An Insight into the Anticancer Activities of Ru(II)-Based Metallocompounds Using Docking Methods

Abstract
:1. Introduction
2. Results and Discussion
2.1. General Features of the Binding Activities of the Metallocompounds
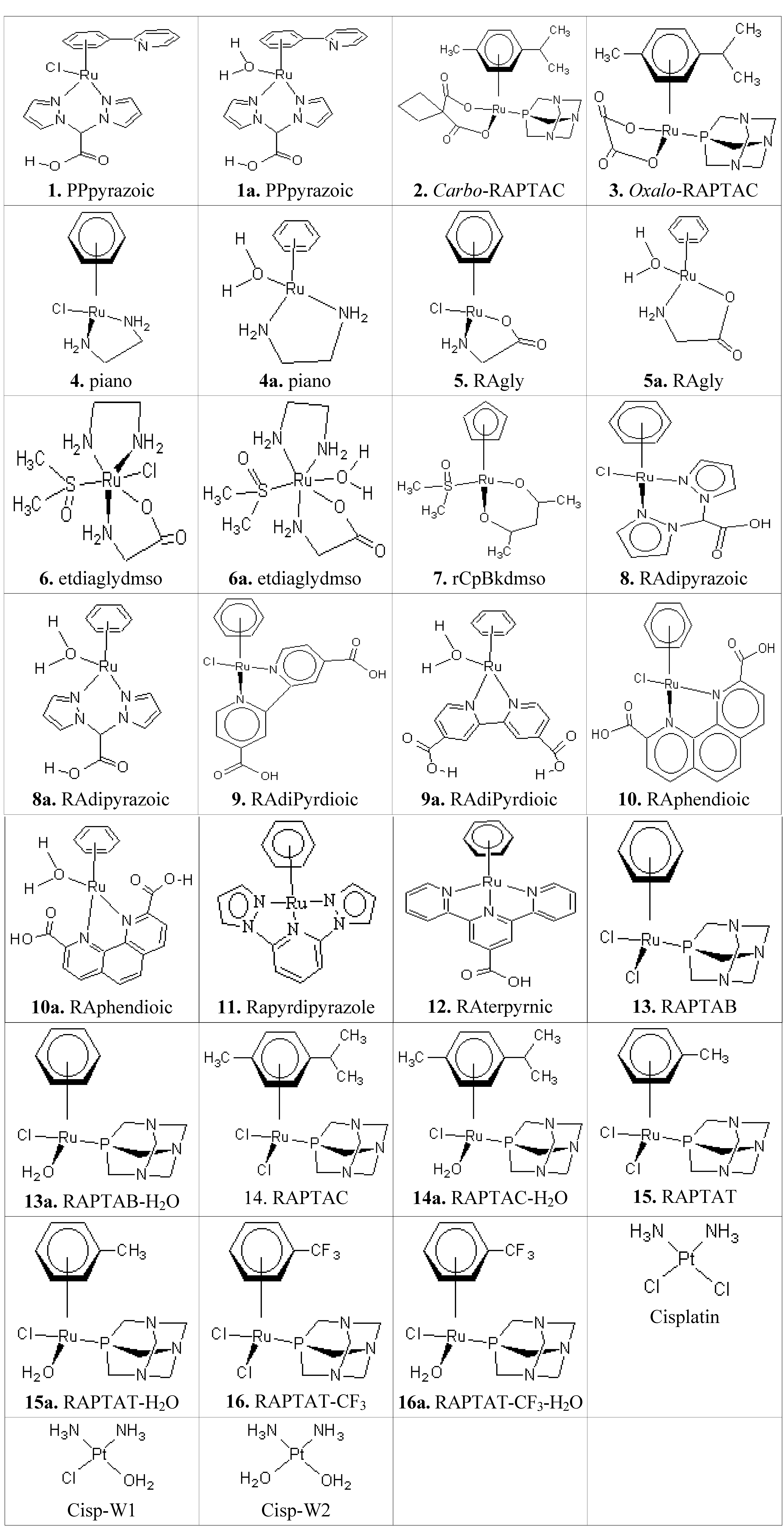
2.2. The Interacting Poses of the Best Two Metallocompounds in the Receptor Binding Sites
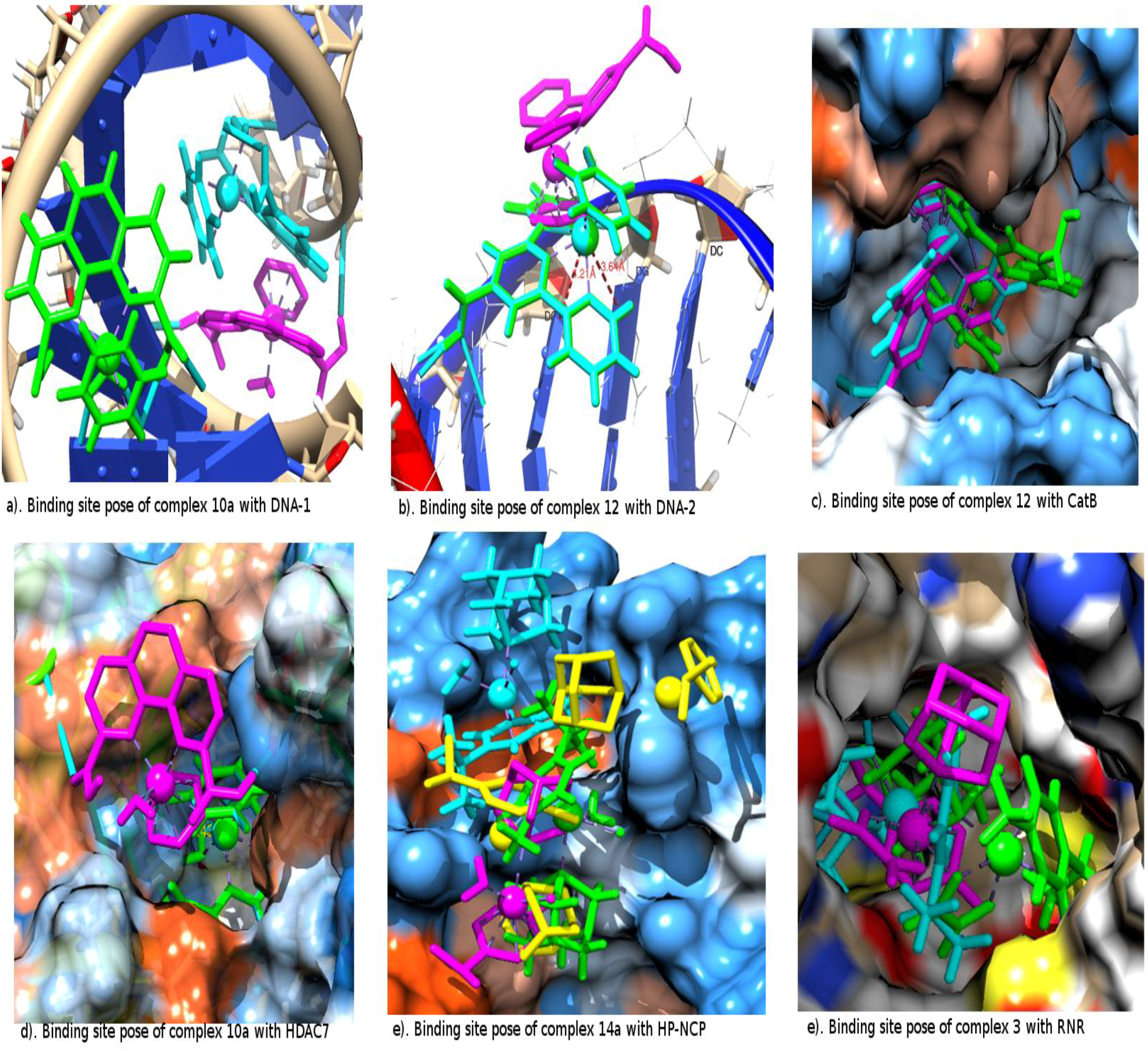

{kind=link}
{kind=link}
| DNA-1 | Molegro | metallocompound
10a {[HB: 2.39 O(COOH) i H(DC 18.B)], [HB: 1.91 H(COOH) ii N7(DG 16.B)]}; |
| metallocompound 12 {[HB: 2.32 O(COOH) H(DA 17.B)], [HB: 1.44 H(COOH) O6(DG 7.A)]} | ||
| Molegro-Constrained | metallocompound 10a {[HB: 1.71 O(COOH) i H(DC 2.A)], [HB: 1.89 H(COOH) i O6(DG 23.B)], [HB: 1.66 H(H2O) O6(DG 23.B)], [HB: 2.01 H(H2O) N7(DG 23.B)], [MR 4.49 N7(DG 34.B)], [MR 4.60 N7(DA 22.B)]}; | |
| metallocompound 9a {[HB: 2.05 H(COOH) i O6(DG 23.B)], [HB: 2.02 H(COOH) iiOP2(DG 23.B)], [HB: 2.11 H(H2O) N7(DA 22.B)], [MR 4.60 N7(DA 22.B)]}; | ||
| Autodock | metallocompound 9a {[HB: 2.00 H(COOH) i OP1(DT 5.A)], [HB: 1.65 H(COOH) ii OP2(DA 15.B)]} | |
| metallocompound 4a {[HB: 1.81 H(NH2) i OP1(DG 6.A)], [HB: 1.70 H(NH2)ii OP2(DG 7.A)], [MR 3.32 O(DG 6.A)], [MR 3.88 O(DG 6.A)]} | ||
| DNA-2 | Molegro | Metallocompound 10a {[HB: 1.92 O(COOH) i H(DG 7.T)], [HB: 1.76 H(COOH) i O4(DT 8.T)], [HB: 2.16 H(COOH) ii O3(DC 9.T)]}; |
| metallocompound 1a {[HB: 1.75 H(COOH) O2(DC 8.P)], [HB: 2.12 H(H2O) O4(DT 8.T)]} | ||
| Molegro-Constrained | metallocompound
12 {[HB: 1.57 H(COOH) O6(DG 5.P)], [HB: 2.50 O(COOH) H(DC 9.T)], [HB: 2.80 O(COOH) H(DA 6.P)], [MR 3.64 N7(DG 6.T)], [MR 4.21 N7(DG 7.T)]}; | |
| metallocompound 1 {[HB: 1.95 N(arene) H(DA 6.P)], [MR 3.91 N7(DG 7.T)]} | ||
| Autodock | metallocompound 1a {[HB: 1.79 H(COOH) OP2(DG 6.T)]}; | |
| metallocompound 4a {[HB: 1.76 H(H2O) OP2(DG 7.T)], [HB: 1.96 H(NH2) i O3(DG 6.T)], [HB: 1.84 H(NH2) i OP1(DG 6.T)]} | ||
| CatB | Molegro | metallocompound 12 {[HB: 1.66 H(COOH) O(GLY 24 D)]} |
| metallocompound 1 {[HB: 2.21 H(COOH) O(MET 196 E)]} | ||
| Molegro-Constrained | metallocompound 1a {[HB: 3.28 N(arene) SG(CYS 29D)], [HB: 2.61 H(COOH) O(GLY 198E)], [MR 4.60 SG(CYS 29D)]} | |
| metallocompound 8a {[MR 4.60 SG(CYS 29D)]} | ||
| Autodock | metallocompound 10a {[HB: 2.99 O(COOH) i SG(CYS 29 D)], [HB: 2.76 O(COOH) i H(GLN 23 D)], [HB: 1.70 H(COOH)i OE1(GLN 23D)], [HB: 2.14 H(COOH) ii OE2(GLU 122D)]}; | |
| metallocompound 1a {[HB: 1.72 H(COOH) OE1(GLU 122 E)], HB: 1.72 H(COOH) OE2(GLU 122E)]} | ||
| Gyrase | Molegro | metallocompound 10 {[HB: 2.17 H(COOH) O(ASP 73A)]} |
| metallocompound 11 {[none]} | ||
| Molegro-Constrained | metallocompound 10a {[HB: 2.24 H(COOH) i OD2(ASP 49A)], [HB: 1.92 H(COOH) ii O(GLY 117A)], MR: 4.60 ND2(ASP 46 A)]} | |
| metallocompound 9a {[HB: 1.50 H(H2O) OD1(ASP 46A)], [HB: 2.08 H(COOH) i O(ASP 46A)], [HB: 1.84 H(COOH) ii O(ASP 45A)], MR: 4.27 ND2(ASP 46 A)]}; | ||
| Autodock | metallocompound 12 {[HB: 2.11 H(COOH) O(ASP 73A)], [HB: 1.81 H(COOH) O(ASP 73A)], [HB: 2.00 O(COOH) H(GLY 77A)]}; | |
| metallocompound 9a {[HB: 1.90 O(H2O) HD22(ASN 46A)], [HB: 1.63 H(COOH) i O(LYS 103 A)], [HB: 2.05 O(COOH) ii H(ALA 100 A)], [HB: 1.65 H(COOH) ii O(ILE 94 A)], [HB: 2.14 O(COOH) ii H(SER 121 A)]} | ||
| HDAC7 | Molegro | metallocompound 1 {[HB: 1.82 N(arene) H(imi@HIS 709A)], [HB: 1.87 O(COOH) H(imi@HIS 669A)]}; |
| metallocompound 12 {[HB: 2.36 H(COOH) O(ASP 707A)]} | ||
| Molegro-Constrained | metallocompound 10a {[HB: 1.89 H(COOH) i O(GLY 678A)], [HB: 2.43 O(COOH) i H(CYS 680A)], [HB: 1.99 O(COOH) i HE2(HIS 669A)], [MR: 3.43 NE2(HIS 709A)]} | |
| metallocompound 11 {[MR: 3.60 N(HIS 709A)]} | ||
| Autodock | metallocompound 1a {[HB: 1.63 H(COOH) OD1(ASP 626A)], [HB: 1.90 H(H2O) OD1(ASP 626A)]}; | |
| metallocompound 9a {[HB: 1.80 O(COOH)i H(PHE 738A)], [HB: 1.79 H(COOH) i O(PRO 809 A)]} | ||
| HP-NCP | Molegro | metallocompound 1a {[none]} |
| metallocompound 11 {[MR: 3.38 OE(GLU 56G)]} | ||
| Molegro-Constrained | metallocompound 10a {[HB: 1.75 H(COOH) i OE1(GLU 92 G)], [[HB: 2.17 H(COOH) ii NE2(HIS 106 H)], [MR: 4.36 OE1(GLU 61 H)], MR: 4.44 NE2(HIS 106 H)]} | |
| metallocompound 11 {[MR: 4.36 OE1(GLU 61 H)], [MR: 4.60 NE2(HIS 106 H)]} | ||
| Autodock | metallocompound 4a {[HB: 1.67 H(H2O) OE2(GLU 61 G)], [HB: 1.67 H(H2O) OE1(GLU 64G)]}; | |
| metallocompound 5a {[MR: 2.90 OE2(GLU 61 G)]} | ||
| BRAF Kinase | Molegro | metallocompound 1a {[HB: 2.09 H(COOH) O(ASN 579A)]}; |
| metallocompound 12 {[HB: 2.59 H(COOH) O(CYS 531A)], [HB: 1.66 O(COOH) H(CYS 531A)]} | ||
| Molegro-Constrained | metallocompound 1 {[HB: 2.04 N(arene) H(SER 535A)], [MR: 4.61 SG(CYS 531 A)]} | |
| metallocompound 8 {[MR: 4.61 SG(CYS 531 A)]} | ||
| Autodock | metallocompound 4a {[HB: 1.91 H(NH2) i OD2(ASP 478A)], [HB: 2.03 H(H2O) OD2(ASP 478A)], [HB: 1.84 H(NH2) ii OE2(GLU 532A)], [HB: 1.94 H(H2O) OE2(GLU 532A)]}; | |
| metallocompound 5a {[none]} | ||
| rHA | Molegro | metallocompound 10 {[HB: 2.24 O(COOH)i H(ARG 117A)], [HB: 2.05 O(COOH)ii H(ARG 186A)]}; |
| metallocompound 12 {[HB: 2.16 H(COOH) O(ASP 108A)], [MR: 4.27 O(SER 193A)]} | ||
| Molegro-Constrain | metallocompound 10a {[HB: 2.12 H(COOH) i OE2(GLU 37A)], [HB: 1.68 O(COOH) ii H(ARG 144A)], [HB: 1.82 H(COOH) ii O(GLN 33A)], [MR: 4.59 NH1(ARG 144A)]} | |
| metallocompound 2 {[HB: 1.91 O(COO)i H(ARG 144A)], [HB: 1.91 O(COO) i H(ARG 144A)], [MR: 4.59 NH1(ARG 144A)]}; | ||
| Autodock | metallocompound 10 {[MR: 1.78 NZ(LYS 195A)], [MR: 4.52 NH2(ARG 222A)]}; | |
| metallocompound 4a {[HB: 1.85 H(H2O) OD2(ASP 38A)], [HB: 1.73 H(H2O) OH(TYR 84A)], [HB: 1.85 H(NH2) i OD2(ASP 34A)]} | ||
| RNR | Molegro | metallocompound 1a {[HB: 1.59 H(H2O) O(PRO 621A)], [HB: 1.98 H(COOH) OE1(GLU 441A)], [HB: 2.39 O(COOH) HD22(ASN 437A)]}; |
| metallocompound 3 {[HB: 1.88 O(COO) i H(GLU 623A)], [HB: 1.92 O(COO) i H(SER 625A)], [HB: 2.34 O(COO) ii H(THR 209A)]} | ||
| Molegro-Constrained | metallocompound 1a {[HB: 2.22 H(COOH) O(SER 224B)], [MR: 4.17 SG(CYS 439A)]}; | |
| metallocompound 10 {[HB: 2.27 H(COOH) i O(PRO 621A)], [HB: 2.16 H(COOH) ii O(SER 224A)], [MR: 4.26 S(CYS 439A)]} | ||
| Autodock | metallocompound 1a {[HB: 1.68 H(COOH) OE1(GLU 623A)], [HB: 2.00 O(H2O) H(ARG 639A)], [MR: 3.69 NH2(ARG 639A)]}; | |
| metallocompound 12 {[HB: 2.60 O(COOH) H(GLU 623A)], [HB: 1.95 O(COOH) N(SER 625A)], [HB: 2.02 O(COOH) HG(SER 625A)], [HB: 1.77 H(COOH) OG1(THR 209A)]} | ||
| TopII | Molegro | metallocompound 1 {[HB: 2.15 H(COOH) O(GLN 365B)], [HB: 3.38 O(COOH) O(THR 27A)]} |
| metallocompound 10 {none}; | ||
| Molegro-Constrained | metallocompound 10a {[HB: 2.47 H(COOH) O(ILE 15A)], [MR: 4.60 NE2(HIS 20B)]}; | |
| metallocompound 1a {[MR: 4.60 NE2(HIS 20B)]} | ||
| Autodock | metallocompound 5 {[HB: 2.21 O(COO) H(GLY)], [HB: 2.57 O(COO) NH2(GLN 365A)], [HB: 2.53 O(COO) NH3(LYS 367A)], [HB: 2.09 O(COO) H(ASN 142A)], [HB: 2.37 O(COO) H(ARG 141A)]}; | |
| metallocompound 9a {[HB: 1.90 H(COOH) i OD1(ASN 70A)], [HB: 1.88 O(COOH) i HZ1(LYS 147A)], [HB: 1.85 H(COOH) ii OD1(ASN 129A)], [HB: 2.15 O(COOH) ii HD21(ASN 129A)]} | ||
| TrXR | Molegro | metallocompound 10 {[HB: 1.77 O(COOH) i O(ALA 198A)], [HB: 2.60 O(COOH) iiH(ARG 166A)], [HB: 2.19 O(COOH) ii H(ARG 166A)]}; |
| metallocompound 1 {[HB: 2.14 H(COOH) O(SER 222A)], [HB: 1.75 O(COOH) OH(SER 222A)]} | ||
| Molegro-Constrained | metallocompound 9a {[HB: 2.27 O(COOH)i HZ1(LYS 67A)], [HB: 2.10 H(COOH) ii O(THR 373A)], [MR: 3.59 SG(CYS 64A)], [MR: 4.60 SG(CYS 59A)]} | |
| metallocompound 8a {[HB: 1.63 H(COOH) OD2(ASP 334A)], [HB: 2.11 O(COOH) HH21(ARG 293A)], [HB: 1.85 O(COOH) HE(ARG 293A)], [MR: 3.06 SG(CYS 64A)], [MR: 4.60 SG(CYS 59A)]} | ||
| Autodock | metallocompound 5a {[HB: 1.96 H(NH2) O(GLU 341A)], [HB: 1.89 H(NH2) OH(TYR 200A)], [HB: 2.10 O(COO) H(THR 343A)], [HB: 2.13 O(COO) H(THR 343A)], [HB: 2.02 H(H2O) OD1(ASP 334A)], [HB: 2.48 H(H2O) OD2(ASP 334A)]} | |
| metallocompound 12 {[HB: 1.69 H(COOH) O(GLU 341A)], [HB: 1.98 O(COOH) NH3(LYS 315A)], [MR: 3.47 H(LEU 340A)]}; | ||
| TS | Molegro | metallocompound 12 {[HB: 2.22 H(COOH) O(SER 232A)]}; |
| metallocompound 10 {[HB: 1.53 O(COOH) H(ASN 229A)]} | ||
| Molegro-Constrained | metallocompound 10a {[HB: 2.40 O(COOH) H(CYS 198A)], [HB: 1.86 O(COOH) HH(TYR 146A)], [MR: 4.60 S(CYS 198A)]} | |
| metallocompound 1 {[HB: 2.13 H(COOH) O(SER 219A)], [HB: 2.70 O(COOH) H(ARG 218A)], [HB: 2.39 O(COOH) H(ARG 218A)], [HB: 2.38 O(COOH) H(ARG 23A)], [HB: 1.56 O(COOH) H(ARG 23A)], [MR: 3.55 S(CYS 198A)]}; | ||
| Autodock | metallocompound 9a {[HB: 1.82 H(COOH) i O(SER 219A)], [HB: 1.97 O(COOH) i H(ASP 221A)], [HB: 2.45 O(COOH) i HG(CYS 198A)], [HB: 2.08 O(COOH) ii HD21(ASN 229A)], [HB: 1.63 H(COOH) ii OE2(GLU 60A)]}; | |
| metallocompound 5a {[HB: 2.36 O(COO) HH(TYR 146A)], [HB: 1.71 H(H2O) OE1(GLU 60A)]} |
2.3. The Binding Affinity of the Metallocompounds to DNA
| DNA-1 | DNA-2 | CatB | DNA_Gyrase | HDAC7 | HP-NCP | BRAF KINASE | rHA | RNR | topoII | TrxR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −4.32 | −3.3 | −4.26 | −3.68 | −3.15 | −3.44 | −3.25 | −3.45 | −3.15 | −3 | −3.43 | −3.03 |
| 2 | −2.47 | −3.08 | −3.38 | −3.21 | −2.64 | −3.43 | −3.79 | −4.19 | −3.71 | −2.65 | −3.35 | −4.14 |
| 3 | −2.73 | −2.5 | −3.43 | −3.49 | −2.5 | −3.82 | −3.29 | −3.91 | −3.49 | −3.84 | −3.35 | −3.7 |
| 4 | −4.67 | −3.88 | −3.98 | −3.49 | −3.92 | −4.16 | −3.65 | −3.86 | −2.85 | −3.05 | −3.21 | −2.84 |
| 5 | −2.93 | −2.7 | −3.27 | −3.01 | −3.02 | −3.54 | −3.39 | −3.62 | −3.07 | −4.08 | −3.39 | −3.21 |
| 6 | −2.29 | −1.9 | −1.24 | −1.31 | −1.77 | −2.46 | −2.37 | −2.32 | −2.45 | −1.62 | −2.19 | −1.31 |
| 7 | −2.82 | −2.79 | −3.42 | −3.38 | −3.1 | −2.59 | −3.97 | −3.97 | −3.6 | −3.47 | −2.79 | −2.96 |
| 8 | −2.24 | −3.41 | −2.92 | −3.09 | −2.59 | −3.29 | −3.39 | −3.4 | −3.25 | −3.03 | −3.43 | −2.53 |
| 9 | −4.27 | −3.73 | −4.02 | −3.24 | −3.5 | −3.63 | −3.66 | −3.34 | −3.39 | −3.1 | −3.92 | −3.67 |
| 10 | −5.13 | −4.94 | −5.19 | −4.86 | −3.78 | −5.14 | −4.48 | −5.24 | −4.03 | −3.86 | −4.24 | −4.51 |
| 11 | −5.02 | −3.81 | −4.5 | −3.68 | −4.01 | −3.85 | −3.12 | −2.98 | −2.43 | −2.19 | −2.91 | −3.65 |
| 12 | −5.97 | −4.65 | −5.77 | −5.12 | −5.1 | −4.56 | −4.37 | −4.16 | −4.19 | −3.4 | −4.31 | −5.23 |
| 13 | −2.5 | −2.44 | −3.35 | −2.94 | −2.78 | −2.84 | −3.13 | −3.38 | −2.88 | −2.88 | −2.86 | −2.78 |
| 14 | −3.04 | −3.14 | −3.55 | −3.54 | −2.72 | −3.83 | −3.41 | −4.29 | −3.52 | −3.98 | −3.79 | −3.7 |
| 15 | −2.43 | −2.56 | −3.38 | −3.1 | −2.82 | −3.05 | −3.27 | −3.39 | −3.09 | −3.13 | −2.83 | −2.96 |
| 16 | −1.63 | −1.87 | −2.23 | −2.15 | −1.66 | −2.22 | −2.08 | −2.59 | −2.41 | −2.08 | −2.11 | −2.4 |
| cisplatin | −2.23 | −2.94 | −2.15 | −1.34 | −1.98 | −2.18 | −2.56 | −1.65 | −1.93 | −1.95 | −2.4 | −1.23 |
| co-crystallized compounds | −5.49 | −8.68 | −4.35 | −7.01 | −12.48 | −4.79 | −9.19 |
| DNA-1 | DNA-2 | CatB | DNA_Gyrase | HDAC7 | HP-NCP | BRAF KINASE | rHA | RNR | topoII | TrxR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | −6.53 | −5.65 | −6.47 | −4.67 | −5.97 | −4.95 | −4.97 | −4.04 | −4.39 | −3.16 | −4.26 | −4.9 |
| 4a | −6.85 | −5.87 | −6.29 | −4.82 | −5.55 | −5.58 | −5.27 | −4.54 | −3.16 | −3.47 | −4.23 | −4.72 |
| 5a | −5.58 | −4.75 | −5.65 | −4.55 | −5.08 | −5.26 | −4.98 | −4.33 | −4.13 | −3.96 | −5.35 | |
| 6a | −5.35 | −3.6 | −3.6 | −2.3 | −3.39 | −4.51 | −4.26 | −2.6 | −2.37 | −2.38 | −3.54 | −2.96 |
| 8a | −6.44 | −5.03 | −5.78 | −4.76 | −5.25 | −5.22 | −3.96 | −4.02 | −3.02 | −3.06 | −2.98 | −4.2 |
| 9a | −6.98 | −5.96 | −6.19 | −5.03 | −5.56 | −5.05 | −4.77 | −3.3 | −3.95 | −4.07 | −3.94 | −6.09 |
| 10a | −6.38 | −5.22 | −6.7 | −4.61 | −5.21 | −4.58 | −4.03 | −4.48 | −3.49 | −3.51 | −4.17 | −5.26 |
| 13a | −4.86 | −4.06 | −5.19 | −4.05 | −4.81 | −4.29 | −3.59 | −3.74 | −3.32 | −3.65 | −3.61 | −3.39 |
| 14a | −4.39 | −3.75 | −4.84 | −4.19 | −4.59 | −4.32 | −3.33 | −3.99 | −3.65 | −3.73 | −3.76 | −3.17 |
| 15a | −4.86 | −4.03 | −4.38 | −3.8 | −4.79 | −4.27 | −3.64 | −3.41 | −3.14 | −3.6 | −3.65 | −3.05 |
| 16a | −4.12 | −3.24 | −3.1 | −3.28 | −3.75 | −3.62 | −3.1 | −2.91 | −2.16 | −2.77 | −2.65 | −2.31 |
| Cisp-W1 | −4.31 | −3.98 | −2.1 | −0.99 | −2.55 | −3.37 | −3.25 | −1.73 | −1.04 | −1.15 | −2.63 | −1.45 |
| Cisp-W2 | −4.92 | −4.4 | −2.15 | −0.93 | −2.76 | −3.18 | −3.47 | −1.72 | 0.02 | −0.33 | −1.79 | −1.2 |
| DNA-1 | DNA-2 | CatB | Gyrase | HDAC7 | HP-NCP | BRAF Kinase | rHA | RNR | TopII | TrXR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −137.57 | −133.77 | −136.37 | −131.99 | −166.09 | −111.45 | −118.79 | −142.13 | −126.87 | −154.82 | −137.87 | −127.39 |
| 2 | −113.84 | −106.15 | −124.88 | −110.82 | −127.16 | −94.13 | −110.31 | −113.18 | −98.90 | −121.50 | −103.71 | −114.61 |
| 3 | −116.96 | −97.09 | −114.43 | −103.00 | −110.06 | −91.82 | −105.79 | −135.47 | −124.93 | −118.69 | −104.43 | −111.20 |
| 4 | −77.36 | −77.97 | −72.59 | −79.89 | −83.51 | −68.21 | −67.34 | −85.02 | −74.88 | −74.75 | −75.04 | −75.89 |
| 5 | −71.22 | −82.12 | −78.61 | −73.09 | −90.05 | −68.85 | −74.96 | −86.25 | −83.41 | −84.15 | −82.63 | −79.76 |
| 6 | −86.14 | −81.39 | −88.78 | −84.63 | −95.90 | −75.53 | −84.76 | −97.60 | −88.56 | −92.57 | −100.22 | −79.41 |
| 7 | −87.95 | −84.94 | −93.69 | −94.11 | −102.98 | −75.06 | −99.78 | −100.28 | −92.07 | −97.69 | −106.18 | −83.43 |
| 8 | −119.77 | −111.18 | −118.67 | −110.79 | −107.93 | −96.84 | −117.76 | −126.06 | −116.02 | −115.67 | −121.37 | −111.59 |
| 9 | −124.03 | −122.96 | −125.25 | −126.26 | −131.56 | −101.43 | −115.00 | −119.84 | −106.90 | −129.34 | −119.82 | −118.59 |
| 10 | −141.56 | −126.74 | −135.89 | −142.43 | −147.55 | −107.85 | −114.94 | −147.15 | −121.72 | −129.66 | −138.35 | −129.75 |
| 11 | −143.11 | −142.45 | −131.53 | −140.36 | −157.61 | −117.62 | −114.61 | −133.71 | −114.68 | −128.89 | −121.04 | −127.35 |
| 12 | −153.94 | −129.90 | −144.21 | −138.83 | −160.63 | −104.74 | −122.12 | −142.37 | −113.66 | −129.13 | −120.89 | −135.47 |
| 13 | −62.98 | −67.16 | −83.32 | −70.45 | −90.53 | −63.02 | −73.54 | −73.40 | −74.49 | −82.38 | −70.73 | −77.41 |
| 14 | −82.56 | −85.26 | −100.23 | −85.99 | −113.53 | −71.41 | −89.94 | −97.88 | −91.04 | −102.67 | −89.64 | −85.02 |
| 15 | −66.45 | −67.86 | −89.21 | −70.59 | −101.13 | −62.61 | −74.58 | −78.25 | −81.13 | −84.18 | −78.17 | −84.17 |
| 16 | −73.52 | −76.16 | −95.70 | −80.20 | −94.92 | −71.53 | −87.20 | −87.77 | −80.40 | −93.80 | −84.48 | −87.89 |
| Cisplatin | −47.86 | −44.53 | −38.09 | −46.03 | −45.22 | −46.40 | −39.91 | −43.53 | −40.83 | −43.89 | −38.82 | −39.79 |
| co-crystallized compounds | −117.36 | −162.48 | −70.17 | −121.74 | −152.80 | −192.15 | −160.24 | −106.57 |
| DNA-1 | DNA-2 | CatB | Gyrase | HDAC7 | HP-NCP | BRAF Kinase | rHA | RNR | TopII | TrXR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | −150.05 | −145.76 | −128.02 | −136.31 | −106.38 | −129.59 | −122.79 | −137.56 | −125.01 | −84.25 | −131.96 | −127.11 |
| 4a | −94.36 | −98.02 | −77.65 | −81.91 | −81.50 | −76.86 | −61.37 | −80.77 | −73.66 | −75.59 | −87.09 | −79.70 |
| 5a | −88.43 | −96.33 | −82.01 | −76.01 | −86.05 | −75.74 | −68.95 | −87.31 | −80.07 | −82.79 | −87.95 | −86.37 |
| 6a | −100.04 | −95.69 | −91.02 | −89.94 | −96.65 | −73.28 | −85.05 | −104.25 | −86.02 | −81.42 | −99.80 | −82.86 |
| 8a | −146.26 | −127.46 | −126.15 | −136.28 | −129.26 | −113.66 | −113.13 | −129.33 | −113.82 | −116.42 | −111.76 | −118.85 |
| 9a | −144.54 | −125.87 | −123.34 | −120.42 | −90.58 | −106.68 | −109.38 | −127.80 | −106.99 | −44.91 | −131.76 | −120.86 |
| 10a | −161.33 | −145.84 | −131.71 | −142.22 | −158.93 | −113.82 | −110.56 | −121.54 | −120.44 | −79.33 | −122.11 | −126.24 |
| 13a | −77.52 | −81.55 | −90.06 | −75.72 | −94.57 | −71.10 | −71.20 | −74.88 | −80.32 | −88.55 | −71.37 | −79.81 |
| 14a | −96.88 | −106.30 | −105.80 | −99.03 | −110.38 | −80.03 | −95.53 | −86.14 | −94.44 | −106.7 | −94.05 | −86.26 |
| 15a | −84.66 | −86.84 | −91.02 | −82.81 | −101.74 | −74.49 | −74.40 | −78.79 | −83.59 | −95.58 | −80 | −75.33 |
| 16a | −89.62 | −89.80 | −98.32 | −85.32 | −106.94 | −81.19 | −85.06 | −90.37 | −82.83 | −99.96 | −81.1 | −84.77 |
| Cisp-W1 | −62.38 | −58.09 | −46.27 | −50.24 | −44.07 | −44.94 | −35.32 | −44.08 | −41.17 | −42.72 | −44.55 | −42.56 |
| Cisp-W2 | −84.39 | −70.95 | −45.17 | −57.47 | −43.45 | −42.73 | −28.77 | −37.91 | −43.82 | −42.74 | −44.54 | −41.68 |
| DNA-1 | DNA-2 | CatB | Gyrase | HDAC7 | HP-NCP | BRAF Kinase | rHA | RNR | TopII | TrXR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −116.11 | −128.23 | −99.45 | −107.80 | −133.48 | −95.08 | −105.04 | −27.44 | −121.13 | −43.60 | −67.44 | −110.86 |
| 2 | −97.80 | −85.91 | −82.34 | −84.02 | −74.26 | −72.24 | −10.28 | −86.36 | −99.26 | −8.38 | −14.82 | −98.92 |
| 3 | −78.05 | −72.06 | −74.09 | −72.94 | −66.77 | −76.14 | −58.45 | 10.16 | −90.08 | −72.78 | −40.92 | −81.43 |
| 4 | −68.50 | −61.59 | −67.53 | −50.22 | −71.28 | −61.87 | −63.35 | −43.68 | −60.08 | −38.15 | −39.06 | −54.89 |
| 5 | −61.04 | −58.83 | −63.33 | −57.28 | −76.44 | −58.64 | −70.82 | 26.31 | −65.84 | −44.97 | −41.88 | −61.54 |
| 6 | −71.93 | −66.06 | −72.19 | −62.16 | −94.46 | −61.01 | −65.64 | −70.29 | −71.03 | −43.94 | −47.54 | −69.91 |
| 7 | −70.49 | −65.18 | −75.95 | −67.76 | −96.59 | −67.18 | −69.16 | 15.13 | −75.00 | −63.31 | −53.38 | −70.06 |
| 8 | −90.84 | −88.44 | −79.93 | −82.66 | −83.25 | −81.53 | −96.36 | −2.29 | −96.45 | −49.40 | −63.45 | −102.19 |
| 9 | −111.80 | −117.06 | −99.06 | −110.65 | −87.96 | −78.92 | −79.57 | −49.97 | −104.49 | −70.13 | −62.38 | −92.44 |
| 10 | −122.46 | −117.40 | −90.07 | −90.46 | −141.61 | −102.69 | −74.46 | −88.52 | −113.02 | −47.87 | −45.13 | −99.57 |
| 11 | −124.47 | −125.50 | −111.86 | −104.70 | −144.86 | −104.53 | −92.96 | −85.74 | −107.52 | −50.71 | −91.01 | −98.64 |
| 12 | −125.54 | −129.90 | −104.35 | −107.26 | −112.43 | −97.12 | −75.51 | −66.29 | −106.43 | −8.28 | −66.59 | −91.48 |
| 13 | −49.78 | −49.53 | −60.05 | −48.08 | −55.13 | −46.22 | −59.27 | −47.94 | −62.99 | −56.55 | −28.91 | −58.53 |
| 14 | −66.71 | −68.14 | −79.05 | −65.58 | −66.86 | −51.48 | −50.64 | 19.86 | −77.96 | 0.00 | −52.96 | −78.21 |
| 15 | −54.61 | −55.65 | −64.40 | −58.80 | −58.83 | −53.29 | −63.23 | −55.84 | −64.54 | −57.33 | −36.73 | −65.11 |
| 16 | −65.29 | −64.79 | −70.31 | −68.07 | −72.82 | −58.20 | −58.63 | −66.45 | −73.20 | −37.72 | −35.54 | −71.26 |
| Cisplatin | −38.97 | −34.23 | −43.49 | −33.31 | −49.14 | −29.06 | −36.20 | −26.49 | −30.79 | −35.58 | −33.00 | −30.63 |
| co-crystallized compounds | −132.12 | −120.11 | −73.24 | −30.80 | −94.68 | −126.37 | −115.68 | −144.33 | −95.40 |
| DNA-1 | DNA-2 | CatB | Gyrase | HDAC7 | HP-NCP | BRAF Kinase | rHA | RNR | TopII | TrXR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | −126.70 | −122.83 | −127.33 | −117.35 | −99.20 | −104.06 | −86.78 | −7.23 | −113.75 | −81.83 | −101.44 | −108.38 |
| 4a | −85.71 | −72.54 | −69.40 | −65.33 | −81.49 | 29.93 | −59.98 | 36.01 | −59.88 | −39.37 | −63.58 | −53.49 |
| 5a | −77.64 | −71.78 | −68.57 | −63.59 | −86.11 | −57.75 | −67.38 | 29.87 | −63.13 | −47.71 | −67.64 | −64.37 |
| 6a | −94.75 | −84.34 | −81.49 | −74.57 | −96.64 | −66.72 | −64.99 | 24.66 | −76.07 | −54.41 | −69.31 | −73.20 |
| 8a | −128.21 | −111.69 | −110.41 | −112.41 | −129.27 | −99.85 | −92.82 | −78.11 | −110.43 | −69.52 | −103.76 | −94.83 |
| 9a | −134.05 | −125.46 | −103.79 | −119.20 | −65.16 | −81.50 | −74.88 | −52.63 | −106.48 | −68.35 | −118.89 | −100.43 |
| 10a | −135.60 | −126.76 | −116.86 | −120.84 | −158.92 | −115.24 | −72.50 | −91.28 | −110.44 | −82.45 | −84.59 | −113.85 |
| 13a | −63.52 | −63.46 | −69.71 | −57.52 | −73.39 | −60.84 | −58.70 | 35.82 | −67.58 | −59.59 | −33.43 | −55.79 |
| 14a | −84.65 | −78.01 | −92.77 | −69.23 | −74.84 | −60.37 | −54.22 | 23.16 | −82.70 | −5.71 | −11.37 | −78.60 |
| 15a | −71.57 | −68.76 | −75.77 | −66.29 | −72.91 | −66.99 | −64.27 | −37.41 | −72.72 | −66.08 | −36.32 | −62.97 |
| 16a | −74.70 | −74.07 | −73.81 | −74.55 | −72.88 | −69.08 | −60.26 | −34.70 | −76.95 | −46.59 | −42.19 | −66.71 |
| Cisp-W1 | −62.37 | −45.20 | −40.91 | −36.93 | −43.65 | −35.62 | −32.23 | −31.06 | −35.91 | −29.93 | −34.13 | −29.65 |
| Cisp-W2 | −78.38 | −55.98 | −34.03 | −38.27 | −43.46 | −40.96 | −29.51 | −24.29 | −32.68 | −25.48 | −32.34 | −26.43 |
2.4. The Binding Affinities of the Metallocompounds to CatB and TrxR
2.5. The Binding Affinity of the Metallocompounds with Gyrase, HDAC7, HP-NCP, BRAF Kinase, rHA, RNR, TopII and TS
| CatB | ||||
|---|---|---|---|---|
| Experiment | Moldock-non | Moldock-cons | Autodock | |
| RAPTATh2o | 1.5 | −91.0243 | −75.7655 | −4.38 |
| RAPTACh2o | 2.5 | −105.798 | −92.7676 | −4.84 |
| CRAPTAC | 5 | −124.877 | −82.3353 | −3.38 |
| ORAPTAC | 200 | −114.427 | −74.0878 | −3.43 |
| RAPTABh2o | 200 | −90.0599 | −69.7078 | −5.19 |
| TrXR | ||||
| CRAPTAC | 4.6 | −103.714 | −14.8246 | −3.35 |
| ORAPTAC | 32.5 | −104.425 | −40.9218 | −3.35 |
| RAPTACh2o | 37.1 | −94.0454 | −52.9618 | −3.76 |
| RAPTATh2o | 144 | −80.0042 | −36.727 | −3.65 |
| RAPTABh2o | 200 | −71.3701 | −28.9068 | −3.61 |
2.6. Metal-Receptor Residues Covalent Interaction Using Constrained Molegro Docking
2.7. The Correlation of Factors that Determined the Binding Activities of the Metallocompounds
| 4DL7 | CatB | Gyrase | HDAC7 | HP-NCP | BRAF Kinase | rHA | RNR | TopII | TrXR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| E.Inter.protein.ligand. | 0.28 | 0.91 | 0.93 | 0.93 | 0.89 | 0.89 | 0.93 | 0.92 | 0.81 | 0.90 | 0.90 |
| E.Intertotal | 0.84 | 0.91 | 0.93 | 0.93 | 0.89 | 0.89 | 0.93 | 0.92 | 0.81 | 0.90 | 0.90 |
| E.Intra.steric. | 0.12 | 0.42 | 0.39 | 0.46 | 0.68 | 0.49 | 0.66 | 0.58 | 0.45 | 0.31 | 0.55 |
| E.Intra.tors. | 0.26 | −0.01 | −0.11 | 0.07 | 0.20 | 0.05 | 0.15 | −0.11 | −0.10 | −0.53 | 0.14 |
| E.Intra.tors.ligandatoms. | 0.16 | 0.37 | 0.30 | 0.43 | 0.65 | 0.46 | 0.63 | 0.51 | 0.36 | −0.01 | 0.52 |
| E.Intra.vdw. | −0.91 | −0.77 | −0.82 | −0.79 | −0.67 | −0.65 | −0.69 | −0.50 | −0.32 | −0.58 | −0.74 |
| Electro | −0.35 | 0.22 | 0.45 | −0.14 | −0.07 | −0.04 | −0.12 | 0.61 | −0.19 | 0.26 | 0.07 |
| ElectroLong | −0.76 | 0.20 | 0.31 | −0.08 | −0.21 | −0.06 | −0.29 | 0.36 | −0.20 | 0.53 | 0.51 |
| HBond | −0.47 | 0.20 | 0.17 | −0.44 | 0.03 | −0.14 | 0.11 | 0.34 | 0.13 | 0.71 | 0.31 |
| HeavyAtoms | −0.71 | −0.91 | −0.82 | −0.84 | −0.80 | −0.85 | −0.76 | −0.78 | −0.52 | −0.82 | −0.84 |
| LE1 | −0.13 | −0.50 | −0.20 | −0.39 | −0.21 | −0.39 | −0.11 | −0.27 | −0.23 | −0.22 | −0.30 |
| LE3 | −0.11 | −0.63 | −0.39 | −0.45 | −0.43 | −0.41 | −0.42 | −0.27 | −0.40 | −0.52 | −0.11 |
| MW | −0.51 | −0.69 | −0.65 | −0.64 | −0.60 | −0.67 | −0.54 | −0.53 | −0.13 | −0.71 | −0.56 |
| N | −0.51 | −0.57 | −0.35 | −0.63 | −0.59 | −0.49 | −0.47 | −0.63 | −0.60 | −0.64 | −0.53 |
| NoHBond90 | −0.35 | 0.19 | 0.15 | −0.15 | 0.08 | 0.01 | 0.22 | 0.64 | 0.45 | 0.74 | 0.31 |
| PoseEnergy | 0.80 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 1.00 | 0.99 | 0.99 | 0.99 | 0.99 |
| RerankScore | 0.81 | 0.69 | 0.48 | 0.08 | 0.52 | 0.54 | 0.32 | 0.85 | −0.23 | 0.38 | 0.53 |
| Steric | 0.48 | 0.85 | 0.90 | 0.94 | 0.89 | 0.87 | 0.92 | 0.77 | 0.80 | 0.85 | 0.86 |
| Torsions | −0.31 | −0.31 | −0.55 | −0.20 | −0.41 | −0.45 | −0.38 | −0.65 | −0.27 | −0.70 | −0.37 |
| VdW.LJ12.6. | 0.32 | −0.01 | −0.17 | −0.30 | −0.11 | 0.04 | −0.29 | 0.35 | −0.34 | −0.37 | 0.20 |
| halogen | 0.26 | 0.38 | 0.50 | 0.29 | 0.40 | 0.40 | 0.45 | 0.50 | 0.51 | 0.50 | 0.42 |
| 4DL7 | CatB | Gyrase | HDAC7 | HP-NCP | BRAF Kinase | rHA | RNR | TopII | TrXR | TS | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| E.Inter.protein.ligand. | −0.43 | 0.87 | 0.87 | 0.94 | 0.83 | −0.17 | 0.31 | 0.89 | 0.67 | 0.52 | 0.85 |
| E.Intertotal | 0.82 | 0.87 | 0.87 | 0.94 | 0.83 | −0.17 | 0.31 | 0.89 | 0.67 | 0.52 | 0.85 |
| E.Intra.steric. | 0.15 | 0.32 | 0.25 | 0.67 | 0.71 | −0.39 | 0.29 | 0.53 | −0.04 | −0.18 | 0.61 |
| E.Intra.tors. | 0.30 | 0.14 | −0.01 | 0.46 | 0.35 | −0.05 | −0.21 | −0.21 | −0.25 | −0.51 | −0.04 |
| E.Intra.tors.ligandatoms | 0.21 | 0.31 | 0.22 | 0.68 | 0.70 | −0.37 | 0.22 | 0.44 | −0.09 | −0.28 | 0.54 |
| E.Intra.vdw. | −0.71 | −0.55 | −0.79 | −0.55 | −0.65 | −0.55 | −0.43 | −0.58 | 0.00 | −0.71 | −0.51 |
| E.SoftConstraintPenalty | 0.14 | 0.15 | 0.46 | −0.01 | −0.37 | 0.76 | 0.89 | NA | 0.33 | 0.41 | −0.45 |
| Electro | 0.05 | 0.57 | −0.33 | −0.24 | −0.07 | −0.24 | −0.10 | 0.19 | 0.06 | −0.01 | 0.31 |
| ElectroLong | −0.73 | 0.18 | −0.33 | 0.16 | 0.72 | 0.17 | 0.08 | −0.31 | 0.16 | −0.33 | −0.36 |
| HBond | 0.04 | 0.10 | 0.42 | 0.12 | 0.10 | 0.15 | 0.25 | 0.53 | 0.17 | 0.62 | 0.34 |
| HeavyAtoms | −0.53 | −0.71 | −0.81 | −0.47 | −0.65 | −0.37 | −0.71 | −0.84 | 0.15 | −0.67 | −0.82 |
| LE1 | 0.08 | −0.10 | −0.20 | 0.16 | 0.06 | 0.78 | 0.79 | −0.14 | 0.72 | 0.40 | −0.10 |
| LE3 | −0.02 | −0.15 | −0.25 | 0.09 | 0.17 | −0.42 | −0.59 | −0.54 | 0.45 | −0.09 | −0.47 |
| MW | −0.39 | −0.35 | −0.67 | −0.23 | −0.40 | −0.50 | −0.75 | −0.61 | 0.39 | −0.57 | −0.55 |
| N | −0.26 | −0.42 | −0.38 | −0.41 | −0.49 | −0.18 | −0.43 | −0.63 | −0.08 | −0.73 | −0.57 |
| NoHBond90 | 0.36 | 0.40 | 0.47 | 0.30 | 0.13 | 0.21 | 0.24 | 0.70 | 0.18 | 0.70 | 0.31 |
| PoseEnergy | 0.83 | 0.99 | 0.99 | 0.99 | 0.99 | 1.00 | 0.99 | 0.99 | 0.99 | 0.99 | 0.98 |
| RerankScore | 0.07 | 0.05 | 0.10 | 0.25 | 0.66 | −0.40 | −0.45 | 0.34 | 0.46 | −0.03 | −0.02 |
| Steric | −0.20 | 0.81 | 0.85 | 0.92 | 0.72 | −0.19 | 0.25 | 0.87 | 0.64 | 0.54 | 0.79 |
| Torsions | −0.25 | −0.62 | −0.55 | −0.04 | −0.16 | −0.17 | −0.62 | −0.63 | −0.17 | −0.70 | −0.51 |
| VdW.LJ12.6. | −0.11 | −0.20 | −0.23 | 0.08 | 0.48 | −0.38 | −0.44 | −0.36 | 0.41 | −0.17 | −0.36 |
| halogen | 0.37 | 0.44 | 0.38 | 0.35 | 0.45 | −0.32 | 0.14 | 0.44 | 0.17 | 0.33 | 0.38 |
3. Computational Methods
4. Conclusion
Acknowledgments
Conflicts of Interest
References
- Allardyce, C.S.; Dorcier, A.; Scolaro, C.; Dyson, P.J. Development of organometallic (organo-transition metal) pharmaceuticals. Appl. Organometal. Chem. 2005, 19, 1–10. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef]
- Ang, W.H.; Daldini, E.; Scolaro, C.; Scopelliti, R.; Juillerat-Jeannerat, L.; Dyson, P.J. Development of organometallic ruthenium−arene anticancer drugs that resist hydrolysis. Inorg. Chem. 2006, 45, 9006–9013. [Google Scholar] [CrossRef]
- Dyson, P.J.; Sava, G. Metal-based antitumour drugs in the post genomic era. Dalton Trans. 2006, 23, 1929–1933. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium(II)–arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53–JNK pathways. J. Biol. Inorg. Chem. 2008, 13, 1149–1155. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A new redox-active anticancer agent–preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Sava, G.; Bergamoa, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef]
- Page, S. Ruthenium compounds as anticancer agents. Education in Chemistry, January 2012. [Google Scholar]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platin. Met. Rev. 2001, 45, 62–69. [Google Scholar]
- Hu, X.; Shelver, W.H. Docking studies of matrix metalloproteinase inhibitors: Zinc parameter optimization to improve the binding free energy prediction. J Mol Graph Model. 2003, 22, 115–126. [Google Scholar] [CrossRef]
- Ciancetta, A.; Genheden, S.; Ryde, U. A QM/MM study of the binding of RAPTA ligands to cathepsin B. J. Comput. Aid. Molec. Design 2011, 25, 729–742. [Google Scholar] [CrossRef]
- Adeniyi, A.A.; Ajibade, P.A. Inhibitory activities and possible anticancer targets of Ru(II)-based complexes using computational docking method. J. Mol. Graph. Model. 2012, 38, 60–69. [Google Scholar] [CrossRef]
- Adeniyi, A.A.; Ajibade, P.A. Comparing the suitability of autodock, gold and glide for the docking and predicting the possible targets of ru(ii)-based complexes as anticancer agents. Molecules 2013, 18, 3760–3778. [Google Scholar] [CrossRef]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging protein targets for anticancer metallodrugs: inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar] [CrossRef]
- Wu, B.; Ong, M.S.; Groessl, M.; Adhireksan, Z.; Hartinger, C.G.; Dyson, P.J.; Davey, C.A. A Ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core. Chem-Eur. J. 2011, 17, 3562–3566. [Google Scholar]
- Xie, P.; Streu, C.; Qin, J.; Bregman, H.; Pagano, N.; Meggers, E.; Marmorstein, R. The crystal structure of BRAF in complex with an organoruthenium inhibitor reveals a mechanism for inhibition of an active form of BRAF kinase. Biochemistry 2009, 48, 5187–5198. [Google Scholar] [CrossRef]
- Stepanenko, I.N.; Casini, A.; Edafe, F.; Novak, M.S.; Arion, V.B.; Dyson, P.J.; Jakupec, M.A.; Keppler, B.K. Conjugation of organoruthenium(II) 3-(1H-Benzimidazol-2-yl)pyrazolo[3,4-b]pyridines and Indolo[3,2-d]benzazepines to recombinant human serum albumin: A strategy to enhance cytotoxicity in cancer cells. Inorg. Chem. 2011, 50, 12669–12679. [Google Scholar]
- Hanif, M.; Henke, H.; Meier, S.M.; Martic, S.; Labib, M.; Kandioller, W.; Jakupec, M.A.; Arion, V.B.; Kraatz, H.; Keppler, B.K.; et al. Is the reactivity of M(II)-arene complexes of 3-hydroxy-2(1H)-pyridones to biomolecules the anticancer activity determining parameter? Inorg. Chem. 2010, 49, 7953–7963. [Google Scholar] [CrossRef]
- Zhuang, W.; Wu, X.; Zhou, Y.; Liu, G.; Wu, T.; Yao, X.; Du, L.; Wei, M. Polymorphisms of thymidylate synthase in the 5'- and 3' -untranslated regions and gastric cancer. Dig. Dis. Sci. 2009, 54, 1379–1385. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Gopal, Y.N.V.; Jayaraju, D.; Kondapi, A.K. Inhibition of topoisomerase II catalytic activity by two ruthenium compounds: A ligand-dependent mode of action. Biochemistry 1999, 38, 4382–4388. [Google Scholar] [CrossRef]
- Arpino, G.; Ciocca, D.R.; Weiss, H.; Allred, D.C.; Daguerre, P.; Vargas-Roig, L.; Leuzzi, M.; Gago, F.; Elledge, R.; and Mohsin, S.K. Predictive value of apoptosis, Proliferation, HER-2, and topoisomerase IIa for anthracycline chemotherapy in locally advanced breast cancer. Breast Cancer Res. Treat. 2005, 92, 69–75. [Google Scholar] [CrossRef]
- Holdgate, G.A.; Tunnicliffe, A.; Ward, W.H.J.; Weston, S.A.; Rosenbrock, G.; Barth, P.T.; Taylor, I.W.F.; Pauptit, R.A.; Timms, D. The entropic penalty of ordered water accounts for weaker binding of the antibiotic novobiocin to a resistant mutant of DNA gyrase: A thermodynamic and crystallographic study. Biochemistry 1997, 36, 9663–9673. [Google Scholar] [CrossRef]
- Ghaneya, S.; Hassan, G.S.; Nahla, A; Farag, N.A.; Gehan, H.; Hegazy, G.H.; Reem, K.; Arafa, R.K. Design and synthesis of novel benzopyran-2-one derivatives of expected antimicrobial activity through DNA gyrase-B inhibition. Arch. Pharm. 2008, 341, 725–733. [Google Scholar] [CrossRef]
- Caballero, N.A.; Meléndez, F.J.; Niño, A.; Muñoz-Caro, C. Molecular docking study of the binding of aminopyridines within the K+ channel. J. Mol. Model. 2007, 13, 579–586. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Egger, A.E.; Hartinger, C.G.; Renfrew, A.K.; Dyson, P.J. Metabolization of [Ru(η6-C6H5CF3)(pta)Cl2]: A cytotoxic RAPTA-type complex with a strongly electron withdrawing arene ligand. J. Biol. Inorg. Chem. 2010, 15, 919–927. [Google Scholar] [CrossRef]
- Wang, F.; Habtemariam, A.; van der Geer, E.P.L.; Fernandez, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.A.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. 2005, 102, 18269–18274. [Google Scholar] [CrossRef]
- Hu, W; Luo, Q; Ma, X; Wu, K; Liu, J; Chen, Y; Xiong, S; Wang, J; Sadler, P.J; Wang, F. Arene Control over thiolate to sulfinate oxidation in albumin by organometallic ruthenium anticancer complexes. Chem-Eur. J. 2009, 15, 6586–6594. [Google Scholar]
- Caruso, F.; Rossi, M.; Benson, A.; Opazo, C.; Freedman, D.; Monti, E.; Gariboldi, M.B.; Shaulky, J.; Marchetti, F.; Pettinari, R.; et al. Ruthenium−arene complexes of curcumin: X-ray and density functional theory structure, Synthesis, and spectroscopic characterization, in vitro antitumor activity, and DNA docking studies of (p-cymene)Ru(curcuminato)chloro. J. Med. Chem. 2012, 55, 1072–1081. [Google Scholar]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N.; et al. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: The structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar]
- Eriksson, M.; Uhlin, U.; Ramaswamy, S.; Ekberg, M.; Regnström, K.; Sjöberg, B.M.; Eklund, H. Binding of allosteric effectors to ribonucleotide reductase protein R1: Reduction of active-site cysteines promotes substrate binding. Structure 1997, 5, 1077–1092. [Google Scholar] [CrossRef]
- Sandalova, T.; Zhong, L.; Lindqvist, Y.; Holmgren, A.; Schneider, G. Three-dimensional structure of a mammalian thioredoxin reductase: implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc. Natl. Acad. Sci. 2001, 98, 9533–9538. [Google Scholar] [CrossRef]
- Newby, Z.; Lee, T.T.; Morse, R.J.; Liu, Y.; Liu, L.; Venkatraman, P.; Santi, D.V.; Finer-Moore, J.S.; Stroud, R.M. The role of protein dynamics in thymidylate synthase catalysis: variants of conserved 2'-deoxyuridine 5'-monophosphate (dUMP)-binding Tyr-261. Biochemistry 2006, 45, 7415–7428. [Google Scholar] [CrossRef]
- Schuetz, A.; Min, J.; Allali-Hassani, A.; Schapira, M.; Shuen, M.; Loppnau, P.; Mazitschek, R.; Kwiatkowski, N.P.; Lewis, T.A.; Maglathin, R.L.; et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 2008, 283, 11355–11363. [Google Scholar] [CrossRef]
- Classen, S.; Olland, S.; Berger, J.M. Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc. Natl. Acad. Sci. 2003, 100, 10629–10634. [Google Scholar] [CrossRef]
- Zhao, Y.; Biertümpfel, C.; Gregory, M.T.; Hua, Y.J.; Hanaoka, F.; Yang, W. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. 2012, 109, 7269–7274. [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Available online: http://www.R-project.org (accessed 1 May 2012).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian, Inc.: Pittsburgh, PA, USA, 2003.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J., J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N.J.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar]
- Becke, A.D. Density–functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dobbs, K.D.; Hehre, W.J. Molecular orbital theory of the properties of inorganic and organometallic compounds 5. Extended basis sets for first-row transition metals. J. Comput. Chem. 1987, 6, 861–879. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- ADL: Parameters for docking with metal ions in receptor. Available online: http://autodock.1369657.n2.nabble.com/ADL-Parameters-for-docking-with-metal-ions-in-receptor-td2505649.html (accessed on 6 May 2012).
- Sapre, N.S; Gupta, S.; Sapre, N. Assessing ligand efficiencies using template-based molecular docking and Tabu-clustering on tetrahydroimidazo-[4,5,1-jk][1,4]-benzodiazepin-2(1H)-one and -thione (TIBO) derivatives as HIV-1RT inhibitors. J. Chem. Sci. 2008, 120, 395–404. [Google Scholar]
- Research collaboratory for structural bioinformatics (RCSB) online pdb database. Available online: http://www.rcsb.org (accessed 30 April 2013).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sample Availability: Details computational data of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adeniyi, A.A.; Ajibade, P.A. An Insight into the Anticancer Activities of Ru(II)-Based Metallocompounds Using Docking Methods. Molecules 2013, 18, 10829-10856. https://doi.org/10.3390/molecules180910829
Adeniyi AA, Ajibade PA. An Insight into the Anticancer Activities of Ru(II)-Based Metallocompounds Using Docking Methods. Molecules. 2013; 18(9):10829-10856. https://doi.org/10.3390/molecules180910829
Chicago/Turabian StyleAdeniyi, Adebayo A., and Peter A. Ajibade. 2013. "An Insight into the Anticancer Activities of Ru(II)-Based Metallocompounds Using Docking Methods" Molecules 18, no. 9: 10829-10856. https://doi.org/10.3390/molecules180910829
APA StyleAdeniyi, A. A., & Ajibade, P. A. (2013). An Insight into the Anticancer Activities of Ru(II)-Based Metallocompounds Using Docking Methods. Molecules, 18(9), 10829-10856. https://doi.org/10.3390/molecules180910829