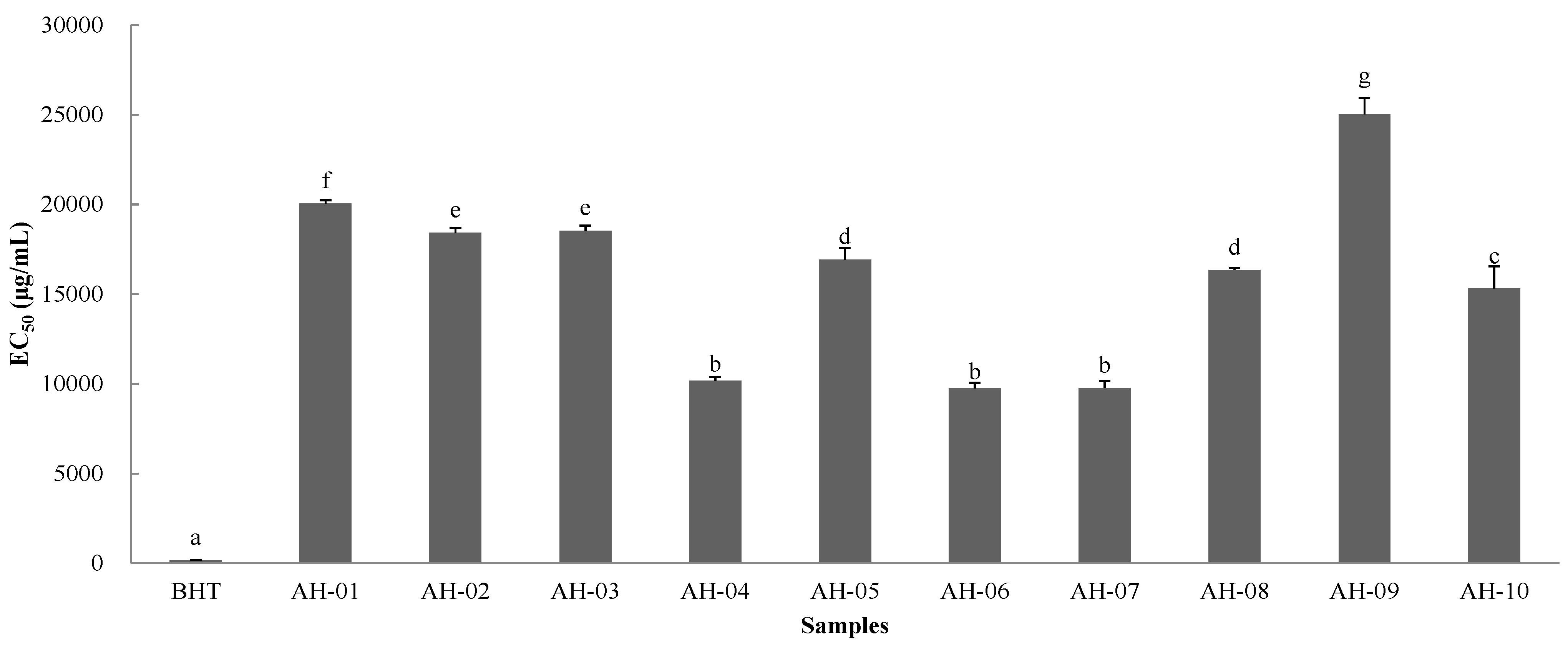
2.8.2. FRAP Assay
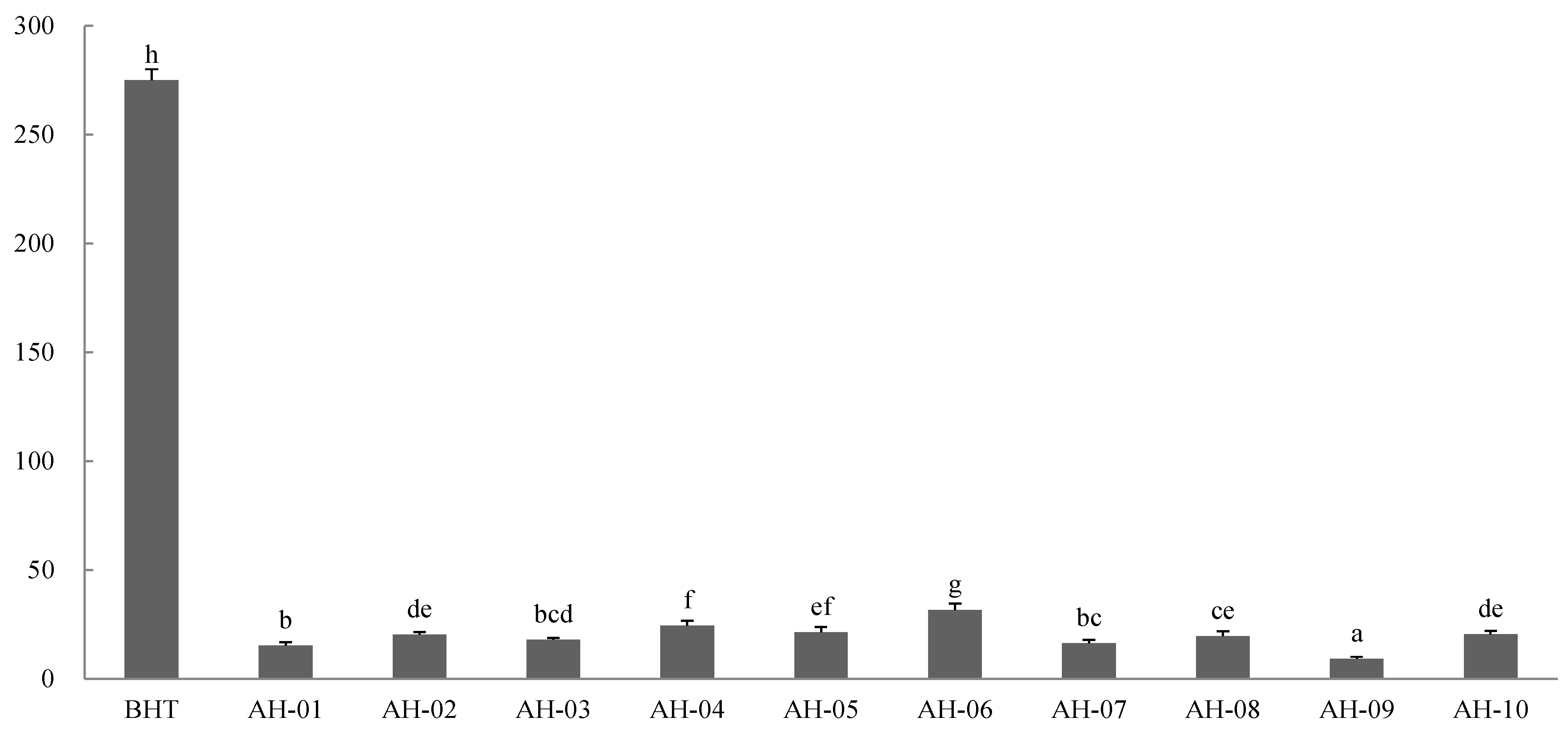
2.8.3. Correlation Analysis
| Peak No. | DPPH Free Radical Scavenging Activity | Ferric Reducing Capacity |
|---|---|---|
| Spearman’s Correlation | Spearman’s Correlation | |
| Peak 01 | 0.236 | 0.236 |
| Peak 02 | −0.127 | −0.127 |
| Peak 03 | 0.285 | 0.261 |
| Peak 05 | 0.297 | 0.370 |
| Peak 06 | −0.018 | 0.018 |
| Peak 07 | 0.212 | 0.297 |
| Peak 08 | −0.503 | −0.467 |
| Peak 09 | −0.224 | −0.261 |
| Peak 10 | 0.115 | 0.079 |
3. Experimental Section
3.1. Chemicals, Solvents and Herbal Materials
3.2. Sample and Reference Preparation
3.3. HPLC Analysis
3.4. HPLC-ESI-MS Analysis
3.5. Antioxidant Activities
3.5.1. DPPH Assay
3.5.2. FRAP Assay
3.6. Data Analysis
3.6.1. Similarity Analysis
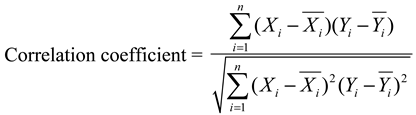
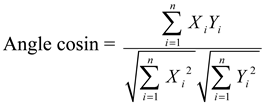
3.6.2. PCA
3.6.3. One-way ANOVA and Correlation Analyses
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chinese Pharmacopoeia Commission. Pharmacopeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2010; Volume 1, pp. 32–33. [Google Scholar]
- Malahubban, M.; Alimon, A.R.; Sazili, A.Q.; Fakurazi, S.; Zakry, F.A. Phytochemical analysis of Andrographis paniculata and Orthosiphon stamineus leaf extracts for their antibacterial and antioxidant potential. Trop. Biomed. 2013, 30, 467–480. [Google Scholar] [PubMed]
- Zein, U.; Fitri, L.E.; Saragih, A. Comparative study of antimalarial effect of sambiloto (Andrographis paniculata) extract, chloroquine and artemisinin and their combination against Plasmodium falciparum in vitro. Acta Med. Indones. 2013, 45, 38–43. [Google Scholar] [PubMed]
- Sheeja, K.; Guruvayoorappan, C.; Kuttan, G. Antiangiogenic activity of Andrographis paniculata extract and andrographolide. Int. Immunopharmacol. 2007, 7, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Sheeja, K.; Shihab, P.K.; Kuttan, G. Antioxidant and anti-inflammatory activities of the plant Andrographis paniculata Nees. Immunopharmacol. Immunotoxicol. 2006, 28, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Tan, B.K. Anti-diabetic property of ethanolic extract of Andrographis paniculata in streptozotocin-diabetic rats. Acta Pharmacol. Sin. 2000, 21, 1157–1164. [Google Scholar]
- Subramanian, R.; Asmawi, M.Z.; Sadikun, A. In vitro α-glucosidase and α-amylase enzyme inhibitory effects of Andrographis paniculata extract and andrographolide. Acta Biochim. Pol. 2008, 55, 391–398. [Google Scholar] [PubMed]
- Medforth, C.J.; Chang, R.S.; Chen, G.Q.; Olmstead, M.M.; Smith, K.M. A conformational study of diterpenoid lactones isolated from the chinese medicinal herb Andrographis paniculata. J. Chem. Soc. Perkin Trans. 2 1990, 6, 1011–1016. [Google Scholar] [CrossRef]
- Chao, W.W.; Lin, B.F. Isolation and identification of bioactive compounds in Andrographis paniculata (Chuanxinlian). Chin. Med. 2010, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.J.; Li, H.W.; Liu, G.W.; Qu, X.H.; Tian, B.; Yan, W.; Lin, Z.; Tang, T.T.; Qin, A.; Dai, K.R. Andrographolide suppresses RANKL-induced osteoclastogenesis in vitro and prevents inflammatory bone loss in vivo. Br. J. Pharmacol. 2014, 171, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Tseng, C.K.; Young, K.C.; Sun, H.Y.; Wang, S.W.; Chen, W.C.; Lin, C.K.; Wu, Y.H. Andrographolide exerts anti-hepatitis C virus activity by up-regulating haeme oxygenase-1 via the p38 MAPK/Nrf2 pathway in human hepatoma cells. Br. J. Pharmacol. 2014, 171, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Yang, W.S.; Yi, Y.S.; Sung, G.H.; Rhee, M.H.; Poo, H.; Kim, M.Y.; Kim, K.W.; Kim, J.H.; Cho, J.Y. AP-1/IRF-3 targeted anti-inflammatory activity of andrographolide isolated from Andrographis paniculata. Evid. Based Complement. Altern. Med. 2013. [CrossRef]
- Basak, A.; Cooper, S.; Roberge, A.G.; Banik, U.K.; Chretien, M.; Seidah, N.G. Inhibition of proprotein convertases-1, -7 and furin by diterpines of Andrographis paniculata and their succinoyl esters. Biochem. J. 1999, 338, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Tan, B.K.H. Effects of 14-deoxyandrographolide and 14-deoxy-11,12-didehydroandrographolide on nitric oxide production in cultured human endothelial cells. Phytother. Res. 1999, 13, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z.T.; Ji, L.L.; Ge, B.X. Inhibitory effects of neoandrographolide on nitric oxide and prostaglandin E-2 production in LPS-stimulated murine macrophage. Mol. Cell. Biochem. 2007, 298, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.J.; Zhang, Z.J.; Yu, J.; Liu, Y.; Xu, F.G. Chemical fingerprinting of Andrographis paniculata (Burm. f.) Nees by HPLC and hierarchical clustering analysis. J. Chromatogr. Sci. 2009, 47, 931–935. [Google Scholar]
- Li, W.K.; Fitzloff, J.F. HPLC-PDA determination of bioactive diterpenoids from plant materials and commercial products of Andrographis paniculata. J. Liquid Chromatogr. Relat. Technol. 2004, 27, 2407–2420. [Google Scholar] [CrossRef]
- Pholphana, N.; Rangkadilok, N.; Saehun, J.; Ritruechai, S.; Satayavivad, J. Changes in the contents of four active diterpenoids at different growth stages in Andrographis paniculata (Burm.f.) Nees (Chuanxinlian). Chin. Med. 2013, 8. [Google Scholar] [CrossRef]
- Xu, T.; Pan, J.; Zhao, L.L. Simultaneous determination of four andrographolides in Andrographis paniculata Nees by silver ion reversed-phase high-performance liquid chromatography. J. Chromatogr. Sci. 2008, 46, 747–750. [Google Scholar] [CrossRef]
- Ding, L.; Luo, X.B.; Tang, F.; Yuan, J.B.; Guo, M.L.; Yao, S.Z. Quality control of medicinal herbs Fructus gardeniae, common Andrographis herb and their preparations for their active constituents by high-performance liquid chromatography-photodiode array detection-electro spray mass spectrometry. Talanta 2008, 74, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.J.; Zhang, L.G.; Hou, J.; Shi, W.Z.; Guo, M.L. A strategy for evaluating antipyretic efficacy of Chinese herbal medicines based on UV spectra fingerprints. J. Ethnopharmacol. 2009, 124, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, J.H.; Yu, L.L.; Chen, P. Chromatographic and mass spectrometric fingerprinting analyses of Angelica sinensis (Oliv.) Diels-derived dietary supplements. Anal. Bioanal. Chem. 2013, 405, 4477–4485. [Google Scholar] [CrossRef]
- Zhao, Y.; Kao, C.P.; Chang, Y.S.; Ho, Y.L. Quality assessment on Polygoni Multiflori Caulis using HPLC/UV/MS combined with principle component analysis. Chem. Cent. J. 2013, 7. [Google Scholar] [CrossRef]
- Sun, J.H.; Chen, P. Chromatographic fingerprint analysis of yohimbe bark and related dietary supplements using UHPLC/UV/MS. J. Pharm. Biomed. Anal. 2012, 6, 142–149. [Google Scholar] [CrossRef]
- Song, Y.X.; Liu, S.P.; Jin, Z.; Qin, J.F.; Jiang, Z.Y. Qualitative and quantitative analysis of Andrographis paniculata by rapid resolution liquid chromatography/time-of-flight mass spectrometry. Molecules 2013, 18, 12192–12207. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.H.; Moore, J.; Yu, L.L. High-throughput relative DPPH radical scavenging capacity assay. J. Agric. Food Chem. 2006, 54, 7429–7436. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, Z.H.; Niu, Y.G.; Shi, H.M.; Chen, P.; Yu, L.L. Chemical compositions, HPLC/MS fingerprinting profiles and radical scavenging properties of commercial Gynostemma pentaphyllum (Thunb.) Makino samples. Food Chem. 2012, 134, 180–188. [Google Scholar] [CrossRef]
- Yang, Z.D.; Zhai, W.W. Identification and antioxidant activity of anthocyanins extracted from the seed and cob of purple corn (Zea mays L.). Innov. Food Sci. Emerg. Technol. 2010, 11, 169–176. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: AH samples and the reference compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Kao, C.-P.; Wu, K.-C.; Liao, C.-R.; Ho, Y.-L.; Chang, Y.-S. Chemical Compositions, Chromatographic Fingerprints and Antioxidant Activities of Andrographis Herba. Molecules 2014, 19, 18332-18350. https://doi.org/10.3390/molecules191118332
Zhao Y, Kao C-P, Wu K-C, Liao C-R, Ho Y-L, Chang Y-S. Chemical Compositions, Chromatographic Fingerprints and Antioxidant Activities of Andrographis Herba. Molecules. 2014; 19(11):18332-18350. https://doi.org/10.3390/molecules191118332
Chicago/Turabian StyleZhao, Yang, Chun-Pin Kao, Kun-Chang Wu, Chi-Ren Liao, Yu-Ling Ho, and Yuan-Shiun Chang. 2014. "Chemical Compositions, Chromatographic Fingerprints and Antioxidant Activities of Andrographis Herba" Molecules 19, no. 11: 18332-18350. https://doi.org/10.3390/molecules191118332
APA StyleZhao, Y., Kao, C.-P., Wu, K.-C., Liao, C.-R., Ho, Y.-L., & Chang, Y.-S. (2014). Chemical Compositions, Chromatographic Fingerprints and Antioxidant Activities of Andrographis Herba. Molecules, 19(11), 18332-18350. https://doi.org/10.3390/molecules191118332