Influence of Sample Processing on the Analysis of Carotenoids in Maize


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Improvements in the Extraction Process
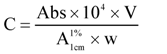
 = absorption coefficient, which is defined as the theoretical absorbance of a solution of 1% (w/v) concentration (i.e., g in 100 mL) in a cuvette with a path length of 1 cm. Lutein and zeaxanthin are the major carotenoids in maize. Therefore, an average value for A
= absorption coefficient, which is defined as the theoretical absorbance of a solution of 1% (w/v) concentration (i.e., g in 100 mL) in a cuvette with a path length of 1 cm. Lutein and zeaxanthin are the major carotenoids in maize. Therefore, an average value for A  of 2,332 was used [15]. C = total carotenoid content (μg/g) in a given sample on dry weight basis. Abs = absorbance measured at 450 nm. V = volume (mL). W = weight of sample (g). 104 = conversion factor to obtain the concentration in units of μg/g.
of 2,332 was used [15]. C = total carotenoid content (μg/g) in a given sample on dry weight basis. Abs = absorbance measured at 450 nm. V = volume (mL). W = weight of sample (g). 104 = conversion factor to obtain the concentration in units of μg/g.
{kind=link}
{kind=link}
{kind=link}
| Modified method | Solvents | Ref. | Replicates | Output Factor c (μg/g DW) | % Non-extracted carotenoids |
|---|---|---|---|---|---|
| 1 | EtOH | [ 16,17] | 2 | 111.8 ± 7.10 | 21.3 |
| 2 | Acetone | [ 18,19] | 3 | 106.5 ± 2.61 | 25.0 |
| 3 | Acetone a | [ 20] | 3 | 126.3 ± 2.50 | 11.10 |
| 4 | Acetone–EtOH–hexane (1:1:2, v/v) | [ 21] | 2 | 120.9 ± 3.64 | 14.9 |
| 5 | MeOH–ethyl–acetate (6:4, v/v) b | - | 3 | 141.6 ± 2.12 | 0 |
| Reference | MeOH–THF (1:1, v/v) | [ 13,22,23] | 3 | 142.1 ± 1.94 | 0 |
2.1.1. Effect of Adding BHT to the Extraction Solvents
| Carotenoid | With BHT (μg/g DW) | Without BHT (μg/g DW) | t Calculated | t Critical | Degree of freedom a |
|---|---|---|---|---|---|
| Astaxanthin | 8.18 ± 0.06 | 8.22 ± 0.06 | 0.76 | 2.78 | 4 |
| Adonixanthin | 2.46 ± 0.03 | 2.51 ± 0.11 | 0.80 | 2.78 | 4 |
| Zeax+lut | 2.25 ± 0.11 | 2.37 ± 0.36 | 0.56 | 2.78 | 4 |
| Adonirubin | 1.52 ± 0.02 | 1.52 ± 0.01 | 0.36 | 2.78 | 4 |
| Canthaxanthin | 0.98 ± 0.00 | 0.96 ± 0.05 | 0.47 | 2.78 | 4 |
| Total carotenoids | 15.39 ± 0.16 | 15.84 ± 0.58 | 1.29 | 2.78 | 4 |
2.2. Solubility of Carotenoids
2.2.1. Solubility of the Carotenoids Extracted from Maize in the Injection Solvent
| Injection solvent a | Anther μg/g DW | Adonix μg/g DW | Lut+zeax μg/g DW | α-Crypt μg/g DW |
|---|---|---|---|---|
| Acetone 100% | 0.12 | 1.93 | 2.20 | 0.98 |
| Mobile phase b–acetone 6.7:3.3, v/v | 0.24 | 4.71 | 4.49 | 1.36 |
| Mobile phase b 100% | 0.20 | 4.48 | 3.89 | 1.27 |
| Mobile phase b–acetone 7.5:2.5, v/v | 0.22 | 4.39 | 4.20 | 1.26 |
| ACN–MeOH– iPrOH 8.5:1:0.5, v/v/v | 0.15 | 3.31 | 3.05 | 1.12 |
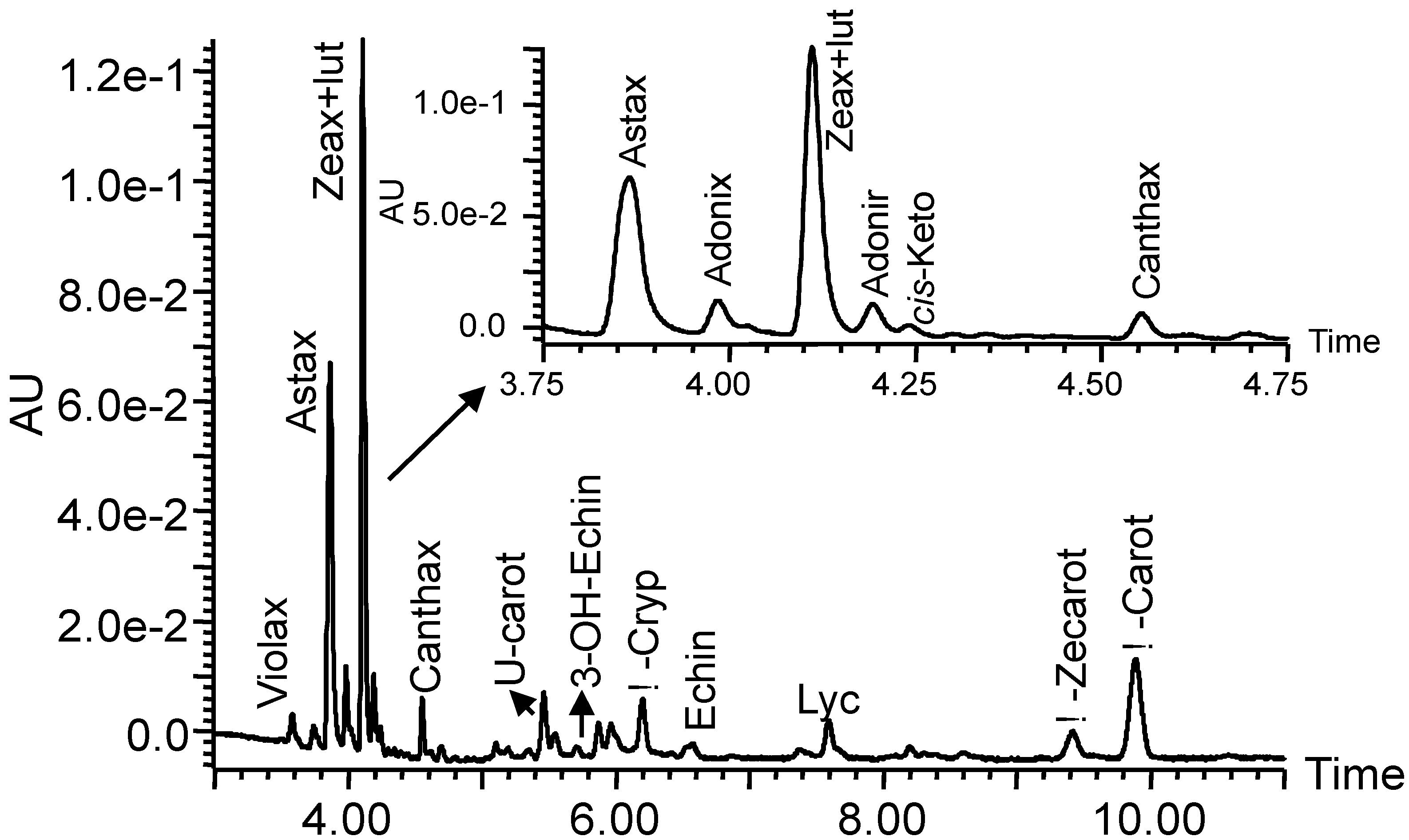
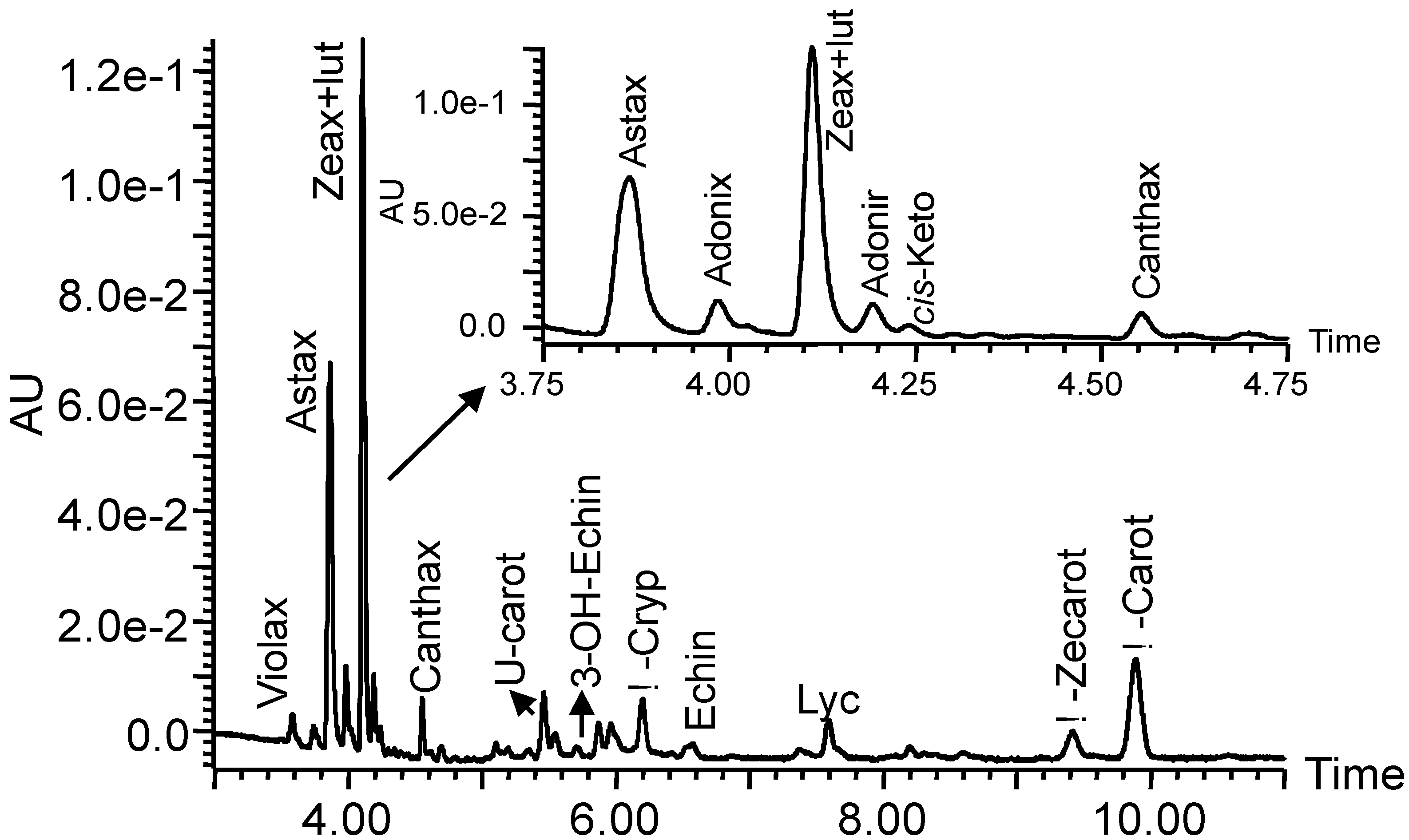
2.2.2. Preparation of the Carotenoid Standards
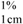 used to quantify each pigment. Working solutions were prepared from stock solutions by sampling an aliquot and diluting it with injection solvent. Solution concentrations were assessed by UHPLC analysis. For those carotenoids dissolved in hexane (canthaxanthin, β-cryptoxanthin, β-carotene, lycopene and phytoene), working solutions were prepared from stock solutions by evaporating an aliquot under nitrogen and diluting it with injection solvent. Calibration curves were obtained from peak area by injecting mixtures of standards. Table 5 shows the linear regression data for each carotenoid standard curve.
used to quantify each pigment. Working solutions were prepared from stock solutions by sampling an aliquot and diluting it with injection solvent. Solution concentrations were assessed by UHPLC analysis. For those carotenoids dissolved in hexane (canthaxanthin, β-cryptoxanthin, β-carotene, lycopene and phytoene), working solutions were prepared from stock solutions by evaporating an aliquot under nitrogen and diluting it with injection solvent. Calibration curves were obtained from peak area by injecting mixtures of standards. Table 5 shows the linear regression data for each carotenoid standard curve.| Carotenoid | Solvent | 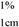 | Stock carotenoid concentration (μg/mL) |
|---|---|---|---|
| cis-Neoxanthin | EtOH | 2,380 | 19.64 a |
| Violaxanthin | EtOH | 2,550 | 16.26 a |
| Antheraxanthin | EtOH | 2,350 | 17.53 a |
| Astaxanthin | EtOH | 2,100 | 1.55 a and 4.96 b |
| Astaxanthin | Injection solvent | - | 5.12 b |
| Zeaxanthin | Acetone | 2,340 | 32.31 a |
| Lutein | EtOH | 2,550 | 21.57 a |
| Canthaxanthin | Hexane | 2,200 | 0.27 a and 4.53 b |
| Canthaxanthin | Injection solvent | - | 5.70 b |
| β-Cryptoxanthin | Hexane | 2,400 | 35.00 a |
| β-Carotene | Hexane | 2,590 | 24.85 a |
| Lycopene | Hexane | 3,450 | 8.26 a |
| cis-Phytoene | Hexane | 915 | 16.16 a |
| Carotenoid | Linear range (μg/mL) | Slope | Intercept | R 2 |
|---|---|---|---|---|
| cis-Neoxanthin | 0.04−19.64 | 2,481 ± 7.57 | −136.62 ± 16.28 | 0.9999 |
| Violaxanthin | 0.03−16.26 | 2,516 ± 5.59 | −121.93 ± 12.97 | 0.9994 |
| Antheraxanthin | 0.03−15.53 | 2,509 ± 22.13 | −413.83 ± 4.13 | 0.9970 |
| Astaxanthin | 0.04−5.12 | 1,825 ± 6.43 | −50.37 ± 10.51 | 0.9999 |
| Astaxanthin a | − | 14,149 | 4414.1 | 0.7741 |
| Lutein | 0.02−17.25 | 2,475 ± 81.74 | −626.20 ± 35.78 | 0.9952 |
| Zeaxanthin | 0.03−17.23 | 2,578 ± 38.04 | −86.96 ± 25.8 | 0.9996 |
| Canthaxanthin | 0.02−5.70 | 1,787 ± 4.24 | −43.96 ± 16.13 | 0.9995 |
| Canthaxanthin b | − | 9,613 | 1583.1 | 0.9320 |
| β-Cryptoxanthin | 0.04−18.67 | 2,379 ± 0.35 | −444.17 ± 31.46 | 0.9988 |
| Lycopene | 0.3−3.11 | 1,398 ± 104.40 | −121.72 ± 24.88 | 0.9998 |
| β-Carotene | 0.1−24.85 | 1,484 ± 27.22 | −189.02 ± 29.80 | 0.9998 |
| cis-Phytoene | 0.08−16.16 | 1,990 ± 285.46 | −259.82 ± 37.32 | 0.9989 |
3. Experimental
3.1. Chemicals
3.2. Plant Material
3.3. Methods
3.3.1. Reference Method
3.3.2. Blank Solution
3.3.3. Extraction Using BHT
3.3.4. Chromatographic Analysis
| Time (min) | Flow Rate (mL/min) | A (%, v/v) | B (%, v/v) | Curve |
|---|---|---|---|---|
| Initial | 0.4 | 85 | 15 | Linear |
| 2.0 | 0.4 | 85 | 15 | Linear |
| 3.0 | 0.4 | 100 | 0 | Linear |
| 7.0 | 0.4 | 100 | 0 | Linear |
| 8.0 | 0.6 | 100 | 0 | Linear |
| 11.6 | 0.6 | 100 | 0 | Linear |
| 12.6 | 0.4 | 85 | 15 | Linear |
| 15.0 | 0.4 | 85 | 15 | Linear |
3.4. Ultraviolet and Visible (UV-vis) Spectroscopy
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Kimura, M.; Rodriguez-Amaya, D.B. Sources of errors in the quantitative analysis of food carotenoids by HPLC. Arch. Latinoam. Nutr. 1999, 49 (Suppl. 1), 58S–66S. [Google Scholar]
- Meléndez-Martínez, A.J.; Vicario, I.M.; Heredia, F.J. Review: Analysis of carotenoids in orange juice. J. Food Compos. Anal. 2007, 20, 638–649. [Google Scholar] [CrossRef]
- Oliver, J.; Palou, A. Chromatographic determination of carotenoids in foods. J. Chromatogr. A 2000, 881, 543–555. [Google Scholar] [CrossRef]
- Rodriguez-Amaya, D.B.; Kimura, M. Harvestplus Handbook for Carotenoid Analysis, HarvestPlus Technical Monograph Series 2; IFPRI: Washington, DC, USA; CIAT: Cali, Colombia, 2004. [Google Scholar]
- Rodríguez-Bernaldo de Quirós, A.; Costa, H.S. Analysis of carotenoids in vegetable and plasma samples: A review. J. Food Compos. Anal. 2006, 19, 97–111. [Google Scholar] [CrossRef]
- Su, Q.; Rowley, K.G.; Balazs, N.D.H. Carotenoids: Separation methods applicable to biological samples. J. Chromatogr. B 2002, 781, 393–418. [Google Scholar] [CrossRef]
- Howe, J.A.; Tanumihardjo, S.A. Evaluation of analytical methods for carotenoid extraction from biofortified maize (Zea mays sp.). Agric. Food. Chem. 2006, 54, 7992–7997. [Google Scholar]
- Azevedo-Meleiro, C.H.; Rodriguez-Amaya, D.B. Confirmation of the identity of the carotenoids of tropical fruits by HPLC-DAD and HPLC-MS. J. Food Compos. Anal. 2004, 17, 385–396. [Google Scholar] [CrossRef]
- Berardo, N.; Brenna, O.V.; Amato, A.; Valoti, P.; Pisacane, V.; Motto, M. Carotenoids concentration among maize genotypes measured by near infrared reflectance spectroscopy (NIRS). IFSET 2004, 5, 393–398. [Google Scholar]
- Scott, C.E.; Eldridge, A.L. Comparison of carotenoid content in fresh, frozen and canned corn. J. Food Compos. Anal. 2005, 18, 551–559. [Google Scholar] [CrossRef]
- Menkir, A.; Liu, W.; White, W.S.; Maziya-Dixon, B.; Rocheford, T. Carotenoid diversity in tropical-adapted yellow maize inbred lines. Food Chem. 2008, 109, 521–529. [Google Scholar] [CrossRef]
- Muzhingi, T.; Yeum, K.J.; Russell, R.M.; Johnson, E.J.; Qin, J.; Tang, G. Determination of carotenoids in Yellow Maize, the effects of saponification and food preparations. Int. J. Vitam Nutr. Res. 2008, 78, 112–120. [Google Scholar] [CrossRef]
- Naqvi, S. Metabolic Engineering of Complex Pathways in Plants by Combinatorial Genetic Transformation. Doctoral Thesis, University of Lleida, Lleida, Spain, 2009. [Google Scholar]
- Most of the methods used to remove peroxides require highly reactive reagents, are time consuming, and call for extreme caution throughout the peroxide elimination process.
- Mishra, P.; Singh, N. Spectrophotometric and TLC based characterization of kernel carotenoids in short duration maize. Maydica 2010, 55, 95–100. [Google Scholar]
- Moros, E.E.; Darnoko, D.; Cheryan, M.; Perkins, E.G.; Jerrell, J. Analysis of xanthophylls in corn by HPLC. J. Agric. Food Chem. 2002, 50, 5787–5790. [Google Scholar]
- Howard, L.A.; Wong, A.D.; Perry, A.K.; Klein, B.P. β-Carotene and ascorbic acid retention in fresh and processed vegetables. J. Food Sci. 1999, 64, 929–936. [Google Scholar] [CrossRef]
- Matthews, P.D.; Luo, R.; Wurtzel, E.T. Maize phytoene desaturase and ζ-carotene desaturase catalyse a poly-Z desaturation pathway: Implications for genetic engineering of carotenoid content among cereal crops. J. Exp. Bot. 2003, 54, 2215–2230. [Google Scholar] [CrossRef]
- Rodrigues, P.; Morais, H.; Mota, T.; Olivera, S.; Forgács, E.; Cserháti, T. Use of HPLC and multivariate methods for the evaluation of the stability of colour pigments of paprika (Capsicum annuum) powder. Anal. Chim. Acta 1998, 372, 411–416. [Google Scholar] [CrossRef]
- Kimura, M.; Kobori, C.N.; Rodriguez-Amaya, D.B.; Nestel, P. Screening and HPLC methods for carotenoids in sweetpotato, cassava and maize for plant breeding trials. Food Chem. 2007, 100, 1734–1746. [Google Scholar] [CrossRef]
- Sérino, S.; Gomez, L.; Costagliola, G.U.Y.; Gautier, H. HPLC assay of tomato carotenoids: Validation of a rapid microextraction technique. J. Agric. Food Chem. 2009, 57, 8753–8760. [Google Scholar] [CrossRef]
- Burkhard, S.; Böhm, V. Development of a new method for the complete extraction of carotenoids from cereals with special reference to durum wheat (Triticum durum Desf.). J. Agric. Food Chem. 2007, 55, 8295–8301. [Google Scholar]
- Aluru, M.; Xu, Y.; Guo, R.; Wang, Z.; Li, S.; White, W.; Wang, K.; Rodermel, S. Generation of transgenic maize with enhanced provitamin A content. J. Exp. Bot. 2008, 59, 3551–3562. [Google Scholar] [CrossRef]
- FAO. Lycopene Extract from Tomate. Available online: http://www.fao.org/fileadmin/templates/agns/pdf/jecfa/cta/71/lycopene_extract_from_tomato.pdf (accessed on 29 February 2012).
- Lucini, L.; Pellizzoni, M.; Baffi, C.; Molinari, G.P. Rapid determination of lycopene and β-carotene in tomato by liquid chromatography/electrospray tandem mass spectrometry. J. Sci. Food Agric. 2012, 92, 1297–1303. [Google Scholar] [CrossRef]
- Strati, I.F.; Oreopoulou, V. Process optimisation for recovery of carotenoids from tomato waste. Food Chem. 2011, 129, 747–752. [Google Scholar] [CrossRef]
- Rodriguez-Amaya, D.B. A Guide to Carotenoid Analysis in Foods; International Life Sciences Inst: Washington, DC, USA, 1999. [Google Scholar]
- Kurilich, A.C.; Juvik, J.A. Quantification of carotenoid and tocopherol antioxidants in Zea mays. J. Agric. Food Chem. 1999, 47, 1948–1955. [Google Scholar] [CrossRef]
- Rivera, S.; Vilaró, F.; Canela, R. Determination of carotenoids by liquid chromatography/mass spectrometry: Effect of several dopants. Anal. Bioanal. Chem. 2011, 400, 1339–1346. [Google Scholar] [CrossRef]
- Waters Corporation. Acquity UPLC BEH Column Care and Use Instructions. Available online: http://www.waters.com/webassets/cms/support/docs/715001371.pdf (accessed on 3 March 2012).
- Britton, G.; Liaaen-Jensen, S.; Pfander, H. Carotenoids Handbook, 1st ed.; Birkhäuser: Basel, Switzerland, 2004; pp. 3–404. [Google Scholar]
- Craft, N.E.; Scares, J.H.Jr. Relative solubility, stability, and absorptivity of lutein and β-carotene in organic solvents. J. Agric. Food Chem. 1992, 40, 431–434. [Google Scholar] [CrossRef]
- Shi, J.; le Maguer, M. Lycopene in tomatoes: Chemical and physical properties affected by food processing. Crit. Rev. Biotechnol. 2000, 20, 293–334. [Google Scholar] [CrossRef]
- Fratianni, A.; Cinquanta, L.; Panfili, G. Degradation of carotenoids in orange juice during microwave heating. LWT-Food Sci. Technol. 2010, 43, 867–871. [Google Scholar]
- Konings, E.J.M.; Roomans, H.H.S. Evaluation and validation of an LC method for the analysis of carotenoids in vegetables and fruit. Food Chem. 1997, 59, 599–603. [Google Scholar] [CrossRef]
- Rosati, C.; Aquilani, R.; Dharmapuri, S.; Pallara, P.; Marusic, C.; Tavazza, R.; Bouvier, F.; Camara, B.; Giuliano, G. Metabolic engineering of beta-carotene and lycopene content in tomato fruit. Plant J. 2000, 24, 413–419. [Google Scholar] [CrossRef]
- Fraser, P.D.; Enfissi, E.M.A.; Halket, J.M.; Truesdale, M.R.; Yu, D.; Gerrish, C.; Bramley, P.M. Manipulation of phytoene levels in tomato fruit: Effects on isoprenoids, plastids, and intermediary metabolism. Plant Cell 2007, 19, 3194–3211. [Google Scholar] [CrossRef]
- Lee, M.T.; Chen, B.H. Separation of lycopene and its cis isomers by liquid chromatography. Chromatographia 2001, 54, 613–617. [Google Scholar] [CrossRef]
- Zhu, C.; Naqvi, S.; Breitenbach, J.; Sandmann, G.; Christou, P.; Capell, T. Combinatorial genetic transformation generates a library of metabolic phenotypes for the carotenoid pathway in maize. Proc. Natl. Acad. Sci. USA 2008, 105, 18232–18237. [Google Scholar]
- The amount depends on the color intensity of the samples. For pale maize samples the extraction of 100 mg of sample is recommended. For darker maize samples the extraction of 50 mg of sample is appropriate.
- Sample Availability: Mixture of carotenoids from maiz extracts are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rivera, S.; Canela, R. Influence of Sample Processing on the Analysis of Carotenoids in Maize. Molecules 2012, 17, 11255-11268. https://doi.org/10.3390/molecules170911255
Rivera S, Canela R. Influence of Sample Processing on the Analysis of Carotenoids in Maize. Molecules. 2012; 17(9):11255-11268. https://doi.org/10.3390/molecules170911255
Chicago/Turabian StyleRivera, Sol, and Ramon Canela. 2012. "Influence of Sample Processing on the Analysis of Carotenoids in Maize" Molecules 17, no. 9: 11255-11268. https://doi.org/10.3390/molecules170911255
APA StyleRivera, S., & Canela, R. (2012). Influence of Sample Processing on the Analysis of Carotenoids in Maize. Molecules, 17(9), 11255-11268. https://doi.org/10.3390/molecules170911255