Antibacterial Activity of Salicylanilide 4-(Trifluoromethyl)-benzoates


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
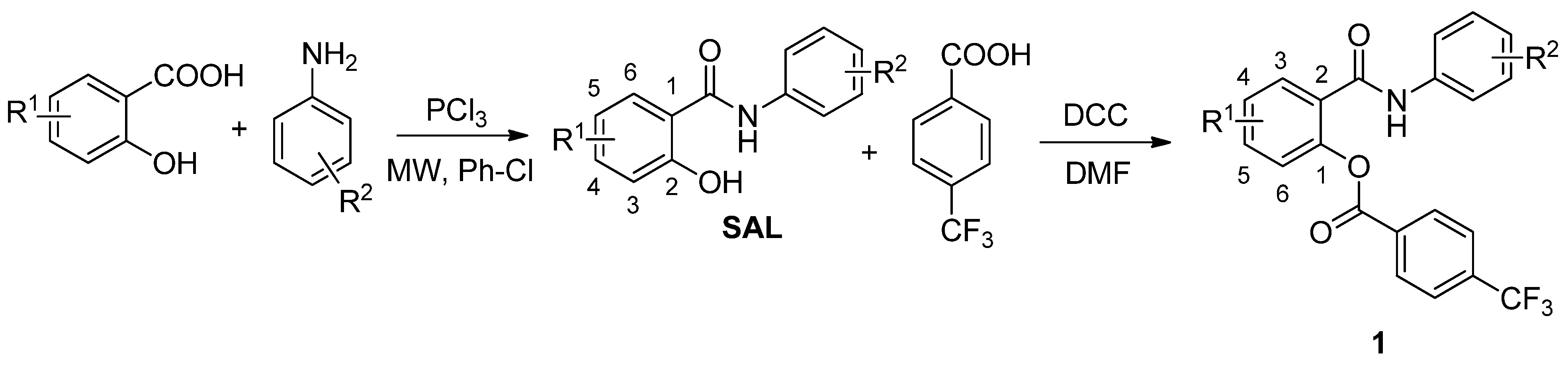
2.2. Antimycobacterial Evaluation

{kind=link}
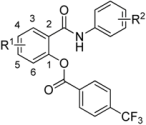 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC [μmol/L] | ||||||||||||||||||
| R1 | R2 | Mtb. 331/88 | M. avium 330/88 | M. kansasii 235/80 | M. kansasii 6509/96 | |||||||||||||
| 14 d | 21 d | 14 d | 21 d | 7 d | 14 d | 21 d | 7 d | 14 d | 21 d | |||||||||
| 1a | 4-Cl | 3-Cl | 2 | 4 | 8 | 16 | 0.5 | 2 | 4 | 2 | 8 | 8 | ||||||
| 1b | 5-Cl | 3-Cl | 4 | 4 | 8 | 8 | 1 | 4 | 4 | 2 | 8 | 8 | ||||||
| 1c | 4-Cl | 4-Cl | 2 | 2 | 4 | 8 | 1 | 2 | 2 | 1 | 4 | 4 | ||||||
| 1d | 5-Cl | 4-Cl | 4 | 4 | 4 | 8 | 1 | 4 | 4 | 1 | 4 | 4 | ||||||
| 1e | 4-Cl | 3,4-diCl | 1 | 1 | 4 | 4 | 1 | 2 | 4 | 1 | 2 | 4 | ||||||
| 1f | 5-Cl | 3,4-diCl | 1 | 2 | 2 | 8 | 1 | 2 | 4 | 1 | 2 | 4 | ||||||
| 1g | 4-Cl | 3-Br | 1 | 2 | 2 | 8 | 2 | 4 | 4 | 1 | 4 | 4 | ||||||
| 1h | 5-Cl | 3-Br | 4 | 8 | 8 | 16 | 4 | 8 | 8 | 2 | 8 | 8 | ||||||
| 1i | 4-Cl | 4-Br | 2 | 2 | 8 | 8 | 1 | 4 | 8 | 2 | 8 | 8 | ||||||
| 1j | 5-Cl | 4-Br | 2 | 4 | 8 | 8 | 1 | 4 | 4 | 2 | 4 | 4 | ||||||
| 1k | 4-Cl | 3-F | 8 | 8 | 16 | 32 | 8 | 16 | 16 | 8 | 16 | 16 | ||||||
| 1l | 5-Cl | 3-F | 4 | 4 | 8 | 16 | 4 | 8 | 8 | 8 | 8 | 16 | ||||||
| 1m | 4-Cl | 4-F | 8 | 8 | 16 | 32 | 4 | 16 | 16 | 8 | 16 | 16 | ||||||
| 1n | 5-Cl | 4-F | 4 | 8 | 4 | 4 | 2 | 8 | 8 | 4 | 8 | 8 | ||||||
| 1o | 4-Cl | 4-CF3 | 1 | 1 | 1 | 4 | 1 | 2 | 2 | 1 | 2 | 2 | ||||||
| 1p | 5-Cl | 4-CF3 | 2 | 2 | 2 | 8 | 2 | 2 | 4 | 1 | 4 | 4 | ||||||
| 1q | 4-Cl | 3-CF3 | 2 | 2 | 4 | 16 | 2 | 4 | 8 | 1 | 4 | 8 | ||||||
| 1r | 4-Br | 4-CF3 | 1 | 1 | 2 | 4 | 1 | 2 | 4 | 1 | 2 | 4 | ||||||
| INH | 0.5–1 | 0.5–1 | ≥250 | ≥250 | >250 | >250 | >250 | 2–4 | 4–8 | 8–16 | ||||||||
| PAS | 62.5 | 62.5 | 32 | 125 | 125 | 1000 | >1000 | 32 | 125 | 500 | ||||||||
| CF3-BA | >1,000 | >1,000 | >1,000 | >1,000 | 1,000 | >1,000 | >1,000 | 250 | 1,000 | 1,000 | ||||||||
| MIC [μmol/L] | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Mtb. 7357/1988 | Mtb. 9449/2006 | Mtb. 53/2009 | Mtb. 234/2005 | Mtb. Praha 1 | Mtb. Praha 131 (XDR-TB) | |||||||
| 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | |||
| 1a | 4-Cl | 3-Cl | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| 1b | 5-Cl | 3-Cl | 8 | 8 | 4 | 4 | 8 | 8 | 8 | 8 | 4 | 4 | 8 | 8 |
| 1c | 4-Cl | 4-Cl | 2 | 2 | 2 | 2 | 4 | 4 | 4 | 4 | 2 | 2 | 2 | 2 |
| 1d | 5-Cl | 4-Cl | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 2 | 4 | 2 | 4 |
| 1e | 4-Cl | 3,4-diCl | 1 | 2 | 2 | 2 | 2 | 4 | 1 | 2 | 1 | 2 | 2 | 2 |
| 1f | 5-Cl | 3,4-diCl | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| 1g | 4-Cl | 3-Br | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| 1i | 4-Cl | 4-Br | 2 | 4 | 2 | 4 | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 2 |
| 1j | 5-Cl | 4-Br | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 1o | 4-Cl | 4-CF3 | 0.5 | 1 | 1 | 1 | 1 | 1 | 0.5 | 1 | 0.5 | 0.5 | 0.5 | 1 |
| 1r | 4-Br | 4-CF3 | 1 | 1 | 1 | 1 | 1 | 2 | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 |
2.3. Isocitrate Lyase Inhibition Assay
| R1 | R2 | % ICL inhibition at 10 μmol/L (± standard deviation) | |
|---|---|---|---|
| 1a | 4-Cl | 3-Cl | 18 ± 1.9 |
| 1b | 5-Cl | 3-Cl | 13 ± 1.7 |
| 1c | 4-Cl | 4-Cl | 12 ± 2.7 |
| 1d | 5-Cl | 4-Cl | 12 ± 2.3 |
| 1e | 4-Cl | 3,4-diCl | 18 ± 1.5 |
| 1f | 5-Cl | 3,4-diCl | 17 ± 1.6 |
| 1g | 4-Cl | 3-Br | 27 ± 0.2 |
| 1h | 5-Cl | 3-Br | 21 ± 1.8 |
| 1i | 4-Cl | 4-Br | 18 ± 1.2 |
| 1j | 5-Cl | 4-Br | 14 ± 4.0 |
| 1k | 4-Cl | 3-F | 25 ± 0.8 |
| 1l | 5-Cl | 3-F | 20 ± 0.5 |
| 1m | 4-Cl | 4-F | 23 ± 1.4 |
| 1n | 5-Cl | 4-F | 17 ± 1.3 |
| 1o | 4-Cl | 4-CF3 | 13 ± 1.2 |
| 1p | 5-Cl | 4-CF3 | 17 ± 2.3 |
| 1q | 4-Cl | 3-CF3 | 19 ± 0.5 |
| 1r | 4-Br | 4-CF3 | 23 ± 0.1 |
| 3-NP | 25 ± 4.1 | ||
| INH | 0 | ||
2.4. Antibacterial Evaluation
| MIC/IC90 [μmol/L] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | S. aureus CCM 4516/08 | MRSA H 5996/08 | S. epidermidis H 6966/08 | Enterococcus sp. J 14365/08 | E. coli CCM 4517 | ||||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |||
| 1b | 5-Cl | 3-Cl | 0.98 | 1.95 | 0.98 | 1.95 | 0.98 | 1.95 | 7.81 | 15.62 | >250 | >250 |
| 1e | 4-Cl | 3,4-diCl | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 3.9 | >500 | >500 |
| 1h | 5-Cl | 3-Br | 0.98 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 31.25 | 250 | >500 | >500 |
| 1j | 5-Cl | 4-Br | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 1.95 | 7.81 | 250 | >250 |
| 1k | 4-Cl | 3-F | 3.9 | 3.9 | 0.98 | 1.95 | 3.9 | 3.9 | 31.25 | >250 | >250 | >250 |
| 1m | 4-Cl | 4-F | 7.81 | 7.81 | 15.62 | 15.62 | 31.25 | >500 | 500 | >500 | >500 | >500 |
| 1o | 4-Cl | 4-CF3 | 3.9 | 7.81 | 3.9 | 7.81 | 3.9 | 7.81 | 7.81 | 62.5 | >125 | >125 |
| 1p | 5-Cl | 4-CF3 | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 3.9 | 31.25 | 250 |
| PNC | 0.98 | 0.98 | 62.5 | 125 | 250 | 250 | 7.81 | 15.62 | >500 | >500 | ||
2.5. Cytotoxicity Evaluation
| R1 | R2 | IC50 [µmol/L] HepG2 | SI for Mtb. H37Rv 331/88 | SI for MDR-TB strains | SI for XDR-TB strain | SI for Staphylococcus sp. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 14 d | 21 d | 14 d | 21 d | 14 d | 14 d | 24 h | 48 h | |||
| 1e | 4-Cl | 3,4-diCl | 3.13 | 3.13 | 3.13 | 1.57–3.13 | 0.78–1.57 | 1.57 | 1.57 | 6.39 | 3.19 |
| 1f | 5-Cl | 3,4-diCl | 4.46 | 4.46 | 2.23 | 2.23–4.46 | 2.23 | 4.46 | 2.23 | - | - |
| 1g | 4-Cl | 3-Br | 2.63 | 2.63 | 1.32 | 1.32–2.63 | 1.32 | 2.63 | 1.32 | - | - |
| 1j | 5-Cl | 4-Br | 4.82 | 2.41 | 1.21 | 2.24 | 2.24 | 4.82 | 2.41 | 9.84 | 4.92 |
| 1o | 4-Cl | 4-CF3 | 1.33 | 1.33 | 1.33 | 1.33–2.66 | 1.33–2.66 | 2.66 | 1.33 | 0.34 | 0.17 |
| 1p | 5-Cl | 4-CF3 | 1.77 | 0.89 | 0.89 | - | - | - | - | 3.61 | 1.81 |
| 1r | 4-Br | 4-CF3 | 2.03 | 2.03 | 2.03 | 2.03–4.06 | 1.02–2.03 | 4.06 | 2.03 | - | - |
| SAL-1 | 5-Cl | 3,4-diCl | 0.84 | 0.21 | 0.10 | - | - | - | - | - | - |
| SAL-2 | 4-Cl | 3,4-diCl | 1.80 | 0.45 | 0.45 | - | - | - | - | - | - |
| SAL-3 | 5-Cl | 3-Br | 1.54 | NT | NT | - | - | - | - | - | - |
| SAL-4 | 4-Cl | 4-Br | 1.98 | 0.5 | 0.5 | - | - | - | - | - | - |
| SAL-5 | 5-Cl | 4-CF3 | 0.36 | 0.18 | 0.18 | - | - | - | - | - | - |
| SAL-6 | 4-Cl | 4-CF3 | 0.69 | 0.17 | 0.17 | - | - | - | - | - | - |
| SAL-7 | 5-Br | 4-CF3 | 1.67 | 1.67 | 1.67 | - | - | - | - | - | - |
| CF3-BA | 563.70 | - | - | - | - | - | - | - | - | ||
3. Experimental
3.1. Chemistry
3.2. Antimycobacterial Evaluation
3.3. Isocitrate Lyase Inhibition Assay (ICL1)
3.4. Antibacterial Evaluation
3.5. Cytotoxicity Evaluation (HepG2 Cells)
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- WHO. 2011/2012 Tuberculosis Global Facts. Available online: http://www.who.int/tb/publications/2011/factsheet_tb_2011.pdf (accessed on 12 December 2012).
- Caminero, J.A. Extensively drug-resistant tuberculosis: Is its definition correct? Eur. Respir. J. 2008, 32, 1413–1415. [Google Scholar] [CrossRef]
- McKinney, J.D.; Bentrup, K.H.; Munoz-Elias, E.J.; Miczak, A.; Chen, B.; Chan, W.T.; Swenson, D.; Sacchettini, J.C.; Jacobs, W.R.; Russell, D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735–738. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Wsól, V.; Trejtnar, F.; Ulmann, V.; Stolaříková, J.; Fernandes, S.; Bhat, S.; Liu, J.O. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis 2012, 92, 434–439. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr. Med. Chem. 2012, 19, 6126–6137. [Google Scholar] [CrossRef]
- Cabrera, C.E.; Gómez, R.F.; Zuñiga, A.E.; Corral, R.H.; López, B.; Chávez, M. Epidemiology of nosocomial bacteria resistant to antimicrobials. Colomb. Med. 2011, 42, 117–125. [Google Scholar]
- Scientific Blueprint for Tuberculosis Drug Development. Available online: http://www.tballiance.org/downloads/publications/TBA_Scientific_Blueprint.pdf (accessed on 12 December 2012).
- Krátký, M.; Vinšová, J. Salicylanilide ester prodrugs as potential antimicrobial agents—A review. Curr. Pharm. Des. 2011, 17, 3494–3505. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Buchta, V.; Horvati, K.; Bösze, S.; Stolaříková, J. New amino acid esters of salicylanilides active against MDR-TB and other microbes. Eur. J. Med. Chem. 2010, 45, 6106–6113. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Rodriguez, N.G.; Stolaříková, J. Antimycobacterial activity of salicylanilide benzenesulfonates. Molecules 2012, 17, 492–503. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Buchta, V. In vitro antibacterial and antifungal activity of salicylanilide benzoates. ScientificWorldJournal 2012, 12. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Buchta, V. In vitro antibacterial and antifungal activity of salicylanilide pyrazine-2-carboxylates. Med. Chem. 2012, 8, 732–741. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Stolaříková, J. Antimycobacterial assessment of salicylanilide benzoates including multidrug-resistant tuberculosis strains. Molecules 2012, 17, 12812–12820. [Google Scholar] [CrossRef]
- Cheng, T.J.R.; Wu, Y.T.; Yang, S.T.; Lo, K.H.; Chen, S.K.; Chen, Y.H.; Huang, W.I.; Yuan, C.H.; Guo, C.W.; Huang, L.Y.; et al. High-throughput identification of antibacterials against methicillin-resistant Staphylococcus aureus (MRSA) and the transglycosylase. Bioorg. Med. Chem. 2010, 18, 8512–8529. [Google Scholar] [CrossRef]
- Garner, A.L.; Gloeckner, C.; Tricoche, N.; Zakhari, J.S.; Samje, M.; Cho-Ngwa, F.; Lustigman, S.; Janda, K.D. Design, synthesis, and biological activities of closantel analogues: Structural promiscuity and its impact on Onchocerca volvulu. J. Med. Chem. 2011, 54, 3963–3972. [Google Scholar] [CrossRef]
- Fomovska, A.; Wood, R.D.; Mui, E.; Dubey, J.P.; Ferreira, L.R.; Hickman, M.R.; Lee, P.J.; Leed, S.E.; Auschwitz, J.M.; Welsh, W.J.; et al. Salicylanilide inhibitors of Toxoplasma gondii. J. Med. Chem. 2012, 55, 8375–8391. [Google Scholar]
- Ding, N.; Zhang, W.; Xiao, H.L.; Wang, P.; Li, Y.X. Synthesis and biological evaluation of a series of novel salicylanilides as inhibitors of EGFR protein tyrosine kinases. Chin. Chem. Lett. 2012, 23, 529–532. [Google Scholar] [CrossRef]
- Steffen, J.D.; Coyle, D.L.; Damodaran, K.; Beroza, P.; Jacobson, M.K. Discovery and structure-activity relationships of modified salicylanilides as cell permeable inhibitors of poly(ADP-ribose) glycohydrolase (PARG). J. Med. Chem. 2011, 54, 5403–5413. [Google Scholar] [CrossRef]
- Zuo, M.; Zheng, Y.W.; Lu, S.M.; Li, Y.; Zhang, S.Q. Synthesis and biological evaluation of N-aryl salicylamides with a hydroxamic acid moiety at 5-position as novel HDAC–EGFR dual inhibitors. Bioorg. Med. Chem. 2012, 20, 4405–4412. [Google Scholar] [CrossRef]
- Zhu, Z.W.; Shi, L.; Ruan, X.M.; Yang, Y.; Li, H.Q.; Xu, S.P.; Zhu, H.L. Synthesis and antiproliferative activities against Hep-G2 of salicylanide derivatives: potent inhibitors of the epidermal growth factor receptor (EGFR) tyrosine kinase. J. Enzym. Inhib. Med. Chem. 2011, 26, 37–45. [Google Scholar] [CrossRef]
- Liu, X.H.; Lv, P.C.; Li, B.; Zhu, H.L.; Song, B.A. Synthesis, structure, and antibacterial activity of novel 5-arylpyrazole derivatives. Aust. J. Chem. 2008, 61, 223–230. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Antifungal activity of salicylanilides and their esters with 4-(trifluoromethyl)benzoic acid. Molecules 2012, 17, 9426–9442. [Google Scholar] [CrossRef]
- Waisser, K.; Bureš, O.; Holý, P.; Kuneš, J.; Oswald, R.; Jirásková, L.; Pour, M.; Klimešová, V.; Kubicová, L.; Kaustová, J. Relationship between the structure and antimycobacterial activity of substituted salicylanilides. Arch. Pharm. Pharm. Med. Chem. 2003, 336, 53–71. [Google Scholar] [CrossRef]
- Imramovský, A.; Vinšová, J.; Férriz, J.M.; Doležal, R.; Jampílek, J.; Kaustová, J.; Kunc, F. New antituberculotics originated from salicylanilides with promising in vitro activity against atypical mycobacterial strains. Bioorg. Med. Chem. 2009, 17, 3572–3579. [Google Scholar] [CrossRef]
- Férriz, J.M.; Vávrová, K.; Kunc, F.; Imramovský, A.; Stolaříková, J.; Vavříková, E.; Vinšová, J. Salicylanilide carbamates: Antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg. Med. Chem. 2010, 18, 1054–1061. [Google Scholar] [CrossRef]
- Liu, X.H.; Song, B.A.; Bhadury, P.S.; Zhu, H.L.; Cui, P.; Hou, K.K.; Xu, H.L. Novel 5-(3-(substituted)-4,5-dihydroisoxazol-5-yl)-2-methoxyphenyl derivatives: Synthesis and anticancer activity. Aust. J. Chem. 2008, 61, 864–869. [Google Scholar] [CrossRef]
- Wada, K.; Ohkoshi, E.; Morris-Natschke, S.L.; Bastow, K.F.; Lee, K.H. Cytotoxic esterified diterpenoid alkaloid derivatives with increased selectivity against a drug-resistant cancer cell line. Bioorg. Med. Chem. Lett. 2012, 22, 249–252. [Google Scholar] [CrossRef]
- Deng, W.; Guo, Z.; Guo, Y.; Feng, Z.; Jiang, Y.; Chu, F. Acryloylamino-salicylanilides as EGFR PTK inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 469–472. [Google Scholar] [CrossRef]
- Liechti, C.; Séquin, U.; Bold, G.; Furet, P.; Meyer, T.; Traxler, P. Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor. Eur. J. Med. Chem. 2004, 39, 11–26. [Google Scholar] [CrossRef]
- Bradford, M.M. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Dixon, G.H.; Kornberg, H.L. Assay methods for key enzymes of the glyoxylate cycle. Biochem. J. 1959, 72, P3. [Google Scholar]
- Sample Availability: Samples of the compounds 1a–r and SAL-1 – SAL-7 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Trejtnar, F.; Stolaříková, J. Antibacterial Activity of Salicylanilide 4-(Trifluoromethyl)-benzoates. Molecules 2013, 18, 3674-3688. https://doi.org/10.3390/molecules18043674
Krátký M, Vinšová J, Novotná E, Mandíková J, Trejtnar F, Stolaříková J. Antibacterial Activity of Salicylanilide 4-(Trifluoromethyl)-benzoates. Molecules. 2013; 18(4):3674-3688. https://doi.org/10.3390/molecules18043674
Chicago/Turabian StyleKrátký, Martin, Jarmila Vinšová, Eva Novotná, Jana Mandíková, František Trejtnar, and Jiřina Stolaříková. 2013. "Antibacterial Activity of Salicylanilide 4-(Trifluoromethyl)-benzoates" Molecules 18, no. 4: 3674-3688. https://doi.org/10.3390/molecules18043674
APA StyleKrátký, M., Vinšová, J., Novotná, E., Mandíková, J., Trejtnar, F., & Stolaříková, J. (2013). Antibacterial Activity of Salicylanilide 4-(Trifluoromethyl)-benzoates. Molecules, 18(4), 3674-3688. https://doi.org/10.3390/molecules18043674