Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases

Abstract
:1. Introduction

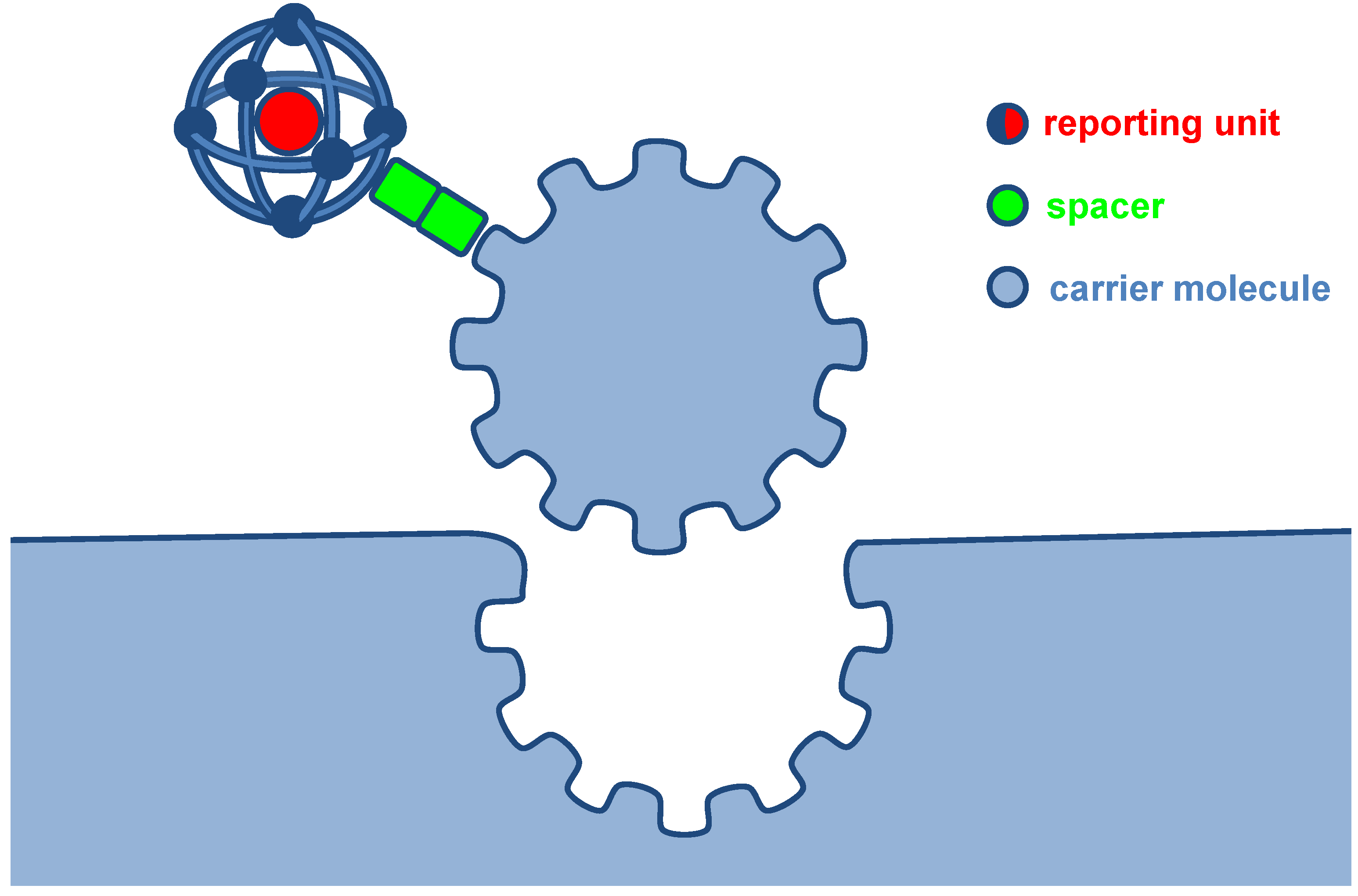{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
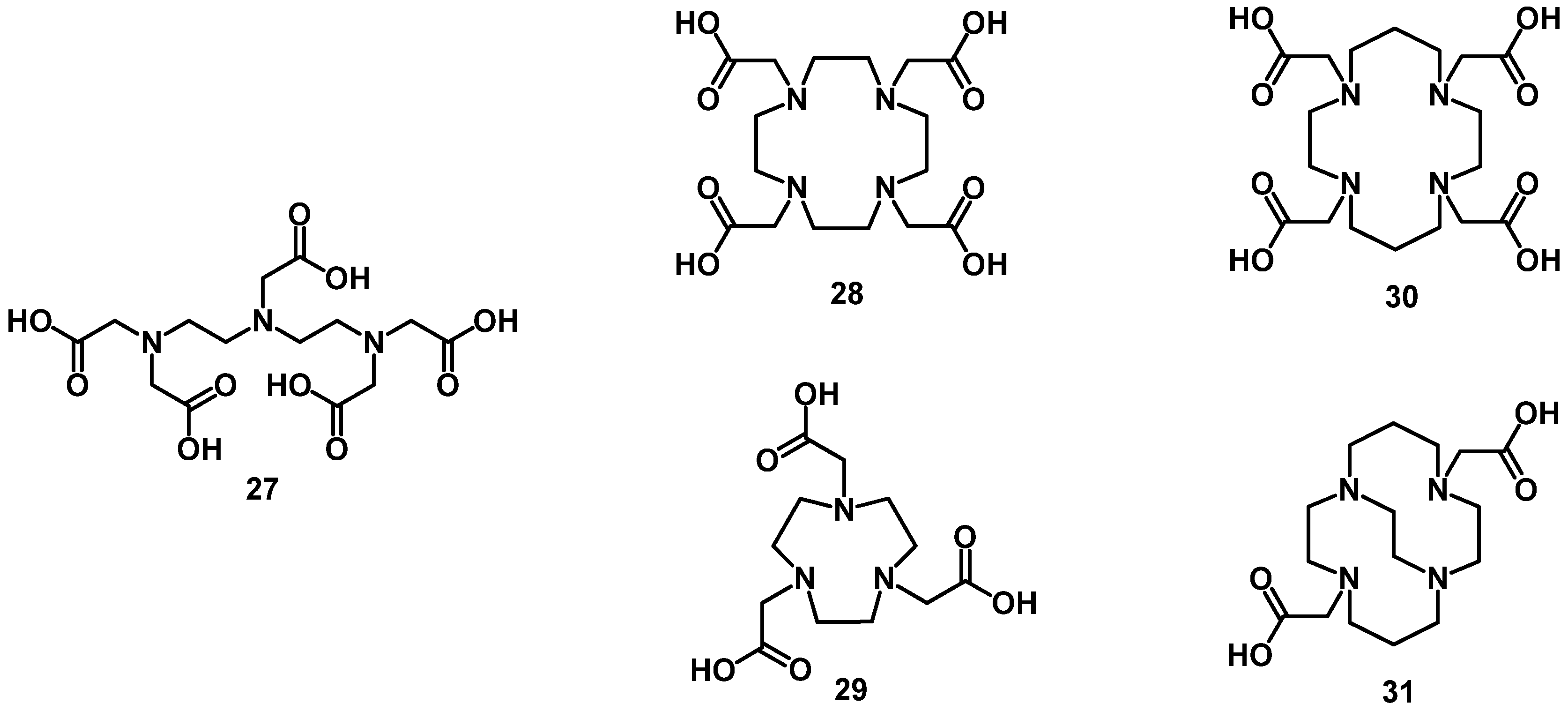{kind=link}
{kind=link}
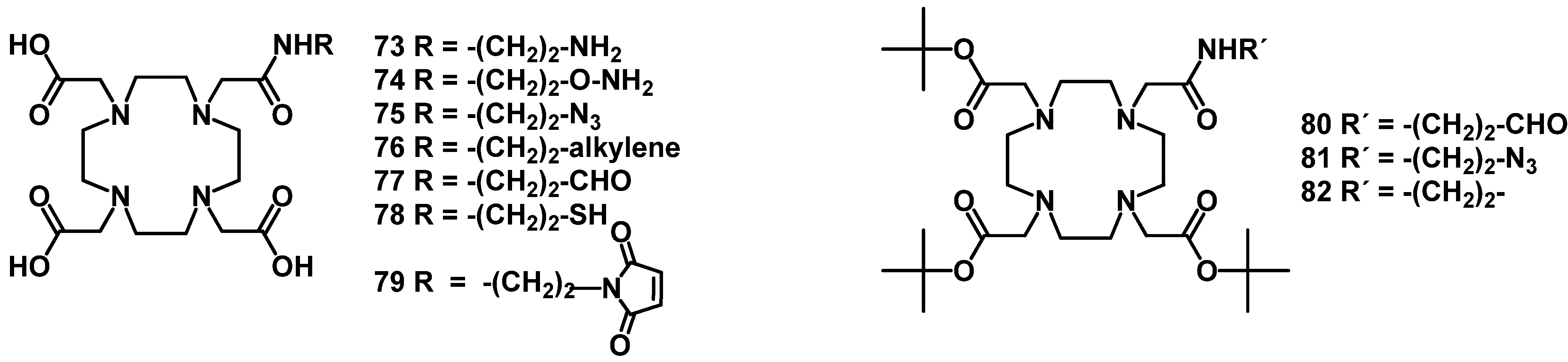{kind=link}
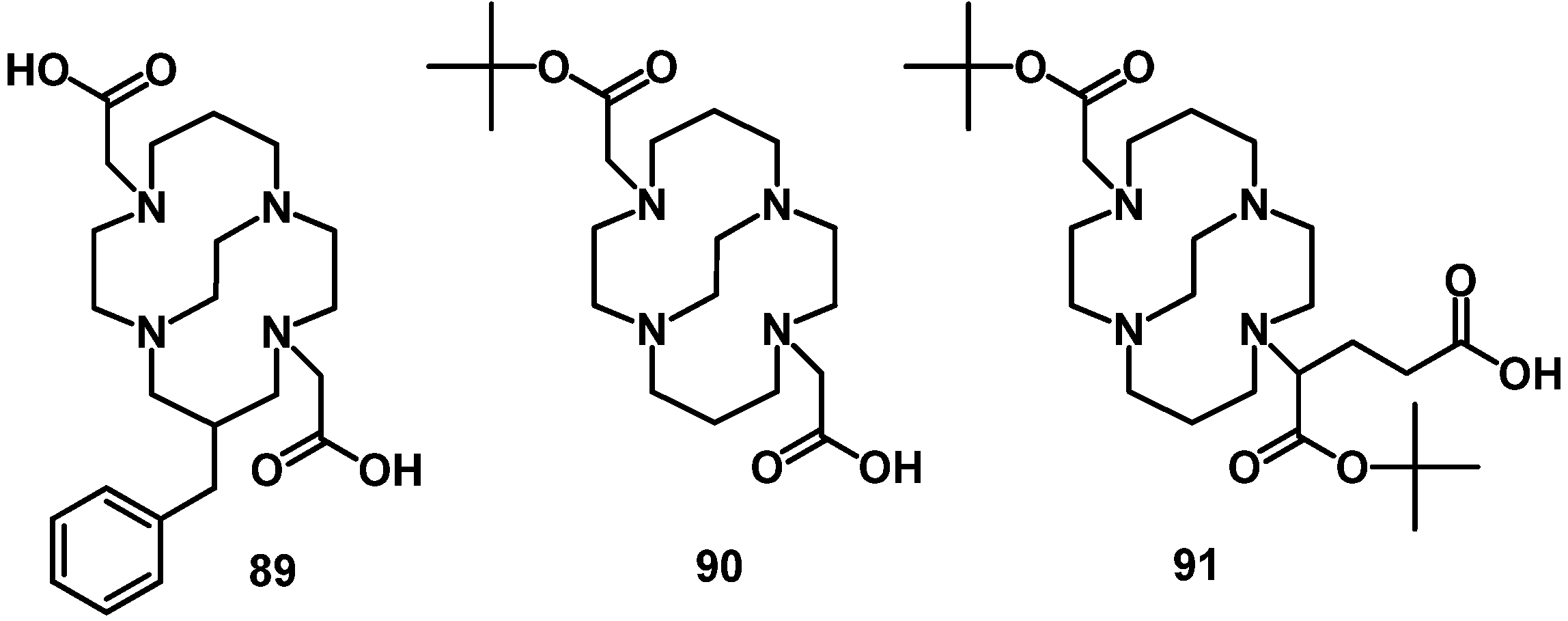{kind=link}
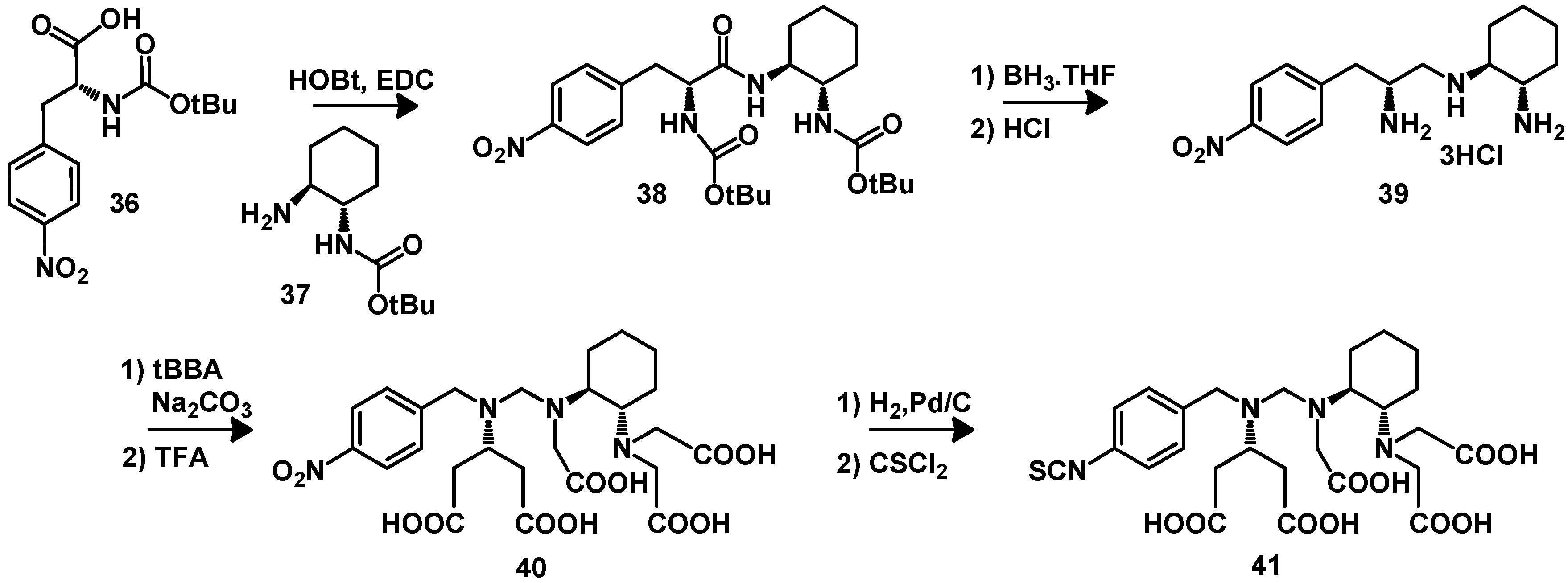{kind=link}
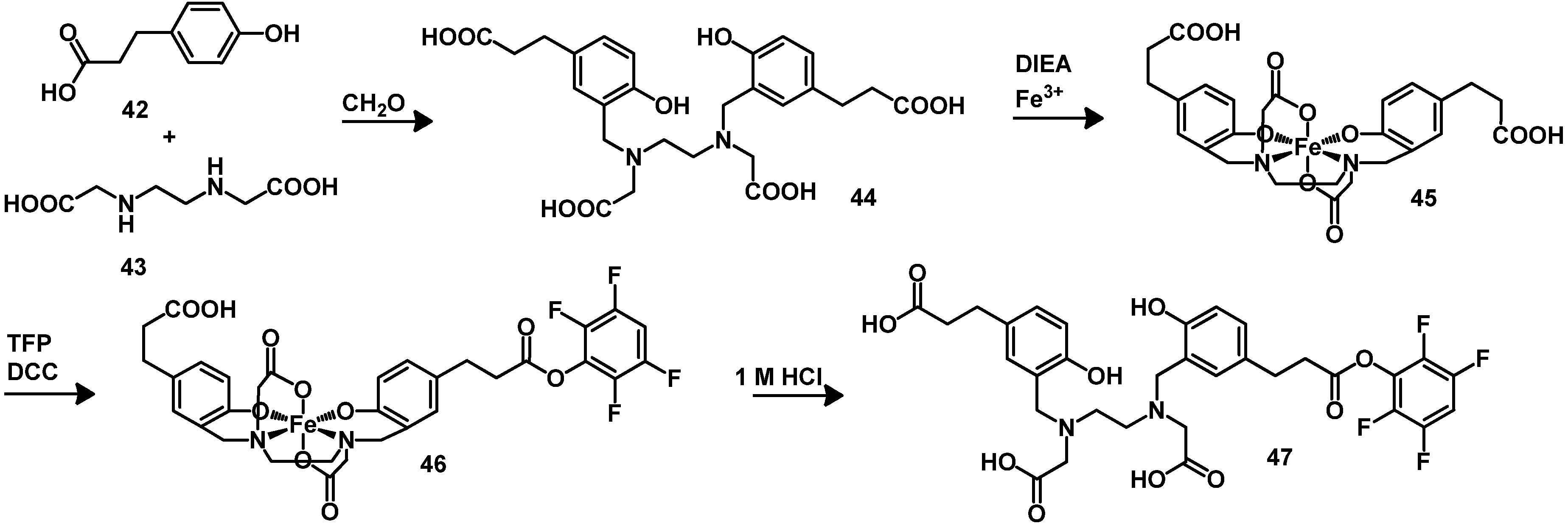{kind=link}
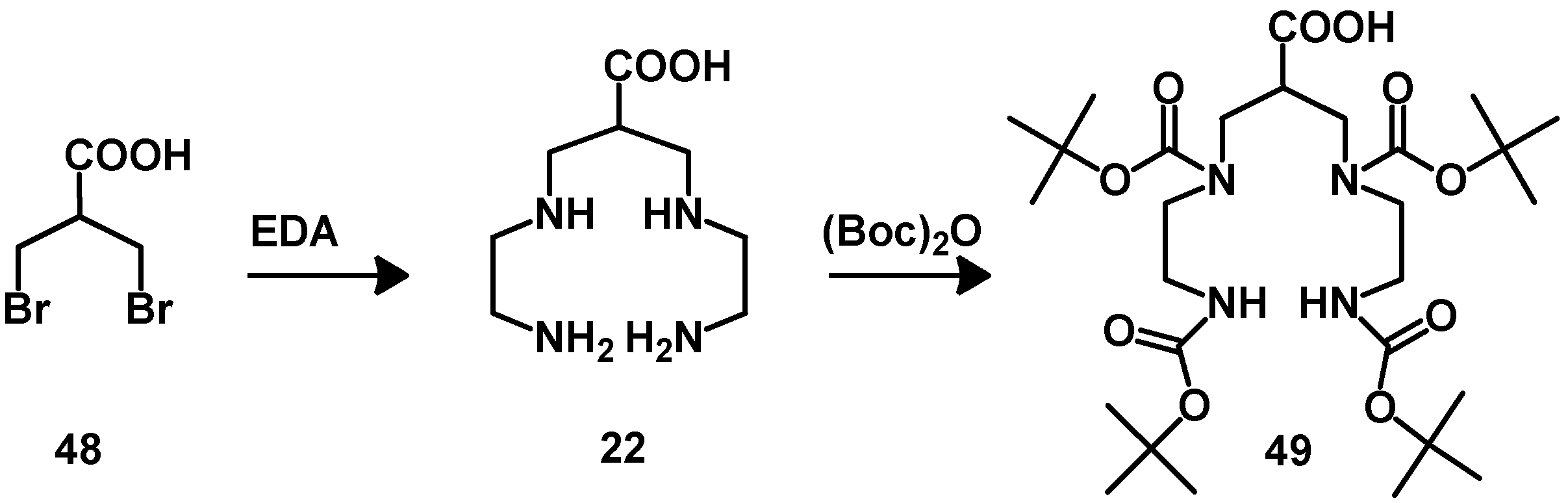{kind=link}
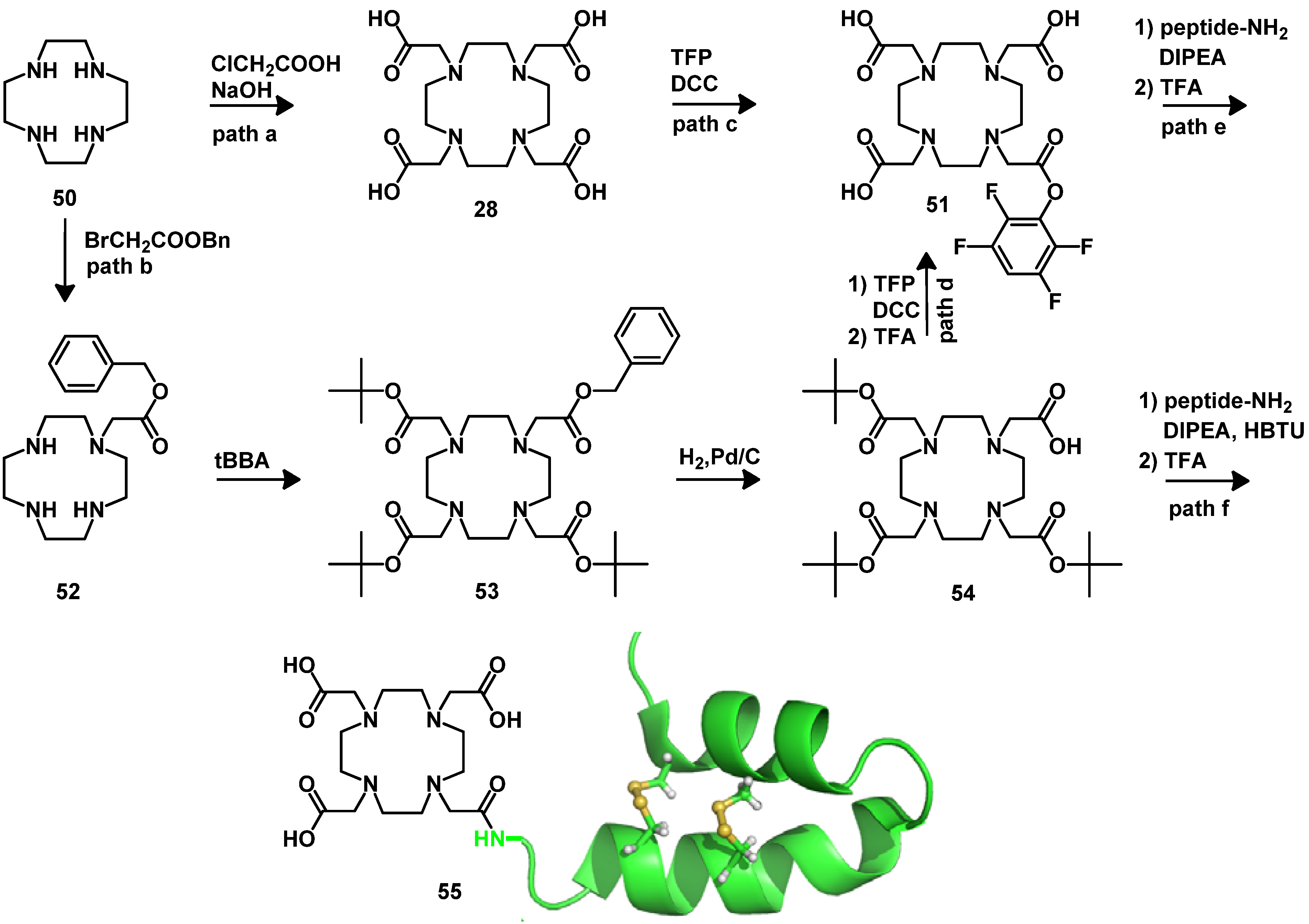{kind=link}
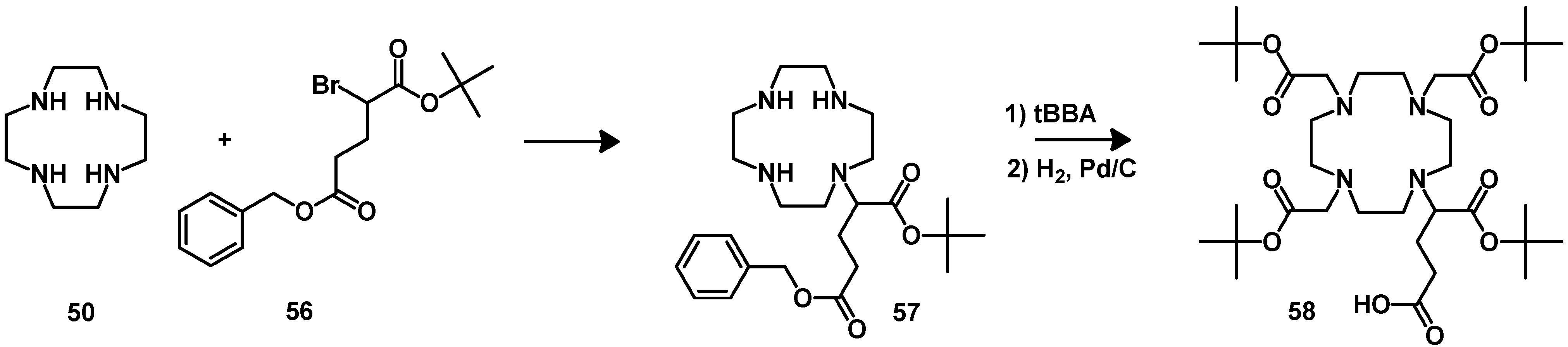{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Perfusion | [15O]H2O |
| Glucose metabolism | [18F]FDG |
| Bone metabolism | [18F]Fluoride |
| Choline metabolism | [18F]Choline |
| DNA synthesis | [18F]FLT |
| Amino acid transport and protein synthesis | [18F]FET, [11C]MET, [18F]FDOPA |
| Receptor binding | [68Ga]-DOTA-TOC |
| Antigen binding | [111In]-anti-CD20 mAb |
| PSMA | [68Ga]-PSMA |
| Angiogenesis | [18F]Galacto-RGD |
| Lipid synthesis | [11C]AcOH |
| Hypoxia | [18F]FAZA, [18F]MISO |
| Apoptosis | [124I]Annexin V |
| Gene expression | [18F]FHBG |
2. Carrier Molecules
2.1. Small Molecules
2.2. Antibodies
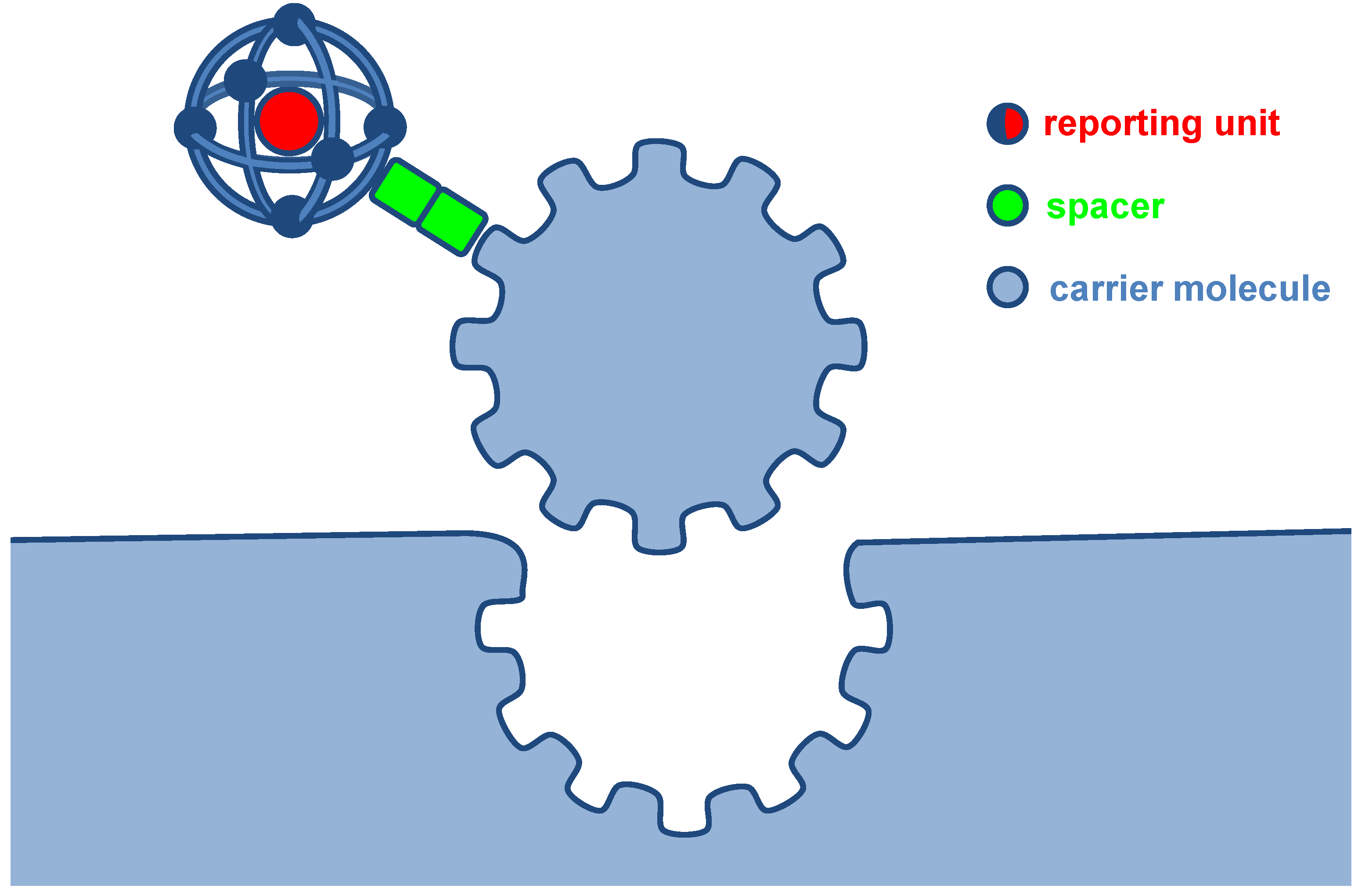
2.3. Peptides

2.3.1. Peptides and Radiopeptides as Targeting Agents
| Peptide | Receptor | Tumor Type | Peptide probe |
|---|---|---|---|
| Somatostatin | sst2 | Gastroenteropancreatic neuro-endocrine tumors | DTPA-octreotide/DOTA-TOC/DOTA-TATE |
| Bombesin | GRPR | Breast, prostate and gastro-intestinal stromal cancer | AMBA/CB-TE2A-AR-06BZH3 |
| RGD | αvβ3 | Melanomas | [18F]Galacto-RGD |
| CCK/gastrin | CCK2R | Medullary thyroid carcinomas | [99mTc]-demogastrin 2 |
| GLP-1/exendin | GLP-1R | Insulinomas | [Lys40(Ahx-DOTA)-NH2]-Exendin-4 |
| α-MSH | MC1R | Melanomas | DOTA-Nle-CycMSHhex DOTA-Re-CCMSH(Arg11) |
| VIP | VIPR | Colorectal cancer | TP3654 |
| substance P | NK-1R | Glioblastoma | DOTAGA-substance P |
2.3.2. Characteristics and Challenges of the Synthesis of Peptide-based Radiopharmaceuticals
3. Label Types
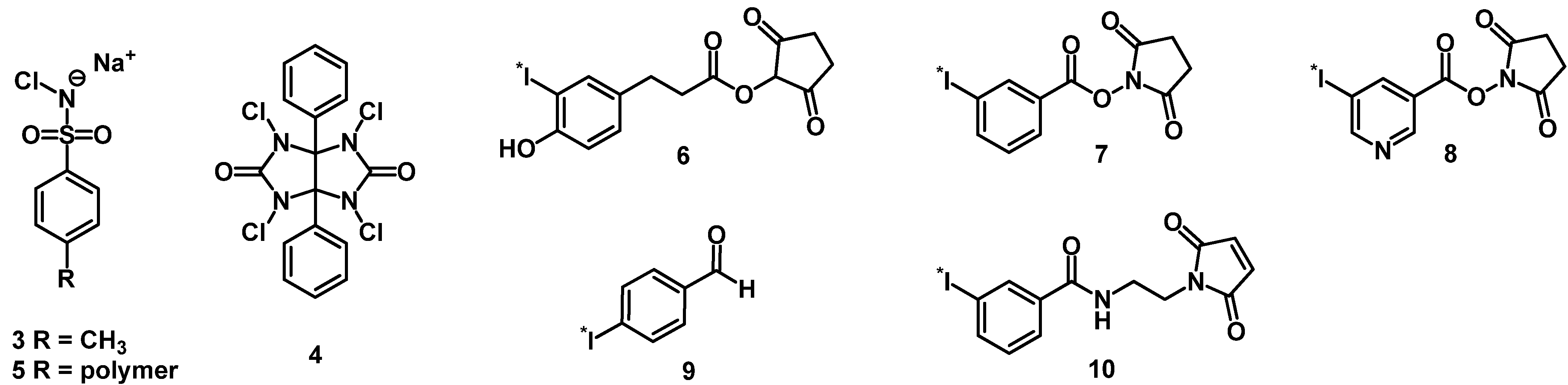
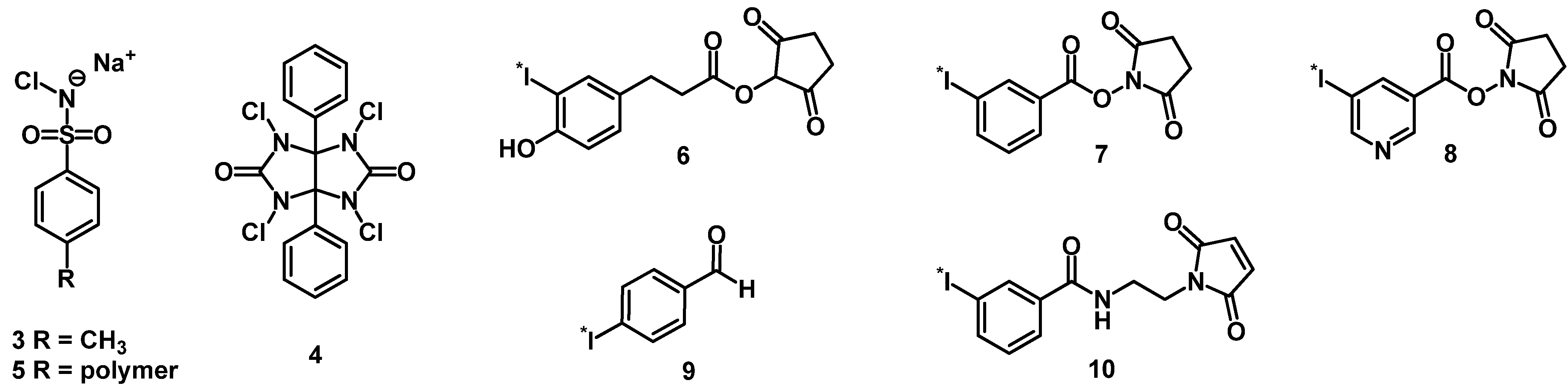
3.1. Iodine-Labeled Peptide Radiopharmaceuticals
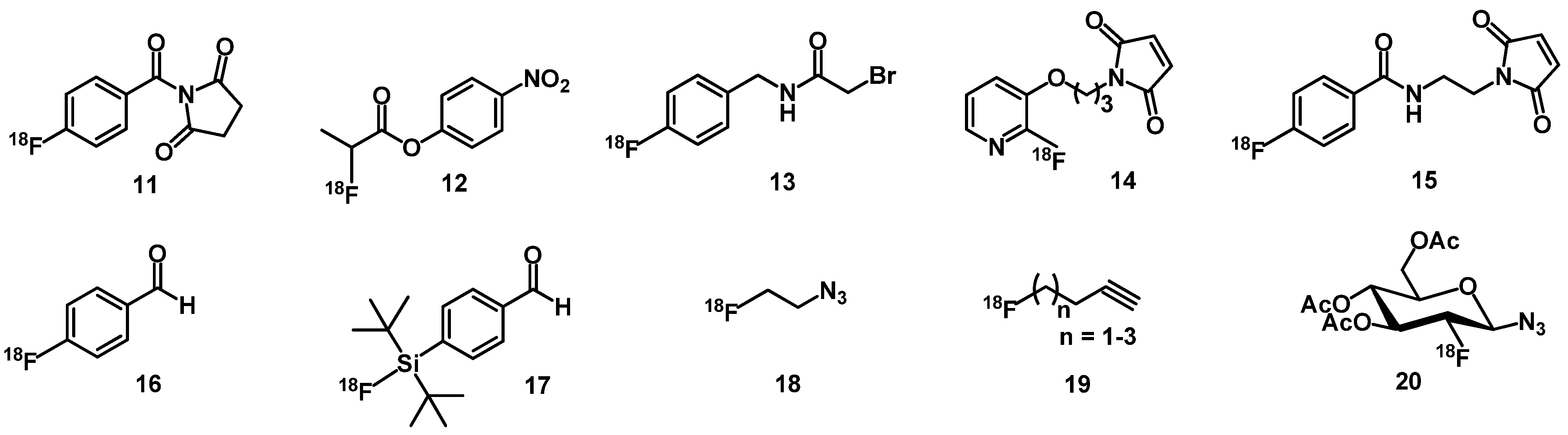
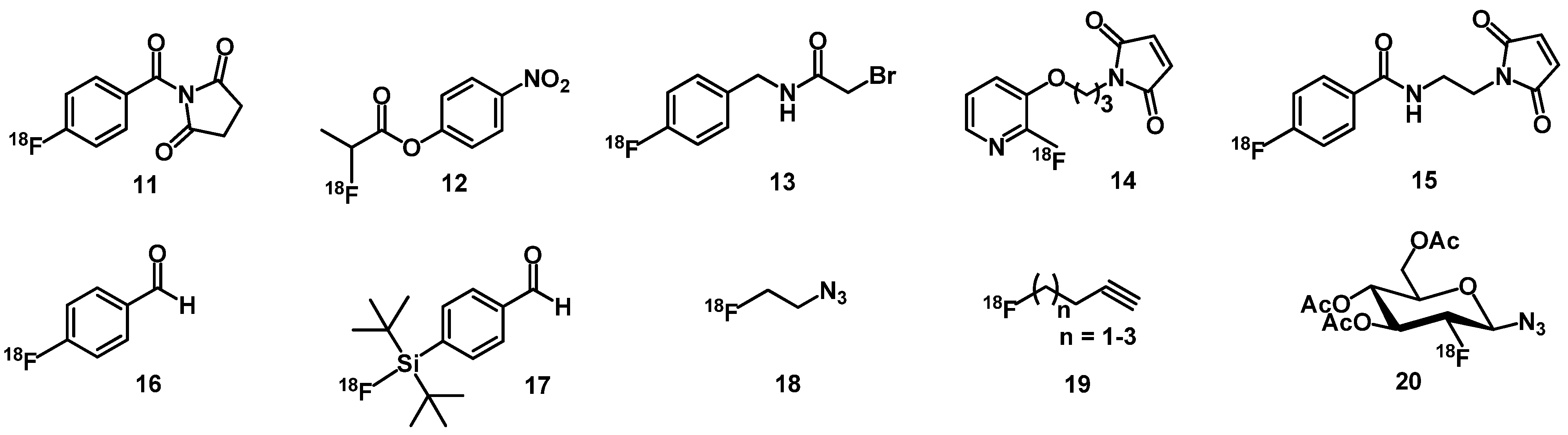
3.2. Fluorine-Labeled Peptide Radiopharmaceuticals
3.3. 99mTc-Labeled Peptide Radiopharmaceuticals
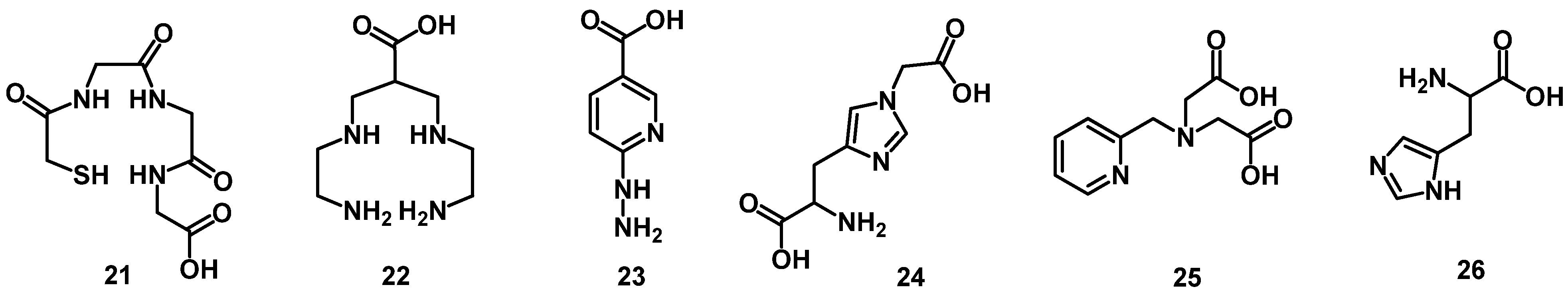
3.4. 111In/67/68Ga/86/90Y/177Lu/64/67Cu-Labeled Peptide Radiopharmaceuticals
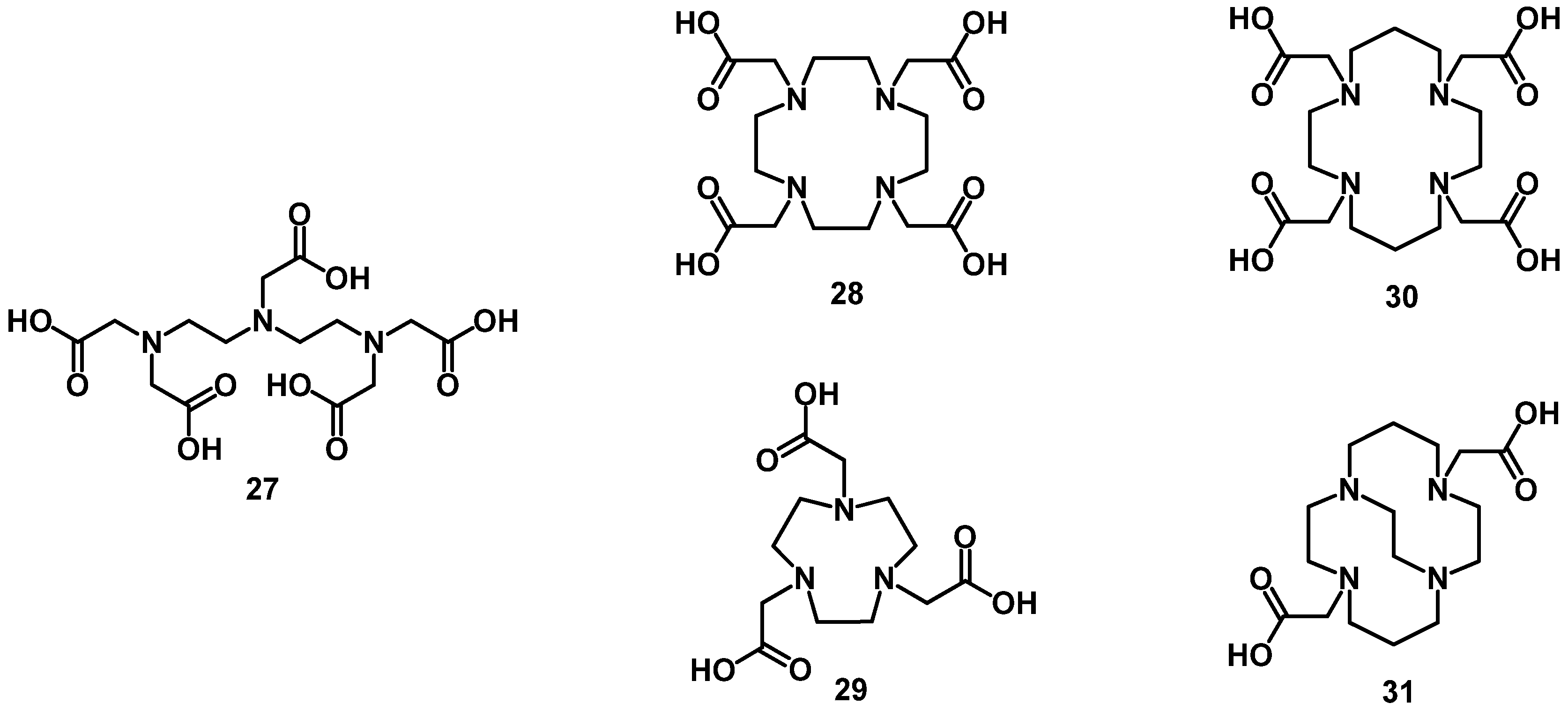
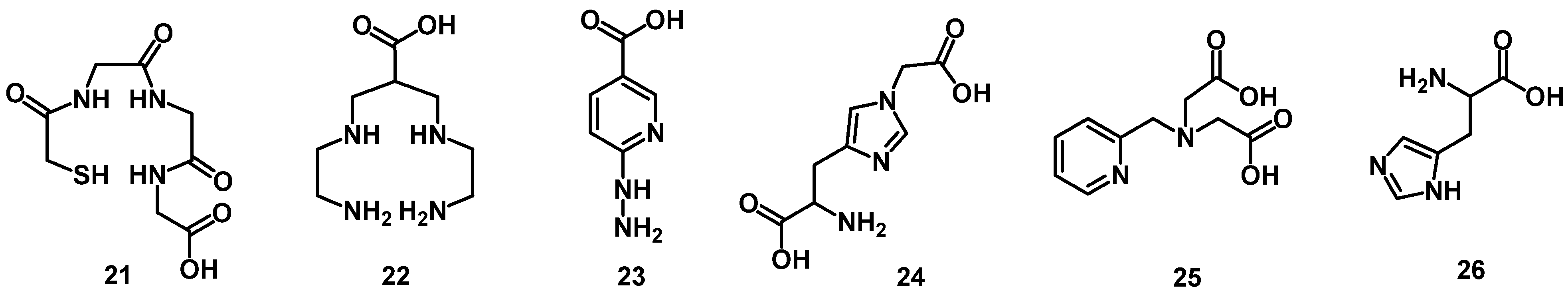
4. Chelator Types
4.1. Acyclic Chelators
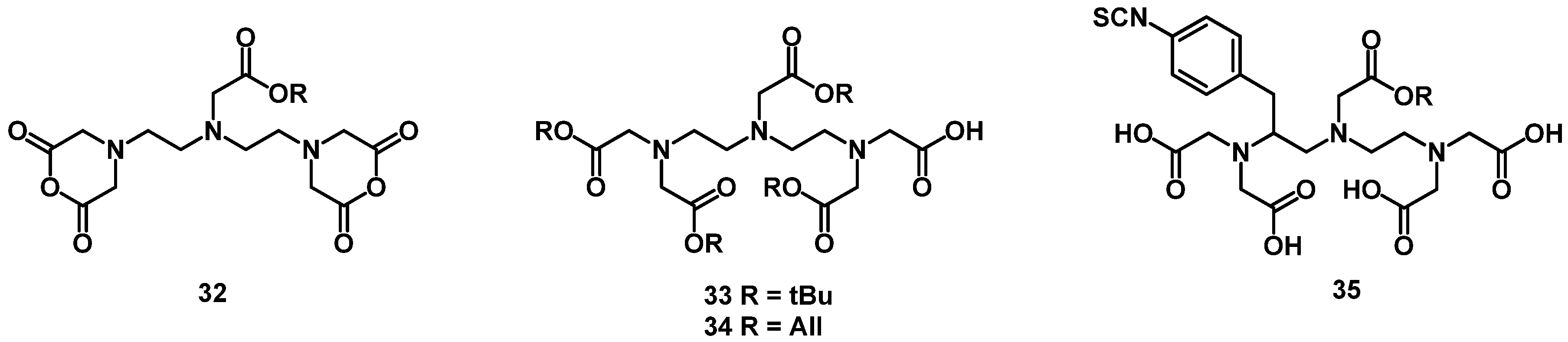
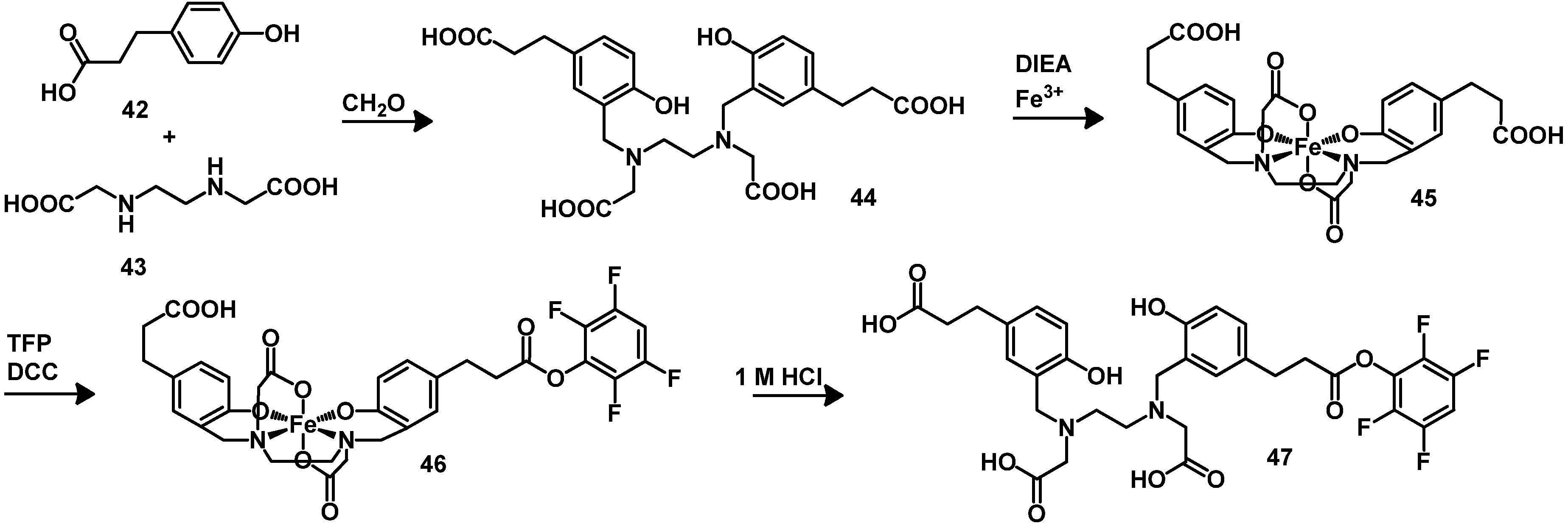
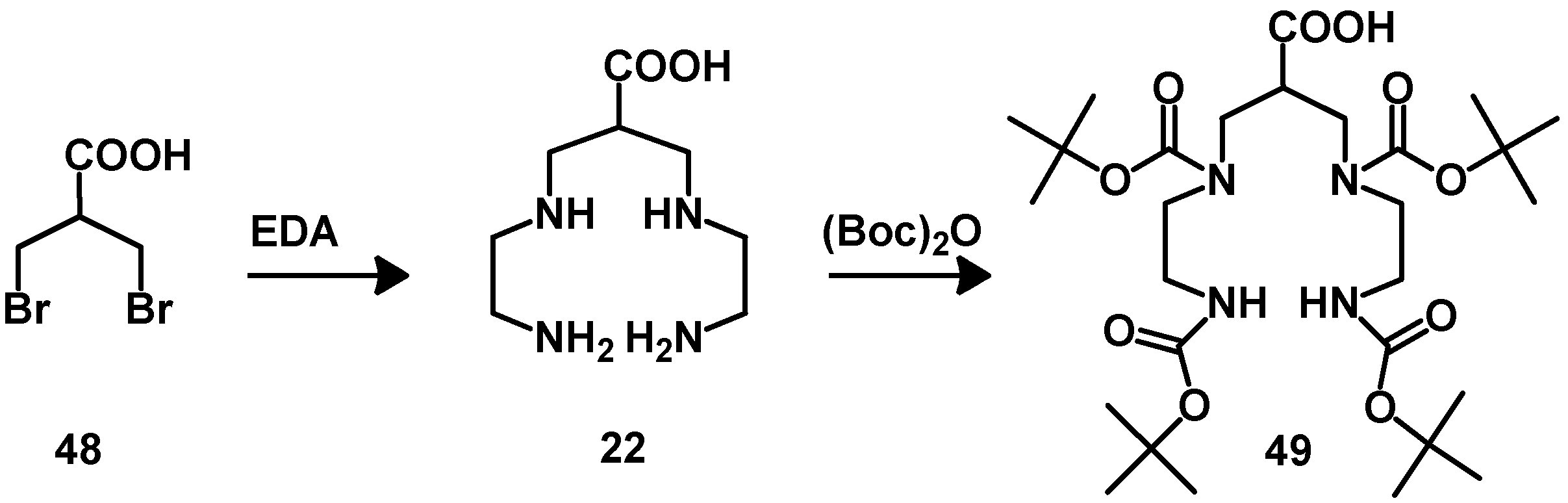
4.2. Macrocyclic Chelators
4.2.1. DOTA and TETA
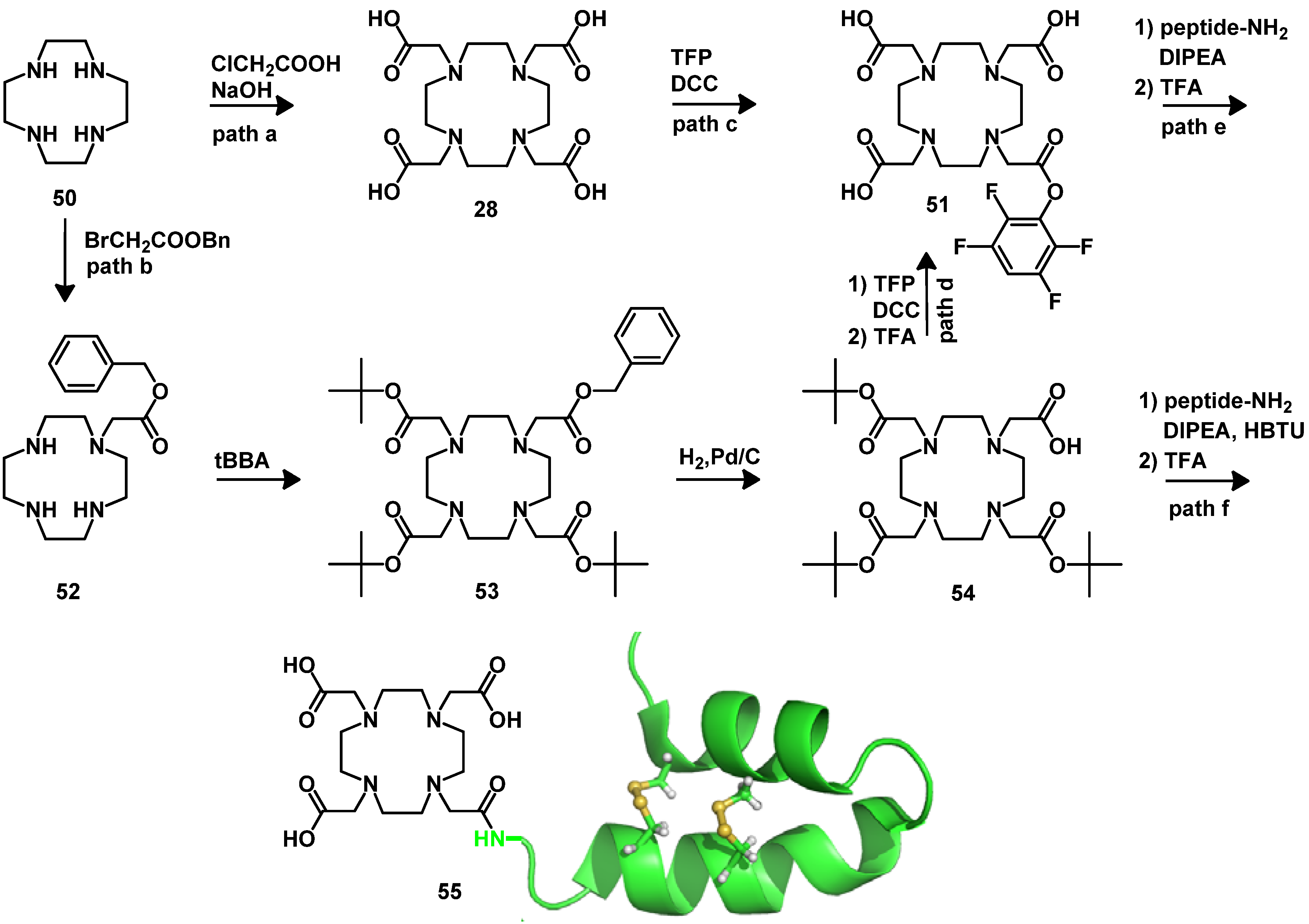
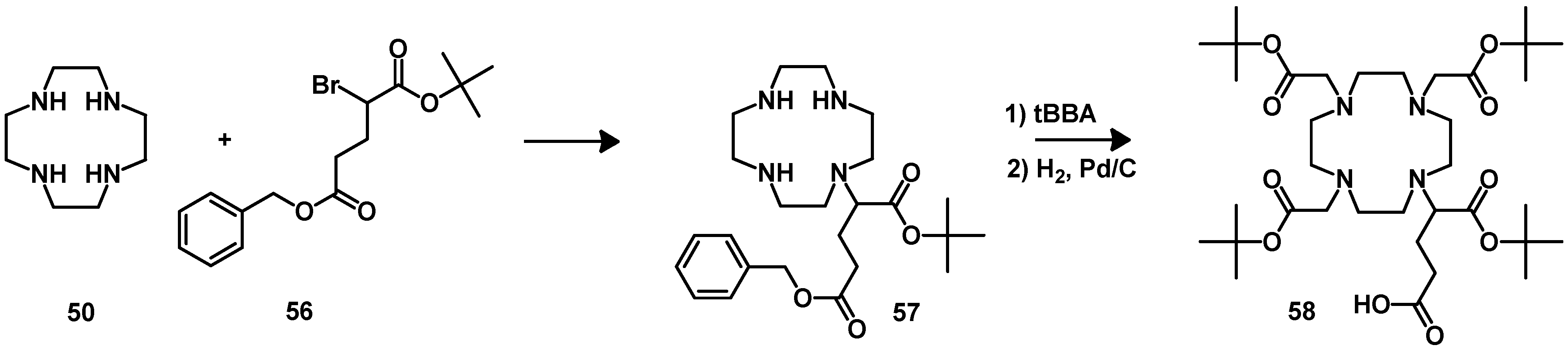
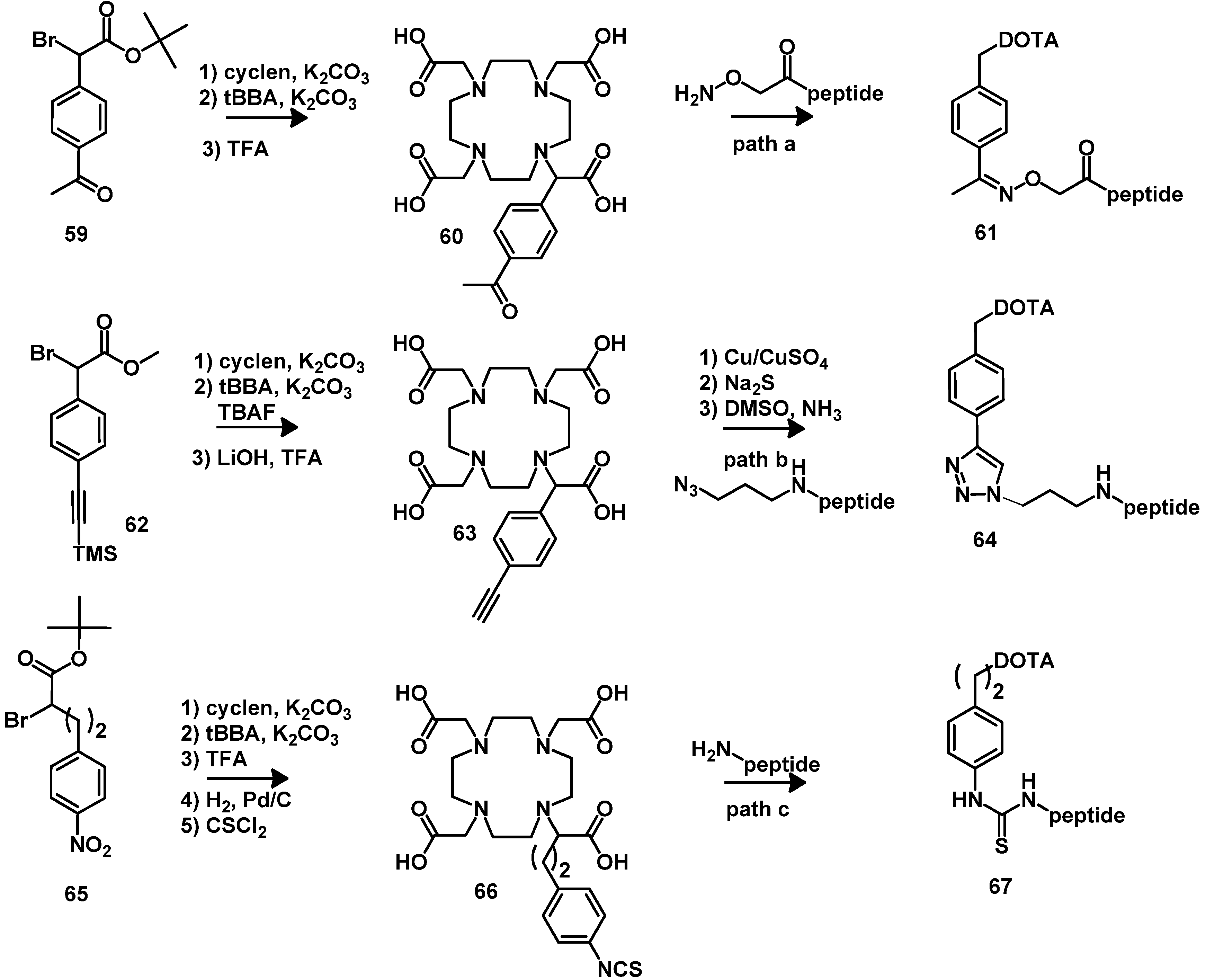
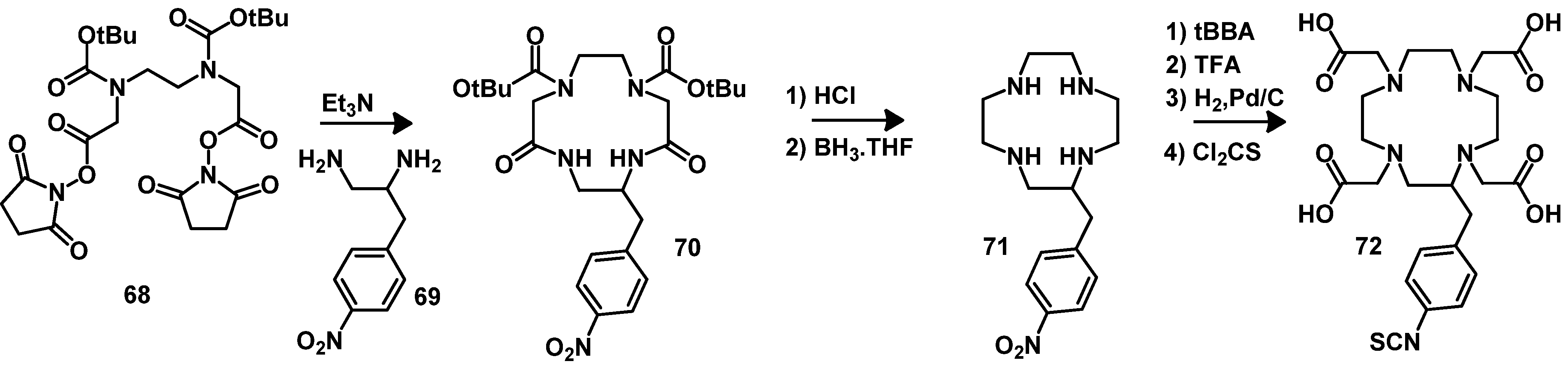
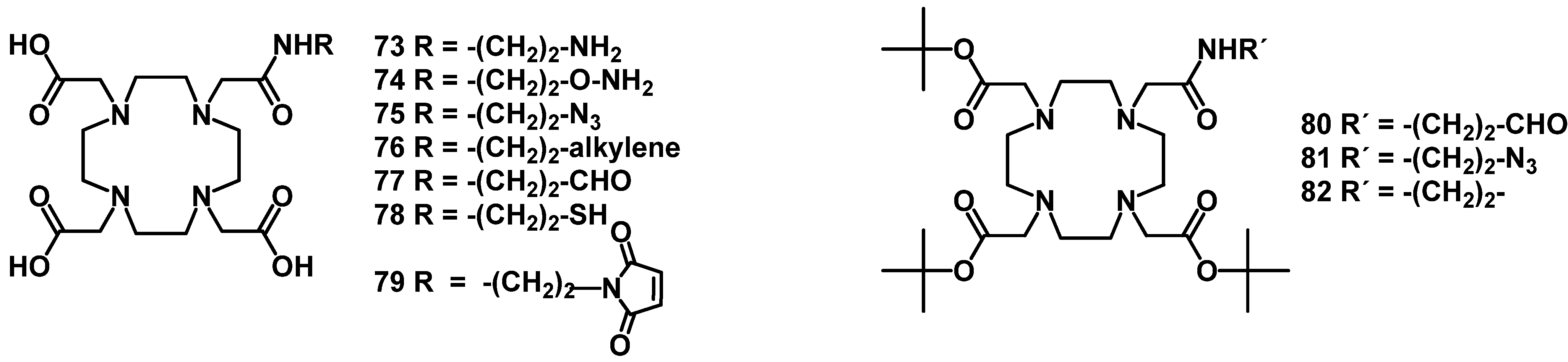
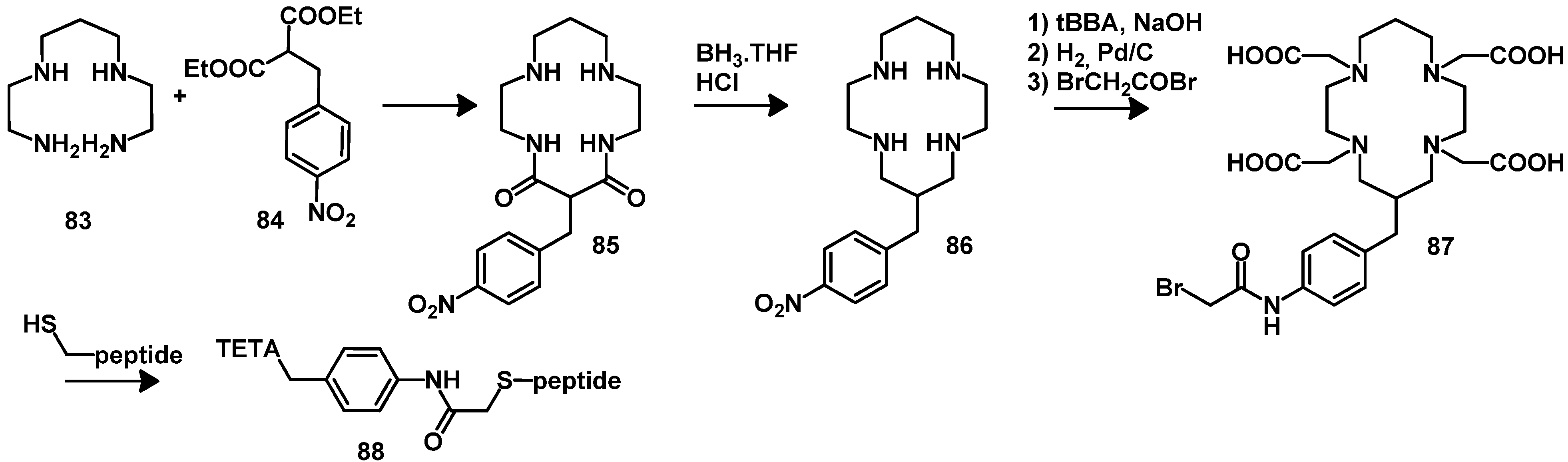
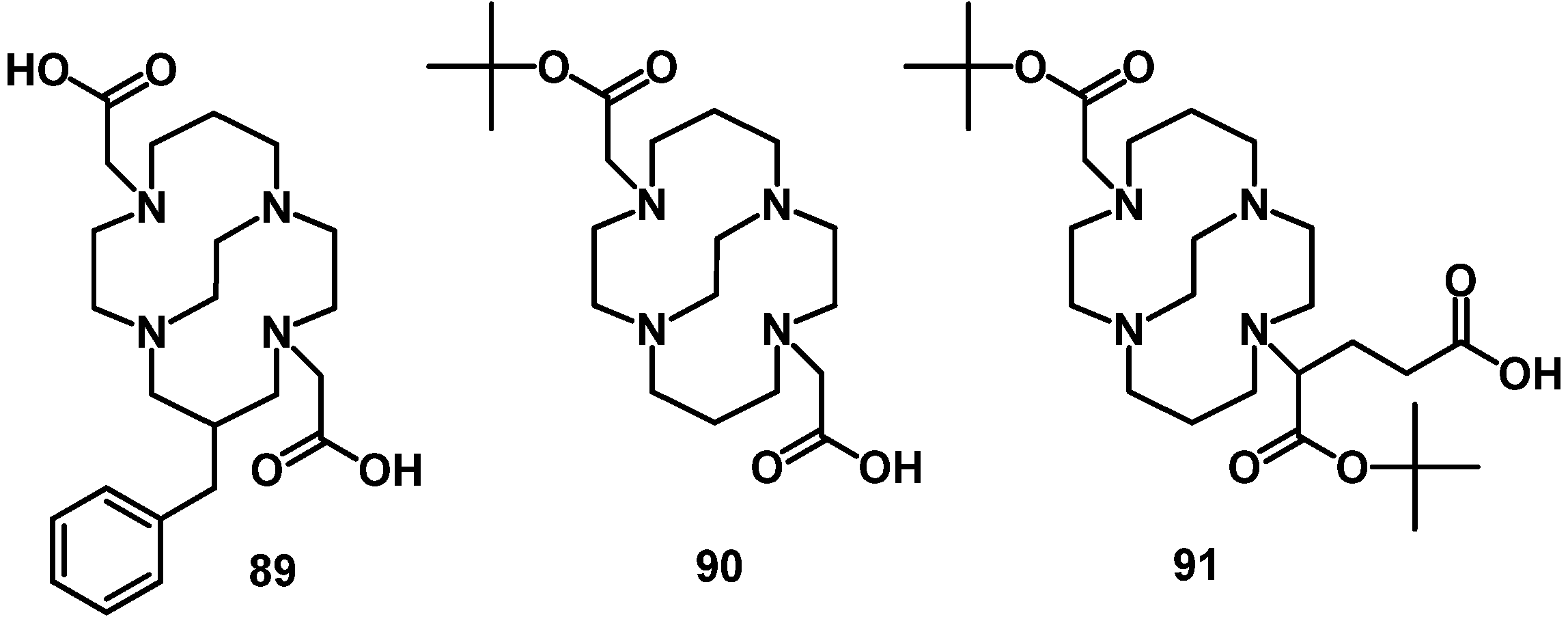
4.2.2. NOTA

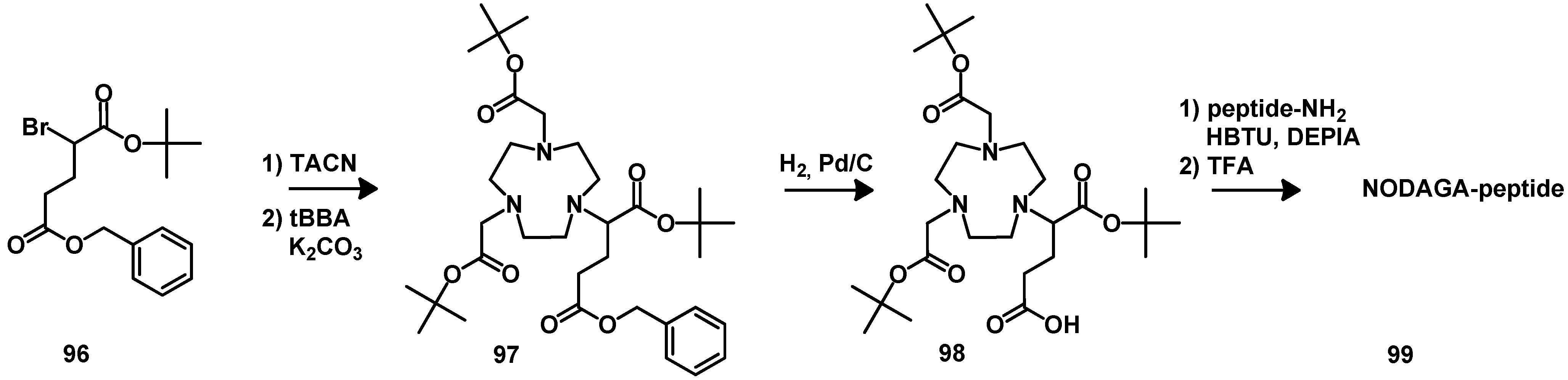
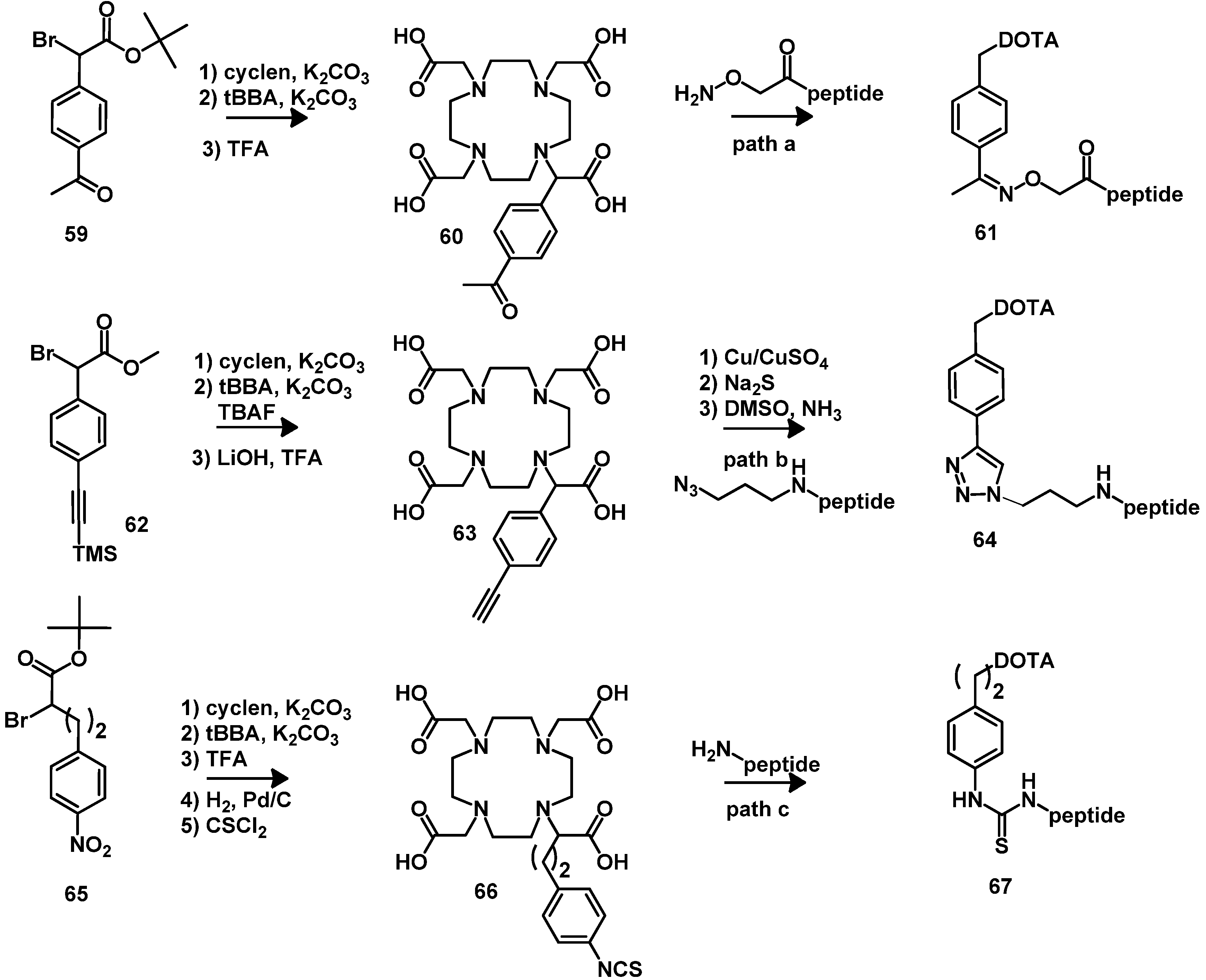
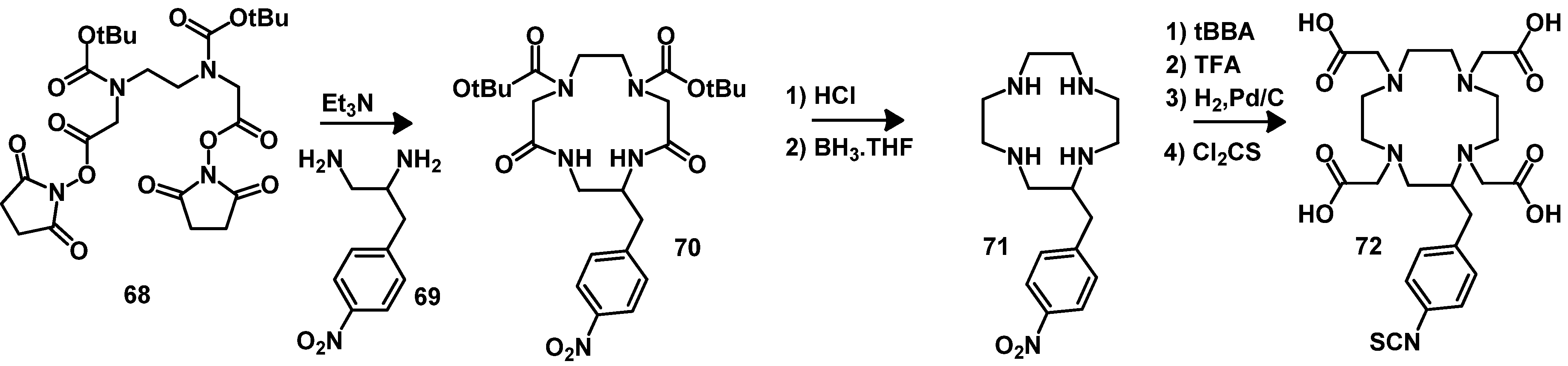
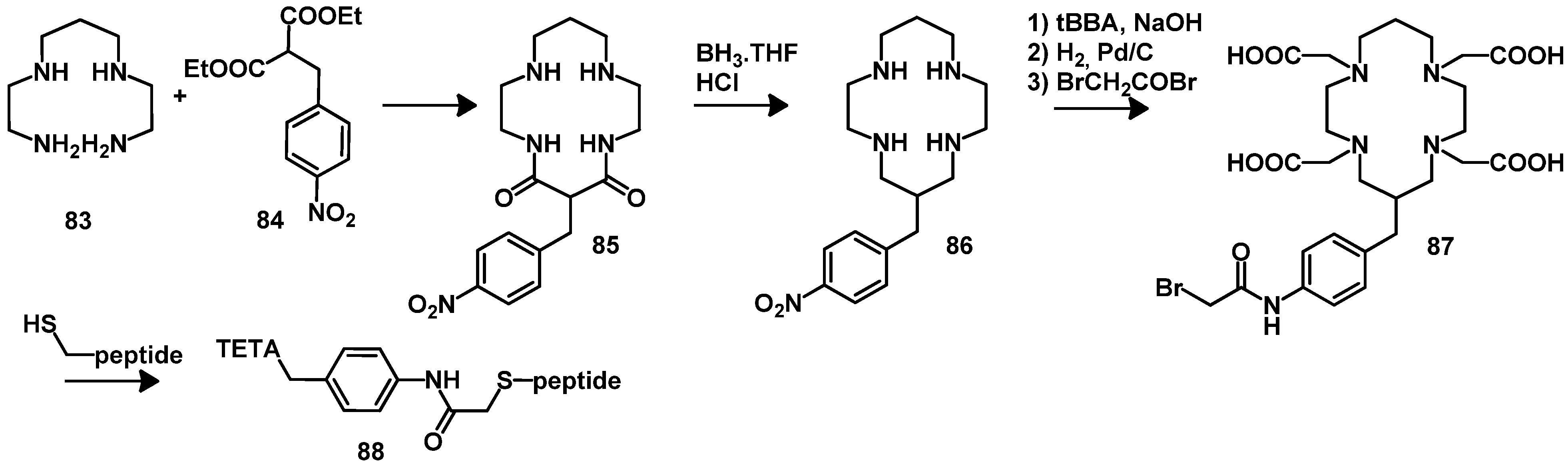
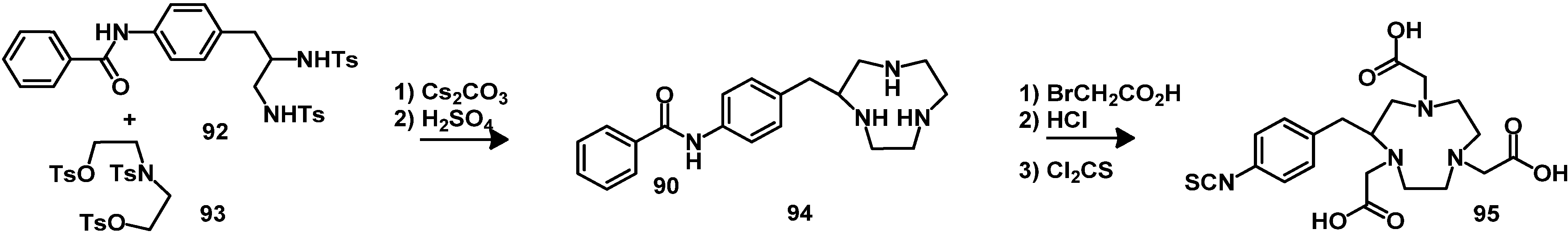
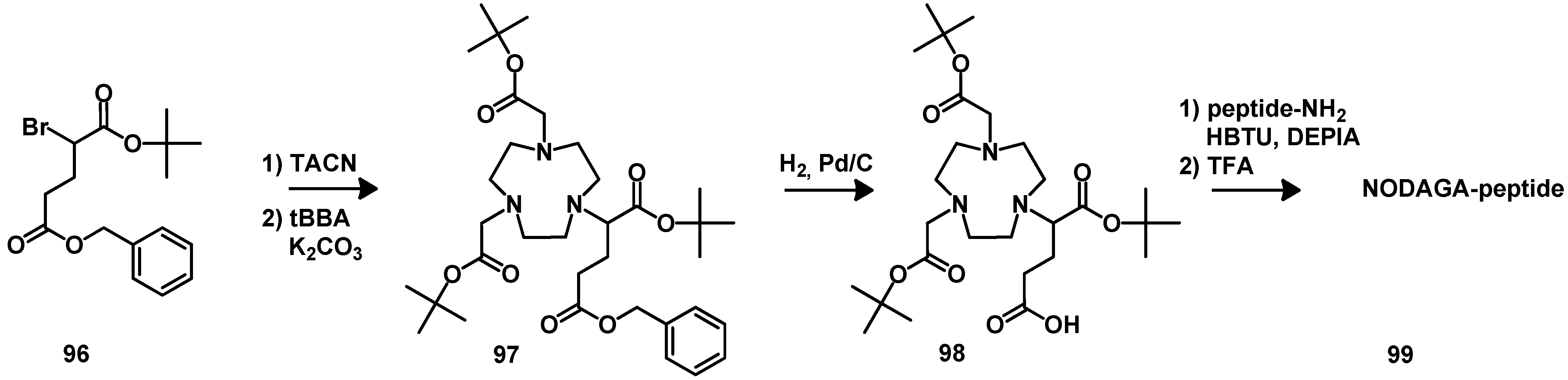
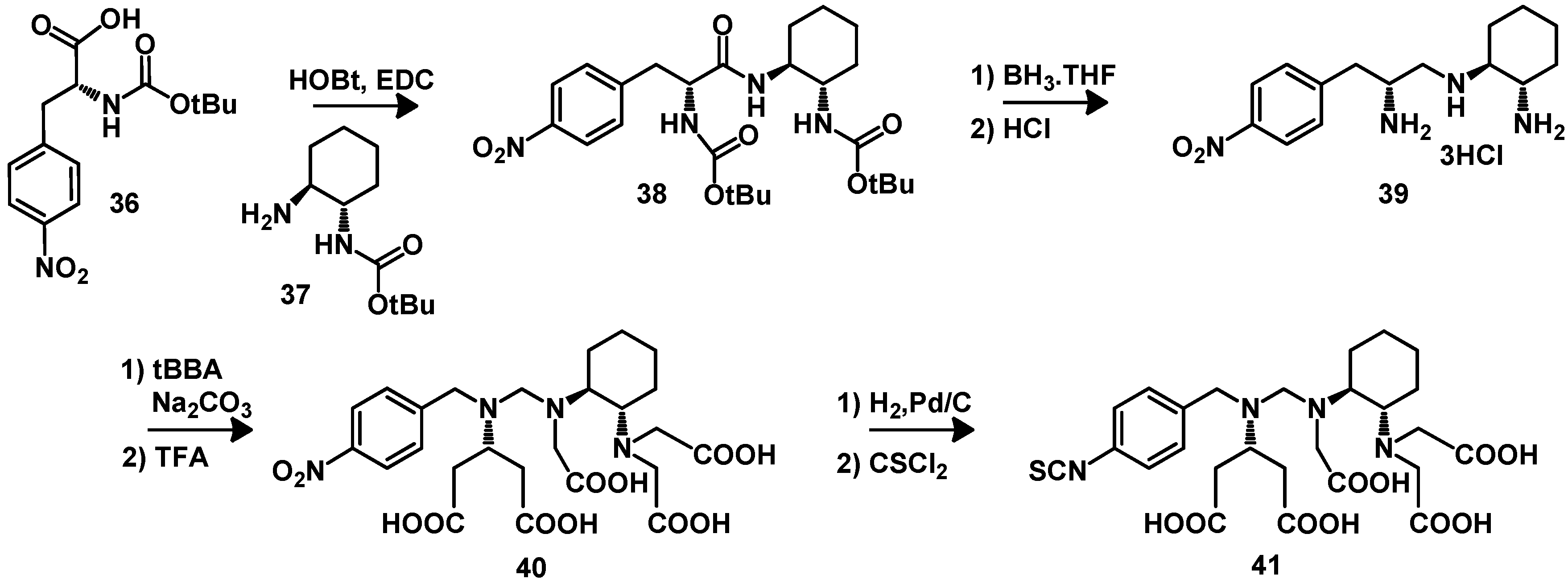
5. Synthesis of Chelator Peptide Conjugates
6. Conclusions
Acknowledgments
References
- Sarko, D.; Eisenhut, M.; Haberkorn, U.; Mier, W. Bifunctional chelators in the design and application of radiopharmaceuticals for oncological diseases. Curr. Med. Chem. 2012, 19, 2667–2688. [Google Scholar] [CrossRef]
- Zoller, F.; Eisenhut, M.; Haberkorn, U.; Mier, W. Endoradiotherapy in cancer treatment-basic concepts and future trends. Eur. J. Pharmacol. 2009, 625, 55–62. [Google Scholar] [CrossRef]
- Smith, M.R.; Li, H.; Gordon, L.; Gascoyne, R.D.; Paietta, E.; Forero-Torres, A.; Kahl, B.S.; Advani, R.; Hong, F.; Horning, S.J. Phase II study of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone immunochemotherapy followed by yttrium-90-ibritumomab tiuxetan in untreated mantle-cell lymphoma: Eastern Cooperative Oncology Group Study E1499. J. Clin. Oncol. 2012, 30, 3119–3126. [Google Scholar] [CrossRef]
- Haberkorn, U.; Eisenhut, M.; Altmann, A.; Mier, W. Endoradiotherapy with peptides—Status and future development. Curr. Med. Chem. 2008, 15, 219–234. [Google Scholar] [CrossRef]
- Wester, H.J. Nuclear imaging probes: From bench to bedside. Clin. Cancer Res. 2007, 13, 3470–3481. [Google Scholar] [CrossRef]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef]
- Morgenroth, A.; Vogg, A.T.; Mottaghy, F.M.; Schmaljohann, J. Targeted endoradiotherapy using nucleotides. Methods 2011, 55, 203–214. [Google Scholar] [CrossRef]
- Witzig, T.E.; Gordon, L.I.; Cabanillas, F.; Czuczman, M.S.; Emmanouilides, C.; Joyce, R.; Pohlman, B.L.; Bartlett, N.L.; Wiseman, G.A.; Padre, N.; et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. 2002, 20, 2453–2463. [Google Scholar] [CrossRef]
- Milenic, D.E.; Brechbiel, M.W. Targeting of radio-isotopes for cancer therapy. Cancer Biol. Ther. 2004, 3, 361–370. [Google Scholar]
- Chamarthy, M.R.; Williams, S.C.; Moadel, R.M. Radioimmunotherapy of non-Hodgkin’s lymphoma: From the ‘magic bullets’ to ‘radioactive magic bullets’. Yale J. Biol. Med. 2011, 84, 391–407. [Google Scholar]
- Reubi, J.C.; Maecke, H.R. Peptide-based probes for cancer imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef]
- Schottelius, M.; Wester, H.J. Molecular imaging targeting peptide receptors. Methods 2009, 48, 161–177. [Google Scholar] [CrossRef]
- Tweedle, M.F. Peptide-targeted diagnostics and radiotherapeutics. Acc. Chem. Res. 2009, 42, 958–968. [Google Scholar] [CrossRef]
- Ambrosini, V.; Fani, M.; Fanti, S.; Forrer, F.; Maecke, H.R. Radiopeptide imaging and therapy in Europe. J. Nucl. Med. 2011, 52 (Suppl. 2), 42S–55S. [Google Scholar]
- Graham, M.M.; Menda, Y. Radiopeptide imaging and therapy in the United States. J. Nucl. Med. 2011, 52 (Suppl. 2), 56S–63S. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Schaer, J.C.; Laissue, J.A. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Nucl Med. 2001, 28, 836–846. [Google Scholar] [CrossRef]
- Rufini, V.; Calcagni, M.L.; Baum, R.P. Imaging of neuroendocrine tumors. Semin. Nucl. Med. 2006, 36, 228–247. [Google Scholar] [CrossRef]
- Lantry, L.E.; Cappelletti, E.; Maddalena, M.E.; Fox, J.S.; Feng, W.; Chen, J.; Thomas, R.; Eaton, S.M.; Bogdan, N.J.; Arunachalam, T.; et al. 177Lu-AMBA: Synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J. Nucl. Med. 2006, 47, 1144–1152. [Google Scholar]
- Schroeder, R.P.; Muller, C.; Reneman, S.; Melis, M.L.; Breeman, W.A.; de Blois, E.; Bangma, C.H.; Krenning, E.P.; van Weerden, W.M.; de Jong, M. A standardised study to compare prostate cancer targeting efficacy of five radiolabelled bombesin analogues. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1386–1396. [Google Scholar] [CrossRef]
- Mansi, R.; Wang, X.; Forrer, F.; Kneifel, S.; Tamma, M.L.; Waser, B.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Evaluation of a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin. Cancer Res. 2009, 15, 5240–5249. [Google Scholar] [CrossRef]
- Mansi, R.; Wang, X.; Forrer, F.; Waser, B.; Cescato, R.; Graham, K.; Borkowski, S.; Reubi, J. C.; Maecke, H.R. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 97–107. [Google Scholar] [CrossRef]
- Abiraj, K.; Mansi, R.; Tamma, M.L.; Fani, M.; Forrer, F.; Nicolas, G.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 2011, 52, 1970–1978. [Google Scholar] [CrossRef]
- Gornik, G.; Mansi, R.; Abiraj, K.; Knippen, S.; Grosu, A.; Maecke, H.; Weber, W. Evaluation of the GRPR radioantagonist Cu-64-CB-TE2A-AR-06 in mice and men. J. Nucl. Med. 2011, 52 (Suppl. 1), 22. [Google Scholar]
- Haubner, R.; Wester, H.J.; Weber, W.A.; Mang, C.; Ziegler, S.I.; Goodman, S.L.; Senekowitsch-Schmidtke, R.; Kessler, H.; Schwaiger, M. Noninvasive imaging of αvβ3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001, 61, 1781–1785. [Google Scholar]
- Haubner, R.; Weber, W.A.; Beer, A.J.; Vabuliene, E.; Reim, D.; Sarbia, M.; Becker, K.F.; Goebel, M.; Hein, R.; Wester, H.J.; et al. Noninvasive visualization of the activated αvβ3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med. 2005, 2, e70. [Google Scholar] [CrossRef]
- Froberg, A.C.; de Jong, M.; Nock, B.A.; Breeman, W.A.; Erion, J.L.; Maina, T.; Verdijsseldonck, M.; de Herder, W.W.; van der Lugt, A.; Kooij, P.P.; et al. Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1265–1272. [Google Scholar] [CrossRef]
- Christ, E.; Wild, D.; Forrer, F.; Brandle, M.; Sahli, R.; Clerici, T.; Gloor, B.; Martius, F.; Maecke, H.; Reubi, J.C. Glucagon-like peptide-1 receptor imaging for localization of insulinomas. J. Clin. Endocrinol. Metab. 2009, 94, 4398–4405. [Google Scholar] [CrossRef]
- Wild, D.; Macke, H.; Christ, E.; Gloor, B.; Reubi, J.C. Glucagon-like peptide 1-receptor scans to localize occult insulinomas. N. Engl. J. Med. 2008, 359, 766–768. [Google Scholar] [CrossRef]
- Wild, D.; Behe, M.; Wicki, A.; Storch, D.; Waser, B.; Gotthardt, M.; Keil, B.; Christofori, G.; Reubi, J.C.; Macke, H.R. [Lys40(Ahx-DTPA-111In)NH2]exendin-4, a very promising ligand for glucagon-like peptide-1 (GLP-1) receptor targeting. J. Nucl. Med. 2006, 47, 2025–2033. [Google Scholar]
- Wicki, A.; Wild, D.; Storch, D.; Seemayer, C.; Gotthardt, M.; Behe, M.; Kneifel, S.; Mihatsch, M.J.; Reubi, J.C.; Macke, H.R.; et al. [Lys40(Ahx-DTPA-111In)NH2]-Exendin-4 is a highly efficient radiotherapeutic for glucagon-like peptide-1 receptor-targeted therapy for insulinoma. Clin. Cancer Res. 2007, 13, 3696–3705. [Google Scholar] [CrossRef]
- Cantorias, M.V.; Figueroa, S.D.; Quinn, T.P.; Lever, J.R.; Hoffman, T.J.; Watkinson, L.D.; Carmack, T.L.; Cutler, C.S. Development of high-specific-activity 68Ga-labeled DOTA-rhenium-cyclized α-MSH peptide analog to target MC1 receptors overexpressed by melanoma tumors. Nucl. Med. Biol. 2009, 36, 505–513. [Google Scholar] [CrossRef]
- Miao, Y.; Owen, N.K.; Fisher, D.R.; Hoffman, T.J.; Quinn, T.P. Therapeutic efficacy of a 188Re-labeled alpha-melanocyte-stimulating hormone peptide analog in murine and human melanoma-bearing mouse models. J. Nucl. Med. 2005, 46, 121–129. [Google Scholar]
- Alshoukr, F.; Prignon, A.; Brans, L.; Jallane, A.; Mendes, S.; Talbot, J.N.; Tourwe, D.; Barbet, J.; Gruaz-Guyon, A. Novel DOTA-neurotensin analogues for 111In scintigraphy and 68Ga PET imaging of neurotensin receptor-positive tumors. Bioconjug. Chem. 2011, 22, 1374–1385. [Google Scholar] [CrossRef]
- Rao, P.S.; Thakur, M.L.; Pallela, V.; Patti, R.; Reddy, K.; Li, H.; Sharma, S.; Pham, H.L.; Diggles, L.; Minami, C.; et al. 99mTc labeled VIP analog: Evaluation for imaging colorectal cancer. Nucl. Med. Biol. 2001, 28, 445–450. [Google Scholar] [CrossRef]
- Kneifel, S.; Cordier, D.; Good, S.; Ionescu, M.C.; Ghaffari, A.; Hofer, S.; Kretzschmar, M.; Tolnay, M.; Apostolidis, C.; Waser, B.; et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance p. Clin. Cancer Res. 2006, 12, 3843–3850. [Google Scholar] [CrossRef]
- Bomanji, J.B.; Papathanasiou, N.D. 111In-DTPA0-octreotide (Octreoscan), 131I-MIBG and other agents for radionuclide therapy of NETs. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S113–S125. [Google Scholar] [CrossRef]
- Reubi, J.C. Peptide receptor expression in GEP-NET. Virchows Arch. 2007, 451 (Suppl. 1), S47–S50. [Google Scholar] [CrossRef]
- Reubi, J.C. Somatostatin and other Peptide receptors as tools for tumor diagnosis and treatment. Neuroendocrinology 2004, 80 (Suppl. 1), 51–56. [Google Scholar] [CrossRef]
- Jackson, A.B.; Nanda, P.K.; Rold, T.L.; Sieckman, G.L.; Szczodroski, A.F.; Hoffman, T.J.; Chen, X.; Smith, C.J. 64Cu-NO2A-RGD-Glu-6-Ahx-BBN(7-14)NH2: A heterodimeric targeting vector for positron emission tomography imaging of prostate cancer. Nucl. Med. Biol. 2012, 39, 377–387. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Z.B.; Cao, Q.; Liu, S.; Wang, F.; Chen, X. Small-animal PET of tumors with 64Cu-labeled RGD-bombesin heterodimer. J. Nucl. Med. 2009, 50, 1168–1177. [Google Scholar] [CrossRef]
- Liu, Z.; Niu, G.; Wang, F.; Chen, X. 68Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1483–1494. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, Y.; Liu, S.; Wang, F.; Chen, X. 18F, 64Cu, and 68Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug. Chem. 2009, 20, 1016–1025. [Google Scholar] [CrossRef]
- Josan, J.S.; Vagner, J.; Handl, H.L.; Sankaranarayanan, R.; Gillies, R.J.; Hruby, V.J. Solid-Phase Synthesis of Heterobivalent Ligands Targeted to Melanocortin and Cholecystokinin Receptors. Int. J. Pept. Res. Ther. 2008, 14, 293–300. [Google Scholar] [CrossRef]
- Vagner, J.; Xu, L.; Handl, H.L.; Josan, J.S.; Morse, D.L.; Mash, E.A.; Gillies, R.J.; Hruby, V.J. Heterobivalent ligands crosslink multiple cell-surface receptors: The human melanocortin-4 and delta-opioid receptors. Angew. Chem. Int. Ed. Engl. 2008, 47, 1685–1688. [Google Scholar]
- Liu, S. The role of coordination chemistry in the development of target-specific radiopharmaceuticals. Chem. Soc. Rev. 2004, 33, 445–461. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled peptides: Valuable tools for the detection and treatment of cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol. Pharm. 2006, 3, 472–487. [Google Scholar] [CrossRef]
- Wester, H.J.; Schottelius, M.; Poethko, T.; Bruus-Jensen, K.; Schwaiger, M. Radiolabeled carbohydrated somatostatin analogs: A review of the current status. Cancer Biother. Radiopharm. 2004, 19, 231–244. [Google Scholar] [CrossRef]
- Chen, X.; Hou, Y.; Tohme, M.; Park, R.; Khankaldyyan, V.; Gonzales-Gomez, I.; Bading, J.R.; Laug, W.E.; Conti, P.S. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor αvβ3-integrin expression. J. Nucl. Med. 2004, 45, 1776–1783. [Google Scholar]
- Garg, S.; Garg, P.K.; Zalutsky, M.R. N-succinimidyl 5-(trialkylstannyl)-3-pyridinecarboxylates: A new class of reagents for protein radioiodination. Bioconjug. Chem. 1991, 2, 50–56. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. Preparation of N-succinimidyl 3-[*I]iodobenzoate: An agent for the indirect radioiodination of proteins. Nat. Protoc. 2006, 1, 707–713. [Google Scholar] [CrossRef]
- Thumshirn, G.; Hersel, U.; Goodman, S.L.; Kessler, H. Multimeric cyclic RGD peptides as potential tools for tumor targeting: Solid-phase peptide synthesis and chemoselective oxime ligation. Chemistry 2003, 9, 2717–2725. [Google Scholar] [CrossRef]
- Bhojani, M.S.; Ranga, R.; Luker, G.D.; Rehemtulla, A.; Ross, B.D.; Van Dort, M.E. Synthesis and investigation of a radioiodinated F3 peptide analog as a SPECT tumor imaging radioligand. PLoS One 2011, 6, e22418. [Google Scholar]
- Zhang, X.; Cai, W.; Cao, F.; Schreibmann, E.; Wu, Y.; Wu, J.C.; Xing, L.; Chen, X. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J. Nucl. Med. 2006, 47, 492–501. [Google Scholar]
- Wester, H.J.; Schottelius, M.; Scheidhauer, K.; Meisetschlager, G.; Herz, M.; Rau, F.C.; Reubi, J.C.; Schwaiger, M. PET imaging of somatostatin receptors: Design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 117–122. [Google Scholar] [CrossRef]
- Guhlke, S.; Wester, H.J.; Bruns, C.; Stocklin, G. 2-[18F]fluoropropionyl-(D)phe1)-octreotide, a potential radiopharmaceutical for quantitative somatostatin receptor imaging with PET: Synthesis, radiolabeling, in vitro validation and biodistribution in mice. Nucl. Med. Biol. 1994, 21, 819–825. [Google Scholar] [CrossRef]
- Wester, H.J.; Hamacher, K.; Stocklin, G. A comparative study of N.C.A. fluorine-18 labeling of proteins via acylation and photochemical conjugation. Nucl. Med. Biol. 1996, 23, 365–372. [Google Scholar] [CrossRef]
- de Bruin, B.; Kuhnast, B.; Hinnen, F.; Yaouancq, L.; Amessou, M.; Johannes, L.; Samson, A.; Boisgard, R.; Tavitian, B.; Dolle, F. 1-[3-(2-[18F]fluoropyridin-3-yloxy)propyl]pyrrole-2,5-dione: Design, synthesis, and radiosynthesis of a new [18F]fluoropyridine-based maleimide reagent for the labeling of peptides and proteins. Bioconjug. Chem. 2005, 16, 406–420. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, X.; Wu, Y.; Chen, X. A thiol-reactive 18F-labeling agent, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide, and synthesis of RGD peptide-based tracer for PET imaging of αvβ3 integrin expression. J. Nucl. Med. 2006, 47, 1172–1180. [Google Scholar]
- Glaser, M.; Arstad, E. “Click labeling” with 2-[18f]fluoroethylazide for positron emission tomography. Bioconjug. Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Poethko, T.; Schottelius, M.; Thumshirn, G.; Hersel, U.; Herz, M.; Henriksen, G.; Kessler, H.; Schwaiger, M.; Wester, H.J. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J. Nucl. Med. 2004, 45, 892–902. [Google Scholar]
- Bruus-Jensen, K.; Poethko, T.; Schottelius, M.; Hauser, A.; Schwaiger, M.; Wester, H.J. Chemoselective hydrazone formation between HYNIC-functionalized peptides and 18F-fluorinated aldehydes. Nucl. Med. Biol. 2006, 33, 173–183. [Google Scholar] [CrossRef]
- Schirrmacher, E.; Wangler, B.; Cypryk, M.; Bradtmoller, G.; Schafer, M.; Eisenhut, M.; Jurkschat, K.; Schirrmacher, R. Synthesis of p-(di-tert-butyl[18F]fluorosilyl)benzaldehyde ([18F]SiFA-A) with high specific activity by isotopic exchange: A convenient labeling synthon for the 18F-labeling of N-amino-oxy derivatized peptides. Bioconjug. Chem. 2007, 18, 2085–2089. [Google Scholar] [CrossRef]
- Marik, J.; Sutcliffe, J.L. Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Schottelius, M.; Poethko, T.; Herz, M.; Reubi, J.C.; Kessler, H.; Schwaiger, M.; Wester, H.J. First 18F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin. Cancer Res. 2004, 10, 3593–3606. [Google Scholar] [CrossRef]
- Meisetschlager, G.; Poethko, T.; Stahl, A.; Wolf, I.; Scheidhauer, K.; Schottelius, M.; Herz, M.; Wester, H.J.; Schwaiger, M. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: Biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J. Nucl. Med. 2006, 47, 566–573. [Google Scholar]
- Lebtahi, R.; Le Cloirec, J.; Houzard, C.; Daou, D.; Sobhani, I.; Sassolas, G.; Mignon, M.; Bourguet, P.; Le Guludec, D. Detection of neuroendocrine tumors: 99mTc-P829 scintigraphy compared with 111In-pentetreotide scintigraphy. J. Nucl. Med. 2002, 43, 889–895. [Google Scholar]
- Van de Wiele, C.; Dumont, F.; Vanden Broecke, R.; Oosterlinck, W.; Cocquyt, V.; Serreyn, R.; Peers, S.; Thornback, J.; Slegers, G.; Dierckx, R. A. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: A feasibility study. Eur. J. Nucl. Med. 2000, 27, 1694–1699. [Google Scholar] [CrossRef]
- Liu, S.; Edwards, D.S.; Looby, R.J.; Poirier, M.J.; Rajopadhye, M.; Bourque, J.P.; Carroll, T.R. Labeling cyclic glycoprotein IIb/IIIa receptor antagonists with 99mTc by the preformed chelate approach: Effects of chelators on properties of [99mTc]chelator-peptide conjugates. Bioconjug. Chem. 1996, 7, 196–202. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Decristoforo, C.; Maina, T.; Nock, B.; Gabriel, M.; Cordopatis, P.; Moncayo, R. 99mTc-Demotate 1: First data in tumour patients-results of a pilot/phase I study. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1211–1219. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.; Nikolopoulou, A.; Sotiriou, P.; Loudos, G.; Maintas, D.; Cordopatis, P.; Chiotellis, E. [99mTc]Demotate, a new 99mTc-based [Tyr3]octreotate analogue for the detection of somatostatin receptor-positive tumours: Synthesis and preclinical results. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 742–753. [Google Scholar] [CrossRef]
- Nock, B.A.; Maina, T.; Behe, M.; Nikolopoulou, A.; Gotthardt, M.; Schmitt, J.S.; Behr, T.M.; Macke, H.R. CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J. Nucl. Med. 2005, 46, 1727–1736. [Google Scholar]
- Nock, B.A.; Nikolopoulou, A.; Galanis, A.; Cordopatis, P.; Waser, B.; Reubi, J.C.; Maina, T. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: A preclinical study. J. Med. Chem. 2005, 48, 100–110. [Google Scholar] [CrossRef]
- Nock, B.; Nikolopoulou, A.; Chiotellis, E.; Loudos, G.; Maintas, D.; Reubi, J.C.; Maina, T. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 247–258. [Google Scholar] [CrossRef]
- Abiraj, K.; Mansi, R.; Tamma, M.L.; Forrer, F.; Cescato, R.; Reubi, J.C.; Akyel, K.G.; Maecke, H.R. Tetraamine-derived bifunctional chelators for technetium-99m labelling: Synthesis, bioconjugation and evaluation as targeted SPECT imaging probes for GRP-receptor-positive tumours. Chemistry 2010, 16, 2115–2124. [Google Scholar] [CrossRef]
- von Guggenberg, E.; Behe, M.; Behr, T.M.; Saurer, M.; Seppi, T.; Decristoforo, C. 99mTc-labeling and in vitro and in vivo evaluation of HYNIC- and (Nα-His)acetic acid-modified [D-Glu1]-minigastrin. Bioconjug. Chem. 2004, 15, 864–871. [Google Scholar] [CrossRef]
- Decristoforo, C.; Mather, S.J.; Cholewinski, W.; Donnemiller, E.; Riccabona, G.; Moncayo, R. 99mTc-EDDA/HYNIC-TOC: A new 99mTc-labelled radiopharmaceutical for imaging somatostatin receptor-positive tumours; first clinical results and intra-patient comparison with 111In-labelled octreotide derivatives. Eur. J. Nucl. Med. 2000, 27, 1318–1325. [Google Scholar] [CrossRef]
- Storch, D.; Behe, M.; Walter, M.A.; Chen, J.; Powell, P.; Mikolajczak, R.; Macke, H.R. Evaluation of [99mTc/EDDA/HYNIC0]octreotide derivatives compared with [111In-DOTA0,Tyr3,Thr8]octreotide and [111In-DTPA0]octreotide: Does tumor or pancreas uptake correlate with the rate of internalization? J. Nucl. Med. 2005, 46, 1561–1569. [Google Scholar]
- King, R.C.; Surfraz, M.B.; Biagini, S.C.; Blower, P.J.; Mather, S.J. How do HYNIC-conjugated peptides bind technetium? Insights from LC-MS and stability studies. Dalton Trans. 2007, 4998–5007. [Google Scholar]
- Decristoforo, C.; Faintuch-Linkowski, B.; Rey, A.; von Guggenberg, E.; Rupprich, M.; Hernandez-Gonzales, I.; Rodrigo, T.; Haubner, R. [99mTc]HYNIC-RGD for imaging integrin αvβ3 expression. Nucl. Med. Biol. 2006, 33, 945–952. [Google Scholar] [CrossRef]
- Schibli, R.; Schubiger, P.A. Current use and future potential of organometallic radiopharmaceuticals. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1529–1542. [Google Scholar] [CrossRef]
- Krenning, E.P.; Kwekkeboom, D.J.; Bakker, W.H.; Breeman, W.A.; Kooij, P.P.; Oei, H.Y.; van Hagen, M.; Postema, P.T.; de Jong, M.; Reubi, J.C.; et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: The Rotterdam experience with more than 1000 patients. Eur. J. Nucl. Med. 1993, 20, 716–731. [Google Scholar]
- Stolz, B.; Smith-Jones, P.; Albert, R.; Tolcsvai, L.; Briner, U.; Ruser, G.; Macke, H.; Weckbecker, G.; Bruns, C. Somatostatin analogues for somatostatin-receptor-mediated radiotherapy of cancer. Digestion 1996, 57 (Suppl. 1), 17–21. [Google Scholar]
- Notni, J.; Pohle, K.; Wester, H. J. Comparative gallium-68 labeling of TRAP-, NOTA-, and DOTA-peptides: Practical consequences for the future of gallium-68-PET. EJNMMI Res. 2012, 2, 28. [Google Scholar]
- Fani, M.; Andre, J.P.; Maecke, H.R. 68Ga-PET: A powerful generator-based alternative to cyclotron-based PET radiopharmaceuticals. Contrast Media Mol. Imaging 2008, 3, 67–77. [Google Scholar]
- Balogh, E.; Tripier, R.; Ruloff, R.; Toth, E. Kinetics of formation and dissociation of lanthanide(III) complexes with the 13-membered macrocyclic ligand TRITA4. Dalton Trans. 2005, 1058–1065. [Google Scholar]
- Frost, A.E. Polyaminopolycarboxylic acids derived from polyethyleneamines. Nature 1956, 178, 322–322. [Google Scholar] [CrossRef]
- Sieving, P.F.; Watson, A.D.; Rocklage, S.M. Preparation and characterization of paramagnetic polychelates and their protein conjugates. Bioconjug. Chem. 1990, 1, 65–71. [Google Scholar] [CrossRef]
- Geraldes, C.F.G.C.; Urbano, A.M.; Alpoim, M.C.; Sherry, A.D.; Kuan, K.T.; Rajagopalan, R.; Maton, F.; Muller, R.N. Preparation, physico-chemical characterization, and relaxometry studies of various gadolinium(III)-DTPA-bis(amide) derivatives as potential magnetic resonance contrast agents. Magn. Reson. Imaging 1995, 13, 401–420. [Google Scholar] [CrossRef]
- Liu, S.; Edwards, D.S. Bifunctional chelators for therapeutic lanthanide radiopharmaceuticals. Bioconjug. Chem. 2001, 12, 7–34. [Google Scholar] [CrossRef]
- Arano, Y.; Uezono, T.; Akizawa, H.; Ono, M.; Wakisaka, K.; Nakayama, M.; Sakahara, H.; Konishi, J.; Yokoyama, A. Reassessment of diethylenetriaminepentaacetic acid (DTPA) as a chelating agent for indium-111 labeling of polypeptides using a newly synthesized monoreactive DTPA derivative. J. Med. Chem. 1996, 39, 3451–3460. [Google Scholar] [CrossRef]
- Lee, J.W.; Lu, J.Y.; Low, P.S.; Fuchs, P.L. Synthesis and evaluation of taxol-folic acid conjugates as targeted antineoplastics. Bioorg. Med. Chem. 2002, 10, 2397–2414. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; Gansow, O.A.; Pippin, C.G.; Rogers, R.D.; Planalp, R.P. Preparation of the novel chelating agent N-(2-aminoethyl)-trans-1,2-diaminocyclohexane- N,N′,N′′-pentaacetic acid (H5CyDTPA), a preorganized analogue of diethylenetriaminepentaacetic acid (H5DTPA), and the structures of BiIII(CyDTPA)2- and BiIII(H2DTPA) complexes. Inorg. Chem. 1996, 35, 6343–6348. [Google Scholar] [CrossRef]
- Wu, C.; Kobayashi, H.; Sun, B.; Yoo, T.M.; Paik, C.H.; Gansow, O.A.; Carrasquillo, J.A.; Pastan, I.; Brechbiel, M.W. Stereochemical influence on the stability of radio-metal complexes in vivo. Synthesis and evaluation of the four stereoisomers of 2-(p-nitrobenzyl)-trans-CyDTPA. Bioorg. Med. Chem. 1997, 5, 1925–1934. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; Gansow, O.A. Backbone-substituted DTPA ligands for yttrium-90 radioimmunotherapy. Bioconjug. Chem. 1991, 2, 187–194. [Google Scholar] [CrossRef]
- Milenic, D.E.; Roselli, M.; Mirzadeh, S.; Pippin, C.G.; Gansow, O.A.; Colcher, D.; Brechbiel, M.W.; Schlom, J. In vivo evaluation of bismuth-labeled monoclonal antibody comparing DTPA-derived bifunctional chelates. Cancer Biother. Radiopharm. 2001, 16, 133–146. [Google Scholar] [CrossRef]
- Roselli, M.; Milenic, D.E.; Brechbiel, M.W.; Mirzadeh, S.; Pippin, C.G.; Gansow, O.A.; Colcher, D.; Schlom, J. In vivo comparison of CHX-DTPA ligand isomers in athymic mice bearing carcinoma xenografts. Cancer Biother. Radiopharm. 1999, 14, 209–220. [Google Scholar] [CrossRef]
- Milenic, D.E.; Garmestani, K.; Chappell, L.L.; Dadachova, E.; Yordanov, A.; Ma, D.; Schlom, J.; Brechbiel, M.W. In vivo comparison of macrocyclic and acyclic ligands for radiolabeling of monoclonal antibodies with 177Lu for radioimmunotherapeutic applications. Nucl. Med. Biol. 2002, 29, 431–442. [Google Scholar] [CrossRef]
- Mathias, C.J.; Sun, Y.Z.; Welch, M.J.; Connett, J.M.; Philpott, G.W.; Martell, A.E. N,N'-bis(2-hydroxybenzyl)-1-(4-bromoacetamidobenzyl)-1,2 -ethylenediamine-N,N'-diacetic acid: A new bifunctional chelate for radiolabeling antibodies. Bioconjug. Chem. 1990, 1, 204–211. [Google Scholar] [CrossRef]
- Schuhmacher, J.; Klivenyi, G.; Hull, W.E.; Matys, R.; Hauser, H.; Kalthoff, H.; Schmiegel, W.H.; Maier-Borst, W.; Matzku, S. A bifunctional HBED-derivative for labeling of antibodies with 67Ga, 111In and 59Fe. Comparative biodistribution with 111In-DPTA and 131I-labeled antibodies in mice bearing antibody internalizing and non-internalizing tumors. Int. J. Rad. Appl. Instrum. B 1992, 19, 809–824. [Google Scholar] [CrossRef]
- Eder, M.; Wangler, B.; Knackmuss, S.; LeGall, F.; Little, M.; Haberkorn, U.; Mier, W.; Eisenhut, M. Tetrafluorophenolate of HBED-CC: A versatile conjugation agent for 68Ga-labeled small recombinant antibodies. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1878–1886. [Google Scholar] [CrossRef]
- Eder, M.; Schafer, M.; Bauder-Wust, U.; Hull, W.E.; Wangler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Haberkorn, U.; Eder, M.; Eisenhut, M.; Zechmann, C.M. [68Ga]Gallium-labelled PSMA ligand as superior PET tracer for the diagnosis of prostate cancer: Comparison with 18F-FECH. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1085–1086. [Google Scholar] [CrossRef]
- Buchmann, I.; Henze, M.; Engelbrecht, S.; Eisenhut, M.; Runz, A.; Schafer, M.; Schilling, T.; Haufe, S.; Herrmann, T.; Haberkorn, U. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1617–1626. [Google Scholar] [CrossRef]
- Stetter, H.; Frank, W. Complex formation with tetraazacycloalkane-N,N′,N″,N′′′-tetraacetic acids as a function of ring size. Angew. Chem. Int. Ed. Engl. 1976, 15, 686–686. [Google Scholar]
- Aime, S.; Cabella, C.; Colombatto, S.; Geninatti Crich, S.; Gianolio, E.; Maggioni, F. Insights into the use of paramagnetic Gd(III) complexes in MR-molecular imaging investigations. J. Magn. Reson. Imaging 2002, 16, 394–406. [Google Scholar] [CrossRef]
- Caravan, P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem. Soc. Rev. 2006, 35, 512–523. [Google Scholar] [CrossRef]
- Heppeler, A.; Froidevaux, S.; Eberle, A.N.; Maecke, H.R. Receptor targeting for tumor localisation and therapy with radiopeptides. Curr. Med. Chem. 2000, 7, 971–994. [Google Scholar] [CrossRef]
- Maecke, H.R.; Hofmann, M.; Haberkorn, U. 68Ga-labeled peptides in tumor imaging. J. Nucl. Med. 2005, 46 (Suppl. 1), 172S–178S. [Google Scholar]
- Pandya, S.; Yu, J.; Parker, D. Engineering emissive europium and terbium complexes for molecular imaging and sensing. Dalton Trans. 2006, 2757–2766. [Google Scholar] [CrossRef]
- Reubi, J.C.; Macke, H.R.; Krenning, E.P. Candidates for peptide receptor radiotherapy today and in the future. J. Nucl. Med. 2005, 46 (Suppl. 1), 67S–75S. [Google Scholar]
- Tanaka, K.; Fukase, K. PET (positron emission tomography) imaging of biomolecules using metal-DOTA complexes: A new collaborative challenge by chemists, biologists, and physicians for future diagnostics and exploration of in vivo dynamics. Org. Biomol. Chem. 2008, 6, 815–828. [Google Scholar] [CrossRef]
- Schottelius, M.; Schwaiger, M.; Wester, H.J. Rapid and high-yield solution-phase synthesis of DOTA-Tyr3-octreotide and DOTA-Tyr3-octreotate using unprotected DOTA. Tetrahedron Lett. 2003, 44, 2393–2396. [Google Scholar] [CrossRef]
- Mier, W.; Hoffend, J.; Kramer, S.; Schuhmacher, J.; Hull, W.E.; Eisenhut, M.; Haberkorn, U. Conjugation of DOTA using isolated phenolic active esters: The labeling and biodistribution of albumin as blood pool marker. Bioconjug. Chem. 2005, 16, 237–240. [Google Scholar] [CrossRef]
- Albericio, F.; Carpino, L.A. Coupling reagents and activation. Methods Enzymol. 1997, 289, 104–126. [Google Scholar] [CrossRef]
- Mier, W.; Graham, K.A.N.; Wang, Q.; Krämer, S.; Hoffend, J.; Eisenhut, M.; Haberkorn, U. Synthesis of peptide conjugated chelator oligomers for endoradiotherapy and MRT imaging. Tetrahedron Lett. 2004, 45, 5453–5455. [Google Scholar] [CrossRef]
- Knor, S.; Modlinger, A.; Poethko, T.; Schottelius, M.; Wester, H.J.; Kessler, H. Synthesis of novel 1,4,7,10-tetraazacyclodecane-1,4,7,10-tetraacetic acid (DOTA) derivatives for chemoselective attachment to unprotected polyfunctionalized compounds. Chemistry 2007, 13, 6082–6090. [Google Scholar] [CrossRef]
- Wängler, B.; Beck, C.; Wagner-Utermann, U.; Schirrmacher, E.; Bauer, C.; Rösch, F.; Schirrmacher, R.; Eisenhut, M. Application of tris-allyl-DOTA in the preparation of DOTA-peptide conjugates. Tetrahedron Lett. 2006, 47, 5985–5988. [Google Scholar] [CrossRef]
- Jaakkola, L.; Ylikoski, A.; Hovinen, J. Simple synthesis of a building block for solid-phase labeling of oligonucleotides with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). Bioconjug. Chem. 2006, 17, 1105–1107. [Google Scholar] [CrossRef]
- Anelli, P.L.; Lattuada, L.; Gabellini, M.; Recanati, P. DOTA tris(phenylmethyl) ester: A new useful synthon for the synthesis of DOTA monoamides containing acid-labile bonds. Bioconjug. Chem. 2001, 12, 1081–1084. [Google Scholar] [CrossRef]
- Jamous, M.; Haberkorn, U.; Mier, W. DOTA-tris(OPp ester) as a bifunctional prochelator for the preparation of DOTA–peptide conjugates. Tetrahedron Lett. 2012, 53, 6810–6814. [Google Scholar] [CrossRef]
- Mukai, T.; Namba, S.; Arano, Y.; Ono, M.; Fujioka, Y.; Uehara, T.; Ogawa, K.; Konishi, J.; Saji, H. Synthesis and evaluation of a monoreactive DOTA derivative for indium-111-based residualizing label to estimate protein pharmacokinetics. J. Pharm. Pharmacol. 2002, 54, 1073–1081. [Google Scholar] [CrossRef]
- Li, C.; Winnard, P., Jr.; Bhujwalla, Z.M. Facile synthesis of 1-(acetic acid)-4,7,10-tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraaza-cyclododecane: A reactive precursor chelating agent. Tetrahedron Lett. 2009, 50, 2929–2931. [Google Scholar] [CrossRef]
- Peterson, J.J.; Pak, R.H.; Meares, C.F. Total solid-phase synthesis of 1,4,7,10-tetraazacyclododecane-N,N', N'',N'''-tetraacetic acid-functionalized peptides for radioimmunotherapy. Bioconjug. Chem. 1999, 10, 316–320. [Google Scholar] [CrossRef]
- Eisenwiener, K.P.; Powell, P.; Macke, H.R. A convenient synthesis of novel bifunctional prochelators for coupling to bioactive peptides for radiometal labelling. Bioorg. Med. Chem. Lett. 2000, 10, 2133–2135. [Google Scholar] [CrossRef]
- Heppeler, A.; Froidevaux, S.; Mäcke, H.R.; Jermann, E.; Béhé, M.; Powell, P.; Hennig, M. Radiometal-labelled macrocyclic chelator-derivatised somatostatin analogue with superb tumour-targeting properties and potential for receptor-mediated internal radiotherapy. Chem. Eur. J. 1999, 5, 1974–1981. [Google Scholar] [CrossRef]
- Kruper, W.J.; Rudolf, P.R.; Langhoff, C.A. Unexpected selectivity in the alkylation of polyazamacrocycles. J. Org. Chem. 1993, 58, 3869–3876. [Google Scholar] [CrossRef]
- Moi, M.K.; Meares, C.F.; Denardo, S.J. The peptide way to macrocyclic bifunctional chelating agents: Synthesis of 2-(p-nitrobenzyl)-1,4,7,10-tetraazacyclododecane-N,N',N",N'''-tetraacetic acid and study of its yttrium(III) complex. J. Am. Chem. Soc. 1988, 110, 6266–6267. [Google Scholar] [CrossRef]
- McMurry, T.J.; Brechbiel, M.; Kumar, K.; Gansow, O.A. Convenient synthesis of bifunctional tetraaza macrocycles. Bioconjug. Chem. 1992, 3, 108–117. [Google Scholar] [CrossRef]
- Wängler, C.; Schäfer, M.; Schirrmacher, R.; Bartenstein, P.; Wängler, B. DOTA derivatives for site-specific biomolecule-modification via click chemistry: Synthesis and comparison of reaction characteristics. Bioorg. Med. Chem. 2011, 19, 3864–3874. [Google Scholar] [CrossRef]
- Barge, A.; Tei, L.; Upadhyaya, D.; Fedeli, F.; Beltrami, L.; Stefania, R.; Aime, S.; Cravotto, G. Bifunctional ligands based on the DOTA-monoamide cage. Org. Biomol. Chem. 2008, 6, 1176–1184. [Google Scholar] [CrossRef]
- Moran, J.K.; Greiner, D.P.; Meares, C.F. Improved synthesis of 6-[p-(bromoacetamido)benzyl]-1,4,8,11-tetraazacyclotetradecane- N,N',N",N"-tetraacetic acid and development of a thin-layer assay for thiol-reactive bifunctional chelating agents. Bioconjug. Chem. 1995, 6, 296–301. [Google Scholar] [CrossRef]
- Fichna, J.; Janecka, A. Synthesis of target-specific radiolabeled peptides for diagnostic imaging. Bioconjug. Chem. 2002, 14, 3–17. [Google Scholar] [CrossRef]
- Prasanphanich, A.F.; Nanda, P.K.; Rold, T.L.; Ma, L.; Lewis, M.R.; Garrison, J.C.; Hoffman, T.J.; Sieckman, G.L.; Figueroa, S.D.; Smith, C.J. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc. Natl. Acad. Sci. USA 2007, 104, 12462–12467. [Google Scholar] [CrossRef]
- Sprague, J.E.; Peng, Y.; Sun, X.; Weisman, G.R.; Wong, E.H.; Achilefu, S.; Anderson, C.J. Preparation and biological evaluation of copper-64-labeled Tyr3-octreotate using a cross-bridged macrocyclic chelator. Clin. Cancer Res. 2004, 10, 8674–8682. [Google Scholar] [CrossRef]
- Lewis, E.A.; Boyle, R.W.; Archibald, S.J. Ultrastable complexes for in vivo use: A bifunctional chelator incorporating a cross-bridged macrocycle. Chem. Commun. 2004, 2212–2213. [Google Scholar]
- Boswell, C.A.; Regino, C.A.; Baidoo, K.E.; Wong, K.J.; Bumb, A.; Xu, H.; Milenic, D.E.; Kelley, J.A.; Lai, C.C.; Brechbiel, M.W. Synthesis of a cross-bridged cyclam derivative for peptide conjugation and 64Cu radiolabeling. Bioconjug. Chem. 2008, 19, 1476–1484. [Google Scholar] [CrossRef]
- Dumont, R.A.; Deininger, F.; Haubner, R.; Maecke, H.R.; Weber, W.A.; Fani, M. Novel 64Cu- and 68Ga-labeled RGD conjugates show improved PET imaging of αvβ3 integrin expression and facile radiosynthesis. J. Nucl. Med. 2011, 52, 1276–1284. [Google Scholar] [CrossRef]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef]
- Cooper, M.S.; Ma, M.T.; Sunassee, K.; Shaw, K.P.; Williams, J.D.; Paul, R.L.; Donnelly, P.S.; Blower, P.J. Comparison of 64Cu-complexing bifunctional chelators for radioimmunoconjugation: Labeling efficiency, specific activity, and in vitro/in vivo stability. Bioconjug. Chem. 2012, 23, 1029–1039. [Google Scholar] [CrossRef]
- Guerin, B.; Ait-Mohand, S.; Tremblay, M.C.; Dumulon-Perreault, V.; Fournier, P.; Benard, F. Total solid-phase synthesis of NOTA-functionalized peptides for PET imaging. Org. Lett. 2010, 12, 280–283. [Google Scholar] [CrossRef]
- Fournier, P.; Dumulon-Perreault, V.; Ait-Mohand, S.; Langlois, R.; Benard, F.; Lecomte, R.; Guerin, B. Comparative study of 64Cu/NOTA-[D-Tyr6,βAla11,Thi13,Nle14]BBN(6-14) monomer and dimers for prostate cancer PET imaging. EJNMMI Res. 2012, 2. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; McMurry, T.J.; Gansow, O.A. A direct synthesis of a bifunctional chelating agent for radiolabeling proteins. Tetrahedron Lett. 1993, 34, 3691–3694. [Google Scholar] [CrossRef]
- Studer, M.; Meares, C.F. Synthesis of novel 1,4,7-triazacyclononane-N,N',N"-triacetic acid derivatives suitable for protein labeling. Bioconjug. Chem. 1992, 3, 337–341. [Google Scholar] [CrossRef]
- McMurry, T.J.; Brechbiel, M.; Wu, C.; Gansow, O.A. Synthesis of 2-(p-thiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid: Application of the 4-methoxy-2,3,6-trimethylbenzenesulfonamide protecting group in the synthesis of macrocyclic polyamines. Bioconjug. Chem. 1993, 4, 236–245. [Google Scholar] [CrossRef]
- Eisenwiener, K.P.; Prata, M.I.; Buschmann, I.; Zhang, H.W.; Santos, A.C.; Wenger, S.; Reubi, J.C.; Macke, H.R. NODAGATOC, a new chelator-coupled somatostatin analogue labeled with [67/68Ga] and [111In] for SPECT, PET, and targeted therapeutic applications of somatostatin receptor (hsst2) expressing tumors. Bioconjug. Chem. 2002, 13, 530–541. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- De Leon-Rodriguez, L.M.; Kovacs, Z.; Dieckmann, G.R.; Sherry, A.D. Solid-phase synthesis of DOTA-peptides. Chemistry 2004, 10, 1149–1155. [Google Scholar] [CrossRef]
- Gallazzi, F.; Wang, Y.; Jia, F.; Shenoy, N.; Landon, L.A.; Hannink, M.; Lever, S.Z.; Lewis, M.R. Synthesis of radiometal-labeled and fluorescent cell-permeating peptide-PNA conjugates for targeting the bcl-2 proto-oncogene. Bioconjug. Chem. 2003, 14, 1083–1095. [Google Scholar] [CrossRef]
- Zoller, F.; Schwaebel, T.; Markert, A.; Haberkorn, U.; Mier, W. Engineering and functionalization of the disulfide-constrained miniprotein min-23 as a scaffold for diagnostic application. Chem. Med. Chem. 2012, 7, 237–247. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jamous, M.; Haberkorn, U.; Mier, W. Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases. Molecules 2013, 18, 3379-3409. https://doi.org/10.3390/molecules18033379
Jamous M, Haberkorn U, Mier W. Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases. Molecules. 2013; 18(3):3379-3409. https://doi.org/10.3390/molecules18033379
Chicago/Turabian StyleJamous, Mazen, Uwe Haberkorn, and Walter Mier. 2013. "Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases" Molecules 18, no. 3: 3379-3409. https://doi.org/10.3390/molecules18033379
APA StyleJamous, M., Haberkorn, U., & Mier, W. (2013). Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases. Molecules, 18(3), 3379-3409. https://doi.org/10.3390/molecules18033379