Preparative Isolation and Purification of Three Sesquiterpenoid Lactones from Eupatorium lindleyanum DC. by High-Speed Counter-Current Chromatography

Abstract
:1. Introduction
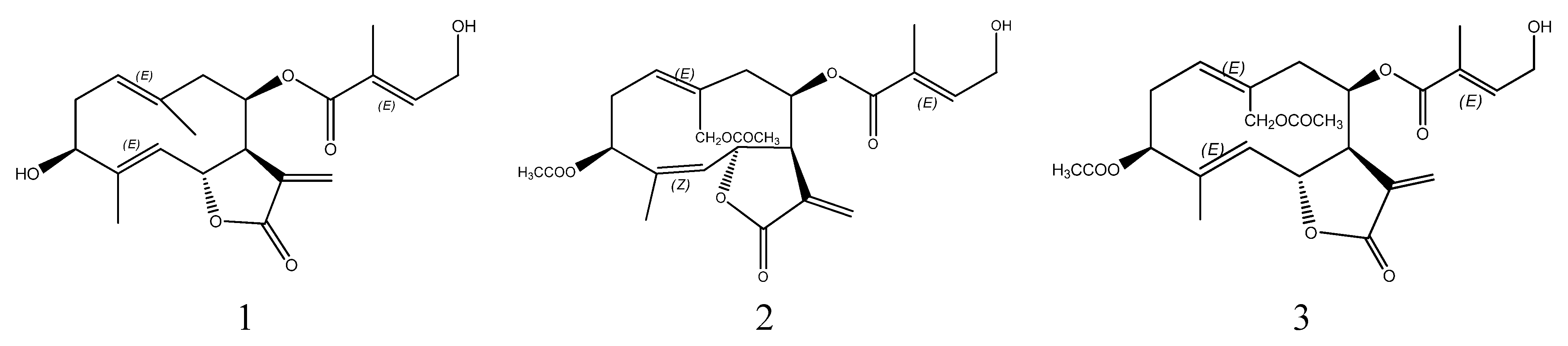
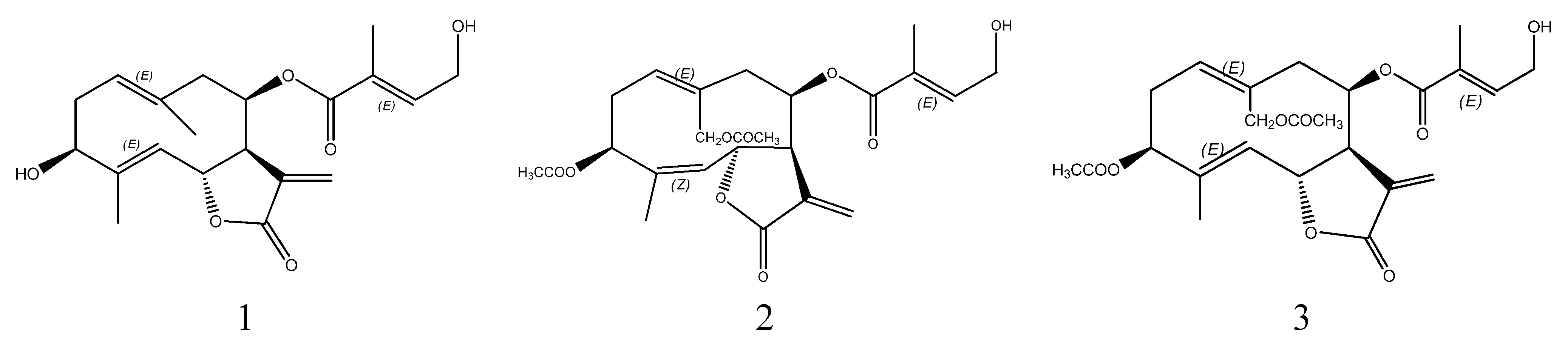
2. Results and Discussion
2.1. Selection of Two-Phase Solvent System and Other Separation Conditions of HSCCC

| n-hexane-ethyl acetate-methanol-water | Partition coefficient (K) | ||
|---|---|---|---|
| (v/v/v/v) | K1 | K2 | K3 |
| 2:4:3:2 | 0.06 | 0.14 | 0.28 |
| 1:4:3:3 | 0.26 | 0.61 | 1.26 |
| 1:4:2:3 | 0.54 | 1.40 | 1.94 |
| 1:8:4:6 | 1.64 | 3.38 | 5.51 |
2.2. HSCCC Separation of Crude Sample
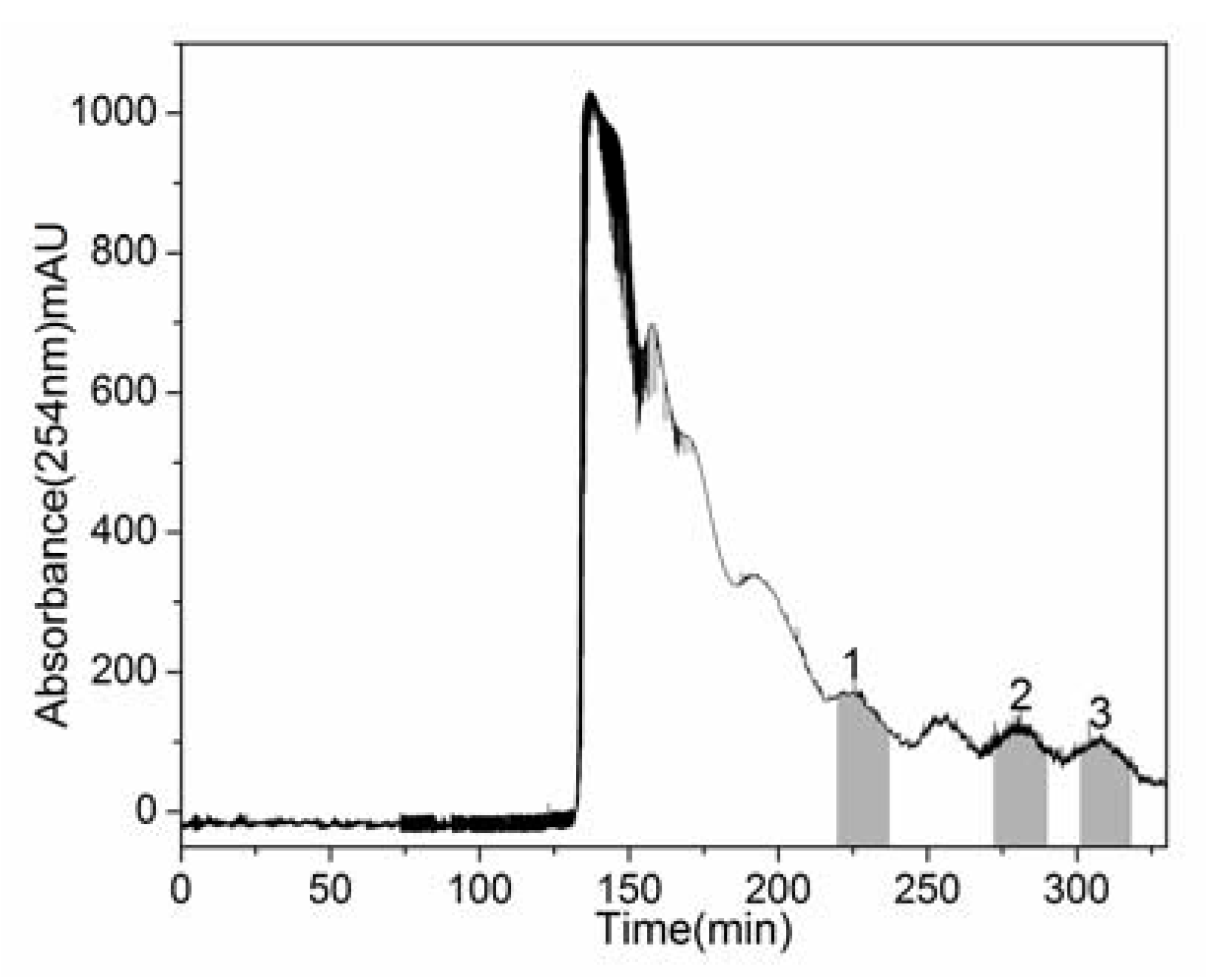
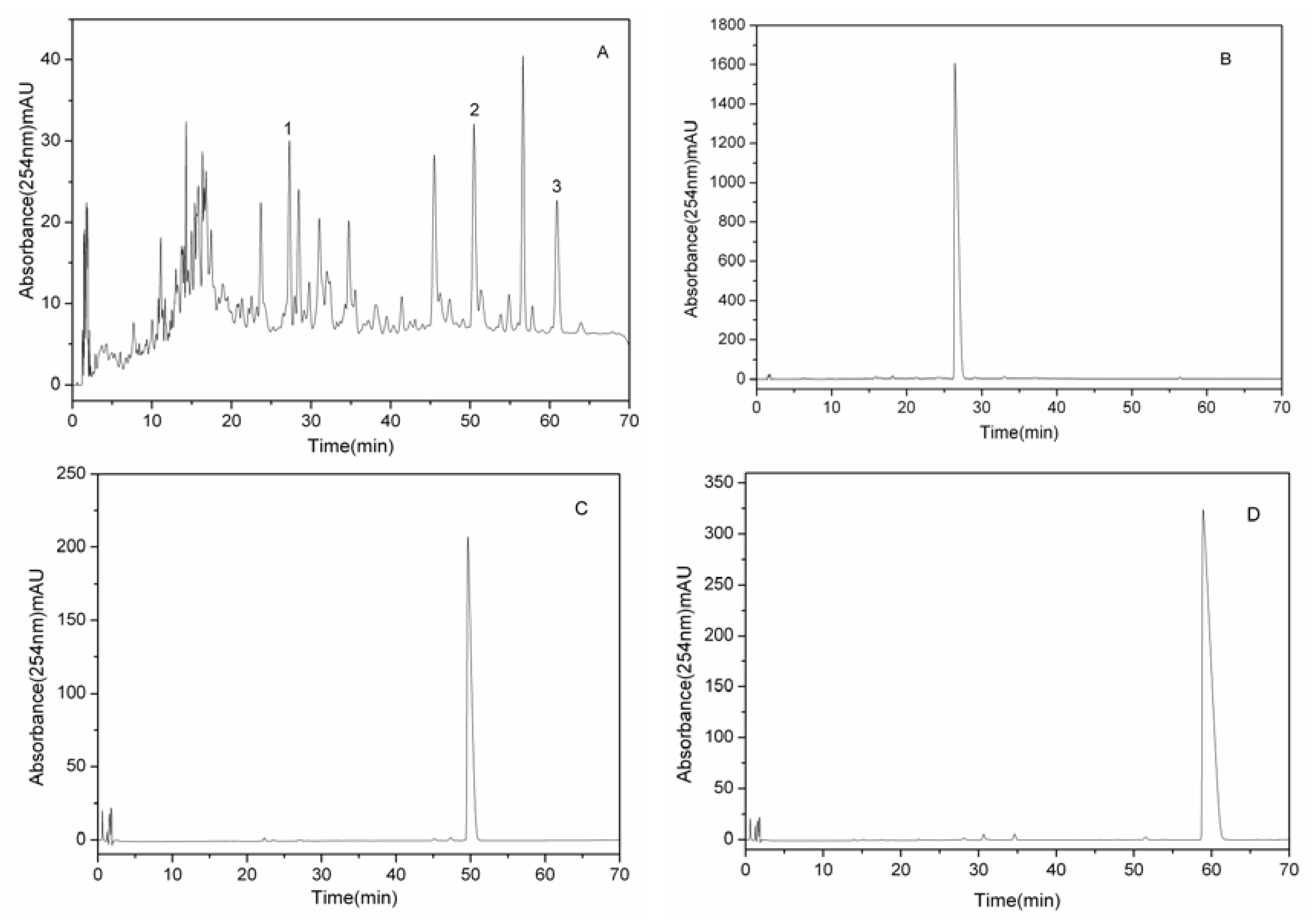
{kind=link}
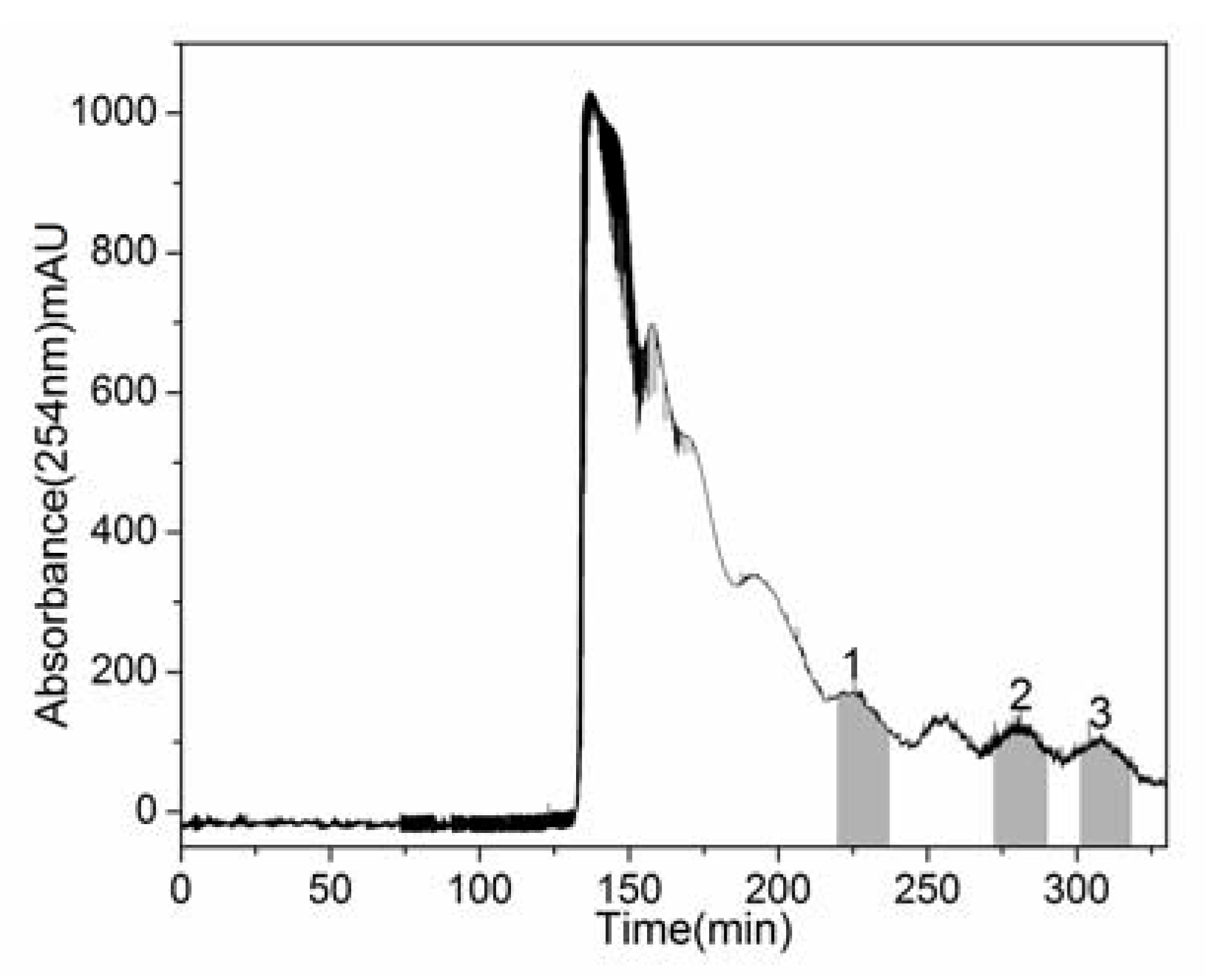{kind=link}
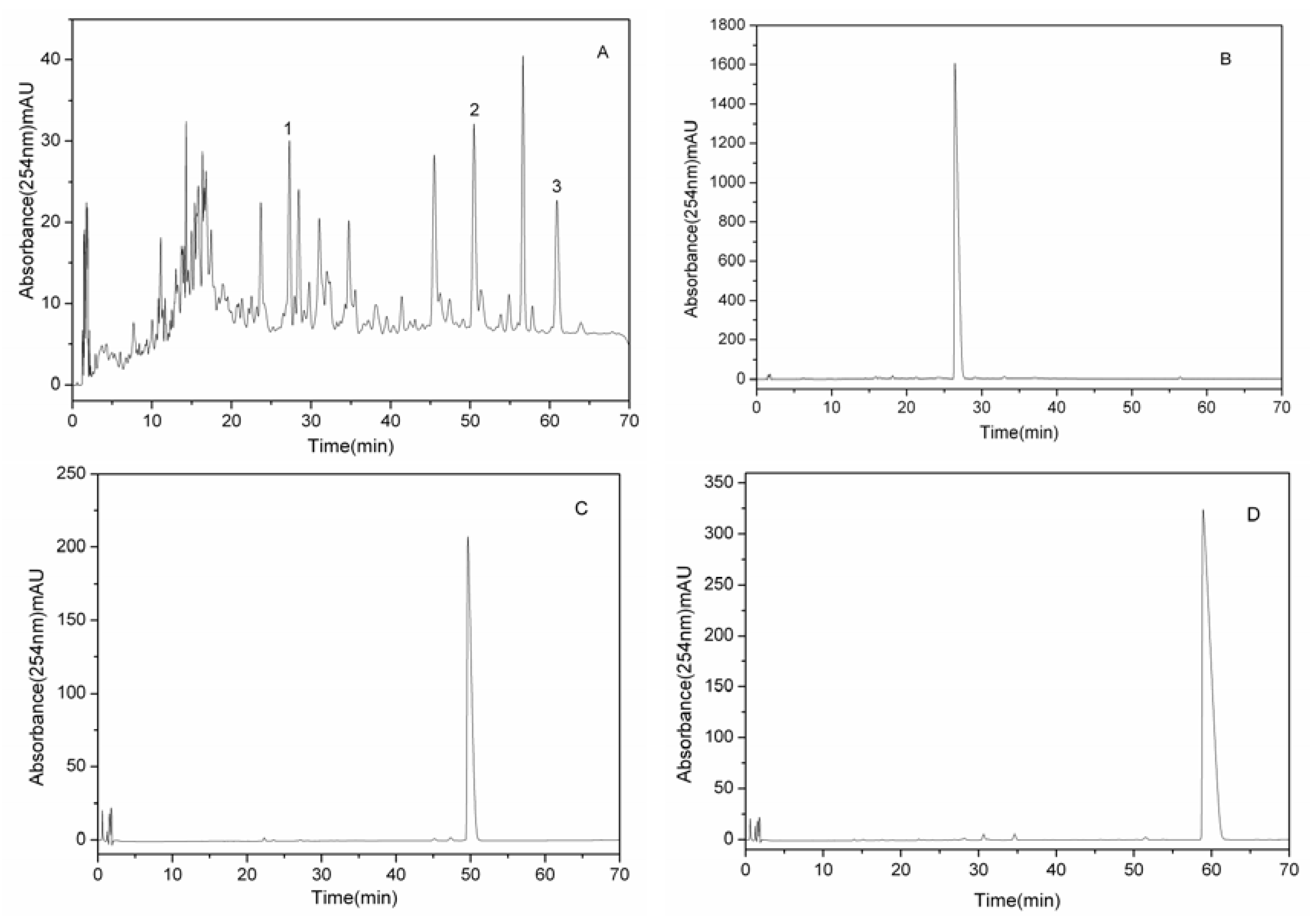{kind=link}
3. Experimental
3.1. Reagents and Materials
3.2. Apparatus
3.3. Plant Material
3.4. Preparation of Samples
3.5. Selection of Two-Phase Solvent System
3.6. Preparation of Two-Phase Solvent System and Sample Solution
3.7. HSCCC Separation Procedure
3.8. Analyses and Identification of HSCCC Peak Fractions
3.9. Structural Identification
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Editorial Committee of the Administration Bureau of Traditional Chinese Medicine. In Chinese Materia Medica; Shanghai Scientific & Technical Publishers: Shanghai, China, 1999; Volume 7, pp. 839–841.
- Ji, L.L.; Luo, Y.M.; Yan, G.L. Studies on the antimicrobial activities of extracts from Eupatorium lindleyanum DC against food spoilage and food-borne pathogens. Food Control 2008, 19, 995–1001. [Google Scholar] [CrossRef]
- Yan, G.L.; Ji, L.L.; Luo, Y.M.; Hu, Y.H. Antioxidant activities of extracts and fractions from Eupatorium lindleyanum DC. Molecules 2011, 16, 5998–6009. [Google Scholar] [CrossRef]
- Huo, J.; Yang, S.P.; Ding, J.; Yue, J.M. Two new cytotoxic sesquiterpenoids from Eupatorium lindleyanum DC. J. Integr. Plant Biol. 2006, 48, 473–477. [Google Scholar] [CrossRef]
- Zhang, M.L.; Wu, M.; Zhang, J.J.; Irwin, D.; Gu, Y.C.; Shi, Q.W. Chemical constituents of plants from the genus Eupatorium. Chem. Biodivers. 2008, 5, 40–55. [Google Scholar] [CrossRef]
- Yang, N.Y.; Duan, J.A.; Shang, E.X.; Tian, L.J. Analysis of sesquiterpene lactones in Eupatorium lindleyanum by HPLC-PDA-ESI-MS/MS. Phytochem. Anal. 2010, 21, 144–149. [Google Scholar]
- Yang, N.Y.; Qian, S.H.; Duan, J.A.; Tian, L.J. Studies on the chemical constituents of Eupatorium lindleyanum. J. China Pharm. Univ. 2003, 34, 220–221. [Google Scholar]
- Zhu, L.C.; Li, H.; Liang, Y.; Wang, X.H.; Xie, H.C.; Zhang, T.Y.; Ito, Y. Application of high-speed counter-current chromatography and preparative high-performance liquid chromatography mode for rapid isolation of anthraquinones from Morinda officinalis How. Sep. Purif. Technol. 2009, 70, 147–152. [Google Scholar] [CrossRef]
- Wei, Y.; Du, S.; Ito, Y. Enantioseparation of lomefloxacin hydrochloride by high-speed counter-current chromatography using sulfated-beta-cyclodextrin as a chiral selector. J. Chromatogr. B 2010, 878, 2937–2941. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y. High-speed counter current chromatography. Crit. Rev. Anal. Chem. 1986, 17, 65–143. [Google Scholar] [CrossRef]
- Gutzeit, D.; Wray, V.; Winterhalter, P.; Jerz, G. Preparative isolation and purification of flavonoids and protocatechuic acid from sea buckthorn juice concentrate (Hippophae rhamnoides L. ssp rhamnoides) by high-speed counter-current chromatography. Chromatographia 2007, 65, 1–7. [Google Scholar]
- Weisz, A.; Ito, Y. Performance comparison of three types of high-speed counter-current chromatographs for the separation of components of hydrophilic and hydrophobic color additives. J. Chromatogr. A 2011, 1218, 6156–6164. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Feng, B.; Zhu, R.; Ma, J.; Wang, W. Preparative isolation of three anthraquinones from Rumex japonicus by high-speed counter-current chromatography. Molecules 2011, 16, 1201–1210. [Google Scholar] [CrossRef]
- He, F.; Bai, Y.; Wang, J.; Wei, J.; Yu, C.; Li, S.; Yang, W.; Han, C. Isolation and purification of oridonin from the whole plant of Isodon rubescens by high-speed counter-current chromatography. Molecules 2011, 16, 7949–7957. [Google Scholar] [CrossRef]
- Schroder, M.; Vetter, W. Investigation of unsaponifiable matter of plant oils and isolation of eight phytosterols by means of high-speed counter-current chromatography. J. Chromatogr. A 2012, 1237, 96–105. [Google Scholar] [CrossRef]
- Wang, J.; Gao, H.; Zhao, J.; Wang, Q.; Zhou, L.; Han, J.; Yu, Z.; Yang, F. Preparative separation of phenolic compounds from Halimodendron halodendron by high-speed counter-current chromatography. Molecules 2010, 15, 5998–6007. [Google Scholar] [CrossRef]
- Jakupovic, J.; Sun, H.; Bohlmann, F.; King, R. Further sesquiterpene lactones from Eupatorium altissimum. Planta Med. 1987, 53, 97–98. [Google Scholar] [CrossRef]
- Yang, N.Y.; Qian, S.H.; Duan, J.A.; Li, P.; Tian, L.J. Two new sesquiterpenes from Eupatotium lindleyanum. Chin. Chem. Lett. 2005, 16, 1223–1226. [Google Scholar]
- Sample Availability: Samples of Eupatorium lindleyanum DC. are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yan, G.; Ji, L.; Luo, Y.; Hu, Y. Preparative Isolation and Purification of Three Sesquiterpenoid Lactones from Eupatorium lindleyanum DC. by High-Speed Counter-Current Chromatography. Molecules 2012, 17, 9002-9009. https://doi.org/10.3390/molecules17089002
Yan G, Ji L, Luo Y, Hu Y. Preparative Isolation and Purification of Three Sesquiterpenoid Lactones from Eupatorium lindleyanum DC. by High-Speed Counter-Current Chromatography. Molecules. 2012; 17(8):9002-9009. https://doi.org/10.3390/molecules17089002
Chicago/Turabian StyleYan, Guilong, Lilian Ji, Yuming Luo, and Yonghong Hu. 2012. "Preparative Isolation and Purification of Three Sesquiterpenoid Lactones from Eupatorium lindleyanum DC. by High-Speed Counter-Current Chromatography" Molecules 17, no. 8: 9002-9009. https://doi.org/10.3390/molecules17089002
APA StyleYan, G., Ji, L., Luo, Y., & Hu, Y. (2012). Preparative Isolation and Purification of Three Sesquiterpenoid Lactones from Eupatorium lindleyanum DC. by High-Speed Counter-Current Chromatography. Molecules, 17(8), 9002-9009. https://doi.org/10.3390/molecules17089002