3-Ishwarone, a Rare Ishwarane Sesquiterpene from Peperomia scandens Ruiz & Pavon: Structural Elucidation through a Joint Experimental and Theoretical Study

Abstract
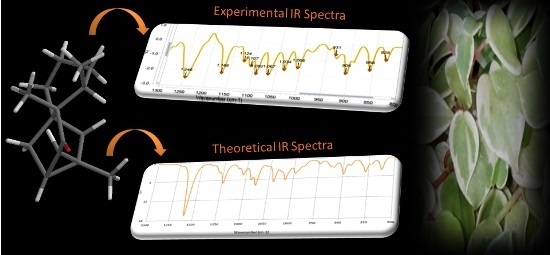
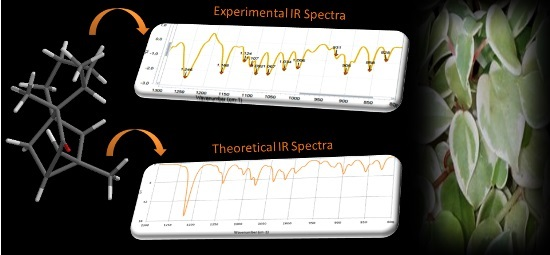
:
1. Introduction
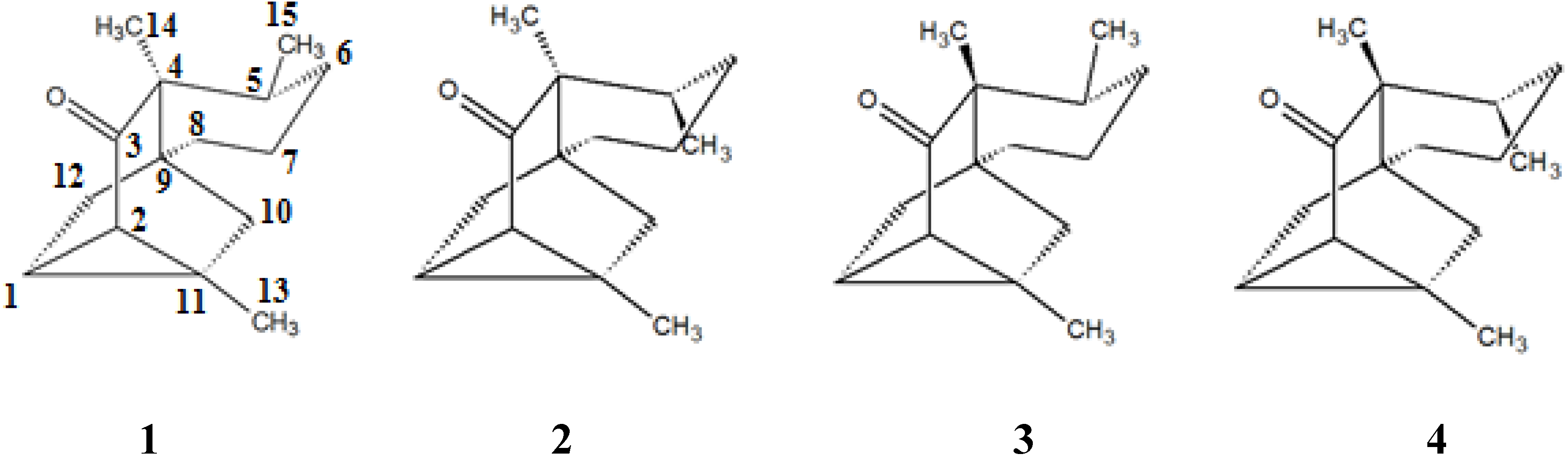
2. Results and Discussion

{kind=link}
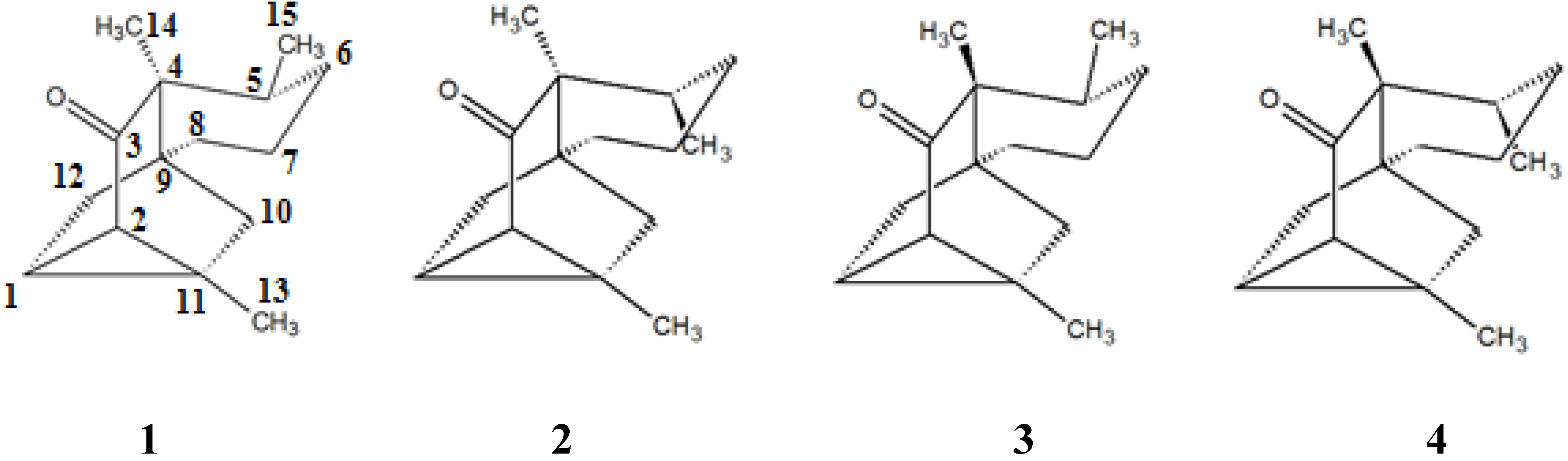{kind=link}
{kind=link}
{kind=link}
| Atom number | C6D6 | ||||
|---|---|---|---|---|---|
| δ 1H (multiplicity, J/Hz) | δ 13C a | δ 13C b | NOESY | δ 1H (multiplicity, J/Hz) | |
| 1 | 1.14 (1H, dd, J = 7.2 and 2.8 Hz) | 29.7 (1) | 29.9 | -- | 1.64 (1H, dd, J = 3,8 and 9,0 Hz) |
| 2 | 1.53 (1H, d, J = 7.3 Hz) | 37.9(1) | 37.9 | -- | 1.51 (1H, d, J = 2,8 Hz) |
| 3 | -- | 212.5 (4) | 213.4 | -- | -- |
| 4 | -- | 49.3 (0) | 49.9 | -- | -- |
| 5 | 1.77 (1H, dqd, J = 18.7, 6.6 and 3.9 Hz) | 31.7 (1) | 31.8 | H-15 | 1.91 (1H. m) |
| 6 | a: 1.05–1.01 (1H, m) b: 1.24–1.17 (1H, m) | 31.5 (2) | 31.5 | -- | a: 1.38 (1H, m) |
| b: 1.22 (1H, m) | |||||
| 7 | a: 1.24–1.21 (1H, m) b: 1.35–1.31 (1H, m) | 23.5 (2) | 23.6 | -- | a: 1.54 (1H, m) |
| b: 1.40 (1H, m) | |||||
| 8 | a: 0.89 (1H, m) b: 1.39–1.35 (1H, m) | 32.2 (2) | 32.2 | H-8b H-8a | a: 1.60 (1H, m) |
| b: 1.19 (1H, m) | |||||
| 9 | -- | 43.0 (0) | 43.1 | -- | -- |
| 10 | a: 1.99 (1H, d, J = 12.3 Hz) b: 0.93 (1H, d, J = 12.3 Hz) | 39.5 (2) | 39.5 | H-10a H-10b | a: 2.27 (1H, d , J = 12,39 Hz) |
| b: 1.35 (1H, d , J = 12,31 Hz) | |||||
| 11 | -- | 29.8 (0) | 30.0 | -- | -- |
| 12 | a: 1.10 (1H, dd, J =12.4, 2.6 Hz) b: 1.66(1H, d, J = 12.4 Hz) | 35.1 (2) | 35.1 | H-12b H-12a; H-14 | a: 1.91 (1H, d , J=12,72 Hz) |
| b: 1.49 (1H, d , J = 12,14 Hz) | |||||
| 13 | 0.95 (3H, s) | 19.4(3) | 19.5 | -- | 1.29 (3H, s) |
| 14 | 0.89 (3H, s) | 11.8(3) | 12.0 | H-12b | 0.91 (3H, s) |
| 15 | 1.27 (3H, d, J = 6.7 Hz) | 17.5 (3) | 17.7 | H-15 | 1.05 (3H, d , J = 6,6 Hz) |
| Atom number | Conf. 1 δesc | Conf. 2 δesc | Conf. 3 δesc | Conf. 4 δesc | EXP |
|---|---|---|---|---|---|
| 1 | 30,69 | 29,54 | 31,05 | 30,43 | 30,4 |
| 2 | 38,10 | 36,57 | 36,74 | 38,24 | 38,1 |
| 3 | 212,46 | 211,57 | 211,70 | 212,35 | 215 |
| 4 | 49,94 | 50,18 | 50,25 | 50,04 | 49,3 |
| 5 | 32,76 | 37,42 | 37,60 | 32,70 | 31,3 |
| 6 | 31,17 | 27,82 | 27,84 | 31,10 | 31,1 |
| 7 | 24,09 | 20,27 | 20,18 | 23,94 | 23,1 |
| 8 | 33,20 | 33,50 | 33,33 | 33,06 | 31,9 |
| 9 | 42,45 | 40,64 | 40,78 | 42,48 | 42,9 |
| 10 | 39,68 | 41,41 | 42,53 | 41,03 | 39,5 |
| 11 | 30,63 | 31,23 | 29,81 | 31,09 | 30,8 |
| 12 | 35,98 | 37,38 | 36,47 | 34,57 | 34,9 |
| 13 | 20,51 | 20,66 | 20,61 | 20,51 | 19,5 |
| 14 | 13,62 | 23,72 | 23,63 | 13,67 | 11,5 |
| 15 | 17,38 | 19,45 | 19,50 | 17,35 | 17,1 |
| MAD | 0,84 | 2,89 | 2,90 | 0,86 | |
| RMS | 1,12 | 4,05 | 4,05 | 1,16 |
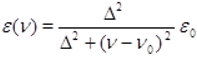


| Exp. (cm−1) | Theor. (cm−1) | Δteor-exp (cm−1) a |
|---|---|---|
| 828 | 824 | 4 [0,5] |
| 858 | 856 | 2 [0,2] |
| 905 | 899 | 6 [0,6] |
| 931 | 930 | 1 [0,1] |
| 1006 | 997 | 9 [0,9] |
| 1034 | 1017 | 17 [1,6] |
| 1067 | 1040 | 27 [2,5] |
| 1092 | 1078 | 14 [1,3] |
| 1107 | 1089 | 18 [1,6] |
| 1124 | 1107 | 17 [1,5] |
| 1168 | 1148 | 20 [1,7] |
| 1246 | 1220 | 26 [1,2] |
| MAD= | 13 [1,2] |
3. Experimental
3.1. General
3.2. GC-FID Analysis
3.3. GC-MS Analysis
3.4. NMR Measurements
3.5. INFRARED Measurements
3.6. Plant Material
3.7. Extraction and Isolation
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Mathieu, G.; Samain, M.-S.; Reynders, M.; Goetghebeur, P. Taxonomy of the Peperomia species (Piperaceae) with pseudo-epiphyllous inflorescences, including four new species. Bot. J. Linn. Soc. 2008, 157, 177–196. [Google Scholar] [CrossRef]
- López, S.N.; Lopes, A.A.; Batista, J.M., Jr.; Flausino, O., Jr.; Bolzani, V.S.; Kato, M.J.; Furlan, M. Geranylation of benzoic acid derivatives by enzymatic extracts from Piper crassinervium (Piperaceae). Bioresour. Technol. 2010, 101, 4251–4260. [Google Scholar] [CrossRef]
- Felippe, L.G.; Batista, J.M., Jr.; Baldoqui, D.C.; Nascimento, I.R.; Kato, M.J.; He, Y.; Nafie, L.A.; Furlan, M. VCD to determine absolute configuration of natural product molecules: secolignans from Peperomia blanda. Org. Biomol. Chem. 2012, 10, 4208–4214. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Tong, A.-P.; Huang, J. Two New Norlignans and a New Lignanamide from Peperomia tetraphylla. Chem. Biodivers. 2012, 9, 769–776. [Google Scholar] [CrossRef]
- Salazar, K.J.M.; Lago, J.H.G.; Guimarães, E.F.; Kato, M.J. Meroterpenes from Peperomia oreophila Hensch. and Peperomia arifolia Miq. J. Braz. Chem. Soc. 2012, 23, 782–785. [Google Scholar] [CrossRef]
- Velozo, L.S.M.; Ferreira, M.J.P.; Santos, M.I.S.; Moreira, D.L.; Emereciano, V.P.; Kaplan, M.A.C. Unusual chromenes from Peperomia blanda. Phytochemistry 2006, 67, 492–496. [Google Scholar] [CrossRef]
- Velozo, L.S.M.; Ferreira, M.J.P.; Santos, M.I.S.; Moreira, D.L.; Guimarães, E.F.; Emerenciano, V.P.; Kaplan, M.A.C. C-glycosyl flavones from Peperomia blanda. Fitoterapia 2009, 80, 119–122. [Google Scholar] [CrossRef]
- Wang, Q.W.; Yu, D.H.; Lin, M.G.; Zhao, M.; Zhu, W.J.; Lu, Q.; Li, G.X.; Wang, C.; Yang, Y.F.; Qin, X.M.; et al. Antiangiogenic Polyketides from Peperomia dindygulensis Miq. Molecules 2012, 17, 4474–4483. [Google Scholar] [CrossRef]
- Aziba, P.I.; Adedeji, A.; Ekor, M.; Adeyemi, O. Analgesic activity of Peperomia pellucida aerial parts in mice. Fitoterapia 2001, 72, 57–58. [Google Scholar] [CrossRef]
- Pinheiro, B.G.; Silva, A.S.B.; Souza, G.E.P.; Figueiredo, J.G.; Cunha, F.Q.; Da Silva, J.K.R.; Maia, J.G.S.; Sousa, P.J.C. Chemical composition, Antinociceptive and anti-inflammatory effects in rodents of the essential oil of Peperomia serpens (Sw.) Loud. J. Ethnopharmacol. 2011, 138, 479–486. [Google Scholar] [CrossRef]
- Holthe, P.A.; Sternberg Lda, S.; Ting, I.P. Developmental Control of CAM in Peperomia scandens. Plant Physiol. 1987, 84, 743–747. [Google Scholar] [CrossRef]
- Lago, J.H.G.; Oliveira, A.; Guimarães, E.F.; Kato, M.J. 3-Ishwarone and 3-Ishwarol, Rare sesquiterpenes in essential oil from leaves of Peperomia oreophila Hensch. J. Braz. Chem. Soc. 2007, 18, 638–642. [Google Scholar] [CrossRef]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2012, 29, 1334–1366. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N.J.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Revision A.01; Gaussian, Inc.: Cillingford, CT, USA, 2009. [Google Scholar]
- Costa, F.L.P.; de Albuquerque, A.C.F.; dos Santos, F.M.; de Amorim, M.B. GIAO-HDFT scaling factor for 13C NMR chemical shifts calculation. J. Phys. Org. Chem. 2010, 23, 972–977. [Google Scholar] [CrossRef]
- Cimino, P.; Gomez-Paloma, L.; Duca, D.; Riccio, R.; Bifulco, G. Comparison of different theory models and basis sets in the calculation of 13C NMR chemical shifts of natural products. Magn. Reson. Chem. 2004, 42, S26–S33. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Haharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Costa, F.L.P.; de Albuquerque, A.C.F.; Borges, R.M.; dos Santos, F.M., Jr.; de Amorim, M.B. High cost-effectiveness ratio: GIAO-MPW1PW91/6-31G(d)//MPW1PW91/6-31G(d) scaling factor for 13C nuclear magnetic resonance chemical shifts calculation. J. Comp. Theor. Nanosci. 2014, 11, 219–225. [Google Scholar] [CrossRef]
- Becke, A.D.J. Density-functional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Sinha, P.; Boesch, S.E.; Changming, G.; Wheeler, R.A.; Wilson, A.K. Harmonic vibrational frequencies: Scaling factors for hf, b3lyp, and mp2 methods in combination with correlation consistent basis sets. J. Phys. Chem. A 2004, 108, 9213–9217. [Google Scholar] [CrossRef]
- Busch, K.W.; Busch, M.A. Chiral Analisys, 1st ed.; Elsevier: Amsterdam, the Netherlands, 2006; pp. 461–504. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dos S. Junior, F.M.; Velozo, L.S.M.; De Carvalho, E.M.; Marques, A.M.; Borges, R.M.; Trindade, A.P.F.; Dos Santos, M.I.S.; De Albuquerque, A.C.F.; Costa, F.L.P.; Kaplan, M.A.C.; et al. 3-Ishwarone, a Rare Ishwarane Sesquiterpene from Peperomia scandens Ruiz & Pavon: Structural Elucidation through a Joint Experimental and Theoretical Study. Molecules 2013, 18, 13520-13529. https://doi.org/10.3390/molecules181113520
Dos S. Junior FM, Velozo LSM, De Carvalho EM, Marques AM, Borges RM, Trindade APF, Dos Santos MIS, De Albuquerque ACF, Costa FLP, Kaplan MAC, et al. 3-Ishwarone, a Rare Ishwarane Sesquiterpene from Peperomia scandens Ruiz & Pavon: Structural Elucidation through a Joint Experimental and Theoretical Study. Molecules. 2013; 18(11):13520-13529. https://doi.org/10.3390/molecules181113520
Chicago/Turabian StyleDos S. Junior, Fernando M., Leosvaldo S. M. Velozo, Erika M. De Carvalho, André M. Marques, Ricardo M. Borges, Ana Paula F. Trindade, Maria Isabel S. Dos Santos, Ana Carolina F. De Albuquerque, Fabio L.P. Costa, Maria Auxiliadora C. Kaplan, and et al. 2013. "3-Ishwarone, a Rare Ishwarane Sesquiterpene from Peperomia scandens Ruiz & Pavon: Structural Elucidation through a Joint Experimental and Theoretical Study" Molecules 18, no. 11: 13520-13529. https://doi.org/10.3390/molecules181113520
APA StyleDos S. Junior, F. M., Velozo, L. S. M., De Carvalho, E. M., Marques, A. M., Borges, R. M., Trindade, A. P. F., Dos Santos, M. I. S., De Albuquerque, A. C. F., Costa, F. L. P., Kaplan, M. A. C., & De Amorim, M. B. (2013). 3-Ishwarone, a Rare Ishwarane Sesquiterpene from Peperomia scandens Ruiz & Pavon: Structural Elucidation through a Joint Experimental and Theoretical Study. Molecules, 18(11), 13520-13529. https://doi.org/10.3390/molecules181113520