Singlet Oxygen in Antimicrobial Photodynamic Therapy: Photosensitizer-Dependent Production and Decay in E. coli



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
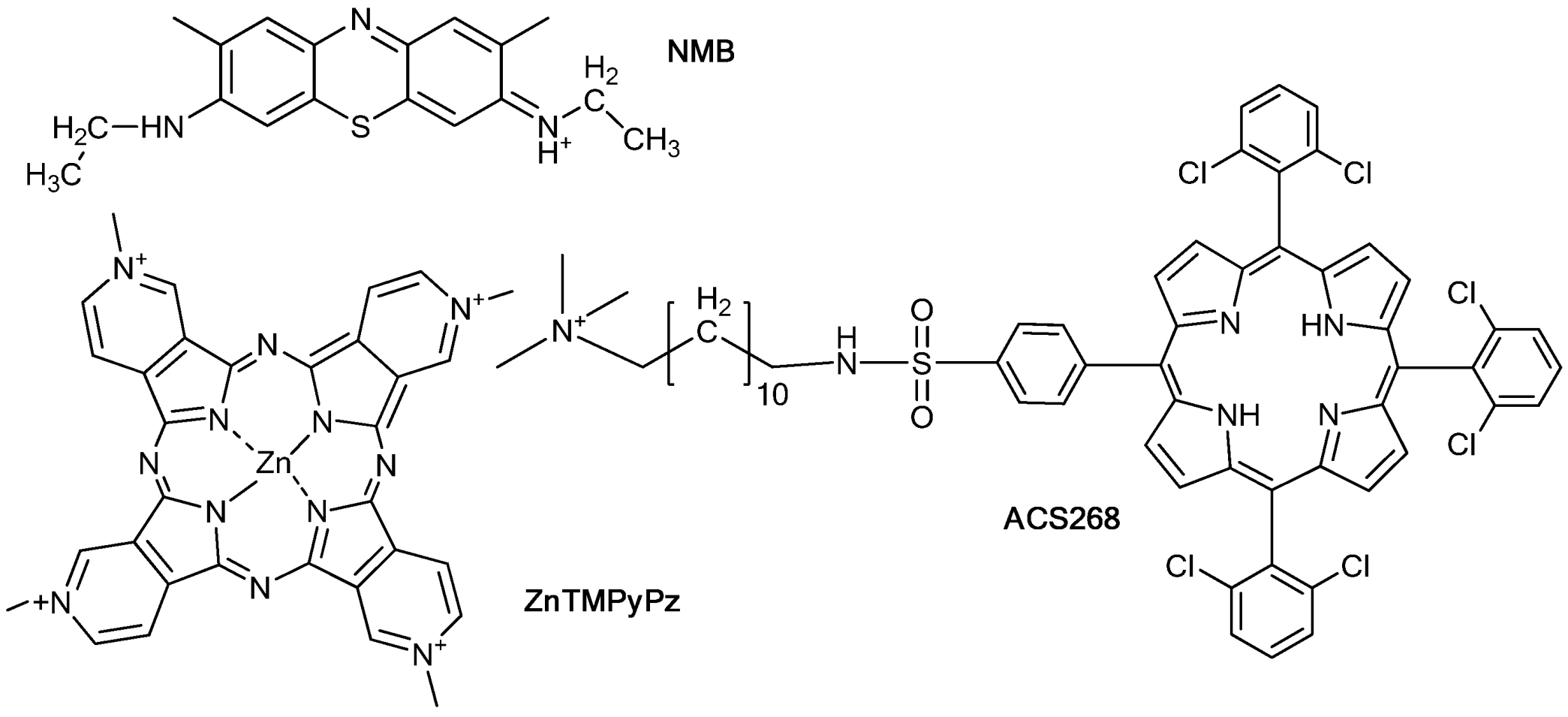
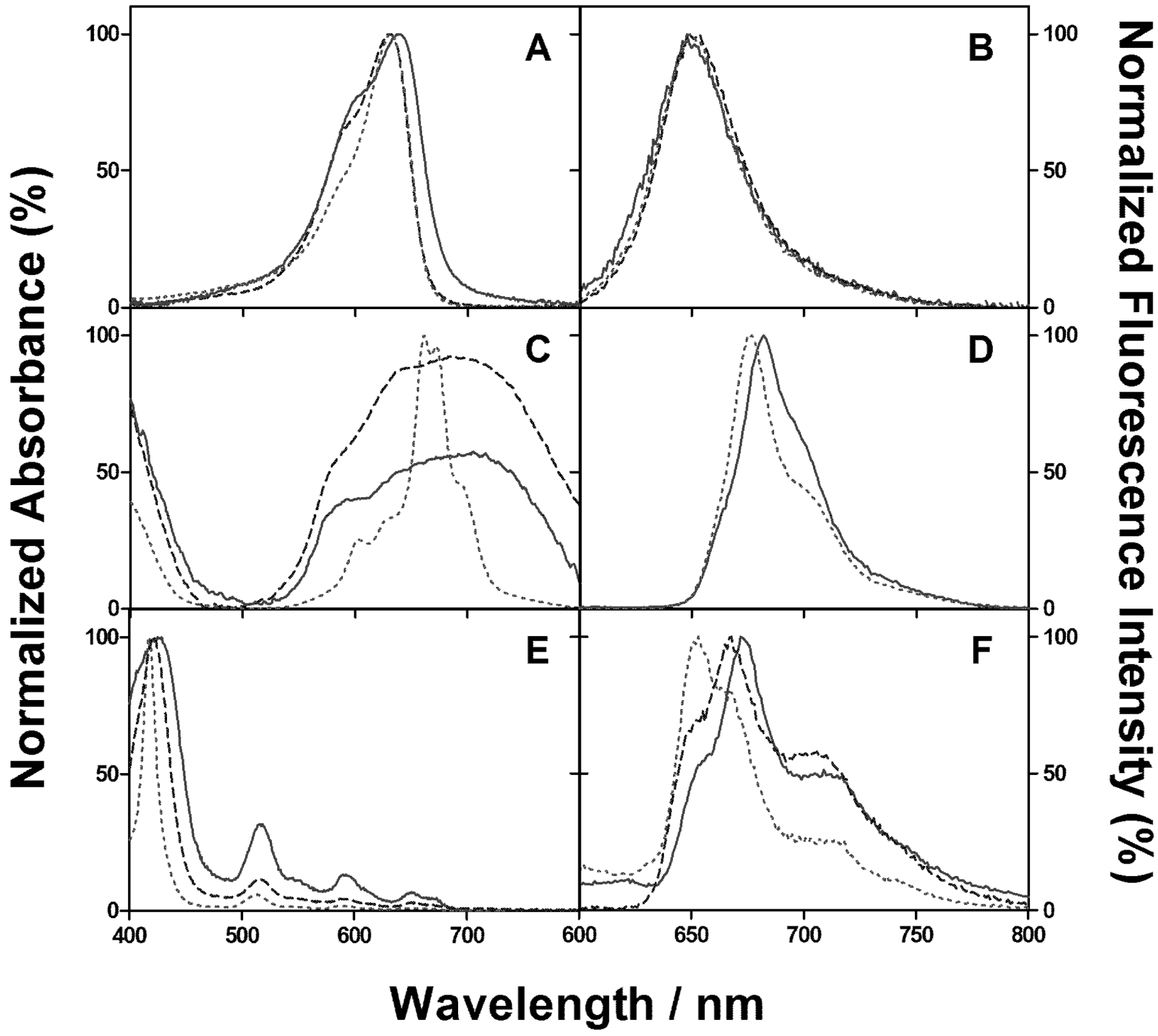
2. Results and Discussion
2.1. PSs Binding to E. coli
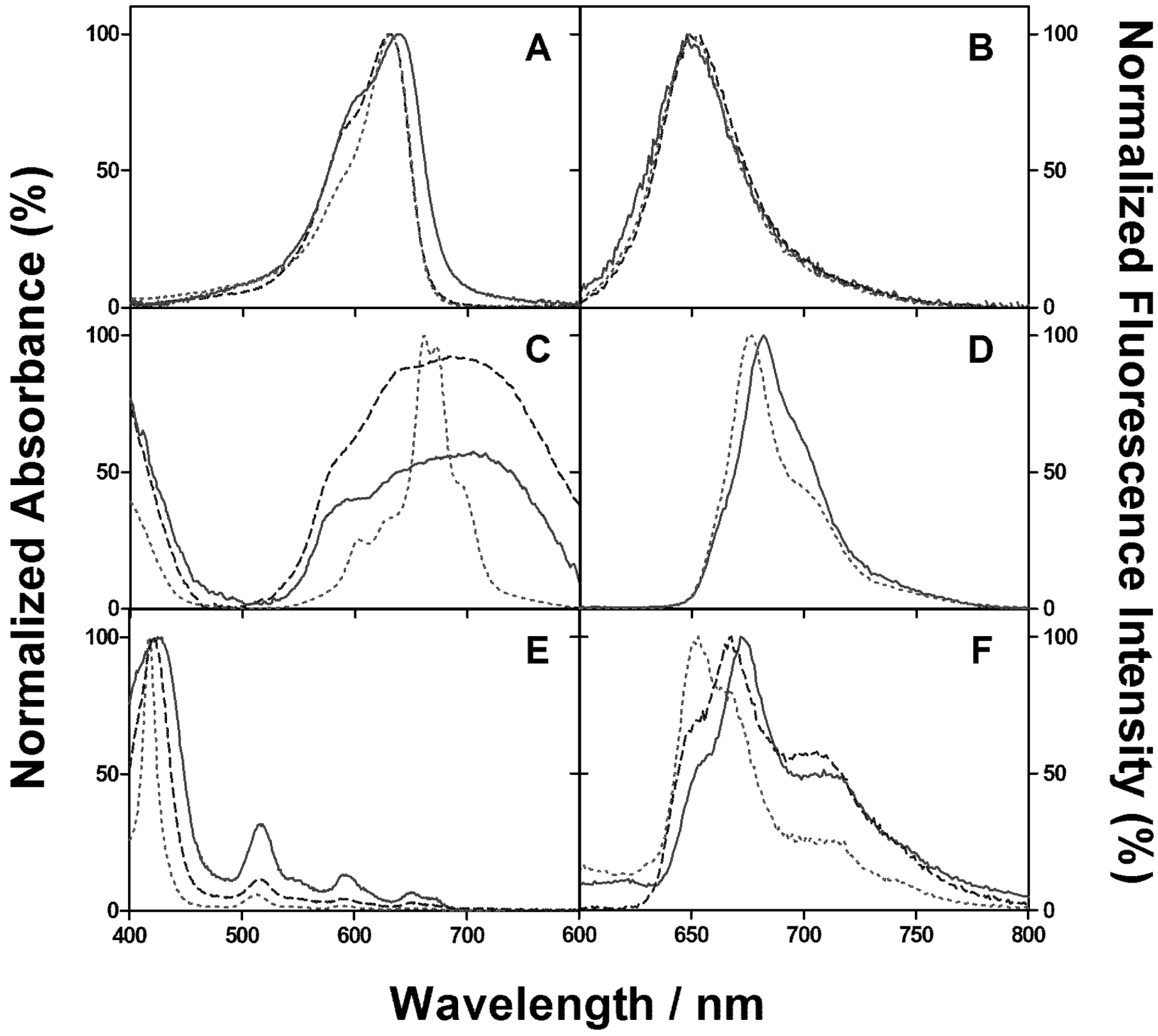
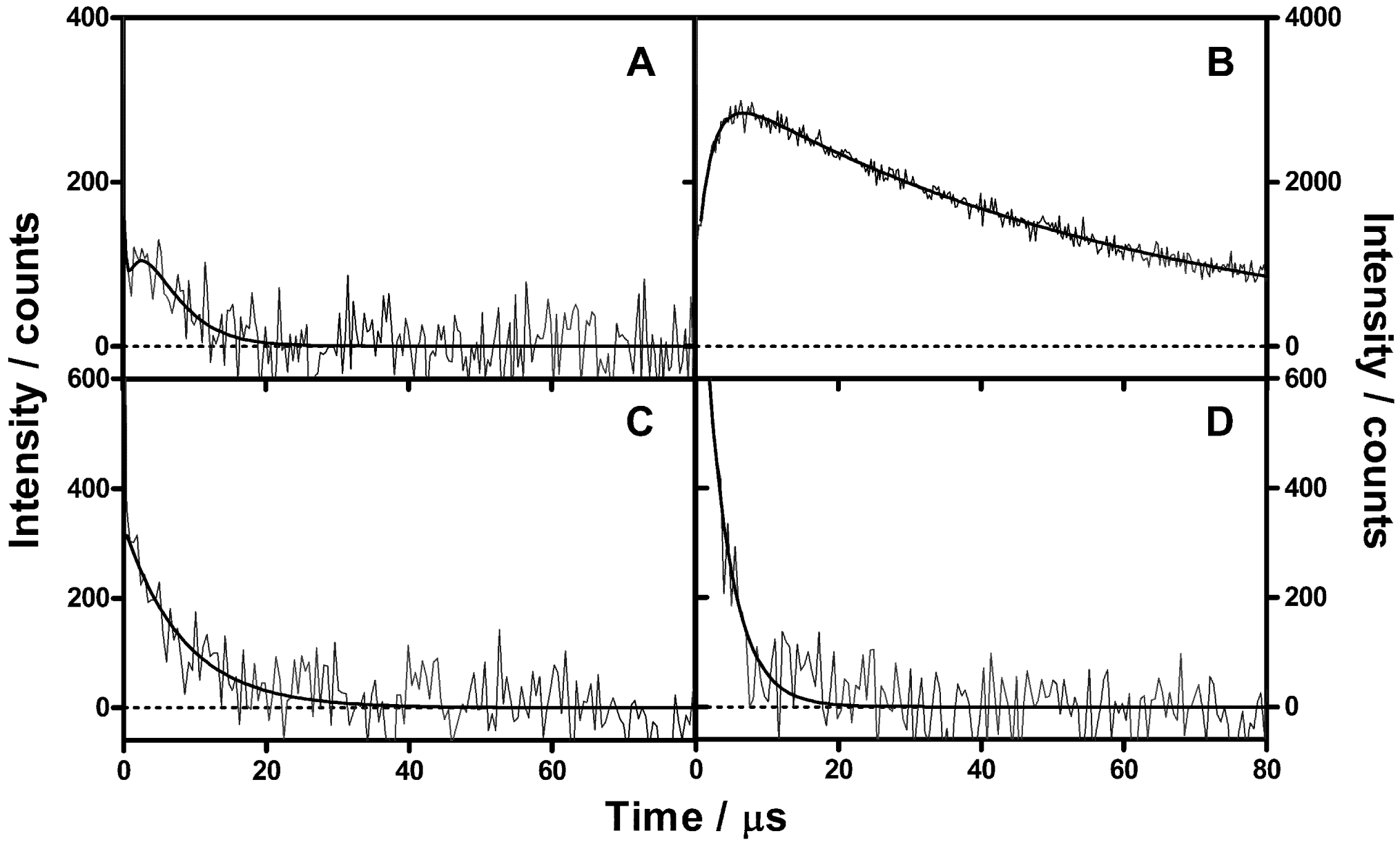
2.2. Singlet Oxygen Kinetics in E. coli Cells with NMB as PS
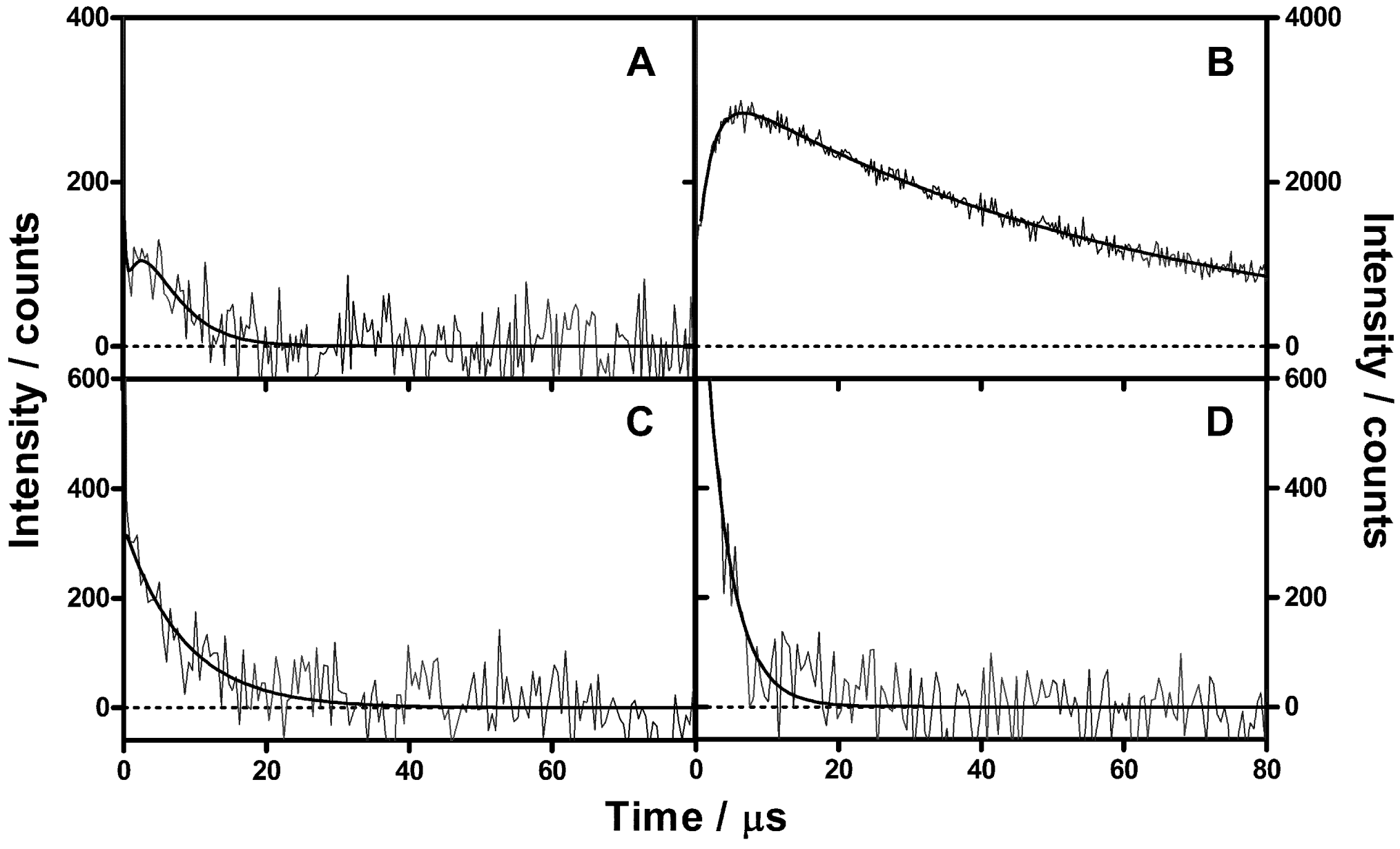
2.3. Singlet Oxygen Kinetics in E. coli Cells with ZnTMPyPz as PS
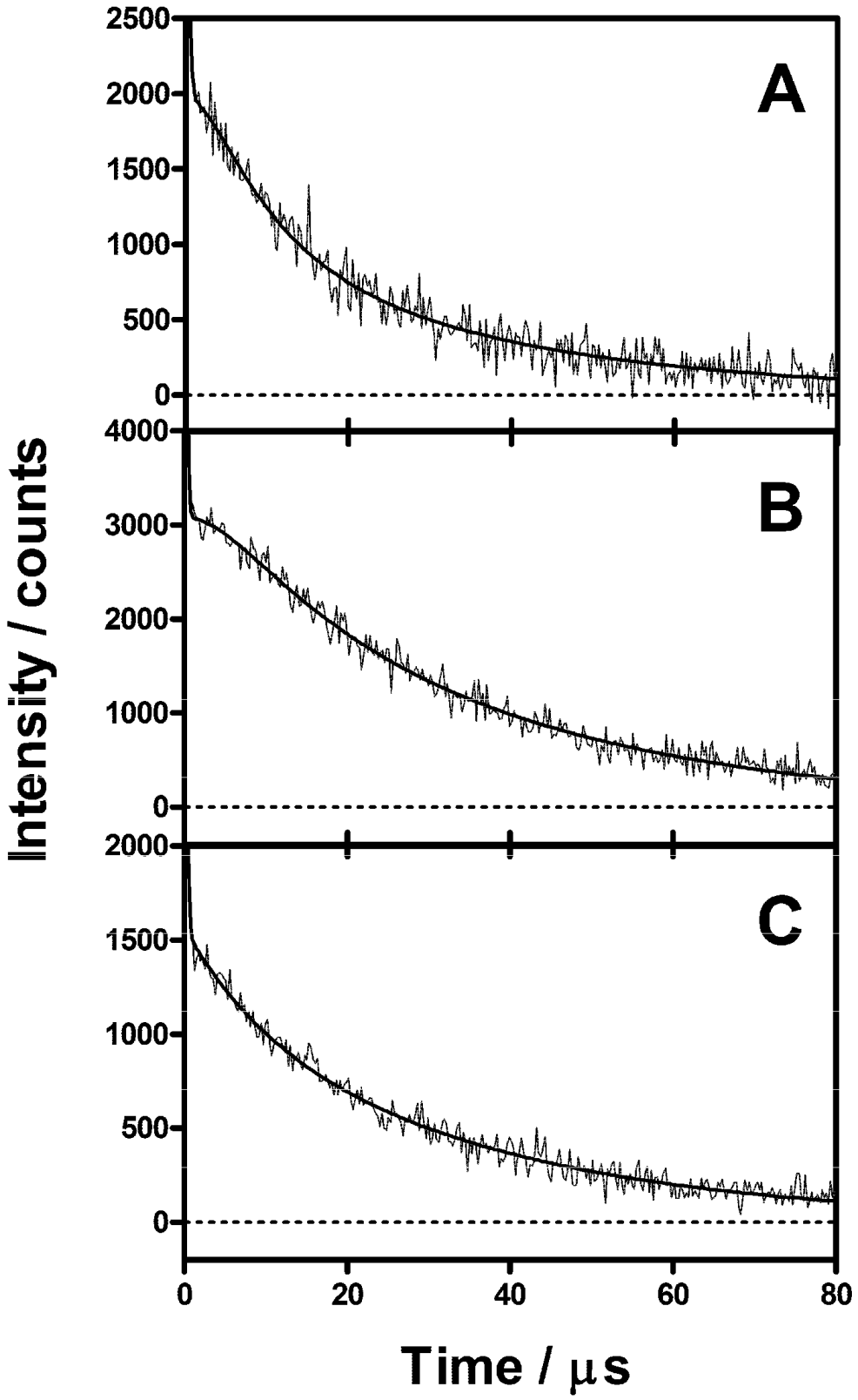
2.4. Singlet Oxygen Kinetics in E. coli Cells with ACS268 as PS
2.5. Discussion



3. Experimental
3.1. Chemicals
3.2. Bacterial Growth
3.3. PS Binding to E. coli
3.4. Spheroplasts Formation
3.5. General Spectroscopic Measurements
3.6. Time-Resolved Singlet Oxygen Measurements
3.7. Measurements in Bacterial Suspensions
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Jori, G.; Fabris, C.; Soncin, M.; Ferro, S.; Coppellotti, O.; Dei, D.; Fantetti, L.; Chiti, G.; Roncucci, G. Photodynamic therapy in the treatment of microbial infections: Basic principles and perspective applications. Lasers Surg. Med. 2006, 38, 468–481. [Google Scholar] [CrossRef]
- Dai, T.; Huang, Y.Y.; Hamblin, M.R. Photodynamic therapy for localized infections—State of the art. Photodiagnosis Photodyn. Ther. 2009, 6, 170–188. [Google Scholar] [CrossRef]
- Wainwright, M. Photoantimicrobials—So what’s stopping us? Photodiagnosis Photodyn. Ther. 2009, 6, 167–169. [Google Scholar] [CrossRef]
- Schweitzer, C.; Schmidt, R. Physical mechanisms of generation and deactivation of singlet oxygen. Chem. Rev. 2003, 103, 1685–1757. [Google Scholar] [CrossRef]
- Garland, M.J.; Cassidy, C.M.; Woolfson, D.; Donnelly, R.F. Designing photosensitizers for photodynamic therapy: Strategies, challenges and promising developments. Future Med. Chem. 2009, 1, 667–691. [Google Scholar] [CrossRef]
- Wainwright, M. Photodynamic antimicrobial chemotherapy (PACT). J. Antimicrob. Chemother. 1998, 42, 13–28. [Google Scholar] [CrossRef]
- Bertoloni, G.; Rossi, F.; Valduga, G.; Jori, G.; Ali, H.; Vanlier, J.E. Photosensitizing activity of water-soluble and lipid-soluble phthalocyanines on prokaryotic and eukaryotic microbial-cells. Microbios 1992, 71, 33–46. [Google Scholar]
- Nitzan, Y.; Gutterman, M.; Malik, Z.; Ehrenberg, B. Inactivation of gram-negative bacteria by photosensitized porphyrins. Photochem. Photobiol. 1992, 55, 89–96. [Google Scholar] [CrossRef]
- Romanova, N.A.; Brovko, L.Y.; Moore, L.; Pometun, E.; Savitsky, A.P.; Ugarova, N.N.; Griffiths, M.W. Assessment of photodynamic destruction of Escherichia coli O157:H7 and Listeria monocytogenes by using ATP bioluminescence. Appl. Environ. Microbiol. 2003, 69, 6393–6398. [Google Scholar] [CrossRef]
- Merchat, M.; Bertolini, G.; Giacomini, P.; Villanueva, A.; Jori, G. Meso-substituted cationic porphyrins as efficient photosensitizers of gram-positive and gram-negative bacteria. J. Photochem. Photobiol. B Biol. 1996, 32, 153–157. [Google Scholar] [CrossRef]
- Minnock, A.; Vernon, D.I.; Schofield, J.; Griffiths, J.; Parish, J.H.; Brown, S.B. Photoinactivation of bacteria. Use of a cationic water-soluble zinc phthalocyanine to photoinactivate both gram-negative and gram-positive bacteria. J. Photochem. Photobiol. B Biol. 1996, 32, 159–164. [Google Scholar]
- Wainwright, M.; Phoenix, D.A.; Marland, J.; Wareing, D.R.A.; Bolton, F.J. A study of photobactericidal activity in the phenothiazinium series. FEMS Immunol. Med. Microbiol. 1997, 19, 75–80. [Google Scholar] [CrossRef]
- Ergaieg, K.; Chevanne, M.; Cillard, J.; Seux, R. Involvement of both type I and type II mechanisms in gram-positive and gram-negative bacteria photosensitization by a meso-substituted cationic porphyrin. Sol. Energy 2008, 82, 1107–1117. [Google Scholar] [CrossRef]
- Ragàs, X.; Agut, M.; Nonell, S. Singlet oxygen in E. coli: New insights for antimicrobial photodynamic therapy. Free Radic. Biol. Med. 2010, 49, 770–776. [Google Scholar]
- Dupouy, E.A.; Lazzeri, D.; Durantini, E.N. Photodynamic activity of cationic and non-charged Zn(II) tetrapyridinoporphyrazine derivatives: Biological consequences in human erythrocytes and Escherichia coli. Photochem. Photobiol. Sci. 2004, 3, 992–998. [Google Scholar] [CrossRef]
- Peleg, A.Y. Optimizing therapy for Acinetobacter baumannii. Semin. Respir. Crit. Care Med. 2007, 28, 662–671. [Google Scholar] [CrossRef]
- Phoenix, D.A.; Sayed, Z.; Hussain, S.; Harris, F.; Wainwright, M. The phototoxicity of phenothiazinium derivatives against Escherichia coli and Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 2003, 39, 17–22. [Google Scholar] [CrossRef]
- Ragàs, X.; Dai, T.; Tegos, G.P.; Agut, M.; Nonell, S.; Hamblin, M.R. Photodynamic inactivation of Acinetobacter baumannii using phenothiazinium dyes: In vitro and in vivo studies. Lasers Surg. Med. 2010, 42, 384–390. [Google Scholar] [CrossRef]
- Snyder, J.W.; Skovsen, E.; Lambert, J.D.C.; Ogilby, P.R. Subcellular, time-resolved studies of singlet oxygen in single cells. J. Am. Chem. Soc. 2005, 127, 14558–14559. [Google Scholar]
- Martí, C.; Nonell, S.; Nicolau, M.; Torres, T. Photophysical properties of neutral and cationic tetrapyridinoporphyrazines. Photochem. Photobiol. 2000, 71, 53–59. [Google Scholar] [CrossRef]
- Redmond, R.W.; Gamlin, J.N. A compilation of singlet oxygen yields from biologically relevant molecules. Photochem. Photobiol. 1999, 70, 391–475. [Google Scholar]
- Kuimova, M.K.; Botchway, S.W.; Parker, A.W.; Balaz, M.; Collins, H.A.; Anderson, H.L.; Suhling, K.; Ogilby, P.R. Imaging intracellular viscosity of a single cell during photoinduced cell death. Nat. Chem. 2009, 1, 69–73. [Google Scholar]
- Schlothauer, J.; Hackbarth, S.; Roder, B. A new benchmark for time-resolved detection of singlet oxygen luminescence—Revealing the evolution of lifetime in living cells with low dose illumination. Laser Phys. Lett. 2009, 6, 216–221. [Google Scholar] [CrossRef]
- Harris, F.; Chatfield, L.K.; Phoenix, D.A. Phenothiazinium based photosensitisers—Photodynamic agents with a multiplicity of cellular targets and clinical applications. Curr. Drug Targets 2005, 6, 615–627. [Google Scholar] [CrossRef]
- Caminos, D.A.; Spesia, M.B.; Durantini, E.N. Photodynamic inactivation of Escherichia coli by novel meso-substituted porphyrins by 4-(3-N,N,N-trimethylammoniumpropoxy)phenyl and 4-(trifluoromethyl)phenyl groups. Photochem. Photobiol. Sci. 2006, 5, 56–65. [Google Scholar] [CrossRef]
- Hamblin, M.R.; O’Donnell, D.A.; Murthy, N.; Rajagopalan, K.; Michaud, N.; Sherwood, M.E.; Hasan, T. Polycationic photosensitizer conjugates: Effects of chain length and gram classification on the photodynamic inactivation of bacteria. J. Antimicrob. Chemother. 2002, 49, 941–951. [Google Scholar] [CrossRef]
- Phoenix, D.A.; Harris, F. Phenothiazinium-based photosensitizers: Antibacterials of the future? Trends Mol. Med. 2003, 9, 283–285. [Google Scholar] [CrossRef]
- Usacheva, M.N.; Teichert, M.C.; Biel, M.A. The role of the methylene blue and toluidine blue monomers and dimers in the photoinactivation of bacteria. J. Photochem. Photobiol. B Biol. 2003, 71, 87–98. [Google Scholar] [CrossRef]
- Chirvony, V.S. Primary photoprocesses in cationic 5,10,15,20-meso-tetrakis(4-N-methylpyridiniumyl)porphyrin and its transition metal complexes bound with nucleic acids. J. Porphyrins Phthalocyanines 2003, 7, 766–774. [Google Scholar] [CrossRef]
- Rodgers, M.A.J. Solvent-induced deactivation of singlet oxygen—Additivity relationships in non-aromatic solvents. J. Am. Chem. Soc. 1983, 105, 6201–6205. [Google Scholar] [CrossRef]
- Borissevitch, I.E.; Tominaga, T.T.; Schmitt, C.C. Photophysical studies on the interaction of two water-soluble porphyrins with bovine serum albumin. Effects upon the porphyrin triplet state characteristics. J. Photochem. Photobiol. A Chem. 1998, 114, 201–207. [Google Scholar] [CrossRef]
- Usacheva, M.N.; Teichert, M.C.; Biel, M.A. The interaction of lipopolysaccharides with phenothiazine dyes. Lasers Surg. Med. 2003, 33, 311–319. [Google Scholar] [CrossRef]
- Usacheva, M.N.; Teichert, M.C.; Sievert, C.E.; Biel, M.A. Effect of Ca2+ on the photobactericidal efficacy of methylene blue and toluidine blue against gram-negative bacteria and the dye affinity for lipopolysaccharides. Lasers Surg. Med. 2006, 38, 946–954. [Google Scholar] [CrossRef]
- Spesia, M.B.; Caminos, D.A.; Pons, P.; Durantini, E.N. Mechanistic insight of the photodynamic inactivation of Escherichia coli by a tetracationic zinc(II) phthalocyanine derivative. Photodiagn. Photodyn. Ther. 2009, 6, 52–61. [Google Scholar] [CrossRef]
- De Paoli, V.M.; de Paoli, S.H.; Borissevitch, L.E.; Tedesco, A.C. Fluorescence lifetime and quantum yield of TMPyPH2 associated with micelles and DNA. J. Alloys Compd. 2002, 344, 27–31. [Google Scholar] [CrossRef]
- Lee, S.; Lee, Y.A.; Lee, H.M.; Lee, J.Y.; Kim, D.H.; Kim, S.K. Rotation of periphery methylpyridine of meso-tetrakis(n-N-methylpyridiniumyl)porphyrin (n = 2, 3, 4) and its selective binding to native and synthetic DNAs. Biophys. J. 2002, 83, 371–381. [Google Scholar] [CrossRef]
- Lee, P.C.; Rodgers, M.A.J. Singlet molecular-oxygen in micellar systems: Distribution equilibria between hydrophobic and hydrophilic compartments. J. Phys. Chem. 1983, 87, 4894–4898. [Google Scholar] [CrossRef]
- Demidova, T.N.; Hamblin, M.R. Photodynamic therapy targeted to pathogens. Int. J. Immunopathol. Pharmacol. 2004, 17, 245–254. [Google Scholar]
- Nitzan, Y.; Balzam-Sudakevitz, A.; Ashkenazi, H. Eradication of Acinetobacter baumannii by photosensitized agents in vitro. J. Photochem. Photobiol. B Biol. 1998, 42, 211–218. [Google Scholar] [CrossRef]
- Wilson, M.; Pratten, J. Lethal photosensitization of Staphylococcus aureus in-vitro—Effect of growth-phase, serum, and preirradation time. Lasers Surg. Med. 1995, 16, 272–276. [Google Scholar] [CrossRef]
- Demidova, T.N.; Hamblin, M.R. Effect of cell-photo sensitizer binding and cell density on microbial photoinactivation. Antimicrob. Agents Chemother. 2005, 49, 2329–2335. [Google Scholar] [CrossRef]
- Merchat, M.; Spikes, J.D.; Bertoloni, G.; Jori, G. Studies on the mechanism of bacteria photosensitization by meso-substituted cationic porphyrins. J. Photochem. Photobiol. B Biol. 1996, 35, 149–157. [Google Scholar] [CrossRef]
- Jimenez-Banzo, A.; Ragàs, X.; Kapusta, P.; Nonell, S. Time-resolved methods in biophysics. 7. Photon counting vs. analog time-resolved singlet oxygen phosphorescence detection. Photochem. Photobiol. Sci. 2008, 7, 1003–1010. [Google Scholar]
- Maisch, T.; Baier, J.; Franz, B.; Maier, M.; Landthaler, M.; Szeimies, R.M.; Bäumler, W. The role of singlet oxygen and oxygen concentration in photodynamic inactivation of bacteria. Proc. Natl. Acad. Sci. USA 2007, 104, 7223–7228. [Google Scholar]
- Sample Availability: Samples of the compound ACS268 is available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ragàs, X.; He, X.; Agut, M.; Roxo-Rosa, M.; Gonsalves, A.R.; Serra, A.C.; Nonell, S. Singlet Oxygen in Antimicrobial Photodynamic Therapy: Photosensitizer-Dependent Production and Decay in E. coli. Molecules 2013, 18, 2712-2725. https://doi.org/10.3390/molecules18032712
Ragàs X, He X, Agut M, Roxo-Rosa M, Gonsalves AR, Serra AC, Nonell S. Singlet Oxygen in Antimicrobial Photodynamic Therapy: Photosensitizer-Dependent Production and Decay in E. coli. Molecules. 2013; 18(3):2712-2725. https://doi.org/10.3390/molecules18032712
Chicago/Turabian StyleRagàs, Xavier, Xin He, Montserrat Agut, Mónica Roxo-Rosa, António Rocha Gonsalves, Arménio C. Serra, and Santi Nonell. 2013. "Singlet Oxygen in Antimicrobial Photodynamic Therapy: Photosensitizer-Dependent Production and Decay in E. coli" Molecules 18, no. 3: 2712-2725. https://doi.org/10.3390/molecules18032712
APA StyleRagàs, X., He, X., Agut, M., Roxo-Rosa, M., Gonsalves, A. R., Serra, A. C., & Nonell, S. (2013). Singlet Oxygen in Antimicrobial Photodynamic Therapy: Photosensitizer-Dependent Production and Decay in E. coli. Molecules, 18(3), 2712-2725. https://doi.org/10.3390/molecules18032712