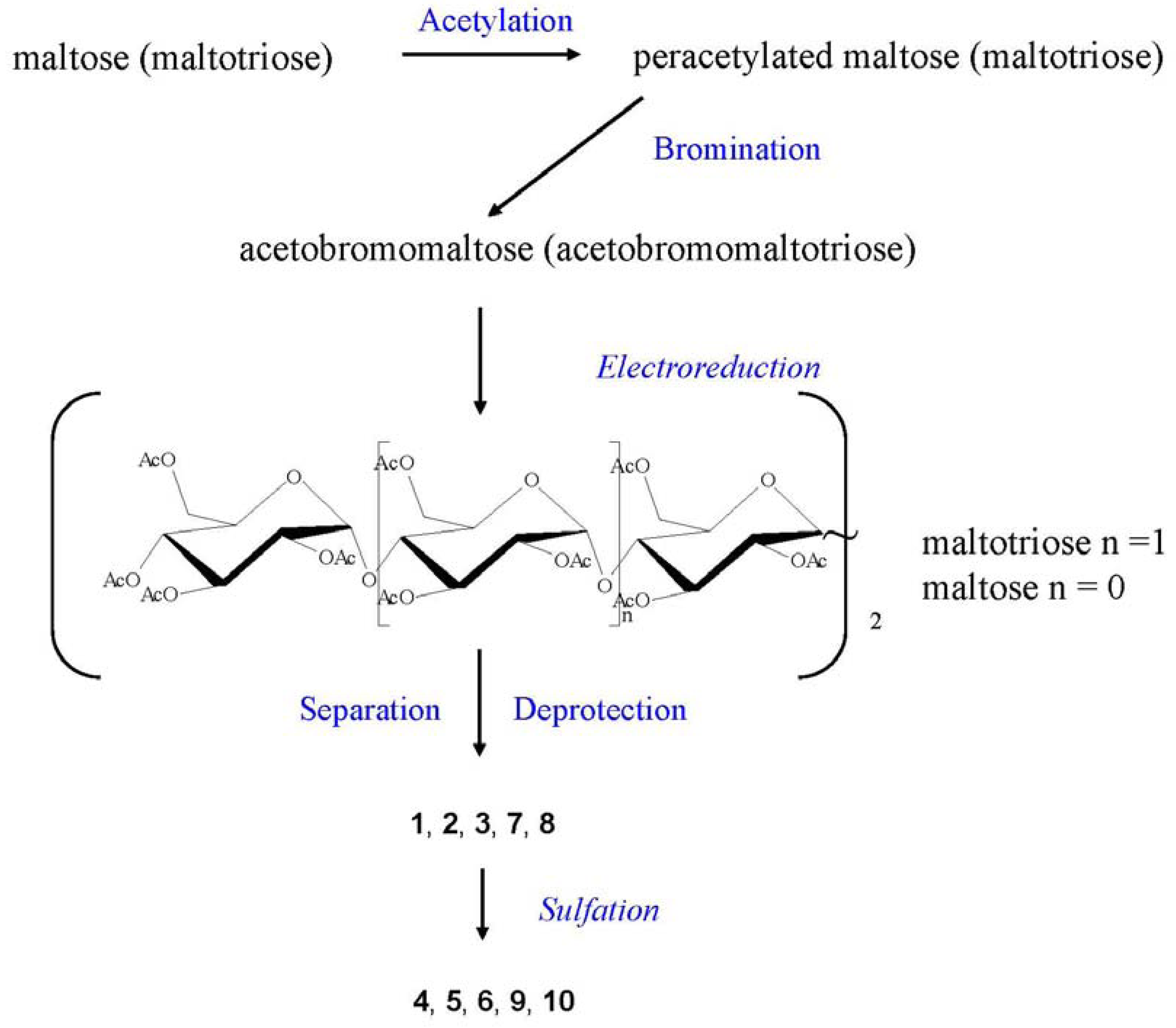
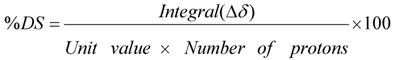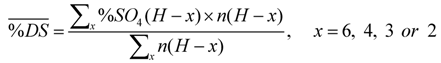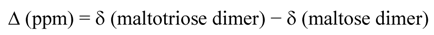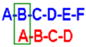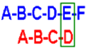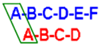Reference [
37] reports an exhaustively overview of sulfation protocols,
i.e., sulfation using sulfuric acid, dicyclohexylcarbodiimide-mediated sulfation and sulfur trioxide amine complexes. Sulfation using sulfur trioxide–amine complexes of SO
3 with organic bases including pyridine (Py), trimethylamine (NMe
3), and triethylamine (NEt
3), or amides such as DMF have typically found extensive usage. NMe
3 and NEt
3 complexes with SO
3 appear to be well suited for sulfation of alcoholic groups present in carbohydrates, steroids, and aliphatic or alicyclic scaffolds.
SO
3–Py complex has been most often used for sulfation of carbohydrate scaffolds from mono-, oligosaccharides to polysaccharides by modulating the sulfation degree (DS), too. For example there is an anti-ulcer agent, sucrose octasulfate prepared by sucrose total sulfation through SO
3–pyridine complex [
38]. DS of sulfated galacto-oligosaccharides (from 3 to 7 sugar units), maltohexaose and heptaose studied for their effects on angiogenesis and prepared also through SO
3–pyridine complex varied from 35 to 64% [
28,
39]. Synthetic heparin oligosaccharides have been obtained by SO
3–pyridine complex [
40]. For the synthesis of galactomannan sulfates, an attempt has been made to correlate the DS with the temperature by using SO
3–Py complex in comparison with chlorosulfonic acid in an organic alkali. Preliminary analysis of the
13C-NMR spectra of the guar derivative has also been reported showing that the sulfate groups primarily substitute the hydroxy groups at C6 of the galactopyranose residues [
41]. Recently, a microwave-based protocol with SO
3–NEt
3 complex has been developed to enhance the rate of sulfation of phenolic structures, especially those with multiple phenolic groups [
42]. It is also interesting to cite that the same microwave-assisted sulfation of heparin oligosaccharides has been recently described to improve yields, although the starting oligosaccharides have only one or two position to sulfate for each sugar units [
43]. In contrast to the above microwave-assisted sulfation, which can be categorised as high temperature, base catalysed reaction, Krylov et al. have recently reported low temperature, acid-catalysed sulfation reaction by SO
3 complexes [
44,
45].
According to the above background we chose the more common and experienced protocol of SO3–Py complex in Py at 80 °C. Noteworthy, both microwave-assisted and acid-catalysed sulfation offer in our opinion very interesting perspectives for our further achievements.
On the other hand the biological activities of
4 and
6 seem to depend on C-C configuration. In fact
4 and
6 that had a quite similar degree of sulfation showed some differences in biological activities as they both are heparanase inhibitors,
6 better than
4, but only
6 inhibits selectin [
29]. The different behavior could be put in relationship with the preferred conformation associated with C-C bond configuration. Compound
4 is weakly bent due to its α,α
anti conformation, while
6 has a huge bend due to the α,β
gauche conformation. Concerning conformation and biological activities,
6 appears quite similar to heparin. Eventually preliminary data showed that
5 acts as very low heparanase and selectin inhibitor, this could be put in relationship both with the low degree of sulfation and with a linear structure suggested by the β,β C-C configuration.
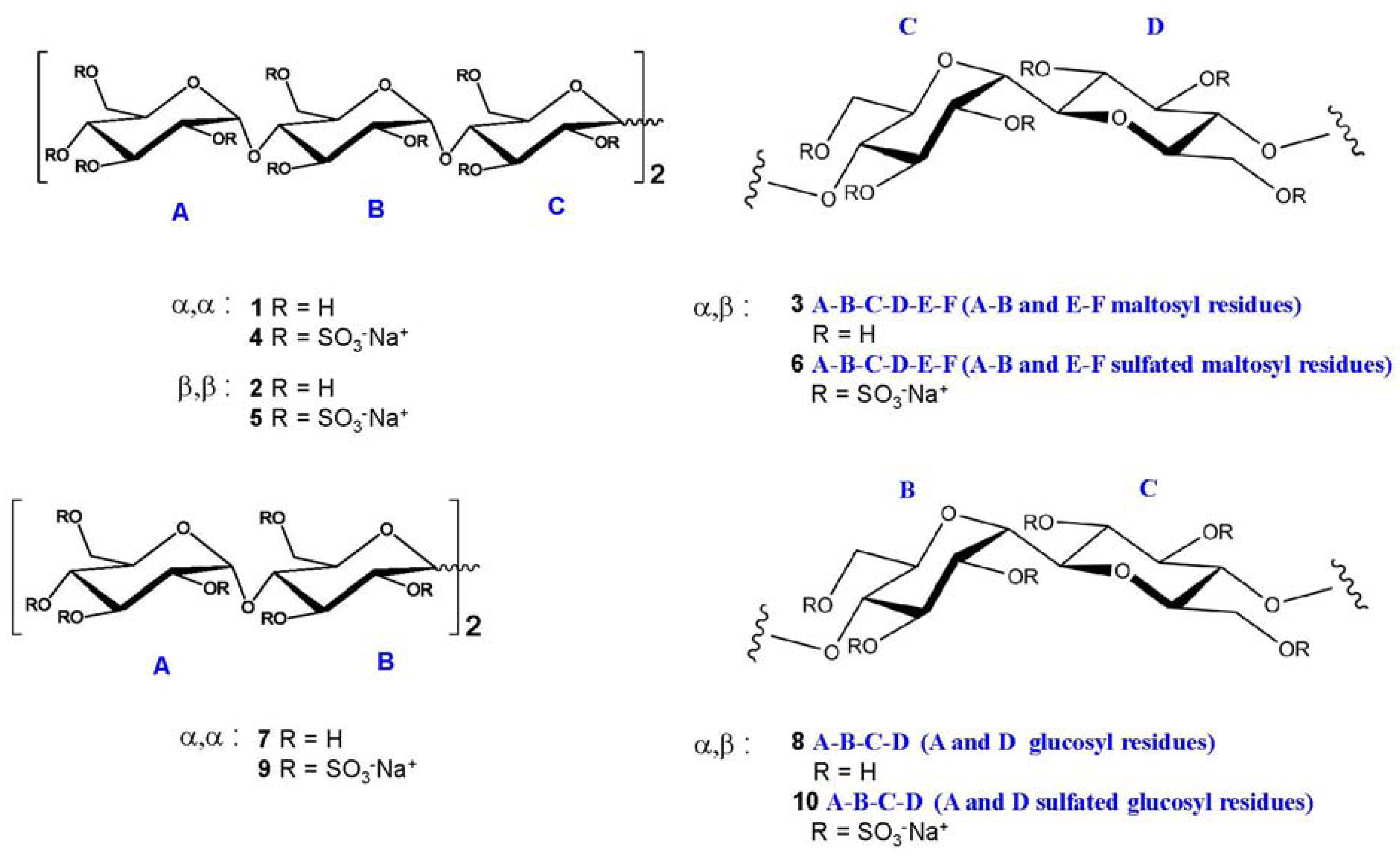
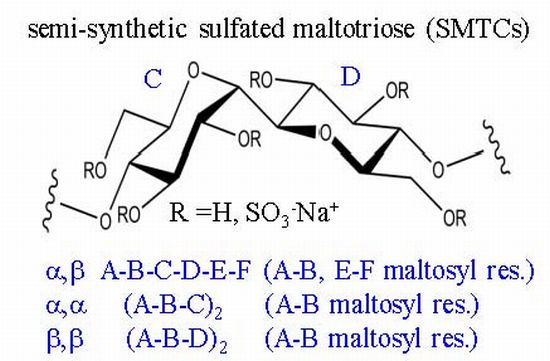
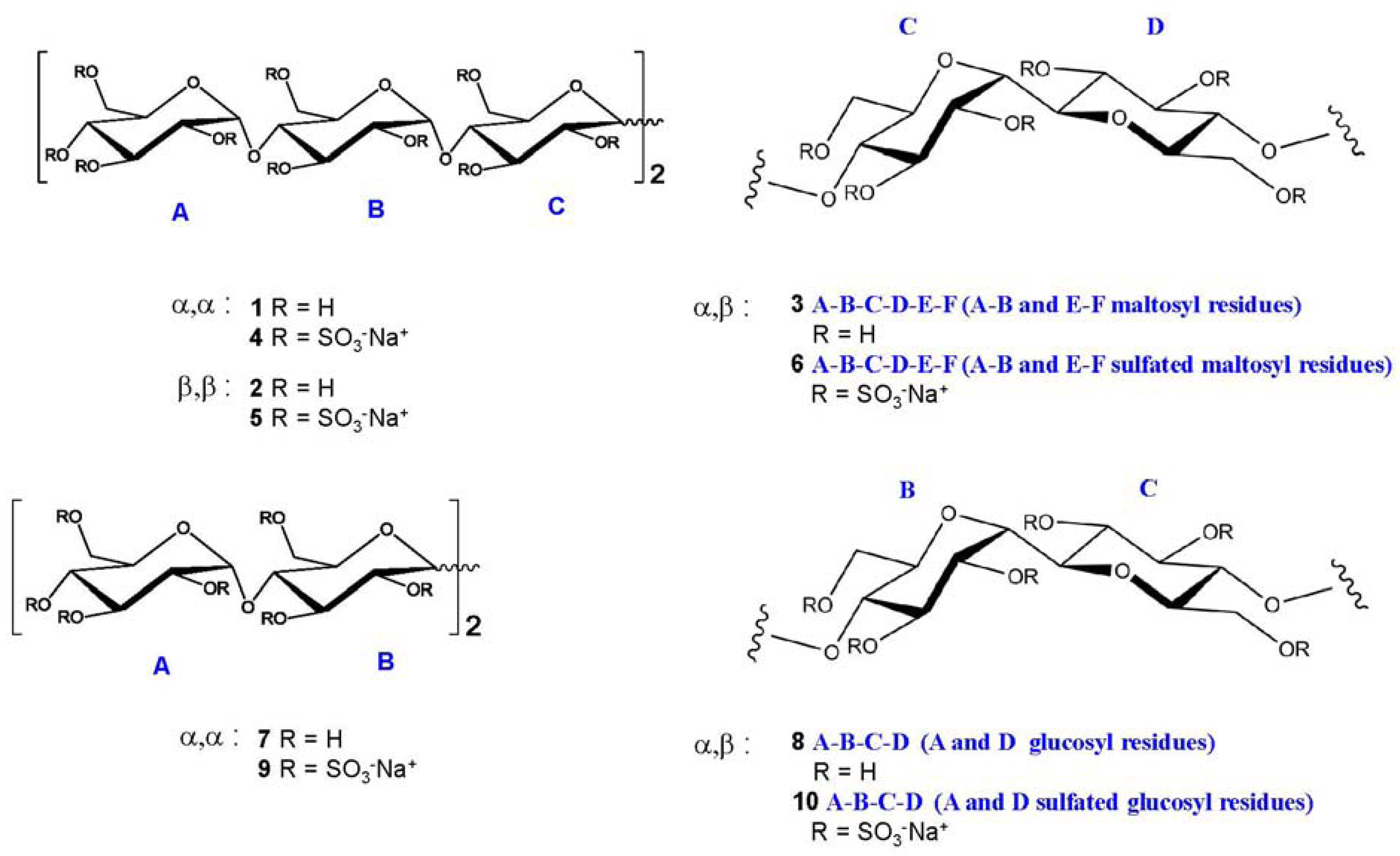
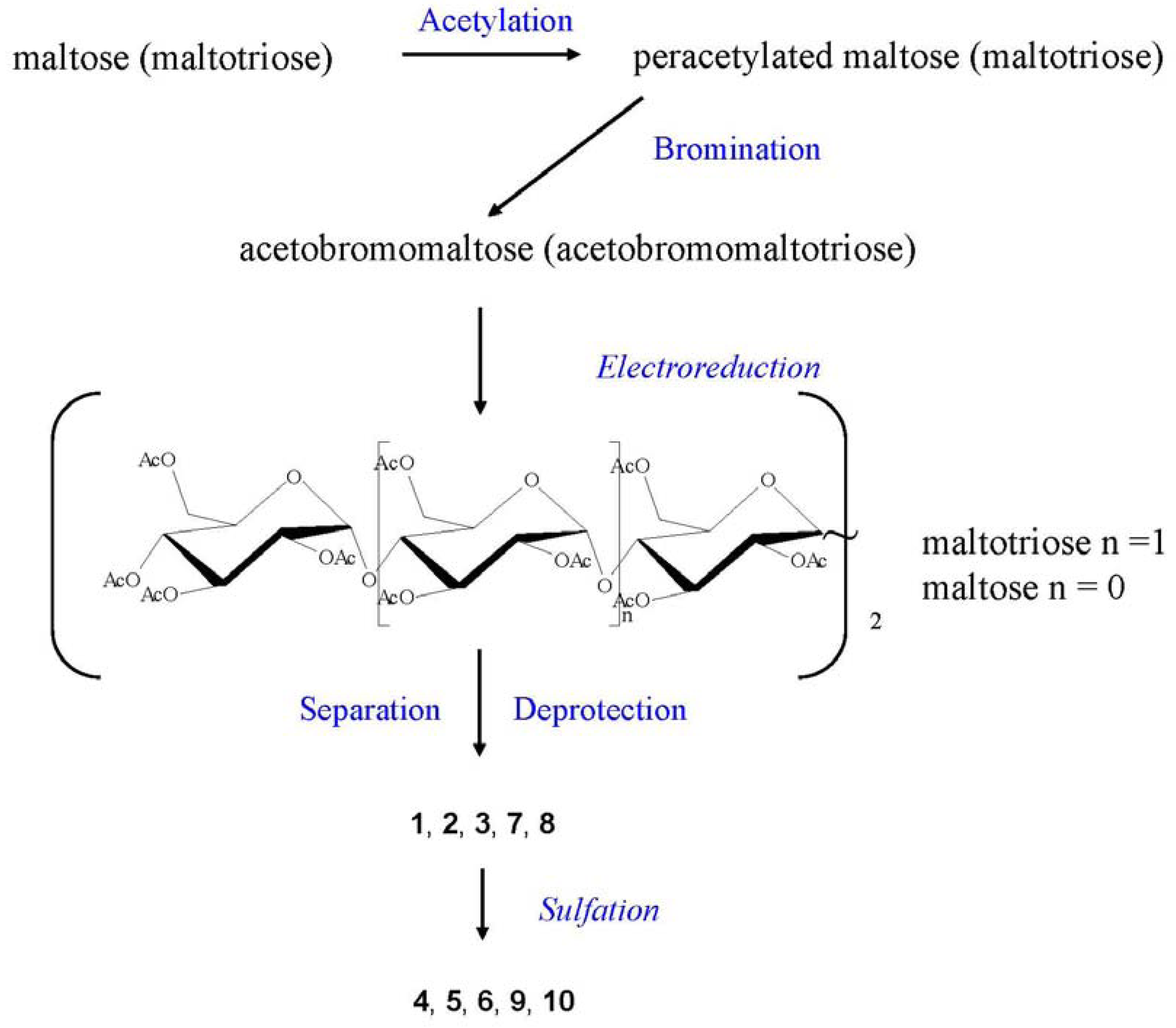
2.2. NMR Characterisation of Sulfated Maltotriose C-C Linked Dimers 4–6
Generally, carbohydrate sulfation induces a significant low field shift of signals in NMR spectra of the proton and the carbon bearing the sulfate ester with respect to the same signals in the non-sulfated forms. Accordingly, the 1H- and 13C-NMR spectrometry (1D-NMR) are rightly considered an informative analytical approach, while signals are dispersed in reason of the both shifts by combining the two techniques in the heteronuclear 1H-13C bidimensional spectroscopy (HSQC NMR).
The discussion of NMR characterisation of
4–
6 deserves a particular attention.
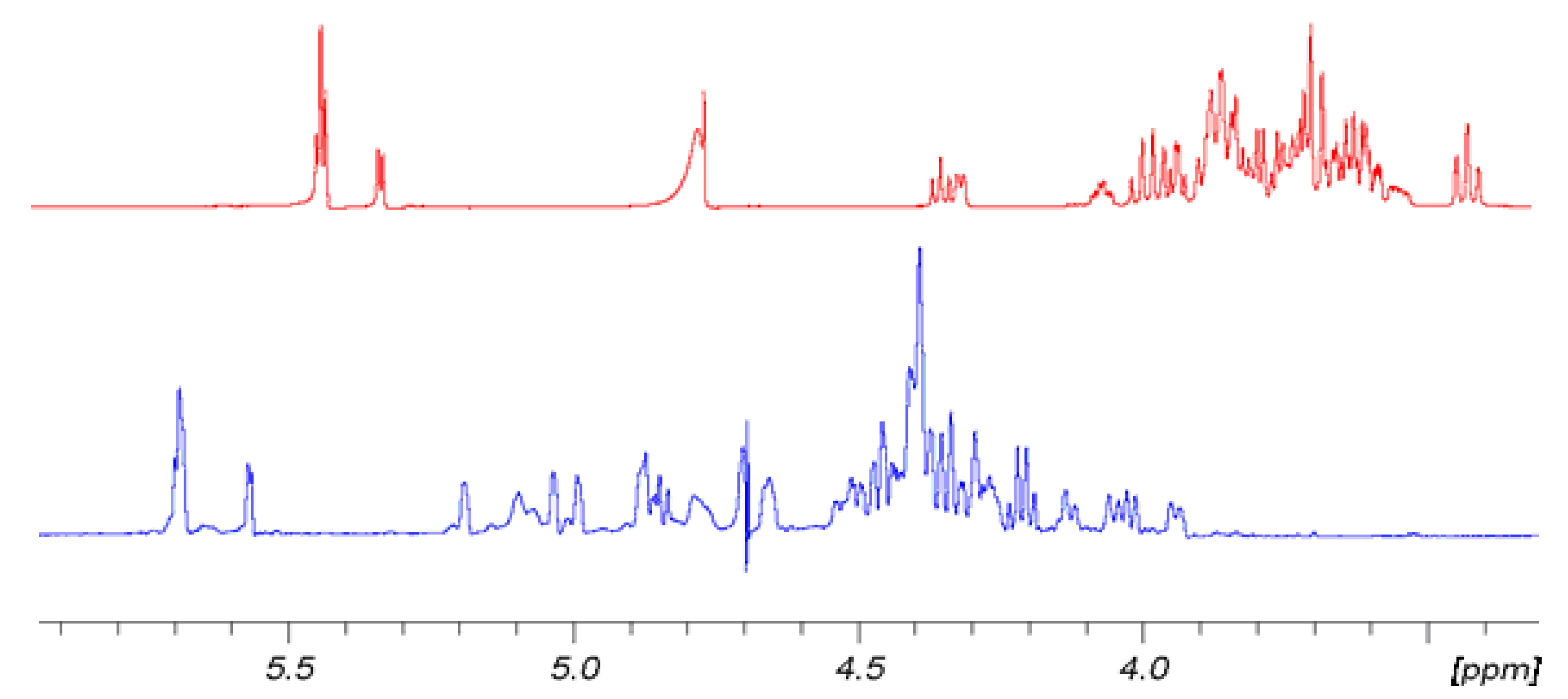
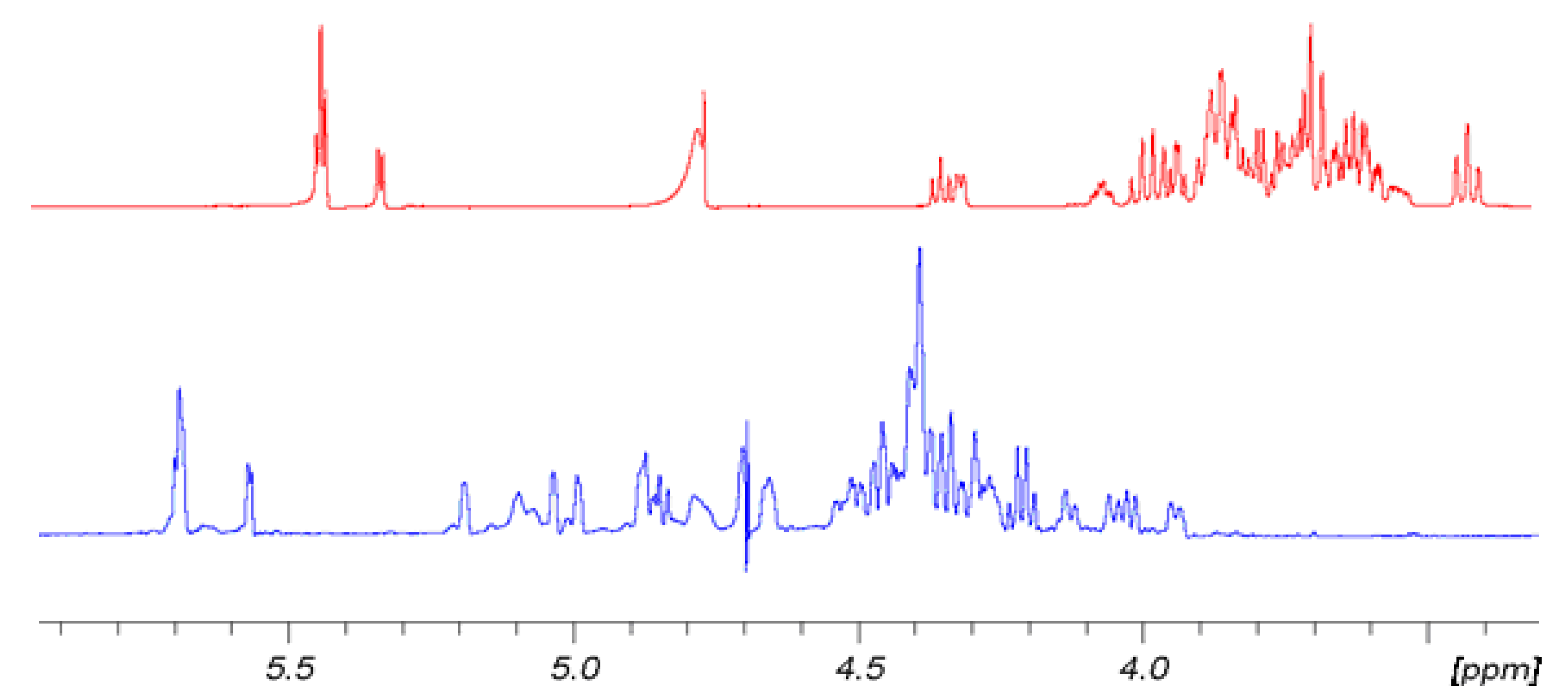
Figure 2 shows for convenience the shift effect on proton spectra of sample
6 with respect to the non-sulfated form sample
3. The trend is the same for
5 with respect to
2 and for
4 with respect to
1. It is evident how the sulfation process modifies the proton signal distribution.
6 shows very low signals in the region between 3.3 and 3.9 ppm where the protons of sample
3 resonate, indicating that non-sulfated structures are present only in trace. Signals assignment was done by using traditional bidimensional NMR techniques as COSY, TOCSY, HSQC, DEPT-HSQC and HMBC. In particular, HMBC was used to assign NMR signals to a specific ring of each oligosaccharide. Results are reported in
Table 1,
Table 2,
Table 3. For convenience, it is anticipated here that in sample
5 two main differently sulfated structures were identified, see
Table 3 for assignment.
Figure 2.
Comparison of 1H-NMR spectra of 6 (blue) at T = 303 K and its hydroxyl precursor 3 (red) at 298 K. 500 MHz in D2O.
Figure 2.
Comparison of 1H-NMR spectra of 6 (blue) at T = 303 K and its hydroxyl precursor 3 (red) at 298 K. 500 MHz in D2O.
Table 1.
1H and
13C NMR assignment of
4. In bracket:
J1 and
J2 values, in Hz. 500 MHz, D
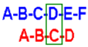
2O. Capital letters indicate the name of the ring, as shown in
Figure 1. Values expressed as ppm or (Hz).
Table 1.
1H and 13C NMR assignment of 4. In bracket: J1 and J2 values, in Hz. 500 MHz, D2O. Capital letters indicate the name of the ring, as shown in Figure 1. Values expressed as ppm or (Hz).
| H | A | B | C |
|---|
| 1H (J) | 13C | 1H (J) | 13C | 1H (J) | 13C |
|---|
| 1 | 5.59 (d, 2.9) | 96.6 | 5.48 (d, 3.4) | 97.8 | 4.33 (d, 1.0) | 69.9 |
| 2 | 4.75 (dd, 2.9, 5.3) | 75.1 | 4.41 (dd, 3.1, 7.4) | 76.3 | 5.01 (dd, 4.1) | 74.0 |
| 3 | 5.04 (dd, 4.4) | 76.9 | 4.87 (dd, 8.0, 9.2) | 79.9 | 5.01 (dd, 2.1) | 72.7 |
| 4 | 4.58 (dd, 3.8, 7.0) | 75.3 | 4.10 (dd, 8.3, 18.6) | 74.7 | 4.17 (dd, 2.5) | 72.4 |
| 5 | 4.21 (ddd, 3.6, 7.1) | 73.9 | 4.06 (ddd, 1.5, 2.1, 7.2) | 72.3 | 4.42 (ddd, 3.3, 8.5) | 77.1 |
| 6 | 4.38–4.33 (dd, 3.4, 10.8) | 68.7 | 4.34–4.31 (dd, 3.4, 10.6) | 68.7 | 4.27; 4.48 (dd, 4.9, 11.3) | 67.6 |
Table 2.
1H and 13C-NMR assignment of 6. Bracket: J1 and J2 values, in Hz. 500 MHz, D2O. Values expressed as ppm or (Hz).
Table 2.
1H and 13C-NMR assignment of 6. Bracket: J1 and J2 values, in Hz. 500 MHz, D2O. Values expressed as ppm or (Hz).
| H | A | B | C | D | E | F |
|---|
| 1H (J) | 13C | 1H (J) | 13C | 1H (J) | 13C | 1H(J) | 13C | 1H (J) | 13C | 1H (J) | 13C |
|---|
| 1 | 5.67 | 96.2 | 5.69 | 97 | 4 | 71.5 | 4.47 | 78.8 | 5.56 | 97.4 | 5.67 | 96.2 |
| (d, 9.5) | (d) | (d, 9.5) | (m) | (d, 3.5) | (d) |
| 2 | 4.77 | 75.4 | 4.51 | 77.1 | 4.86 | 72.9 | 5.18 | 75.7 | 4.49 | 77.1 | 4.78 | 75.4 |
| (m) | (dd) | (t) | (nd) | (dd) | (m) |
| 3 | 5.07 | 77.1 | 4.83 | 80 | 5.02 | 74.2 | 4.98 | 78.2 | 4.86 | 80 | 5.09 | 77.1 |
| (d, 3.0) | (t) | (d, 3.0) | (d) | (t) | (d, 3.0) |
| 4 | 4.66 | 75.3 | 4.19 | 74.1 | 4.28 | 72.5 | 4.33 | 75.3 | 4.21 | 74.1 | 4.65 | 75.3 |
| (dd) | (dd) | (m) | (m) | (dd) | (dd) |
| 5 | 4.25 | 73.4 | 4.11 | 72 | 4.39 | 76.7 | 3.93 | 76.5 | 4.03 | 72 | 4.255 | 73.4 |
| (q) | (t) | (m) | (dt) | (t) | (q) |
| 6 | 4.45–4.36 | 69.5–68.0 | 4.39–4.35 | 69.5–68.0 | 4.47–4.35 | 69.5–68.0 | 4.32 | 70.2 | 4.376 | 69.5–68.0 | 4.43–4.38 | 69.5–68.0 |
| (m) | (m) | (m) | (m) | (m) | (m) |
Table 3.
1H and 13C-NMR assignment of 5 *.
Table 3.
1H and 13C-NMR assignment of 5 *.
| H | A | B | C |
|---|
| 1H (J) | 13C | 1H (J) | 13C | 1H (J) | 13C |
|---|
| 1 | 5.63 (d, 3.5) | 96.5 | 5.56 (d, 3.7) | 97.1 | 3.93 (d, 8.6) | 76.8 |
| 2 | 4.52 | 76.0 | 4.82 | 80.0 | 4.64 | 76.1 |
| 3 | 4.81 | 77.8 | 5.04 | 78.0 | 4.07 | 78.8 |
| 4 | 4.51 | 78.7 | 4.21 | 72.6 | 3.77 | 79.2 |
| 5 | N.A. | N.A. | 4.60 | 75.5 | 4.32 | 78.5 |
| 6 | 4.36 | 70.5–68.4 | 4.39; 4.32 | 70.5–68.4 | 4.38 | 70.5–68.4 |
| H | a | b | c |
| 1H (J) | 13C | 1H (J) | 13C | 1H (J) | 13C |
| 1 | 5.76 (d, 3.8) | 99.2 | 5.60 (d, 3.7) | 96.5 | 3.66 (?) | 78.5 (?) |
| 2 | 4.41 | 77.8 | 4.72 | 79.9 | 3.88 (?) | 78.6 (?) |
| 3 | 4.81 | 79.9 | 5.02 | 77.2 | 4.01 (?) | 78.9 (?) |
| 4 | 4.09 | 74.6 | 4.19 | 73.4 | 3.77 (?) | 79.2 (?) |
| 5 | 4.16 | 72.2 | 4.59 | 75.4 | N.A. | N.A. |
| 6 | 4.35 | 70.5–68.4 | 4.39-4.32 | 70.5–68.4 | N.A. | 70.5–68.4 (?) |
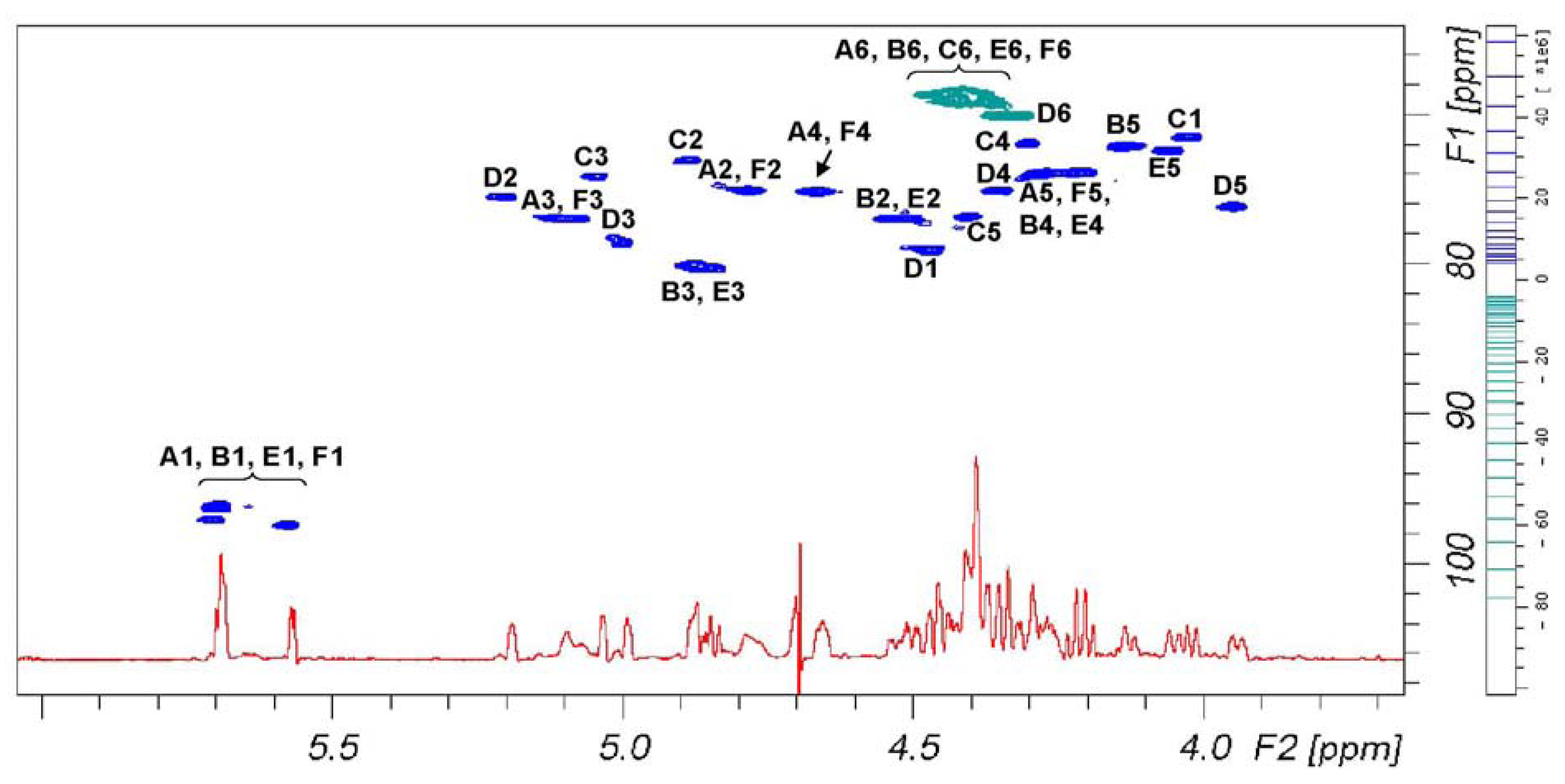
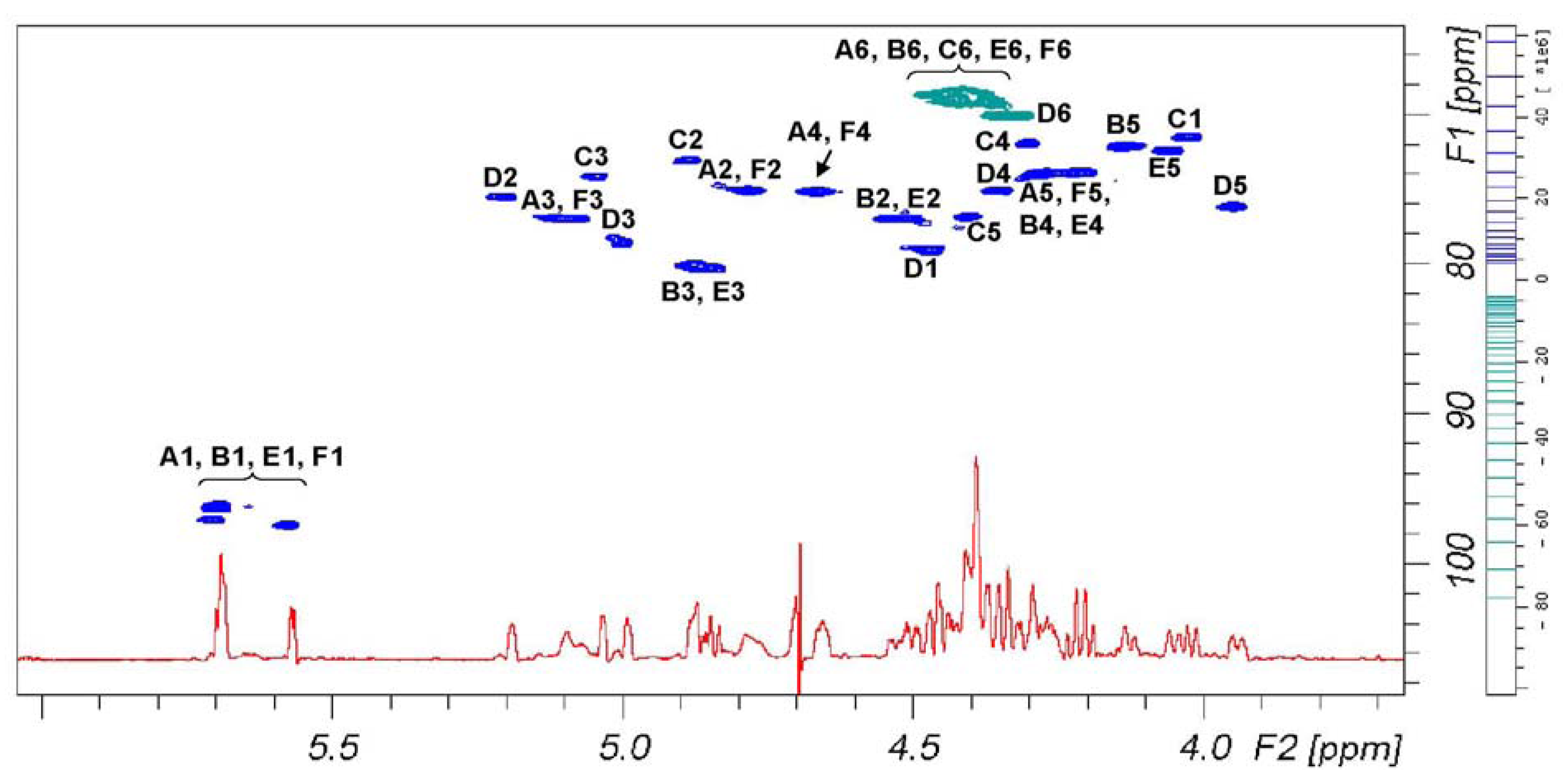
Figure 3 reports signal dispersion in the
1H-
13C DEPT-HSQC spectrum of
6 that discriminates between primary and secondary carbons. According to
Table 2, signals of secondary alcohol involved in the sulfation are comprise in a plane area defined by 4.44 and 5.25 ppm on proton axis and 72.5 and 80.7 ppm on carbon axis, while signals comprise between 4.50–4.28 and 68.0–70.0 ppm belong to primary alcohols. According to these assignments, we can see that primary hydroxyl groups are fully sulfated.
Figure 3.
1D 1H-NMR (red) and 1H-13C DEPT-HSQC NMR spectra (positive phase (blue) and negative phase (green), respectively secondary and primary carbons) of 6.
Figure 3.
1D 1H-NMR (red) and 1H-13C DEPT-HSQC NMR spectra (positive phase (blue) and negative phase (green), respectively secondary and primary carbons) of 6.
2.3. Evaluation of Sulfation Degree of 4–6
Generally speaking, due to the well-known reactivity of hydroxyl groups of sugar units, sulfation likely occurs at first on the primary alcohols, being the most reactive positions, as well as at the non-reducing end of hexamer structures. However, since the various positions of the glucose units reacts differently and the different structure of diastereoisomers seems to play a role, evaluation of the sulfation degree of each compound becomes necessary.
As stated by NMR theory, quantitative evaluation is possible by proton spectra. Because of 1D spectral signal overlapping only an average sulfation degree relative to H-2, H-3 of all rings could be calculated allowing, anyway, to compare the different compounds and to discriminate batch to batch preparations. Furthermore, the signal dispersion given by HSQC spectra is helpful to evaluate the sulfation pattern. In fact, if defined criteria of spectral measurements [
46] are followed, each signal corresponding to single sulfated positions should be quantified. We found that for D ring the H-3 signal volume is not proportional to the other signal volume. So, the following approach was used to give a complete sulfation degree and some information relative to the sulfation pattern of the different diastereoisomers.
At first, the absence of 13C signals, in the range of 61.0–67.0 ppm in HSQC spectra of α,α, α,β and β,β diastereoisomers, attributable to the presence of primary hydroxyl groups, allows us to assume that such positions are totally sulfated, being the limit of detection of the technique better than 2%. Therefore, their 6-O-sulfation degree is around 100%, as already mentioned above. The 1H-NMR spectra of α,α and α,β sulfated diastereoisomers were considered, using the four regular anomeric signals integral value as calibration area. On the base of the NMR assignment, the integral of all H-2, H-3, H-4 non-reducing sites bearing a sulfate were evaluated.
The chemical shift range from 4.55 to 5.25 ppm could be used to measure the content of 12 over 20 possible sulfate residues, being signals of H-2 of sulfated B and E rings partially overlapping other signals. Such evaluation requires to set the residual H-O-D solvent signal in a free signal area of the spectrum, as
Figure 3 shows, and to exclude it from integral area. The presence of residual solvent signals, suppressed by selective proton decoupling, could induce an underestimation of the two vicinal signals.
To consider the contribution to the DS of the two remaining H-2 signals of the B and E ring, the volume integral of the signals was measured in the HSQC spectra recorded according to Guerrini
et al. [
46] and compared to the volume area of the four anomeric signals used as calibrators. The percentage of recovery was confirmed fitting data also with other spectrum signals, in between the signal of two H-5 and two H-4 of rings, A, F, B and E. The average degree of sulfation is obtained with a good level of approximation adding up the three different measured values.
It is to note that intermediate sulfation degree gives complex spectra due to sequence effects. In such a case, spectra usually are affected by small change in chemical shifts, increment of number of signals and signals broadening giving a more signal crowded spectrum. It makes more complex the analysis of HSQC spectra. But being that the sulfate signal distribution remains inside the previous described area, the evaluation of an average sulfation degree is possible by 1D proton spectra that in the case become more potent than the 2D technique. Therefore the quantitative approach based on both 1D and 2D heteronuclear spectroscopy was preferred to the use of the single 2D one.
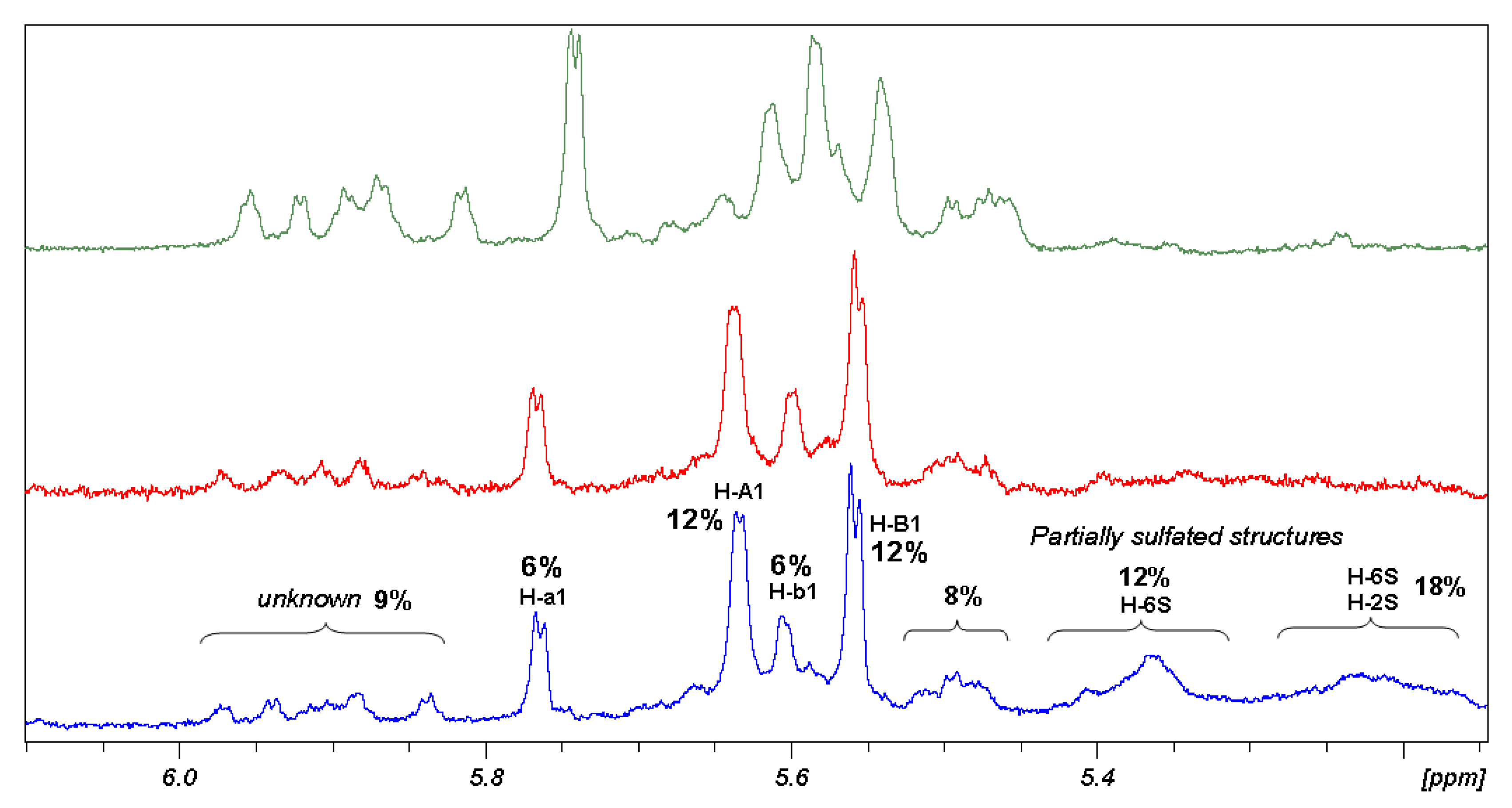
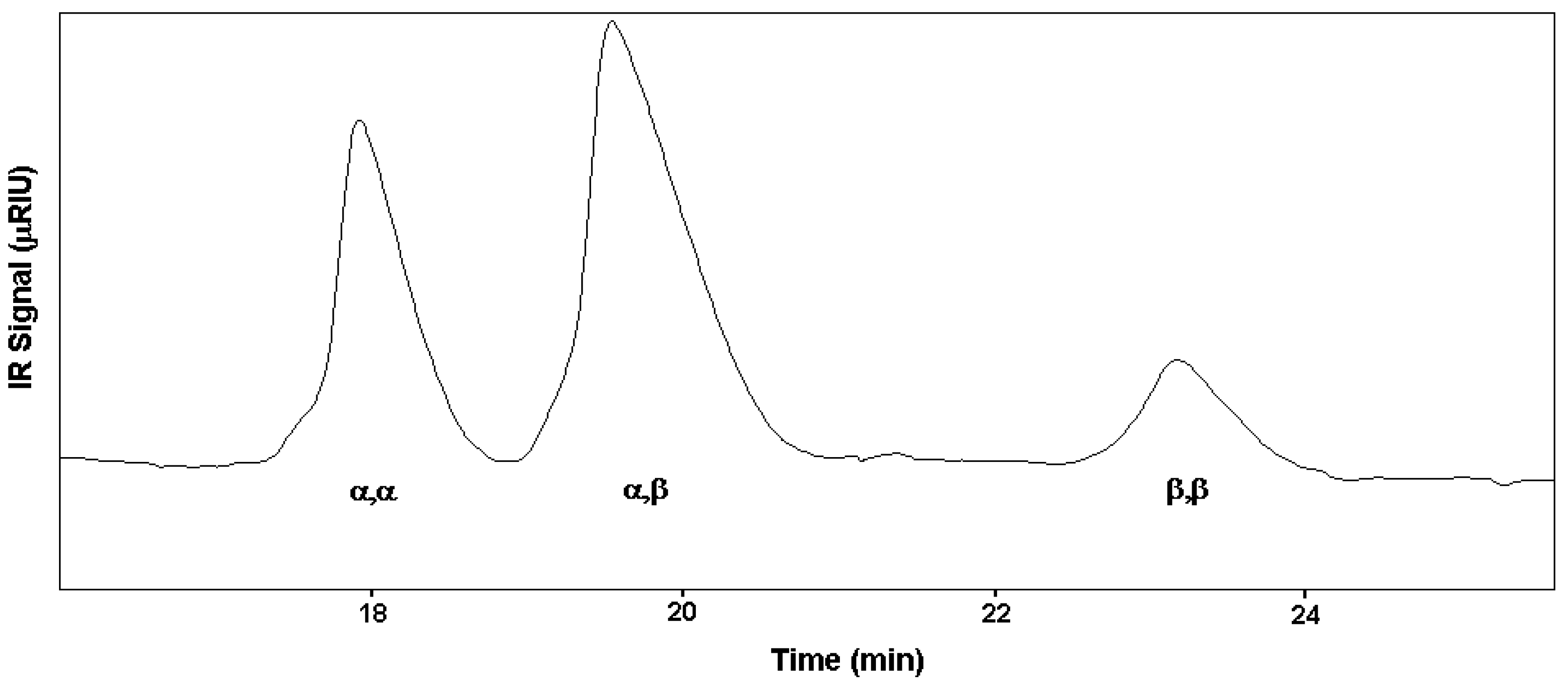
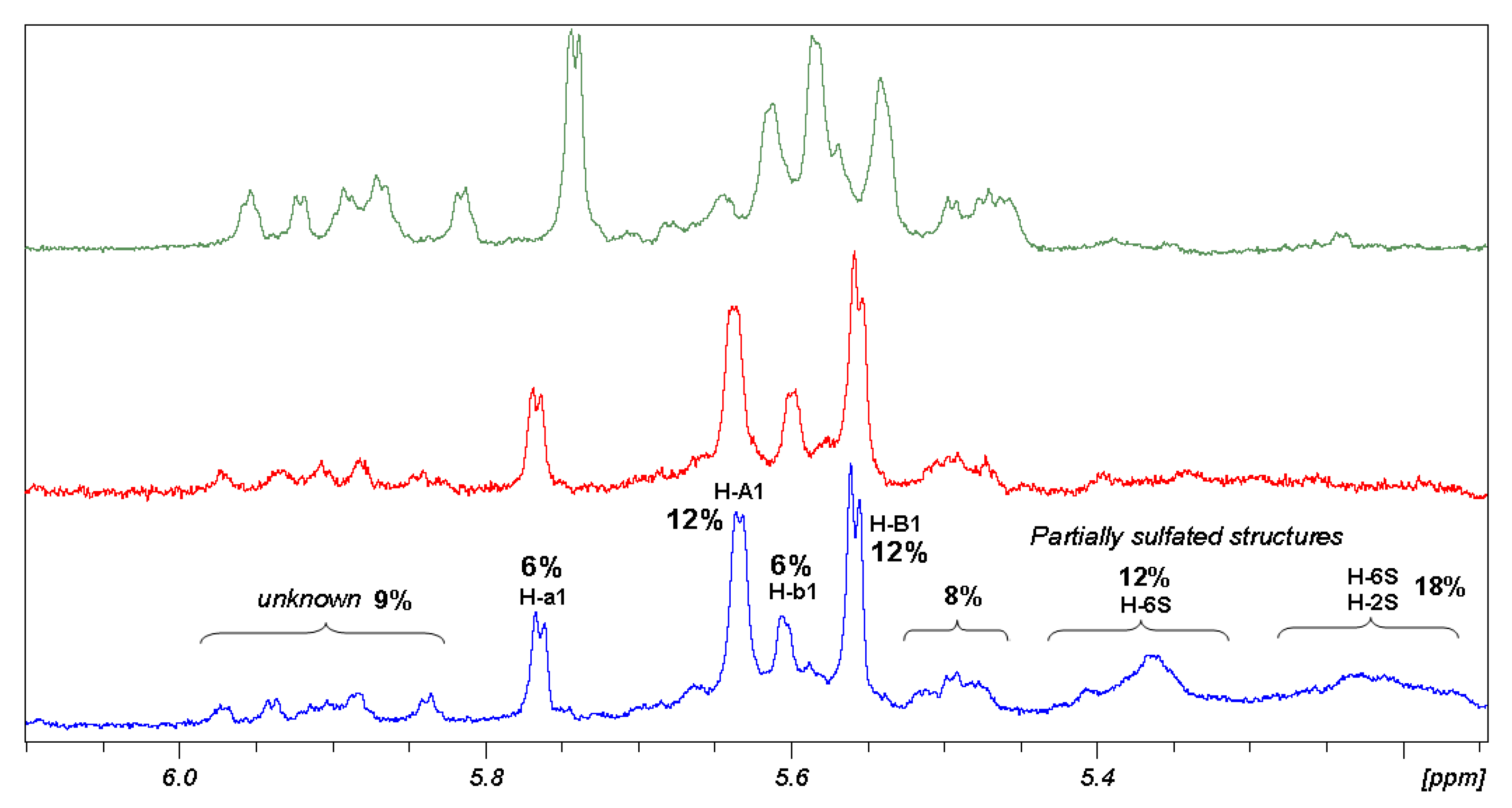
This is the case of β,β diasteroisomer. The range of chemical shift between 5.15 and 6.00 ppm is useful to analyse the composition of the β,β sample. The anomeric signals of proton spectrum compared with their sulfation intermediate (
Figure 4) show the progressive decrease of heterogeneity of the sample without reaching a final homogeneity. Groups of signals have been identified using different correlation techniques. As a result, β,β sample is composed of two main structures having a well-defined NMR pattern, which assignment was reported in
Table 3. Those structures represent more than 70% of the compounds and their ratio is 1:2. The remaining of the sample could not be studied because of structural polydispersion but in any case some 2D signal correlation allows to establish presence of signals of H-6 sulfated β,β hexamer (12%), H-6 and H-2 sulfated β,β hexamer (18%) and 20% of unknown structures of the β,β diastereoisomer.
Figure 4.
Comparison of 1H-NMR regions of anomeric protons of 5, after one (green), two (red) and three (blue) sulfation reactions.
Figure 4.
Comparison of 1H-NMR regions of anomeric protons of 5, after one (green), two (red) and three (blue) sulfation reactions.
The procedure used to calculate sulfation degree of maltotriose dimers interpreting 1D-NMR data is described in detail as follows. In the 5.80–5.35 ppm range of 1H-NMR only signals of anomeric protons of sulfated molecules are present, therefore the integral value divided for the numbers of protons resonating in that region was taken as the unit. The ratio between the integral of each region and the unit value, multiplied for the number of protons responsible for the signals, gives the degree of sulfation of functional groups resonating in that region, according to summarised formula (1):
Due to the complex NMR profiles, only clear assignment regions were selected to calculate the sulfation degree average value, applying formula (2):
where n is the number of protons and x indicates the number of the positions.
Results obtained, collected in
Table 4, show that
5 is the less sulfated structure, even after three sulfation steps (
Figure 4).
Table 4.
Sulfation degree obtained for α,α 4, α,β 6 and β,β 5 samples of C-C linked maltotriose dimers. Value for 5 is given as an average between major and minor products.
Table 4.
Sulfation degree obtained for α,α 4, α,β 6 and β,β 5 samples of C-C linked maltotriose dimers. Value for 5 is given as an average between major and minor products.
| Diastereoisomer | Sulfation degree |
|---|
| α,β 4 | >95% |
| α,β 6 | 84% |
| α,β 5 | 78% |
Percentage of sulfation obtained for α,α and α,β diastereoisomers means that likely 17–19 hydroxyl groups are sulfated among the 20 possible positions. β,β maltotriose dimer appears to have only 15 sulfated positions. To note, positions H-6 are always considered totally sulfated. This is an average value; it does not correspond to the DS of the two defined structures, being those chemical shifts aligned to that of the α,α and α,β diastereoisomers. In fact, assignment of 1H- and 13C-NMR spectra of β,β diastereoisomers reveal that the high degree of sulfation and the difference between the two structures are due to the H-4 site of ring A that in the major product is sulfated, having a signal at 4.51 ppm, while the species present in minor quantity bear a hydroxyl group in the same site. A second difference seems to be linked to site H-2 of C rings but it remain doubtful because of uncertain assignments of the minor structure.
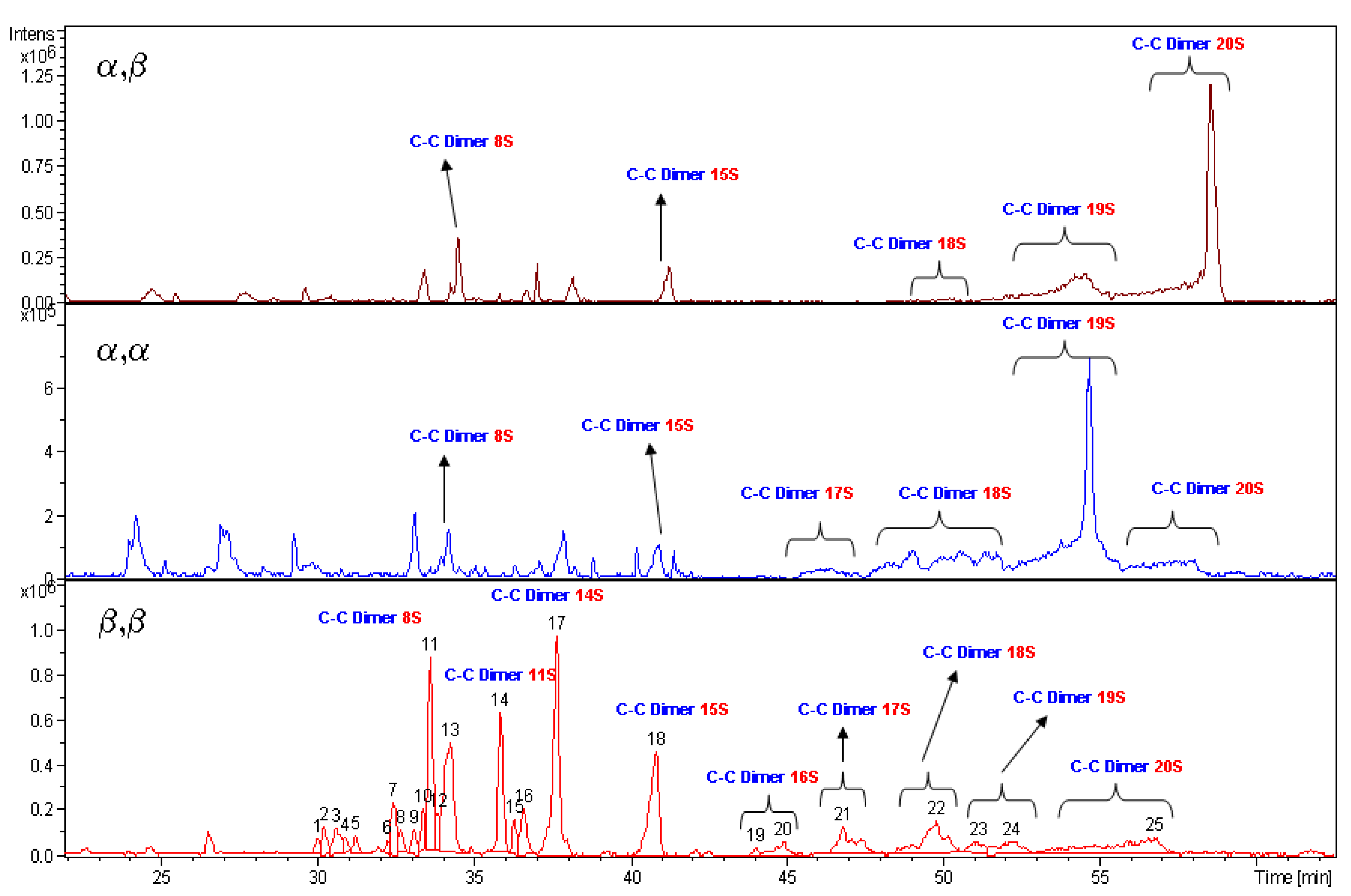
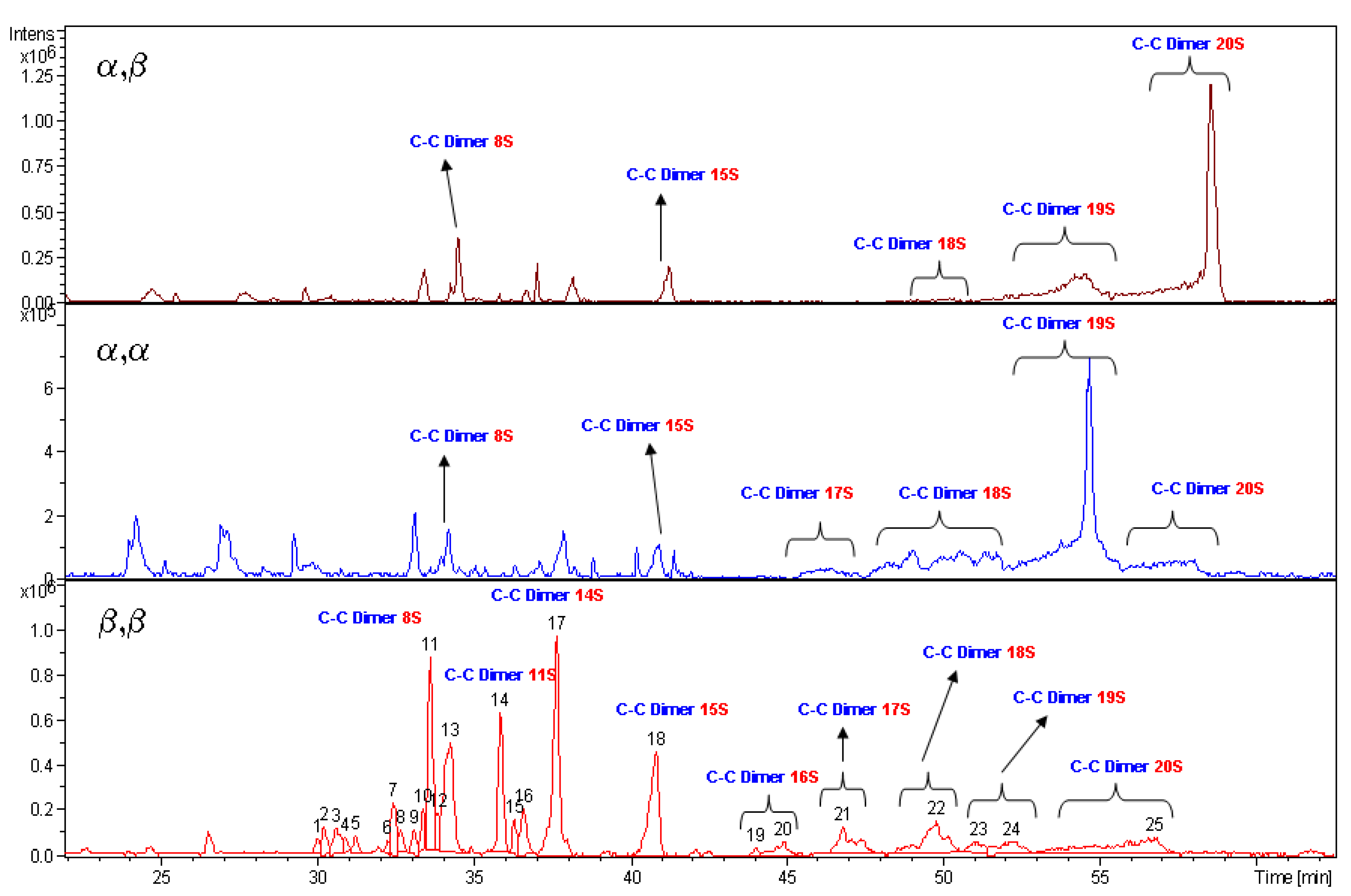
The high value of DS obtained from the above discussed NMR data are supported by LC-MS measurements. The species bearing 20 sulfate groups have been detected by mass spectroscopy for each diastereoisomer. The spectrum profiles, run under same liquid chromatography conditions, are reported in
Figure 5. Unfortunately the MS technique is not a quantitative one. The intensity of mass signals depends from the molecular mass, so the higher is the degree of sulfation of a molecule the lower is the signal intensity. In fact,
5 having a more heterogeneous pattern in the region of low molecular weight species show high signals.
4 and
6 diastereoisomers are characterised by the presence of low signals dispersion. It is relevant to note that, in spite of the previous statement,
4 and
5 show, each one, a high intensity and prevalent signal, respectively for structures bearing 19 and 20 sulfate residues.
2.4. NMR Characterisation of Sulfated Maltose C-C Linked Dimers 9,10. Comparison with 4–6
As evidenced by their NMR characterisation reported in
Table 5 and
Table 6,
9 and
10 were obtained at an almost total sulfation degree.
To evaluate the influence of the chain length,
1H-NMR chemical shift δ values for each position of
9,
10 and
4–
6 have been compared. Their difference was calculated for each position according to Equation 3, and summarised in
Table 7 and
Table 8.
Figure 5.
LC-MS analyses of α,β 6, α,α 4 and β,β 5 compounds. Labels indicate the number of sulfate groups for detected species.
Figure 5.
LC-MS analyses of α,β 6, α,α 4 and β,β 5 compounds. Labels indicate the number of sulfate groups for detected species.
Table 5.
1H and 13C-NMR assignment of 9.
Table 5.
1H and 13C-NMR assignment of 9.
| H | A | B |
|---|
| 1H (J) | 13C | 1H | 13C |
|---|
| 1 | 5.541 (3.4) | 97.9 | 4.376 (1.6) | 70.3 |
| 2 | 4.445 (9.8) | 77.2 | 5.054 (nd) | 72.8 |
| 3 | 4.848 (8.5) | 78.1 | 5.117 (3.4) | 74.2 |
| 4 | 4.501 (9.8) | 76.7 | 4.241 (nd) | 73.6 |
| 5 | 4.125 (m) | 72.2 | 4.472 (m) | 76.1 |
| 6 | 4.387 (3.5;11) | 67.9 | 4.386 (dd) | 69.1 |
| 6 | 4.439 (3;11) | - | 4.437 (dd) | - |
Table 6.
1H and 13C-NMR assignment of 10.
Table 6.
1H and 13C-NMR assignment of 10.
| H | A | B | C | D |
|---|
| 1H (J) | 13C | 1H (J) | 13C | 1H (J) | 13C | 1H (J) | 13C |
|---|
| 1 | 5.668 (3.8) | 97.0 | 4.019 (2.3;9.7) | 72.0 | 4.441 (9.8;1) | 78.8 | 5.555 (3.4) | 97.6 |
| 2 | 4.457 (dd) | 77.1 | 4.884 (2.6;2.4) | 73.3 | 5.189 (m?) | 75.7 | 4.447 (dd) | 77.2 |
| 3 | 4.732 (dd) | 78.3 | 5.109 (1.3;3.3) | 74.6 | 5.026 (1;3.2) | 78.1 | 4.810 (dd) | 78.2 |
| 4 | 4.496 (dd) | 76.7 | 4.275 (dd) | 73.5 | 4.353 (9) | 75.5 | 4.532 (dd) | 76.6 |
| 5 | 4.090 | 72.0 | 4.368 | 76.1 | 3.964 (3.2;3;9) | 76.1 | 4.072 | 72.1 |
| 6 | 4.396 ? | 69.2 | 4.43 | 68.9 | 4.307 (3.4) | 70.3 | 4.43 | 68.9 |
| 6 | - | - | 4.37 | - | - | - | 4.37 (dd) | - |
Table 7.
Arithmetical differences for the 1H-NMR chemical shift of 4 and 9, in D2O.
Table 8.
Arithmetical differences for the 1H-NMR chemical shift of 6 and 10, in D2O.
In general, the chemical shift values for analogous positions of corresponding rings are comparable. In the case of α,αand α,βdiastereoisomers, internal rings bearing the C-C bond show very similar
δ values, indicating that their chemical properties are not influenced by the length of the chain. As to the rings vicinal to the C-C linked ones, it has to be noted that they are external rings in the case of maltose dimers (rings A and D), while they are internal rings in the case of maltotriose dimers (rings B and E). Therefore, ring A of
9 has been compared both with rings A and B of the
4 observing that, ring A of the maltose dimer appears to be more similar to the internal ring B rather than the external one A of the maltotriose dimer, aside from position 4 that is involved in different bonds. Similarly, taking into consideration the α,β conformation,
δ values of external rings A and D of the maltose dimer are respectively similar to the rings B and E of the maltotriose dimer. A plausible explanation for such behaviour could be that the C-C bond acts as an element of rigidity, thus affecting the mobility of the rings, where the number of rotational degrees of freedom is proportional to the distance from the C-C bond. As a consequence, chemical shift values for the maltose dimers’ external rings are influenced by the presence of the C-C bond on the vicinal ring and behave likewise the internal ones B and E of the corresponding maltotriose dimers. The influence of the C-C bond was confirmed by obtaining the
1H-NMR spectrum of
10 and
6 at different temperatures (303 K, 328 K). The change in the chemical shift of the
1H-NMR resonances of H-1B and H-1C of
10 and
6, respectively, is less pronounced than that of anomeric protons of the external rings (
Supplementary Table S1).


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}