A New Benzofuran Derivative from Flemingia philippinensis Merr. et Rolfe

Abstract
:1. Introduction
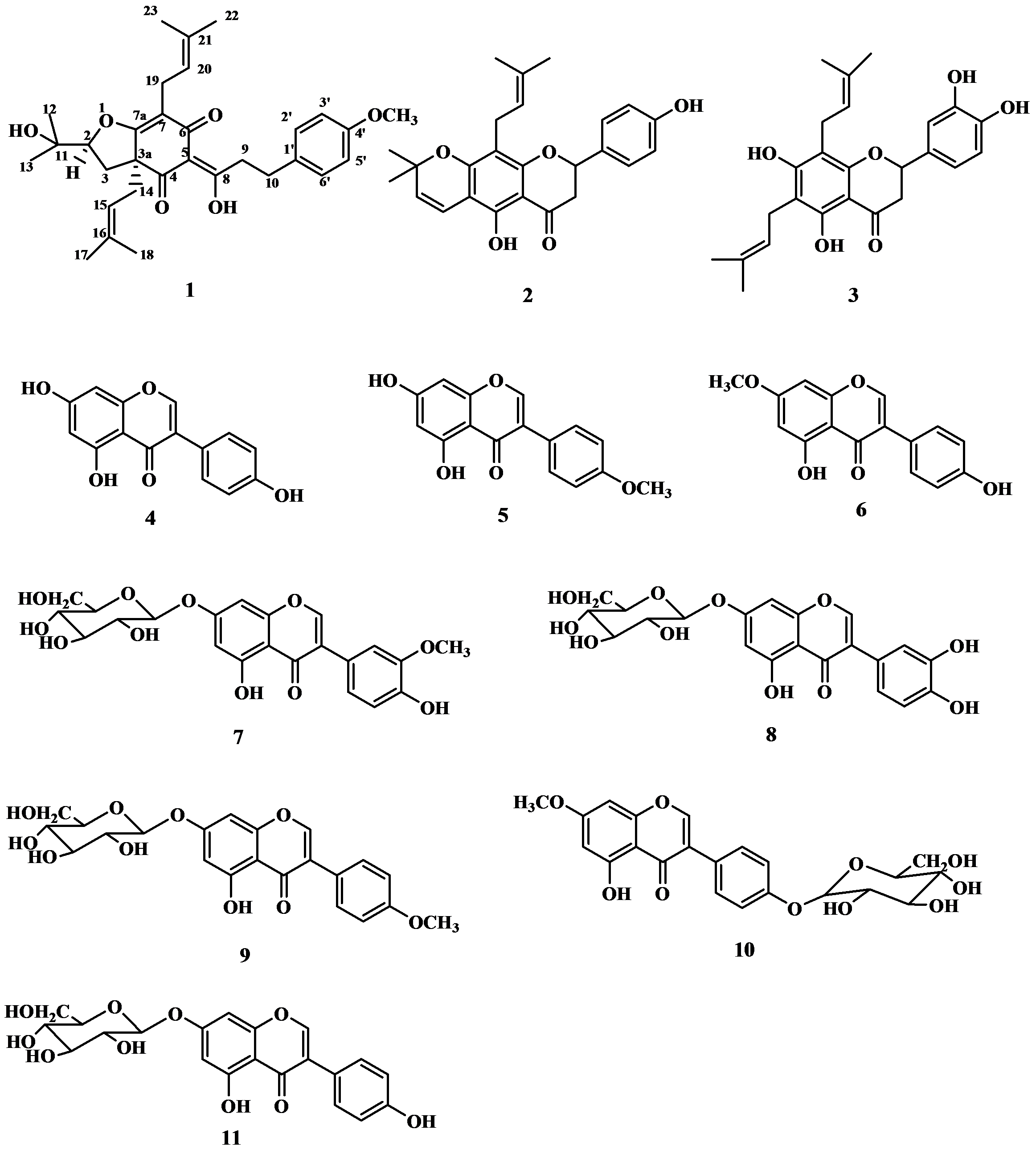
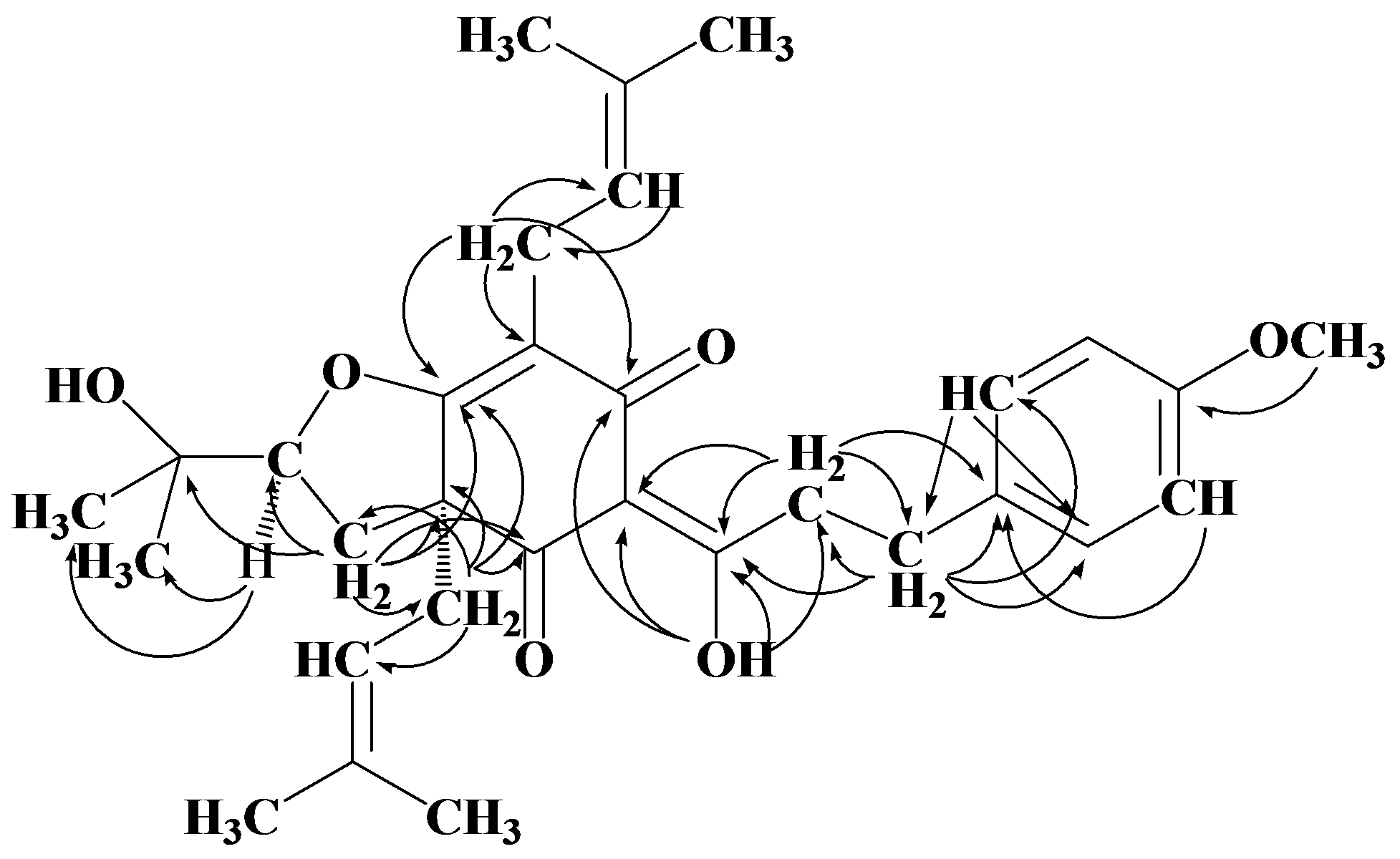
2. Results and Discussion

{kind=link}
{kind=link}
| Position | δH J(Hz) | δC | HMBC | COSY | NOE |
|---|---|---|---|---|---|
| 2 | 4.51 dd (6.5, 10) | 90.5 | C-12, 13 | H-3 | H-3, 12, 13, 14 |
| 3 | 2.16 m | 30.6 | C-2,3a, 4, 7a, 11, 14 | H-2 | H-13 |
| 3a | 60.6 | ||||
| 4 | 175.1 | ||||
| 5 | 107.3 | ||||
| 6 | 192.3 | ||||
| 7 | 106.5 | ||||
| 7a | 195.7 | ||||
| 8(-OH) | 18.84 s | 198.2 | C-6, 7, 8, 9 | ||
| 9 | 3.18 m | 40.0 | C-7, 8, 10, 1′ | ||
| 3.10 m | |||||
| 10 | 2.92 m | 31.0 | C-8, 9, 1′, 2′, 6′ | ||
| 2.85 m | |||||
| 1′ | 133.3 | ||||
| 2′ | 7.19 d (8.5) | 113.7 | C-10, 3′, 4′, 6′ | ||
| 3′ | 6.82 d (8.5) | 129.4 | C-1′, 4′, 5′ | OCH3-4′ | |
| 4′ | 158.0 | ||||
| 5′ | 6.82 d (8.5) | 129.4 | C-1′, 3′, 4′ | OCH3-4′ | |
| 6′ | 7.19 d (8.5) | 113.8 | C-10, 2′, 4′, 5′ | ||
| 11 | 71.2 | ||||
| 12 | 1.33 s | 26.6 | C-2, 11, 13 | H-2 | |
| 13 | 1.16 s | 23.8 | C-2, 11, 12 | H-2 | |
| 14 | 2.50 dd (7.5, 14.0) | 38.7 | C-3a, 4, 7a, 3, 15, 16 | H-15 | H-2 |
| 2.39 dd (7.5, 14.0) | |||||
| 15 | 4.96 t (7.5) | 117.1 | C-17, 18 | H-14, 17, 18 | H-17 |
| 16 | 137.0 | ||||
| 17 | 1.66 s | 25.8 | C-15, 16, 18 | H-15 | H-15 |
| 18 | 1.52 s | 17.8 | C-15, 16, 17 | H-15 | |
| 19 | 3.10 m | 21.5 | C-4, 5, 6, 20, 21 | H-20, 23 | |
| 3.00 dd (7.5, 14.5) | |||||
| 20 | 5.11 t (7.5) | 121.5 | C-19, 22, 23 | H-19, 23 | H-23 |
| 21 | 132.1 | ||||
| 22 | 1.72 s | 17.8 | C-20, 21, 23 | ||
| 23 | 1.68 s | 25.6 | C-20, 21, 22 | H-19, 20 | |
| 4′(-OCH3) | 3.77 s | 55.2 | C-4′ | H-3′, 5′ |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation of Chemical Constituents
3.4. Flemiphilippinone A (1)
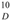 : +0.050° (c 0.10, CHCl3); UV (MeOH) λ max (log εmax): 277 (0.59), 223 (1.40) nm. IR (KBr) ν max: 3455, 2927, 2971, 1669, 1579, 1512, 1453, 1375, 1245, 1180, 1107, 1071, 1035, 826 cm−1. 1H- and 13C-NMR: see Table 1. EI MS m/z: 508 [M] + (13), 439 (100), 383 (15), 305 (31), 278 (11), 249 (29), 177 (13), 121 (100), 69 (48). HR EIMS m/z: 508.2788 (calc. for C31H40O6, 508.2825).
: +0.050° (c 0.10, CHCl3); UV (MeOH) λ max (log εmax): 277 (0.59), 223 (1.40) nm. IR (KBr) ν max: 3455, 2927, 2971, 1669, 1579, 1512, 1453, 1375, 1245, 1180, 1107, 1071, 1035, 826 cm−1. 1H- and 13C-NMR: see Table 1. EI MS m/z: 508 [M] + (13), 439 (100), 383 (15), 305 (31), 278 (11), 249 (29), 177 (13), 121 (100), 69 (48). HR EIMS m/z: 508.2788 (calc. for C31H40O6, 508.2825).4. Conclusions
Supplementary Materials
Acknowledgements
References
- Chen, M.; Lou, S.Q.; Chen, J.H. Two isoflavones from Flemingia philippinensis. Phytochemistry 1991, 30, 3842–3844. [Google Scholar] [CrossRef]
- Chen, M.; Luo, S.Q.; Chen, J.H. Studies on the chemical constituents of Flemingia philippinensis. Acta Pharmacol. Sin. 1990, 26, 42–48. [Google Scholar]
- Ahn, E.M.; Nakamura, N.; Akao, T.; Komatsu, K.; Qui, M.H.; Hattori, M. Prenylated flavonoids from Moghania philippinensis. Phytochemistry 2003, 64, 1389–1394. [Google Scholar]
- Li, H.; Yang, M.H.; Miao, J.H.; Ma, X.J. Prenylated isoflavones from Flemingia philippinensis. Magn. Reson. Chem. 2008, 46, 1203–1207. [Google Scholar]
- Li, H.; Yang, M.H.; Ma, X.J. Flavonoids from roots of Flemingia philippinensis. Chin. J. Chin. Mat. Med. 2009, 34, 724–726. [Google Scholar]
- Li, H.; Yang, M.H.; Si, J.Y.; Miao, J.H.; Ma, X.J. Chemical constituents from roots of Flemingia philippinensis. Chin. Tradit. Herb Drugs 2009, 40, 512–516. [Google Scholar]
- Chulabhorn, M.; Hunsa, P.; Somsak, R. Prenylated flavanones from Derris reticulata. Phytochemistry 1977, 45, 825–829. [Google Scholar]
- Bonaventure, T.N.; Berhanu, M.A.; Etienne, D.; Helene, T.; Kouam, F. Geranylated and prenylated flavonoids from the twigs of Dorstenia mannii. Phytochemistry 1998, 48, 349–354. [Google Scholar]
- Feng, J.; Xiang, C.; Liang, H. Chemical constituents of isoflavones from vine stems of Millettia nitita var. hirsutissima. Chin. J. Chin. Mat. Med. 2007, 32, 321–322. [Google Scholar]
- Huang, S.Y.; Tu, P.F. Isolation and Identification of Isoflavones from Trifolium pratense. Acta Sci. Natur. Univ. Pekinensis 2007, 40, 544–549. [Google Scholar]
- Deng, Y.R.; Wang, T.; He, Y.Z. Studies on chemical constituents of Caragana spinifera. Chin. J. Chin. Mat. Med. 2008, 33, 775–777. [Google Scholar]
- Wang, Y.F.; Mu, T.H.; Chen, J.J.; Luo, S.D. Studies on Chemical Constituents from the Root of Polygonatum kingianum. Chin. J. Chin. Mat. Med. 2003, 28, 524–527. [Google Scholar]
- Huang, M.Z.; Chen, H.S.; Liu, J.G.; Zhou, X.H.; Du, J.L.; Xiang, Z.B. Studies on the chemical constituents of Bidens bipinnata L. Acad. J. Second Military Med. Univ. 2006, 27, 888–891. [Google Scholar]
- Tan, Y.X.; Sun, Y.H.; Chen, R.Y. Studies on chemical constituents in seeds of Cicer arietinum. Chin. J. Chin. Mat. Med. 2007, 32, 1650–1652. [Google Scholar]
- Tang, Y.P.; Lou, F.C.; Ma, W.; Wang, J.H.; Li, Y.F. Isoflavonoid glycosides from the pericarps of Sophora japonica. J. Chin. Pharm. Univ. 2001, 32, 187–189. [Google Scholar]
- Derogis, P.B.; Martins, F.T.; Souza, T.C.; Moreira, M.E.; Souza, F.J.D.; Doriguetto, A.C.; Souza, K.R.; Veloso, M.P.; Santos, M.H. Complete assignment of the 1H and 13C-NMR spectra of garciniaphenone and keto-enol equilibrium statements for prenylated benzophenones. Magn. Reson. Chem. 2008, 46, 278–282. [Google Scholar]
- Hartati, S.; Wang, H.B.; Kardono, L.B.S.; Kosela, S.; Qin, G.W. Chemical constituents of Garcinia maingayii. Chin. J. Nat. Med. 2007, 5, 272–275. [Google Scholar]
- Sample Availability: Samples of the flemiphilippinone A, lupinifolin, 6,8-diprenyleriodictyol, genistein biochanin A, prunetin, 3′-O-methylorobol-7-O-glycoside, luteoloside, sissotrin, prunetin 4′-O-glycoside and genistin are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.; Zhai, F.; Yang, M.; Li, X.; Wang, P.; Ma, X. A New Benzofuran Derivative from Flemingia philippinensis Merr. et Rolfe. Molecules 2012, 17, 7637-7644. https://doi.org/10.3390/molecules17077637
Li H, Zhai F, Yang M, Li X, Wang P, Ma X. A New Benzofuran Derivative from Flemingia philippinensis Merr. et Rolfe. Molecules. 2012; 17(7):7637-7644. https://doi.org/10.3390/molecules17077637
Chicago/Turabian StyleLi, Hua, Fengyan Zhai, Meihua Yang, Xiaowan Li, Pingli Wang, and Xiaojun Ma. 2012. "A New Benzofuran Derivative from Flemingia philippinensis Merr. et Rolfe" Molecules 17, no. 7: 7637-7644. https://doi.org/10.3390/molecules17077637
APA StyleLi, H., Zhai, F., Yang, M., Li, X., Wang, P., & Ma, X. (2012). A New Benzofuran Derivative from Flemingia philippinensis Merr. et Rolfe. Molecules, 17(7), 7637-7644. https://doi.org/10.3390/molecules17077637