Extraction of Betulin, Trimyristin, Eugenol and Carnosic Acid Using Water-Organic Solvent Mixtures

Abstract
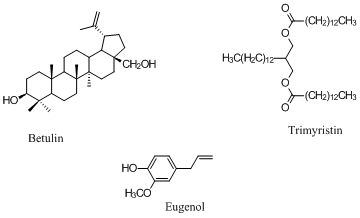
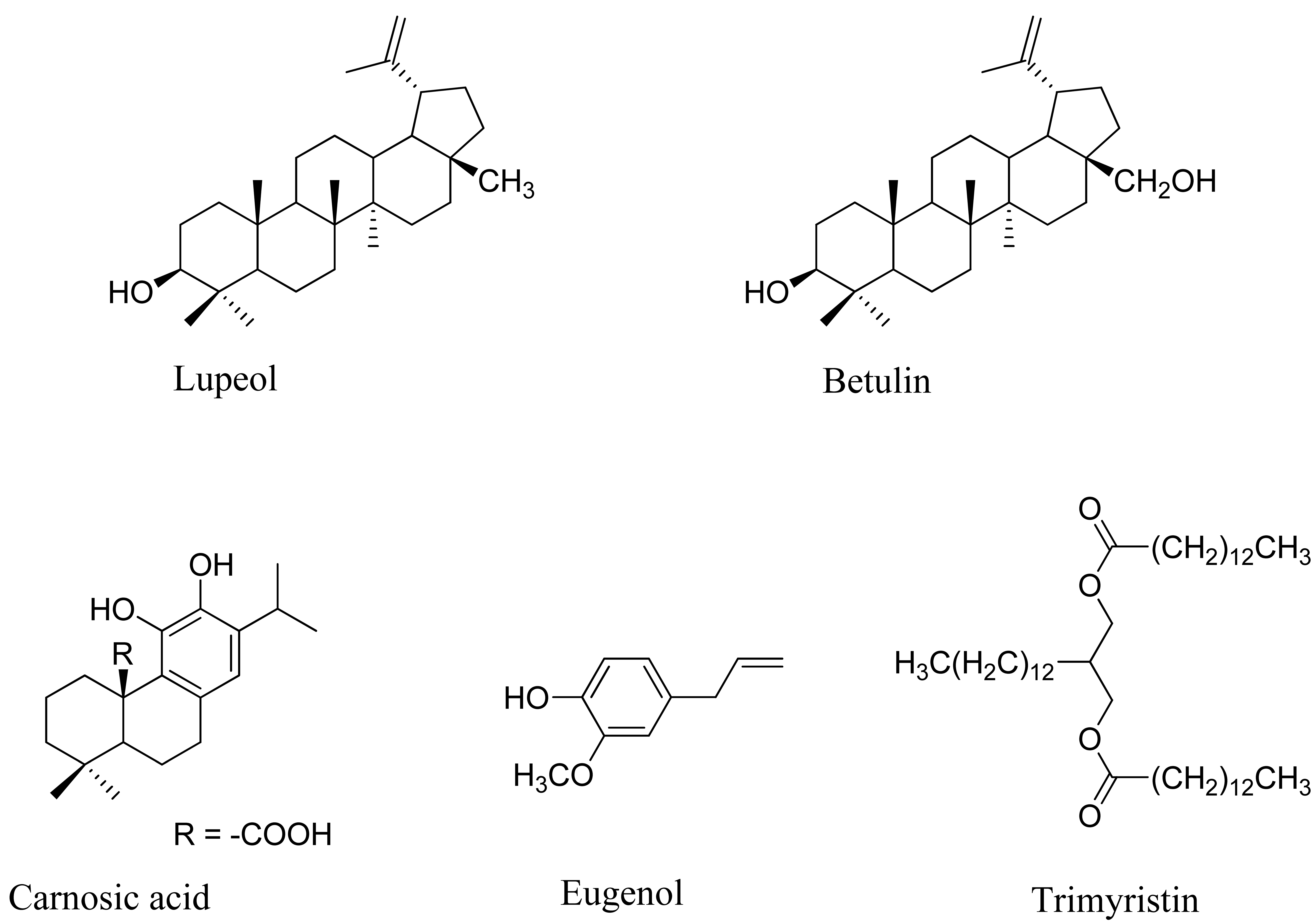
:
1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Solvent system | Extract (g) |
|---|---|
| Ethyl acetate:ethanol:water (4.5:4.5:1,v/v/v) | 4.4 |
| Ethyl acetate and ethanol (1:1,v/v) | 4.5 |
| Ethanol | 2.2 |
| Ethyl acetate | 2.5 |
| Ethyl acetate:ethanol:water (6:3:1) | 3.7 |
| Ethanol:water (1:1) | 2.0 |
| Solvent system | Betulin (g) |
|---|---|
| Ethyl acetate:ethanol:water (4.5:4.5:1, v/v/v) | 2.8 |
| Ethyl acetate:ethanol (1:1, v/v) | 2.2 |
| Hot toluene | 2.7 |
| Solvent | Carnosic acid mg/g |
|---|---|
| Ethyl acetate:ethanol:water | 32.5 |
| Acetone | 26.3 |
| Methanol | 20.5 |
| Plant | First extraction LC50 (mg of phenol/L) | Second extraction LC50 (mg of phenol/L) |
|---|---|---|
| Rosemary | 414.0 | >9409 |
| Sage | 787.9 | >16,530 |
| Thyme | 483.5 | >10,626 |
| Oregano | 591.5 | >12,830 |
| White oak | 879.5 | >19,338 |
3. Experimental Section
3.1. Extraction of Betulin from White Birch Bark
3.2. Extraction of Carnosic Acid from Rosemary and Analysis by HPLC
3.3. Extraction of Trimyristin
3.4. Extraction of Eugenol from Cloves
3.5. Extraction of Plant Material for Determining Free-Radical Scavenging Activity and Total Phenolic Content
3.6. Determination of the Free-Radical Scavenging Activity
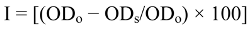
3.7. Determination of Total Phenol
4. Conclusions
Acknowledgments
References
- Rajab, M.S.; Bentley, M.D.; Alford, A.R.; Mendel, M.J. A new limonoid insect antifeedant from the fruit of Melia volkensii. J. Nat. Prod. 1988, 51, 168–171. [Google Scholar] [CrossRef]
- Mayer, R. Calycopterones and Calyflorenones, Novel Biflavonoids from Calycopteris floribunda. J. Nat. Prod. 1999, 62, 1274–1278. [Google Scholar] [CrossRef]
- Green, B.; Bentley, M.B.; Chung, B.Y.; Lynch, N.G.; Jensen, B.L. Allobetulin and its derivatives: Synthesis and biological activity. J. Chem. Educ. 2007, 84, 1985–1987. [Google Scholar] [CrossRef]
- Meksuriyen, D.; Cordell, G.A.; Ruangrungsi, N.; Tantivatana, P. Traditional medicinal plants of thailand, IX. 10-Hydroxy-11-methoxydracaenone and 7,10-Dihydroxy-11-methoxydracaenone from Dracaena loureiri. J. Nat. Prod. 1987, 50, 1118–1125. [Google Scholar] [CrossRef]
- Graber, M.A.; Gerwick, W.H. Antibiotic bisanthraquinones produced by a streptomycete isolated from a cyanobacterium associated with esteinascidia turbinate. J. Nat. Prod. 1998, 61, 677–680. [Google Scholar] [CrossRef]
- Tchouanken, J.-C.; Nyasse, B.; Tsamo, E. 7α,20(S)-Dihydroxy-4,24(28)-ergostadien-3-one from Entandrophragma utile. J. Nat. Prod. 1996, 59, 958–959. [Google Scholar] [CrossRef]
- Toki, M.; Ooi, T.; Kusumi, T. Sesterterpenoids and diterpenoids of the wax excreted by a scale insect, Ceroplastes pseudoceriferus. J. Nat. Prod. 1999, 62, 1504–1509. [Google Scholar] [CrossRef]
- Kikuzaki, H.; Sato, A.; Mayahara, Y.; Nakatani, N. Galloylglucosides from berries of Pimenta dioica. J. Nat. Prod. 2000, 63, 749–752. [Google Scholar] [CrossRef]
- Sree Rama Murthy, M.; Venkata Rao, E. Comparative toxicity assessment of three Tephrosia species on Artemia salina and animal cell lines. J. Nat. Prod. 1985, 48, 967–968. [Google Scholar] [CrossRef]
- Guidoin, M.; Yang, J.; Pichette, A.; Roy, C. Betulin isolation from birch bark by vacuum and atmospheric sublimation. A thermogravimetric study. Themochim. Acta 2003, 398, 153–166. [Google Scholar] [CrossRef]
- Puech, J.-L. Extraction and evolution of lignin products in Armagnac matured in oak. Am. J. Enol. Viticult. 1981, 32, 111–114. [Google Scholar]
- Cadahía, E.; Fernández De Simón, B.; Jalocha, J. Volatile compounds in Spanish, French, and American oak woods after natural seasoning and toasting. J. Agric. Food Chem. 2003, 51, 5923–5932. [Google Scholar] [CrossRef]
- Litchev, V. Changes in the profile of volatile compounds in wines stored under oxygen and their relationship with the browning process. Am. J. Enol. Viticult. 1989, 40, 34–35. [Google Scholar]
- Glabasnia, A.; Hofmann, T. Sensory-directed identification of taste-active ellagitannins in American (Quercus Alba L.) and European oak wood (Quercus robur L.) and quantitative analysis in Bourbon whiskey and oak-matured red wines. J. Agric. Food Chem. 2006, 54, 3380–3390. [Google Scholar] [CrossRef]
- O’Conell, M.M.; Bentley, M.D.; Campbell, C.S.; Cole, B.J.W. Betulin and lupeol in bark from four white-barked birches. Phytochemistry 1988, 27, 2175–2176. [Google Scholar]
- Chen, Q.H.; Fu, M.L.; Liu, J.; Zang, H.F.; He, G.Q.; Ruan, H. Optimization of ultrasonic-assisted extraction (UAE) of betulin from white birch bark using response surface methodology. Ultrason. Sonochem. 2009, 16, 599–604. [Google Scholar] [CrossRef]
- Albu, S.; Joyce, E.; Paniwnyk, L.; Lorimer, J.P.; Mason, T.J. Potential for the use of ultrasound in the extraction of antioxidants from Rosmarinus officinalis for the food and pharmaceutical industry. Ultrason. Sonochem. 2004, 11, 261–265. [Google Scholar] [CrossRef]
- Nakatani, N. Phenolic Compounds in Food and Their Effects on Health II; Juliani, H.R., Welch, C., Asante-Dartey, J., Acquaye, D., Wang, M., Simon, J.E., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1992; Chapter 6. [Google Scholar]
- Tena, M.T.; Valcárcel, M.; Hidalgo, P.J.; Ubera, J.L. Supercritical fluid extraction of natural antioxidants from rosemary: Comparison with liquid solvent sonication. Anal. Chem. 1997, 69, 521–526. [Google Scholar]
- Basile, A.; Jimnez-Carmona, M.M.; Clifford, A.A. Extraction of rosemary by superheated water. J. Agric. Food Chem. 1998, 46, 5205–5209. [Google Scholar] [CrossRef]
- Hidalgo, P.J.; Ubera, J.L.; Tena, M.T.; Valcárcel, M. Determination of the carnosic acid content in wild and cultivated Rosmarinus officinalis. J. Agric. Food Chem. 1998, 46, 2624–2627. [Google Scholar]
- Ibáñez, E.; Oca, A.; de Murga, G.; López-Sebastián, S.; Tabera, J.; Reglero, G. Supercritical fluid extraction and fractionation of different preprocessed Rosemary plants. J. Agric. Food Chem. 1999, 47, 1400–1404. [Google Scholar] [CrossRef]
- Ibáñez, E.; Kubotova, A.; Senorans, F.J.; Cavero, S.; Reglero, G.; Hawthorne, S.B. Subcritical water extraction of antioxidant compounds from rosemary plants. J. Agric. Food Chem. 2003, 51, 375–382. [Google Scholar]
- Miura, K.; Kikuzaki, H.; Nakatani, N. Antioxidant activity of chemical components from Sage (Salvia officinalis L.) and Thyme (Thymus vulgaris L.) measured by the oil stability index method. J. Agric. Food Chem. 2002, 50, 1845–1851. [Google Scholar] [CrossRef]
- Milos, M.; Mastelic, J.; Jerkovic, I. Chemical composition and antioxidant effect of glycosidically bound volatile compounds from oregano (Origanum vulgare L. ssp. hirtum). Food Chem. 2000, 71, 79–83. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant determination by use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Folin, O.; Ciocalteu, V. On tyrosine and tryptophane determination in proteins. J. Biol. Chem. 1927, 73, 627–650. [Google Scholar]
- Lewis, K.G. Triterpene constituents of fruits of Osage orange, Maclura pomifera. J. Chem. Soc. 1959, 73–75. [Google Scholar] [CrossRef]
- Postovskn, I.Ya. Structure of betulin. J. Russ. Phys.-Chem. Soc. 1930, 62, 101–109. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lugemwa, F.N. Extraction of Betulin, Trimyristin, Eugenol and Carnosic Acid Using Water-Organic Solvent Mixtures. Molecules 2012, 17, 9274-9282. https://doi.org/10.3390/molecules17089274
Lugemwa FN. Extraction of Betulin, Trimyristin, Eugenol and Carnosic Acid Using Water-Organic Solvent Mixtures. Molecules. 2012; 17(8):9274-9282. https://doi.org/10.3390/molecules17089274
Chicago/Turabian StyleLugemwa, Fulgentius N. 2012. "Extraction of Betulin, Trimyristin, Eugenol and Carnosic Acid Using Water-Organic Solvent Mixtures" Molecules 17, no. 8: 9274-9282. https://doi.org/10.3390/molecules17089274
APA StyleLugemwa, F. N. (2012). Extraction of Betulin, Trimyristin, Eugenol and Carnosic Acid Using Water-Organic Solvent Mixtures. Molecules, 17(8), 9274-9282. https://doi.org/10.3390/molecules17089274