Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects

Abstract
: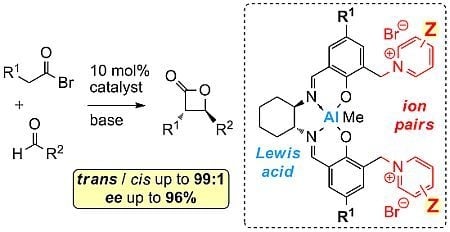
1. Introduction
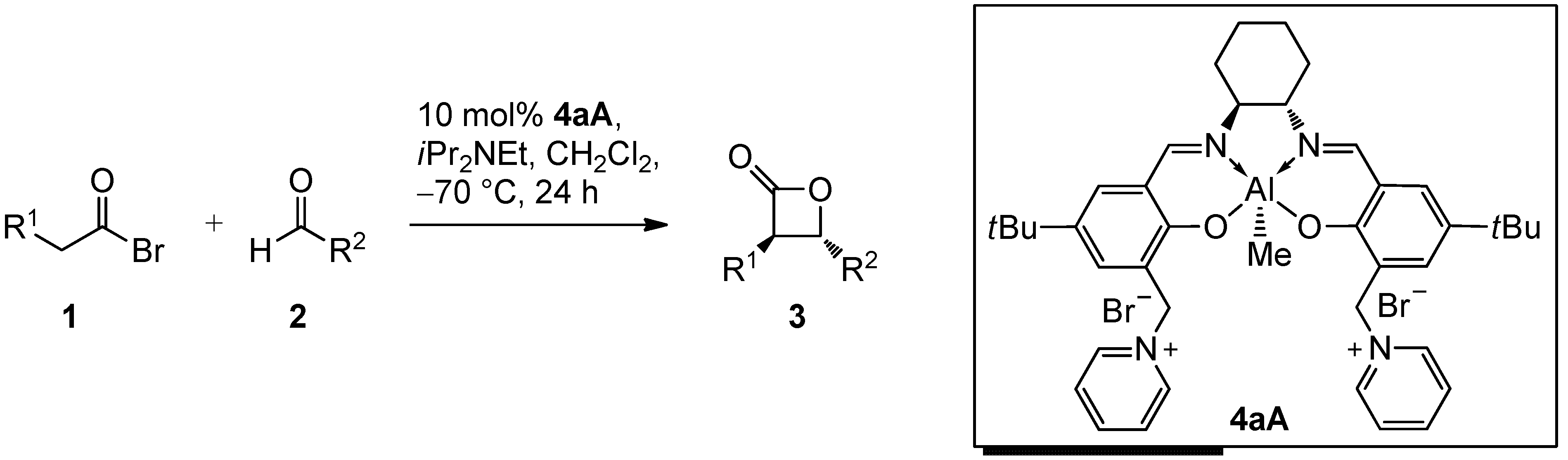
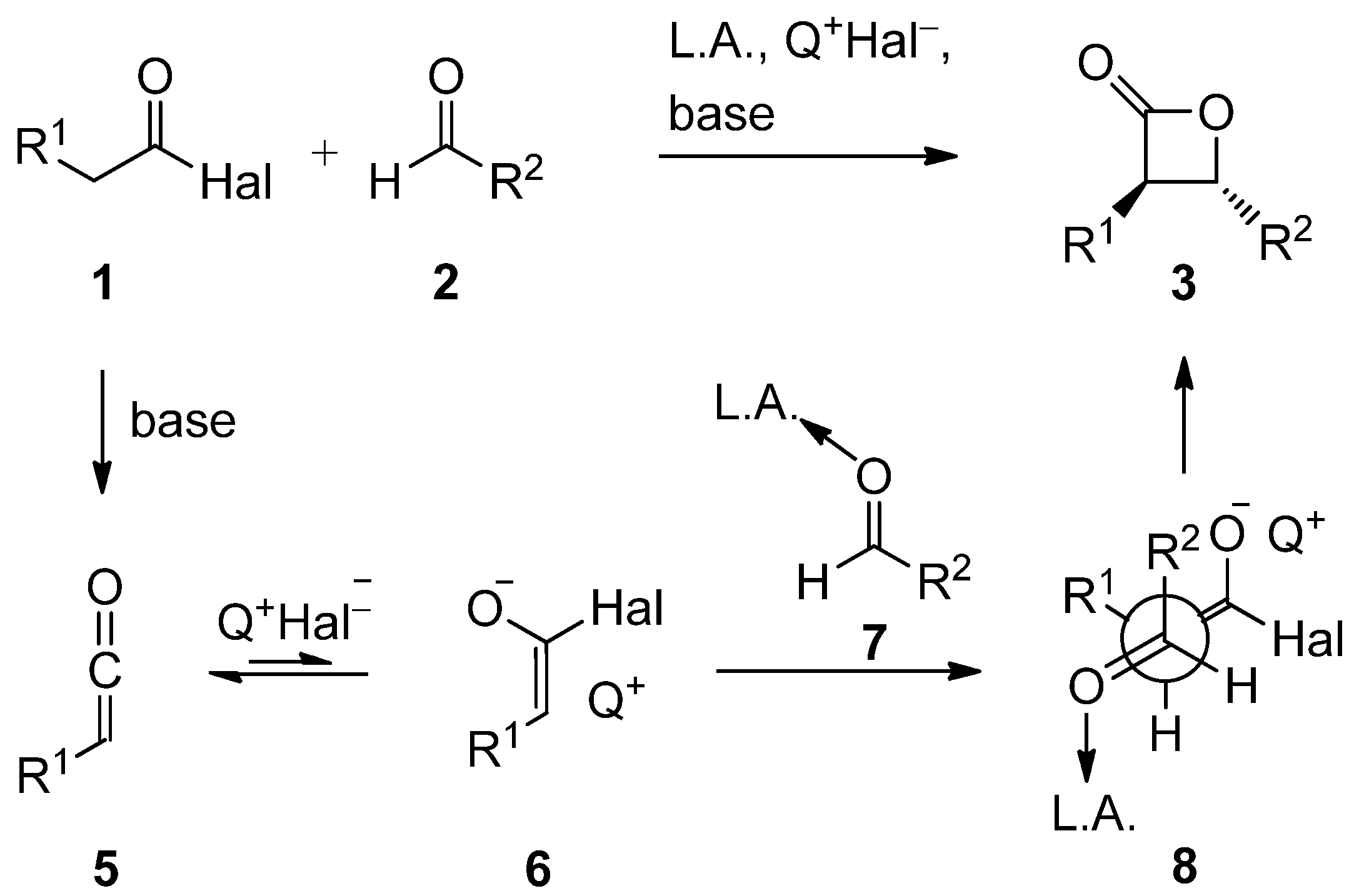
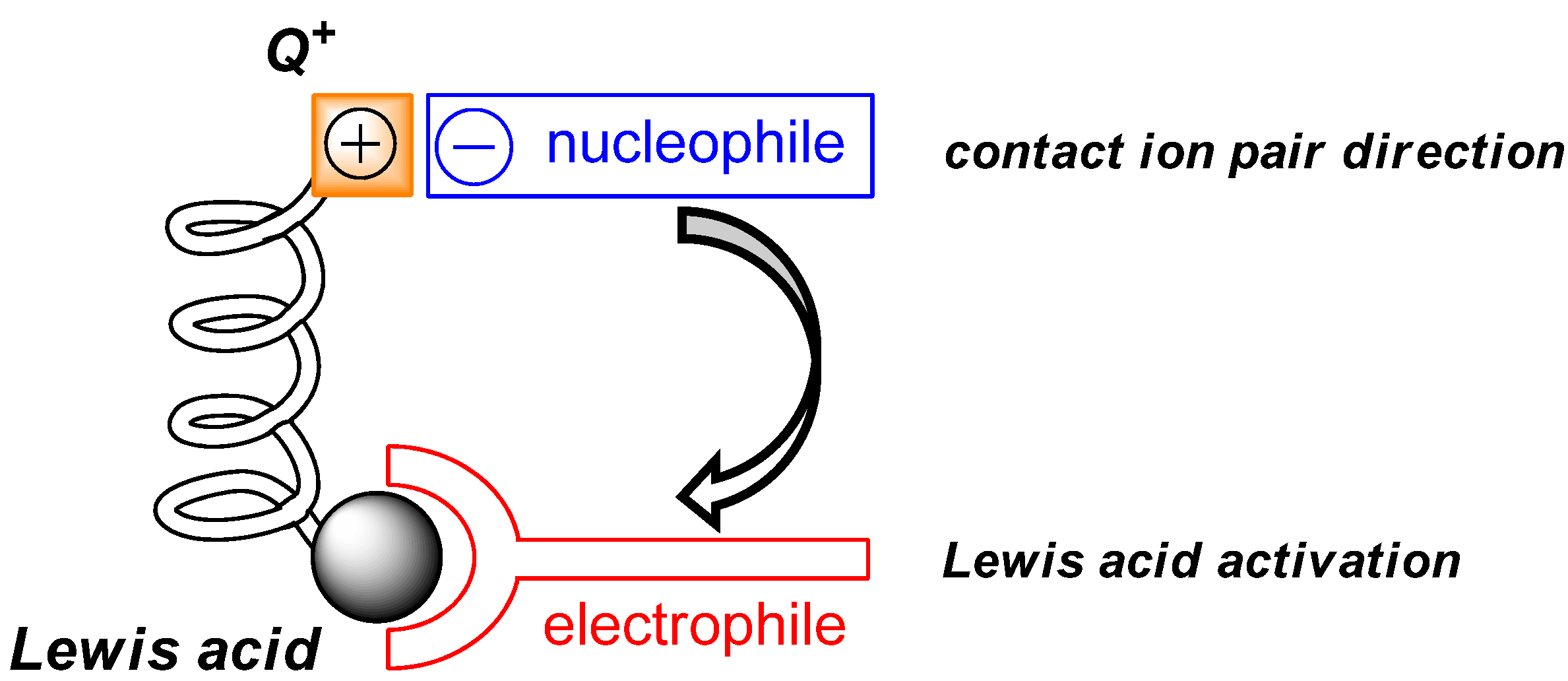

2. Results and Discussion
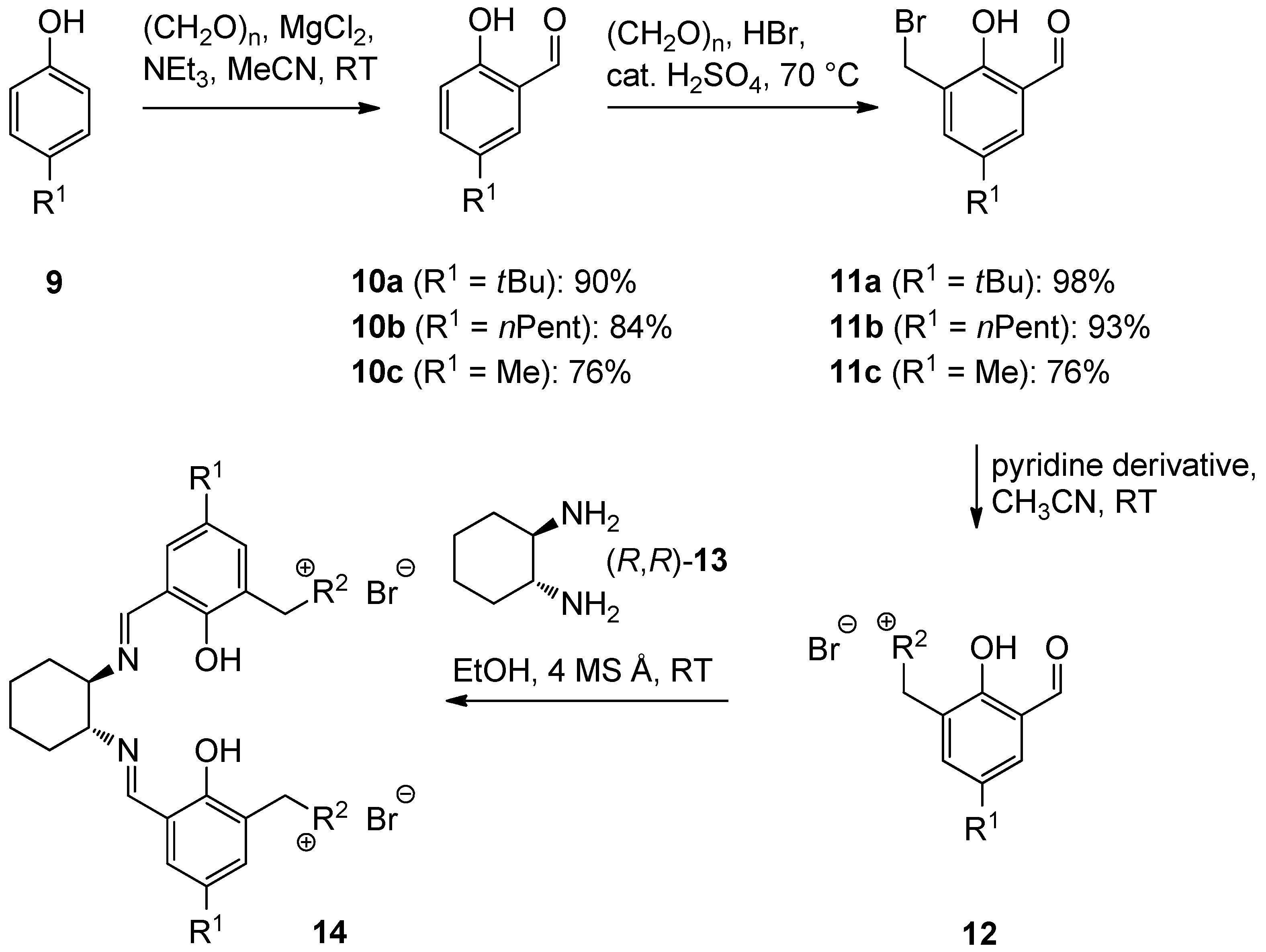
2.1. Ligand Preparation
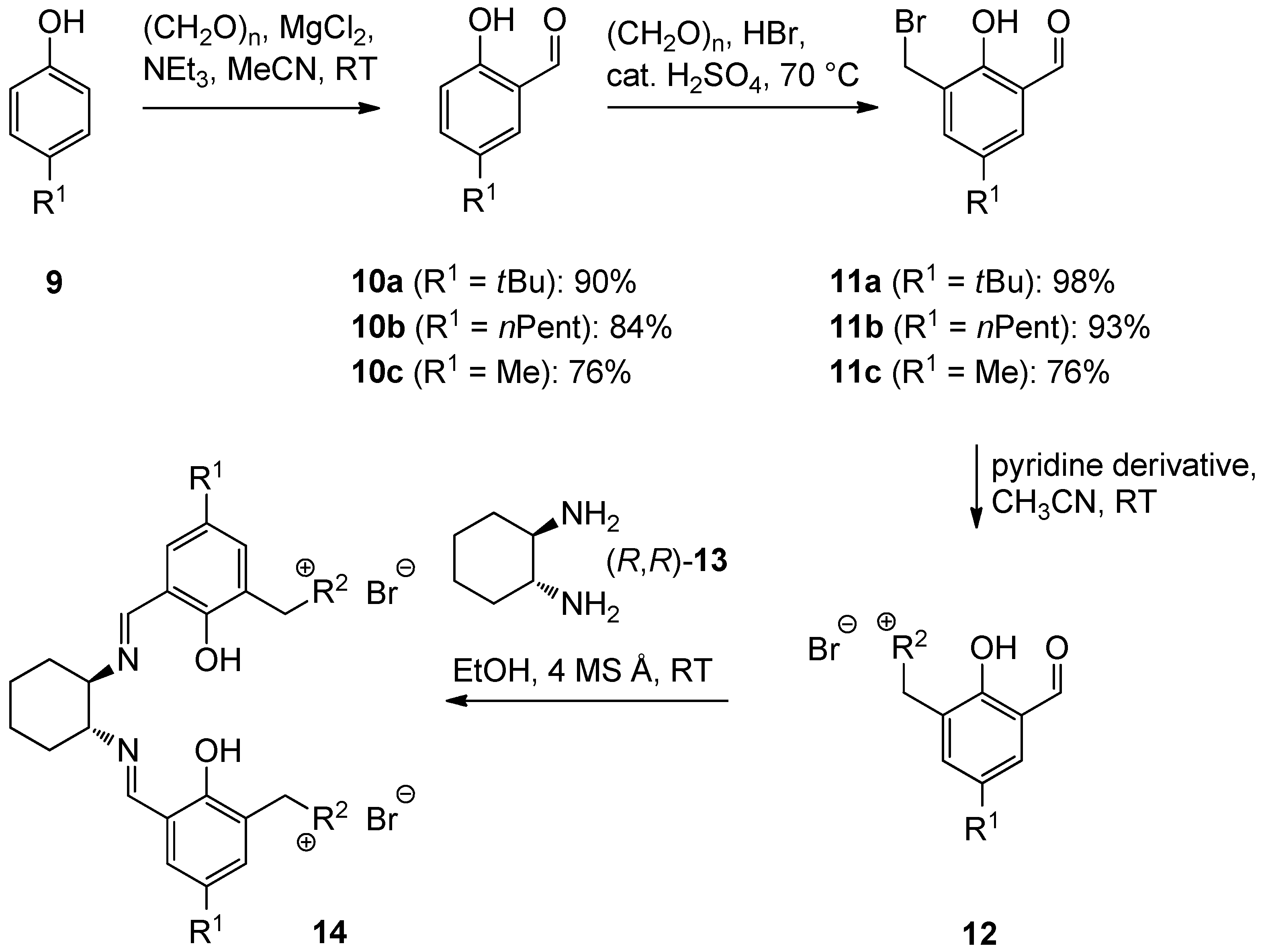

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | R2 | 12 | Yield 12 [%] a | 14 | Yield 14 [%] a |
|---|---|---|---|---|---|---|
| 1 | tBu |  | 12aA | 92 | 14aA | 100 |
| 2 | n-Pent |  | 12bA | 62 | 14bA | 91 |
| 3 | Me |  | 12cA | 79 | 14cA | 84 |
| 4 | t-Bu | 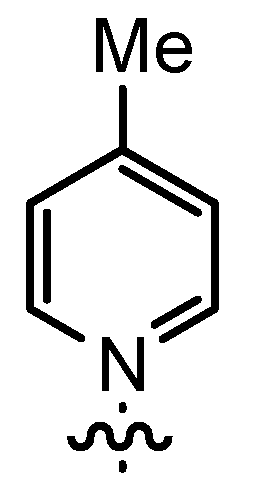 | 12aB | 67 | 14aB | 89 |
| 5 | n-Pent | 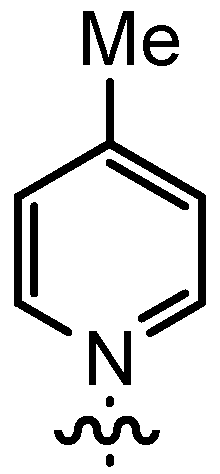 | 12bB | 91 | 14bB | 93 |
| 6 | n-Pent | 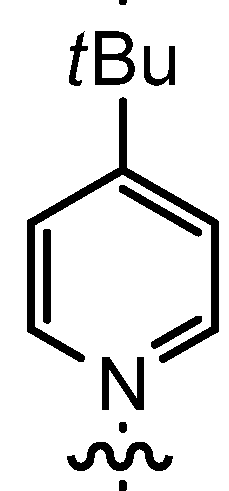 | 12bC | 96 | 14bC | 82 |
| 7 | n-Pent | 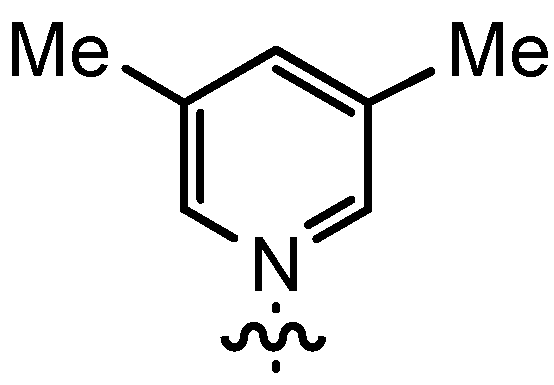 | 12bD | 54 | 14bD | 91 |
| 8 | nPent | 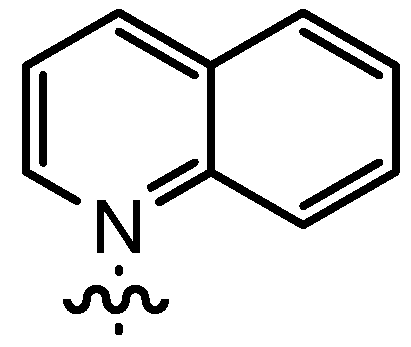 | 12bE | 47 | 14bE | 86 |
| 9 | n-Pent |  | 12bF | 81 | 14bF | 92 |
| 10 | n-Pent | 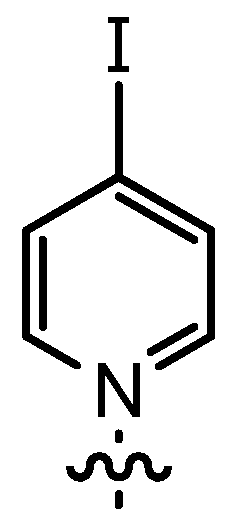 | 12bG | 65 | 14bG | 77 |
| 11 | n-Pent |  | 12bH | 29 | 14bH | 0 |
| 12 | n-Pent | 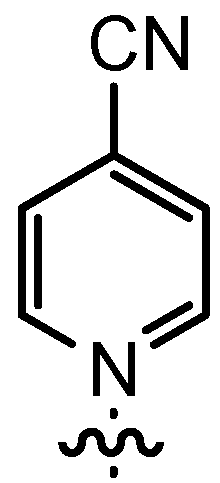 | 12bI | 80 | 14bI | 0 |
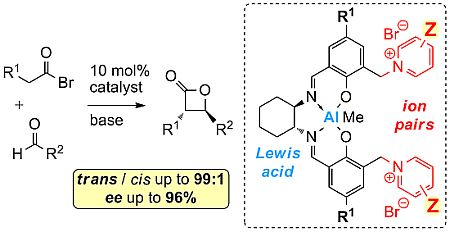
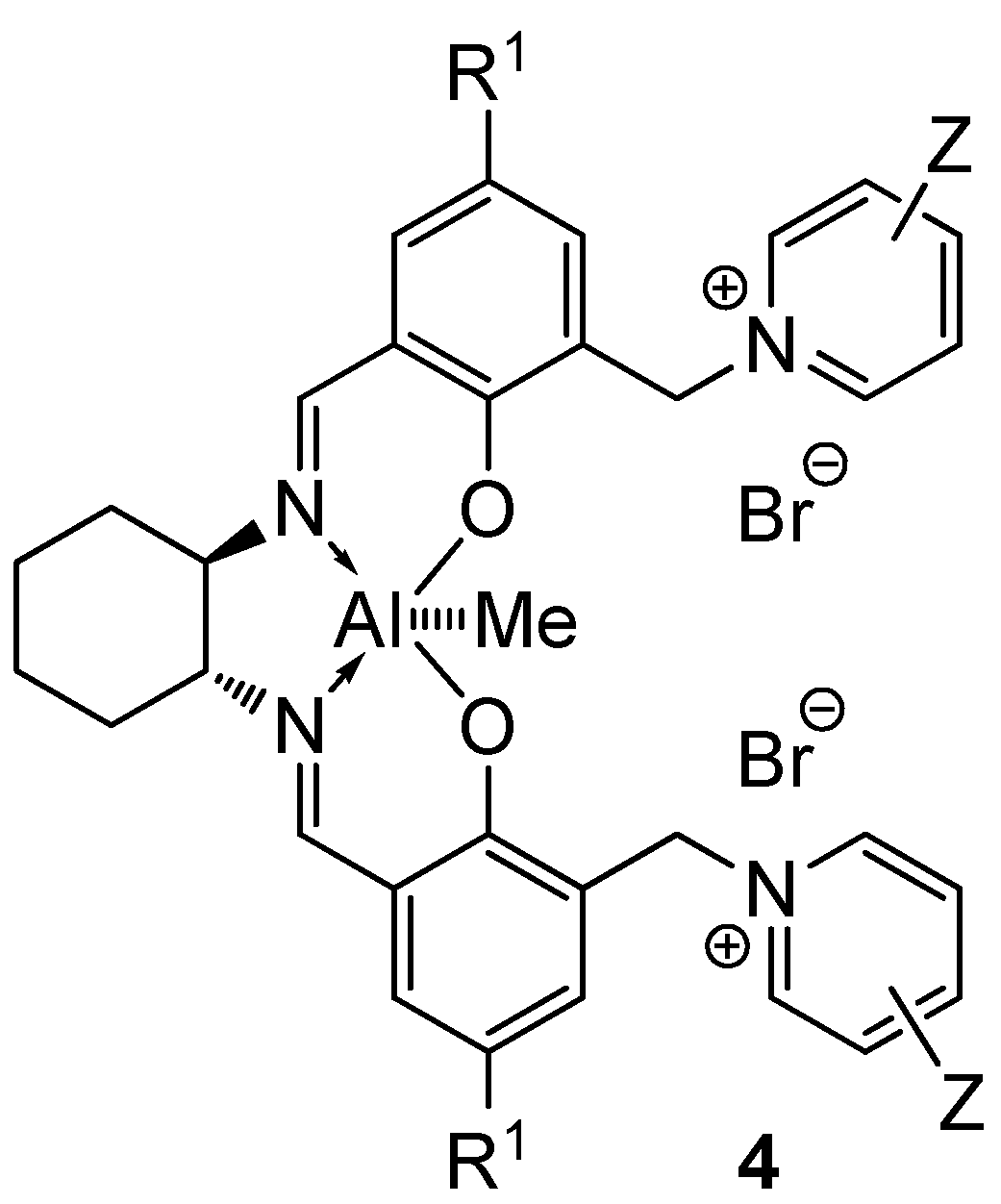
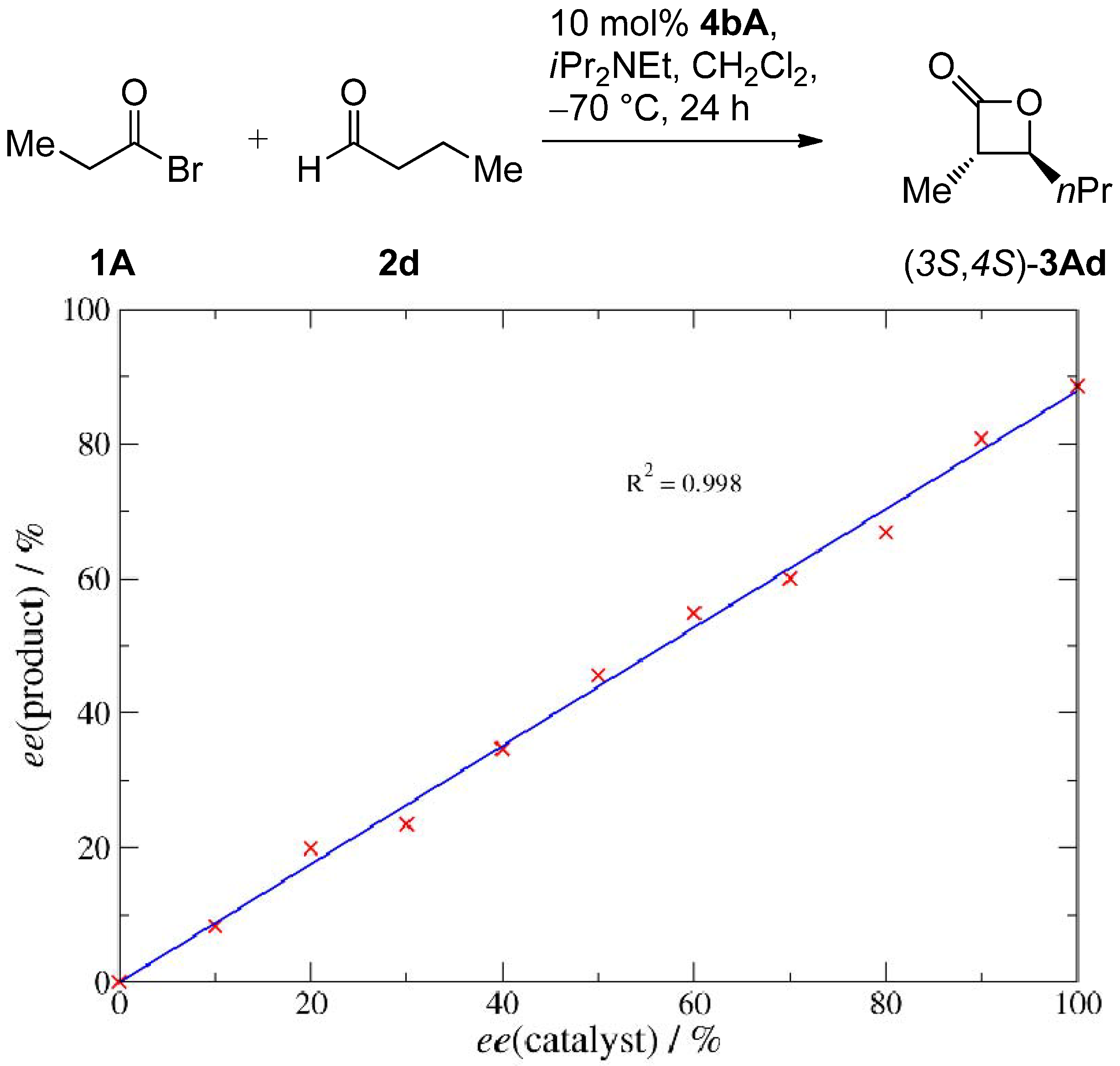
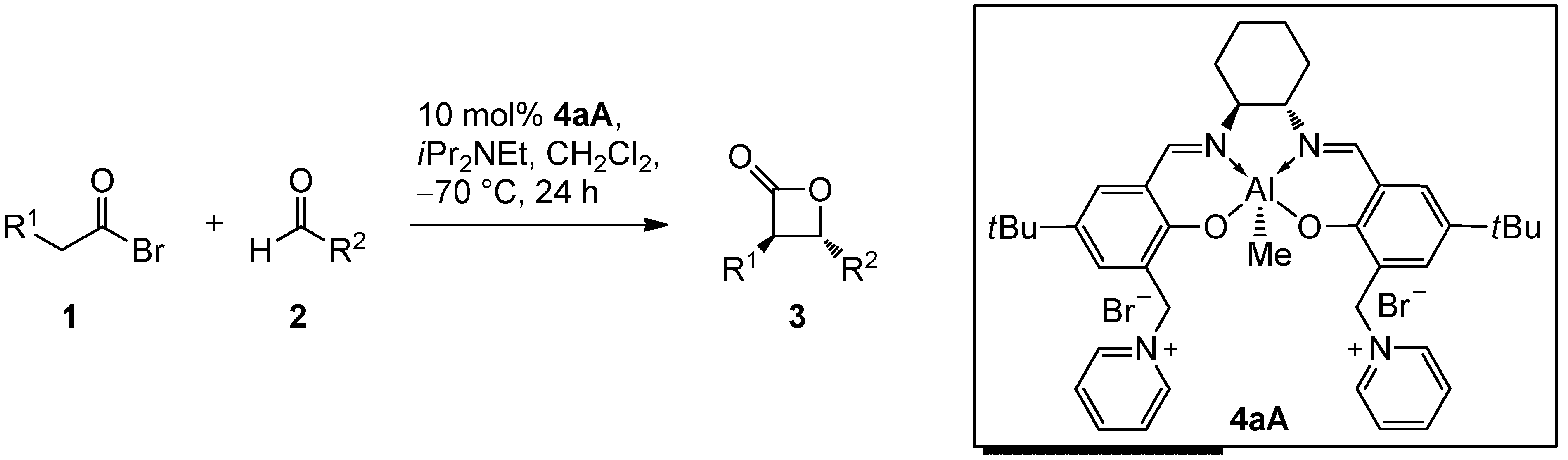
2.2. Catalysis
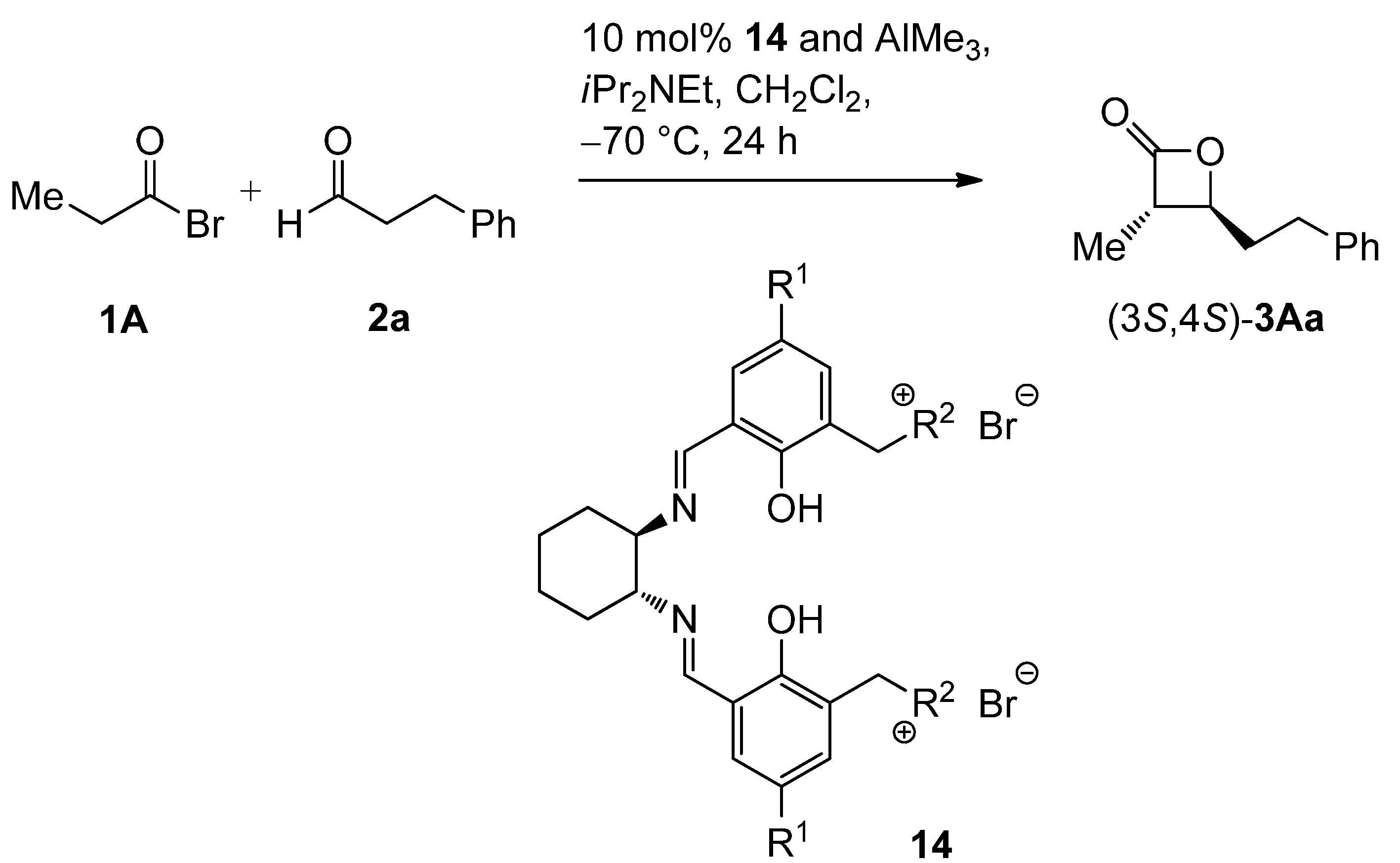
| Entry | 14 | R1 | R2 | Conversion [%] b | Yield 3Aa [%] b | dr 3Aa [trans/cis] c | ee 3Aa [%] d |
|---|---|---|---|---|---|---|---|
| 1 | 14aA | t-Bu | 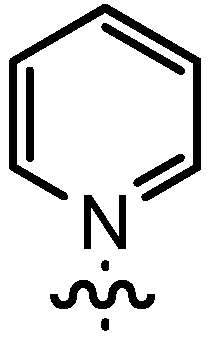 | >95 | 91 | 93:7 | 89 |
| 2 | 14bA | n-Pent |  | >98 | 98 | 93:7 | 88 |
| 3 | 14cA | Me | 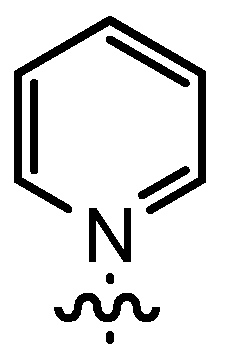 | n.d. | 71 | 91:9 | 88 |
| 4 | 14aB | t-Bu | 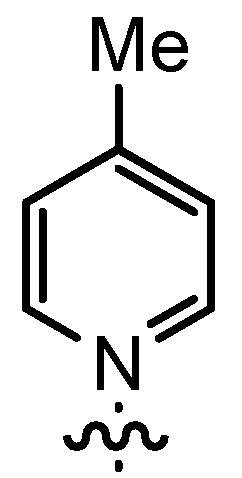 | 71 | 29 | 97:3 | 89 |
| 5 | 14bB | n-Pent | 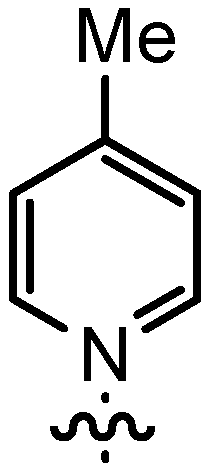 | 88 | 60 | 97:3 | 90 |
| 6 | 14bC | n-Pent | 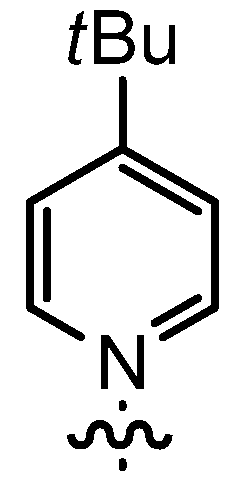 | 75 | 56 | 92:8 | 80 |
| 7 | 14bD | n-Pent |  | 82 | 39 | 88:12 | 80 |
| 8 | 14bE | n-Pent | 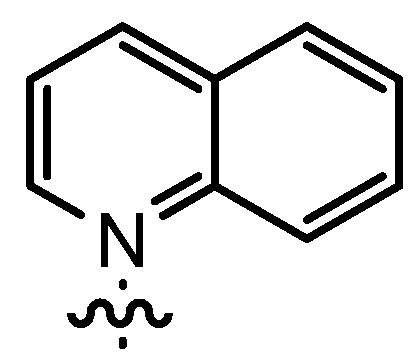 | 62 | 38 | 88:12 | 77 |
| 9 | 14bF | n-Pent |  | 73 | 37 | 94:6 | 82 |
| 10 | 14bG | n-Pent | 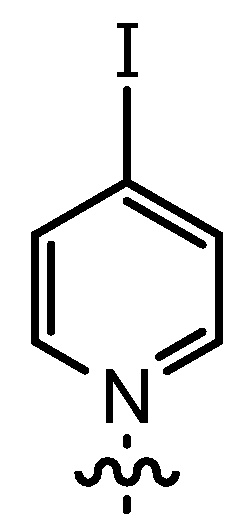 | 78 | 45 | 92:8 | 81 |
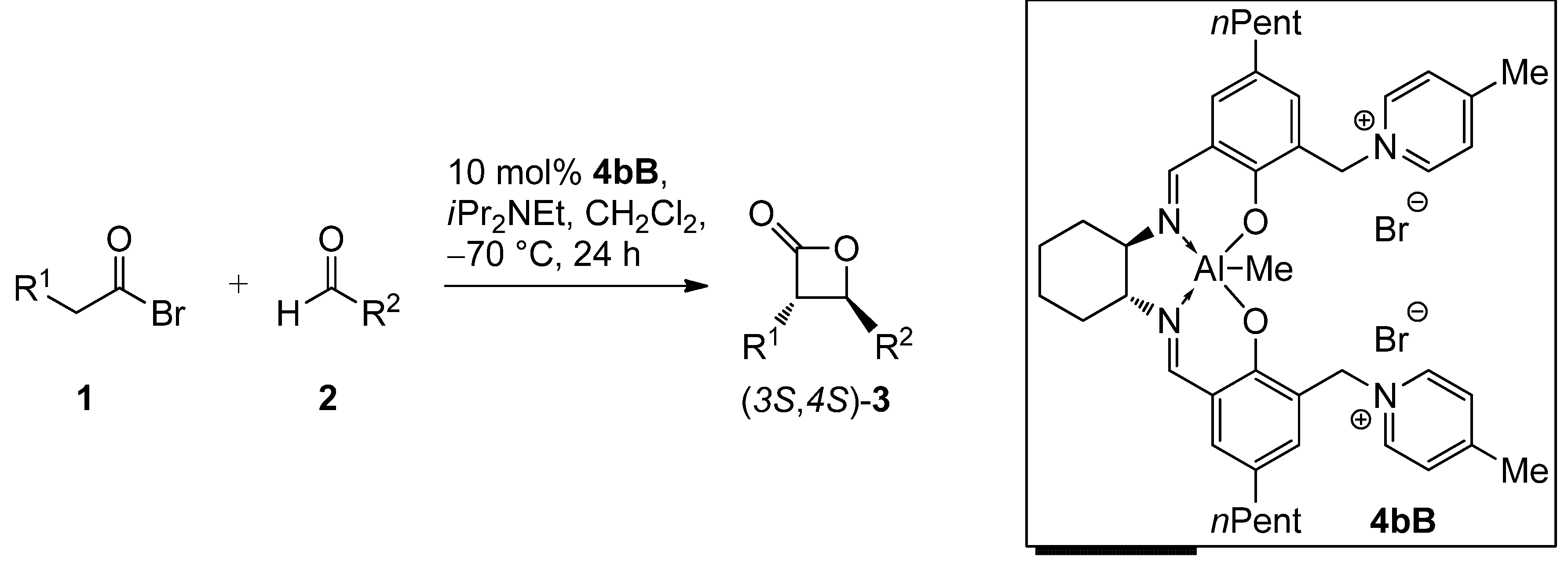
| Entry | 1 | R1 | 2 | R2 | 3 | Yield [%] a | dr [trans/cis] b | ee [%] c |
|---|---|---|---|---|---|---|---|---|
| 1 d | 1A | Me | 2a | (CH2)2Ph | 3Aa | 60 | 97:3 (97:3) | 90 (88) |
| 2 e | 1A | Me | 2a | (CH2)2Ph | 3Aa | 69 | 97:3 (97:3) | 90 (88) |
| 3 d | 1A | Me | 2b | Et | 3Ab | 59 | 98:2 (95:5) | 90 (87) |
| 4 d | 1A | Me | 2c | Ph | 3Ac | 37 | 98:2 (94:6) | 96 (91) |
| 5 d | 1B | n-Pr | 2a | (CH2)2Ph | 3Ba | 59 | 99:1 (98:2) | 95 (94) |
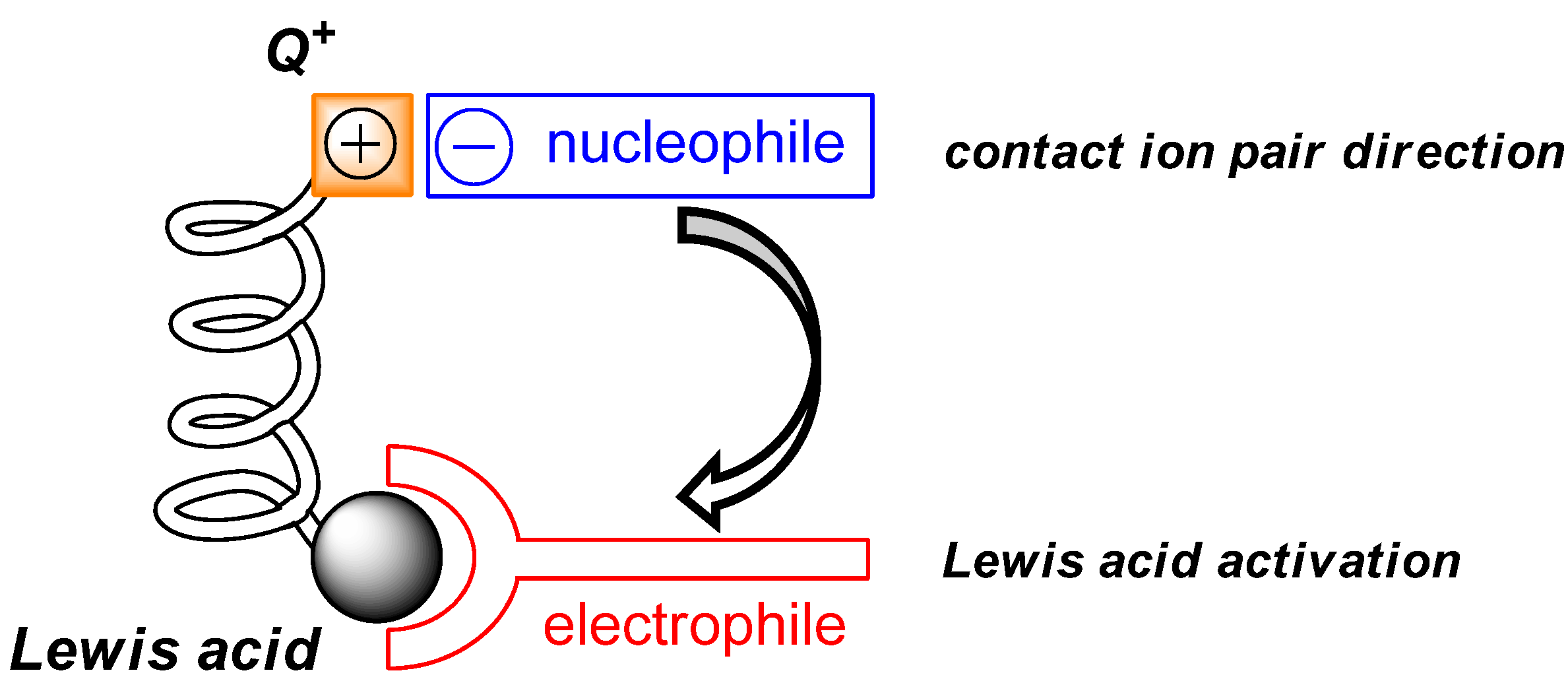
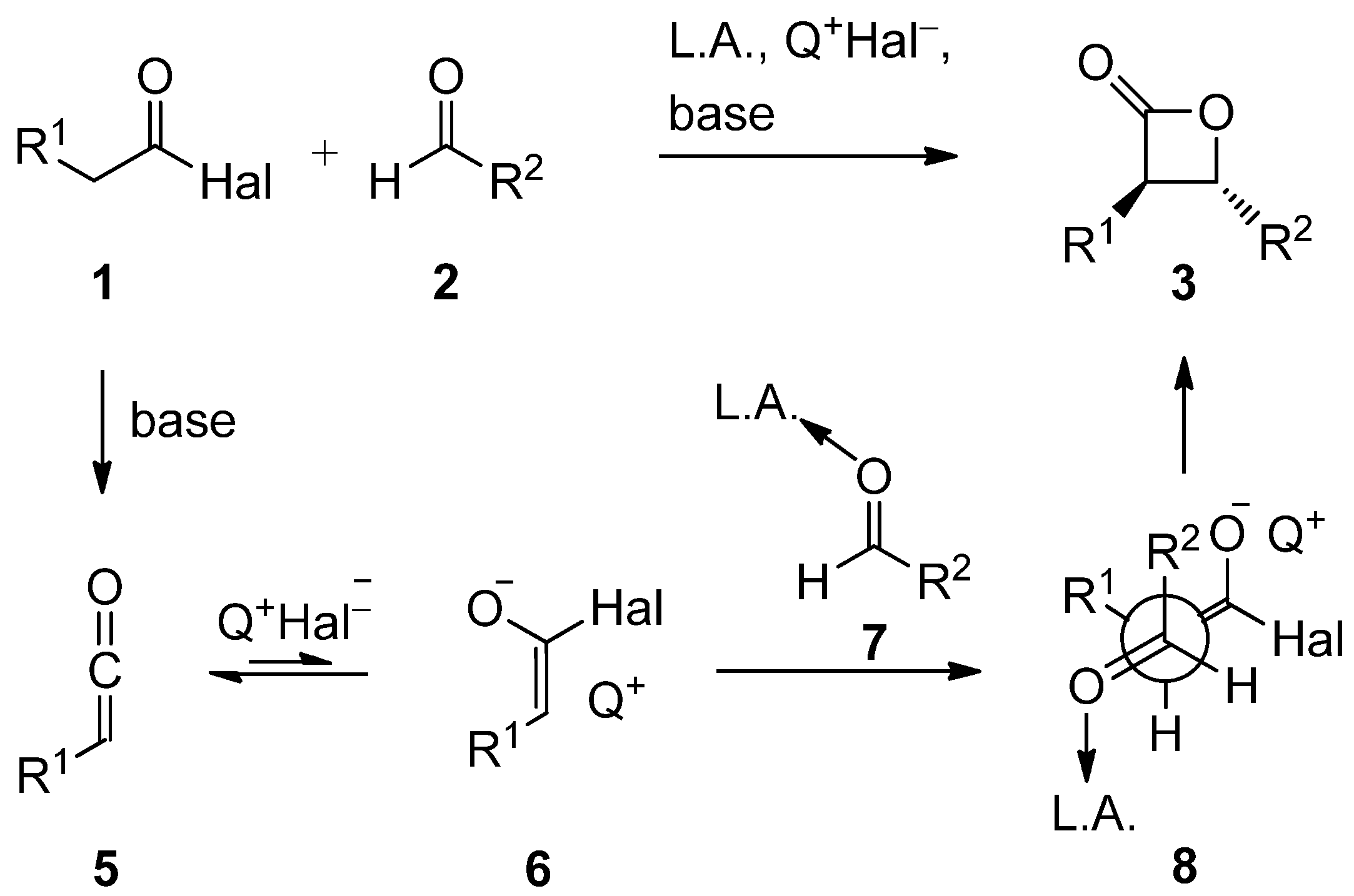
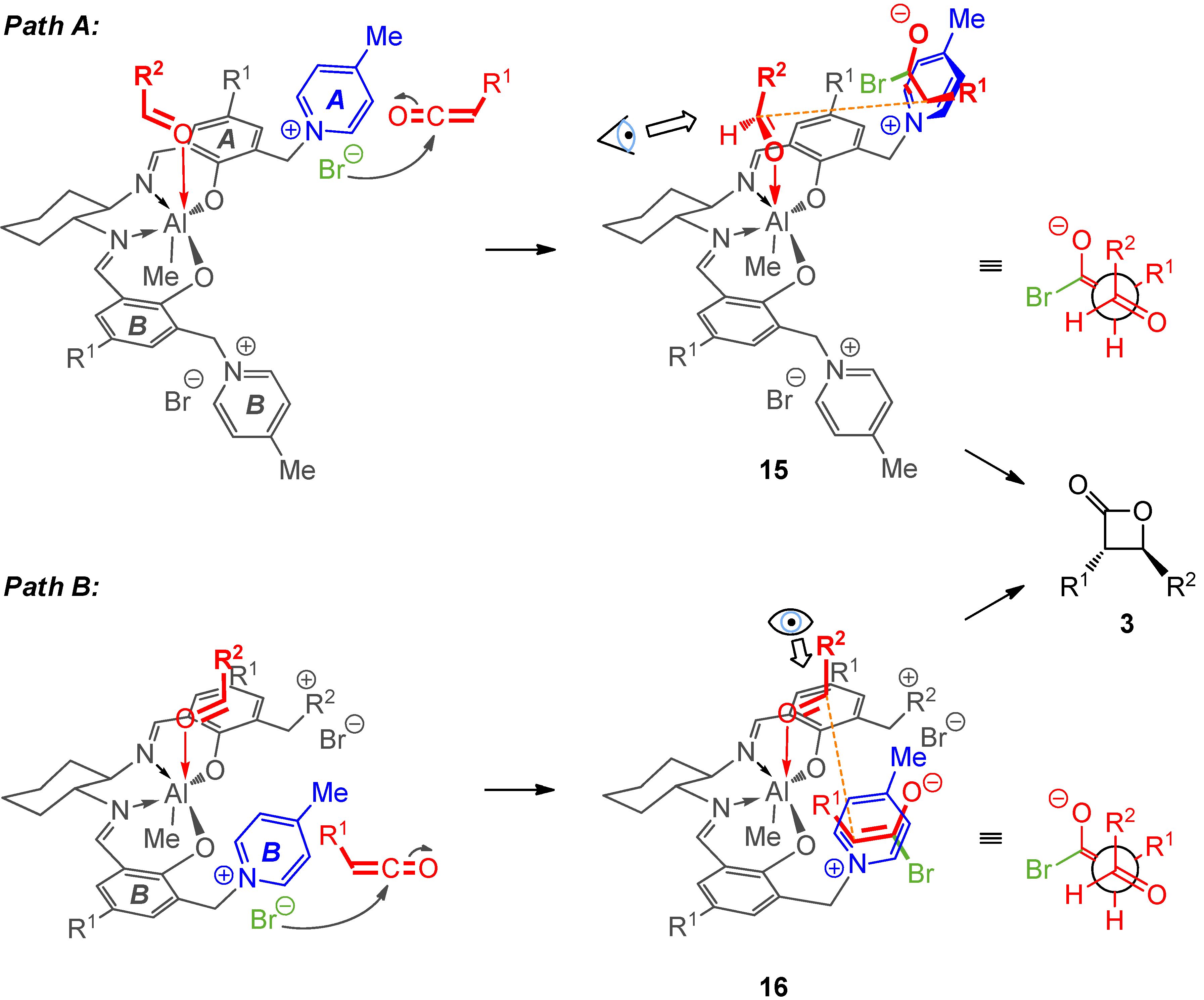
2.3. Mechanistic and Theoretical Investigations
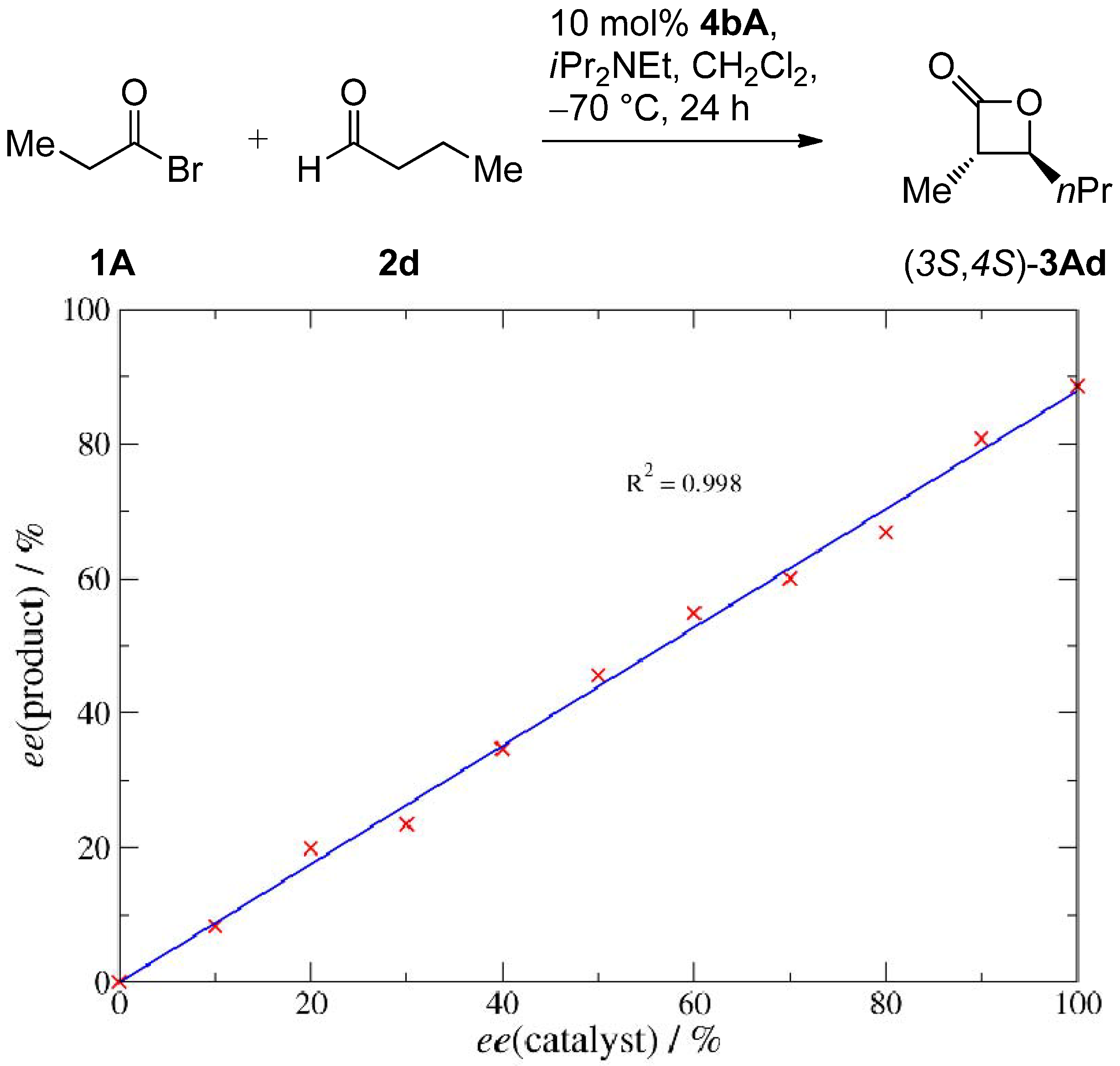
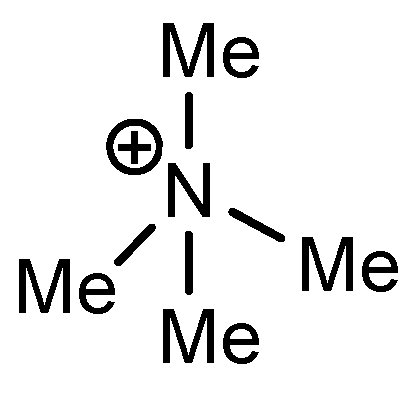 | 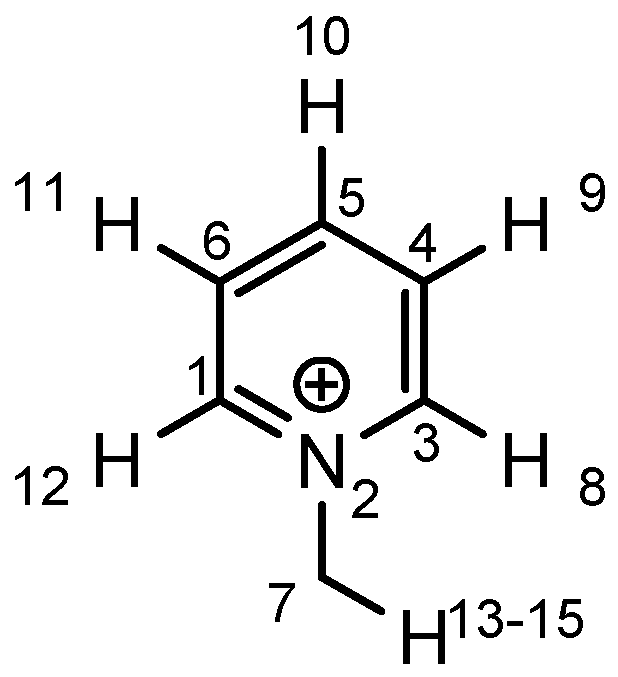 | 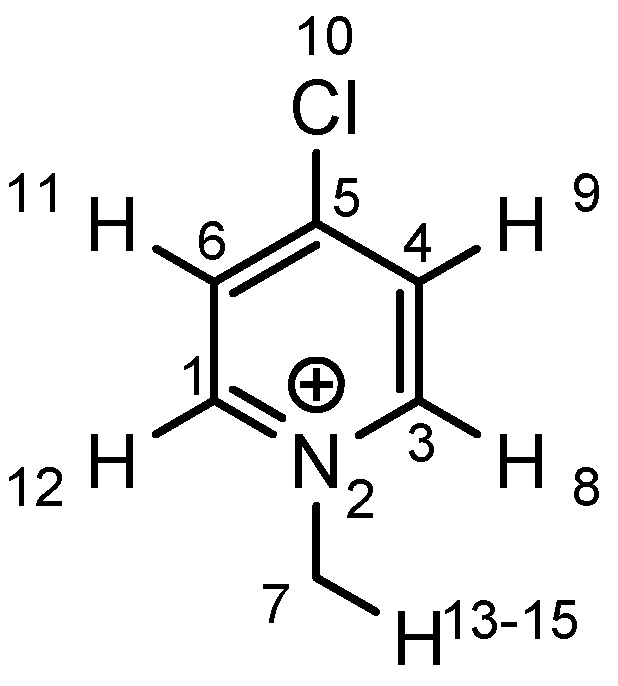 | 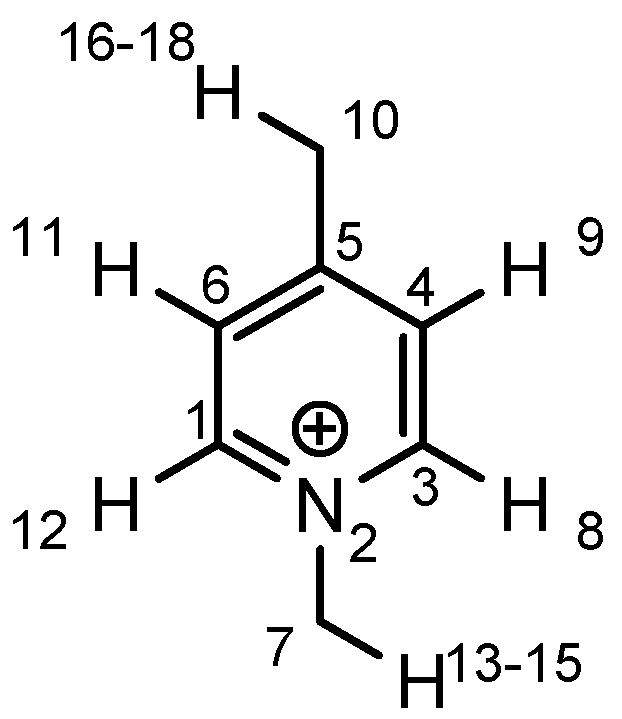 | ||||
| atom | charge | atom | charge | atom | charge | atom | charge |
| N | −0.36 | C (1) | +0.11 | C (1) | +0.12 | C (1) | +0.11 |
| C | −0.41 | N (2) | −0.35 | N (2) | −0.36 | N (2) | −0.36 |
| H | +0.25 | C (3) | +0.11 | C (3) | +0.11 | C (3) | +0.10 |
| C (4) | −0.22 | C (4) | −0.24 | C (4) | −0.22 | ||
| C (5) | −0.12 | C (5) | +0.02 | C (5) | +0.07 | ||
| C (6) | −0.22 | C (6) | −0.24 | C (6) | −0.23 | ||
| C (7) | −0.41 | C (7) | −0.40 | C (7) | −0.41 | ||
| H (8) | +0.26 | H (8) | +0.26 | H (8) | +0.26 | ||
| H (9) | +0.28 | H (9) | +0.29 | H (9) | +0.27 | ||
| H (10) | +0.27 | Cl (10) | +0.14 | C (10) | −0.67 | ||
| H (11) | +0.28 | H (11) | +0.29 | H (11) | +0.27 | ||
| H (12) | +0.26 | H (12) | +0.26 | H (12) | +0.26 | ||
| H (13–15) | +0.25 | H (13–15) | +0.25 | H (13–15) | +0.25 | ||
| H (16–18) | +0.26 | ||||||

3. Experimental
3.1. General
3.2. General Procedure for the Synthesis of 10 (GP1)
3.3. General Procedure for the Synthesis of 11 (GP2)
3.4. General Procedure for the Synthesis of 12 (GP3)
3.5. General Procedure for the Synthesis of 14 (GP4)
3.6. General Procedure for the Catalytic Asymmetric Synthesis of 3 (GP5)
4. Conclusions
Acknowledgements
References and Notes
- Lowe, C.; Vederas, J.C. Naturally occurring β-lactones: Occurrence, syntheses and properties. A review. Org. Prep. Proceed. Int. 1995, 27, 305–346. [Google Scholar] [CrossRef]
- Pommier, A.; Pons, J.-M. The Synthesis of Natural 2-Oxetanones. Synthesis 1995, 729–744. [Google Scholar] [CrossRef]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A Highly Cytotoxic Proteasome Inhibitor from a Novel Microbial Source, a Marine Bacterium of the New Genus Salinospora. Angew. Chem. Int. Ed. Engl. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Masse, C.; Morgan, J.; Adams, J.; Panek, J. Syntheses and Biological Evaluation of (+)-Lactacystin and Analogs. Eur. J. Org. Chem. 2000, 2513–2528. [Google Scholar]
- Kridel, S.J.; Axelrod, F.; Rosenkrantz, N.; Smith, J.W. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Knowles, L.M.; Axelrod, F.; Browne, C.D.; Smith, J.W. A Fatty Acid Synthase Blockade Induces Tumor Cell-cycle Arrest by Down-regulating Skp2. J. Biol. Chem. 2004, 279, 30540–30545. [Google Scholar]
- Yang, P.-Y.; Liu, K.; Hong Ngai, M.; Lear, M.J.; Wenk, M.R.; Yao, S.Q. Activity-Based Proteome Profiling of Potential Cellular Targets of Orlistat—An FDA-Approved Drug with Anti-Tumor Activities. J. Am. Chem. Soc. 2010, 132, 656–666. [Google Scholar] [CrossRef]
- Greenberg, A.; Liebman, J.F. Strained Organic Molecules; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Yang, H.W.; Romo, D. Methods for the synthesis of optically active β-lactones (2-oxetanones). Tetrahedron 1999, 55, 6403–6434. [Google Scholar] [CrossRef]
- Wang, Y.; Tennyson, R.L.; Romo, D. β-Lactones as Intermediates for Natural Product Total Synthesis and New Transformations. Heterocycles 2004, 64, 605–658. [Google Scholar] [CrossRef]
- Nelson, S.G.; Wan, Z. Catalytic Asymmetric Propionate Aldol Reactions via Acyl Halide-Aldehyde Cyclocondensations. Org. Lett. 2000, 2, 1883–1886. [Google Scholar] [CrossRef]
- Zipp, G.G.; Hilfiker, M.A.; Nelson, S.G. Enantioenriched Dihydropyrones from β-Lactone Templates. Org. Lett. 2002, 4, 1823–1826. [Google Scholar] [CrossRef]
- Nelson, S.G.; Cheung, W.S.; Kassick, A.J.; Hilfiker, M.A. A de Novo Enantioselective Total Synthesis of (−)-Laulimalide. J. Am. Chem. Soc. 2002, 124, 13654–13655. [Google Scholar]
- Palomo, C.; Oiarbide, M.; Garcia, J.M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33, 65–75. [Google Scholar]
- Tidwell, T.T. Ketenes; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Tidwell, T.T. Ketene Chemistry after 100 Years: Ready for a New Century. Eur. J. Org. Chem. 2006, 563–576. [Google Scholar] [CrossRef]
- Tidwell, T.T. The First Century of Ketenes (1905–2005): The Birth of a Versatile Family of Reactive Intermediates. Angew. Chem. Int. Ed. Engl. 2005, 44, 5778–5785. [Google Scholar] [CrossRef]
- Paull, D.H.; Weatherwax, A.; Lectka, T. Catalytic, asymmetric reactions of ketenes and ketene enolates. Tetrahedron 2009, 65, 6771–6803. [Google Scholar]
- Orr, R.K.; Calter, M.A. Asymmetric synthesis using ketenes. Tetrahedron 2003, 59, 3545–3565. [Google Scholar]
- Tamai, Y.; Yoshiwara, H.; Someya, M.; Fukumoto, J.; Miyano, S. Asymmetric [2+2] cycloaddition of ketene with aldehydes catalysed by chiral bissulfonamide-trialkylaluminium complexes. J. Chem. Soc. Chem. Commun. 1994, 2281–2282. [Google Scholar]
- Dymock, B.W.; Kocienski, P.J.; Pons, J.-M. 3-(Trimethylsilyl)oxetan-2-ones via enantioselective [2+2] cycloaddition of (trimethylsilyl)ketene to aldehydes catalysed by methylaluminiomidazolines. Chem. Commun. 1996, 1053–1054. [Google Scholar]
- Yang, H.W.; Romo, D. Studies of the asymmetric [2+2] cycloaddition of silylketenes and aldehydes employing Ti-TADDOL catalysts. Tetrahedron Lett. 1998, 39, 2877–2880. [Google Scholar] [CrossRef]
- Nelson, S.G.; Peelen, T.J.; Wan, Z. Catalytic Asymmetric Acyl Halide-Aldehyde Cyclocondensations. A Strategy for Enantioselective Catalyzed Cross Aldol Reactions. J. Am. Chem. Soc. 1999, 121, 9742–9743. [Google Scholar]
- Evans, D.A.; Janey, J.M. C 2-Symmetric Cu(II) Complexes as Chiral Lewis Acids. Catalytic, Enantioselective Cycloadditions of Silyl Ketenes. Org. Lett. 2001, 3, 2125–2128. [Google Scholar]
- Nelson, S.G.; Wan, Z. Catalytic Asymmetric Propionate Aldol Reactions via Acyl Halide−Aldehyde Cyclocondensations. Org. Lett. 2000, 2, 1883–1886. [Google Scholar] [CrossRef]
- Nelson, S.G.; Zhu, C.; Shen, X. Catalytic Asymmetric Acyl Halide-Aldehyde Cyclocondensation Reactions of Substituted Ketenes. J. Am. Chem. Soc. 2004, 126, 14–15. [Google Scholar] [CrossRef]
- Forslund, R.E.; Cain, J.; Colyer, J.; Doyle, M.P. Chiral Dirhodium(II) Carboxamidate-Catalyzed [2+2]-Cycloaddition of TMS-Ketene and Ethyl Glyoxylate. Adv. Synth. Catal. 2005, 347, 87–92. [Google Scholar] [CrossRef]
- Kull, T.; Peters, R. Practical Enantioselective Synthesis of β-Lactones Catalyzed by Aluminum Bissulfonamide Complexes. Adv. Synth. Catal. 2007, 349, 1647–1652. [Google Scholar] [CrossRef]
- Schneider, C. Catalytic, Enantioselective Syntheses of β-Lactones—Versatile Synthetic Building Blocks in Organic Chemistry. Angew. Chem. Int. Ed. 2002, 41, 744–746. [Google Scholar] [CrossRef]
- Borrmann, D.; Wegler, R. Über die Umsetzung von Carbonylverbindungen mit Säurechloriden in Gegenwart tertiärer Amine, II. Optisch aktive β-Lactone. Chem. Ber. 1967, 100, 1575–1575. [Google Scholar]
- Wynberg, H.; Staring, E.G.J. Asymmetric synthesis of (S)- and (R)-malic acid from ketene and chloral. J. Am. Chem. Soc. 1982, 104, 166–168. [Google Scholar]
- Wynberg, H.; Staring, E.G.J. Catalytic asymmetric synthesis of chiral 4-substituted 2-oxetanones. J. Org. Chem. 1985, 50, 1977–1979. [Google Scholar]
- Cortez, G.S.; Tennyson, R.L.; Romo, D. Intramolecular, Nucleophile-Catalyzed Aldol-Lactonization (NCAL) Reactions: Catalytic, Asymmetric Synthesis of Bicyclic β-Lactones. J. Am. Chem. Soc. 2001, 123, 7945–7946. [Google Scholar] [CrossRef]
- Wilson, J.E.; Fu, G.C. Asymmetric Synthesis of Highly Substituted β-Lactones by Nucleophile-Catalyzed [2+2] Cycloadditions of Disubstituted Ketenes with Aldehydes. Angew. Chem. Int. Ed. 2004, 43, 6358–6360. [Google Scholar] [CrossRef]
- Oh, S.H.; Cortez, G.S.; Romo, D. Asymmetric Synthesis of Bicyclic β-Lactones via the Intramolecular, Nucleophile-Catalyzed Aldol Lactonization: Improved Efficiency and Expanded Scope. J. Org. Chem. 2005, 70, 2835–2838. [Google Scholar]
- Henry-Riyad, H.; Lee, C.; Purohit, V.C.; Romo, D. Bicyclic- and Tricyclic-β-lactones via Organonucleophile-Promoted Bis-Cyclizations of Keto Acids: Enantioselective Synthesis of (+)-Dihydroplakevulin. Org. Lett. 2006, 8, 4363–4366. [Google Scholar] [CrossRef]
- Mondal, M.; Ibrahim, A.A.; Wheeler, K.A.; Kerrigan, N.J. Phosphine-Catalyzed Asymmetric Synthesis of β-Lactones from Arylketoketenes and Aromatic Aldehydes. Org. Lett. 2010, 12, 1664–1667. [Google Scholar] [CrossRef]
- Zhu, C.; Shen, X.; Nelson, S.G. Cinchona Alkaloid-Lewis Acid Catalyst Systems for Enantioselective Ketene−Aldehyde Cycloadditions. J. Am. Chem. Soc. 2004, 126, 5352–5353. [Google Scholar] [CrossRef]
- Calter, M.A.; Tretyak, O.A.; Flaschenriem, C. Formation of Disubstituted β-Lactones Using Bifunctional Catalysis. Org. Lett. 2005, 7, 1809–1812. [Google Scholar]
- Gnanadesikan, V.; Corey, E.J. Enantioselective β-Lactone Formation from Ketene and Aldehydes Catalyzed by a Chiral Oxazaborolidine. Org. Lett. 2006, 8, 4943–4945. [Google Scholar] [CrossRef]
- Lin, Y.-M.; Boucau, J.; Li, Z.; Casarotto, V.; Lin, J.; Nguyen, A.N.; Ehrmantraut, J. A Lewis Acid-Lewis Base Bifunctional Catalyst from a New Mixed Ligand. Org. Lett. 2007, 9, 567–570. [Google Scholar]
- Chidara, S.; Lin, Y.-M. Reaction rate acceleration enabled by tethered Lewis acid-Lewis base bifunctional catalysis: A catalytic, enantioselective [2+2] ketene aldehyde cycloaddition reaction. Synlett 2007, 9, 1675–1679. [Google Scholar]
- Denmark, S.E.; Wynn, T.; Beutner, G.L. Lewis Base Activation of Lewis Acids. Addition of Silyl Ketene Acetals to Aldehydes. J. Am. Chem. Soc. 2002, 124, 13405–13407. [Google Scholar]
- Ishitani, H.; Yamashita, Y.; Shimizu, H.; Kobayashi, S. Highly anti-Selective Catalytic Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 5403–5404. [Google Scholar] [CrossRef]
- Denmark, S.E.; Chung, W.-J. Lewis Base Activation of Lewis Acids: Catalytic Enantioselective Glycolate Aldol Reactions. Angew. Chem. Int. Ed. 2008, 47, 1890–1892. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Kumagai, N.; Matsunaga, S.; Moll, G.; Ohshima, T.; Suzuki, T.; Shibasaki, M. Direct Catalytic Asymmetric Aldol Reaction: Synthesis of Either syn- or anti-α,β-Dihydroxy Ketones. J. Am. Chem. Soc. 2001, 123, 2466–2467. [Google Scholar] [CrossRef]
- Lecea, B.; Arrieta, A.; Lopez, X.; Ugalde, J.M.; Cossio, F.P. J. Am. Chem. Soc. 1995, 117, 12314–12321. [CrossRef]
- Pons, J.-M.; Oblin, M.; Pommier, A.; Rajzmann, M.; Liotard, D. Formation of β-Lactones through Lewis Acid-Promoted [2+2] Cycloaddition Reaction. A Theoretical Study. J. Am. Chem. Soc. 1997, 119, 3333–3338. [Google Scholar]
- Singleton, D.A.; Wang, Y.; Yang, H.W.; Romo, D. Mechanism and Origin of Stereoselectivity in Lewis Acid Catalyzed [2+2] Cycloadditions of Ketenes with Aldehydes. Angew. Chem. Int. Ed. Engl. 2002, 41, 1572–1575. [Google Scholar] [CrossRef]
- Shen, X.; Wasmuth, A.S.; Zhao, J.; Zhu, C.; Nelson, S.G. Catalytic Asymmetric Assembly of Stereodefined Propionate Units: An Enantioselective Total Synthesis of (−)-Pironetin. J. Am. Chem. Soc. 2006, 128, 7438–7439, For a trans-diastereoselective Lewis acid-Lewis base catalyzed procedure using chiral enantiopure aldehydes. . [Google Scholar] [CrossRef]
- Mulzer, J.; Kerkmann, T. α Deprotonation of β-lactones—An example of a forbidden β elimination. J. Am. Chem. Soc. 1980, 102, 3620–3622, For trans-selective alkylation of β-lactones. . [Google Scholar] [CrossRef]
- Mulzer, J.; Chucholowski, A.; Lammer, O.; Ibrill, I.; Huttner, G. Diastereoselective Michael additions of β-lactone enolates to dimethyl maleate. J. Chem. Soc. Chem. Commun. 1983, 869–871. [Google Scholar]
- Kull, T.; Peters, R. Contact Ion Pair Directed Lewis Acid Catalysis: Asymmetric Synthesis of trans-Configured β-Lactones. Angew. Chem. Int. Ed. Engl. 2008, 47, 5461–5464. [Google Scholar] [CrossRef]
- Kull, T.; Cabrera, J.; Peters, R. Catalytic Asymmetric Synthesis of trans-Configured β-Lactones: Cooperation of Lewis Acid and Ion Pair Catalysis. Chem. Eur. J. 2010, 16, 9132–9139. [Google Scholar]
- Nelson, S.G.; Peelen, T.J.; Wan, Z. Mechanistic alternatives in Lewis acid-catalyzed acyl halide-aldehyde cyclocondensations. Tetrahedron Lett. 1999, 40, 6541–6543, Mechanistic studies by Nelson et al. support a ketene-aldehyde reaction pathway as the operative mechanism in AlIII-catalyzed acid chloride/aldehyde cyclocondensations. In contrast, for TiIV-catalyzed reactions providing racemic dioxanones acyl halide enolates were suggested as reactive intermediates. . [Google Scholar] [CrossRef]
- Rosenberg, R.E. Enols and Enolates of Carboxylic Acid Derivatives. A G2(MP2) ab Initio Study. J. Org. Chem. 1998, 63, 5562–5567, Species such as H2C=C(O)−Cl proved computationally unstable relative to ketene and chloride.. [Google Scholar]
- Brady, W.T.; Scherubel, G.A. Halogenated ketenes. XXIII. Mechanism of the dehydrohalogenation of acid halides. J. Am. Chem. Soc. 1973, 95, 7447–7449, Ammonium salts of acid chloride enolates can be isolated, if the electron density of the enolate C atom is stabilized by an electron withdrawing group such as a Cl atom. Studies by Brady and Scherubel have shown that the enolate forming step is reversible. In contrast, in the absence of an electron withdrawing substituent, the enolate salts could neither be isolated nor detected spectroscopically as a result of their instability.. [Google Scholar]
- Brady, W.T.; Scherubel, G.A. Halogenated ketenes. XXVII. Mechanism of the dehydrohalogenation of α-halo acid halides. J. Org. Chem. 1974, 39, 3790–3791. [Google Scholar]
- Yoon, T.P.; Jacobsen, E.N. Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef]
- Matsumoto, K.; Saito, B.; Katsuki, T. Asymmetric catalysis of metal complexes with non-planar ONNO ligands: Salen, salalen and salan. Chem. Commun. 2007, 35, 3619–3627. [Google Scholar]
- Myers, J.K.; Jacobsen, E.N. Asymmetric Synthesis of β-Amino Acid Derivatives via Catalytic Conjugate Addition of Hydrazoic Acid to Unsaturated Imides. J. Am. Chem. Soc. 1999, 121, 8959–8960. [Google Scholar] [CrossRef]
- Cabrera, J.; Hellmuth, T.; Peters, R. Synthesis of Densely Substituted Trans-Configured 4-Acylated Piperidine-2,4-diones as 3:1 adducts of Imines and Ketenes. J. Org. Chem. 2010, 75, 4326–4329. [Google Scholar]
- Tiseni, P.S.; Peters, R. Catalytic Asymmetric Formation of d-Lactones from Unsaturated Acyl Halides. Chem. Eur. J. 2010, 16, 2503–2517. [Google Scholar] [CrossRef]
- Tiseni, P.S.; Peters, R. Lewis Acid-Lewis Base Catalyzed Enantioselective Hetero-Diels-Alder Reaction for Direct Access to d-Lactones. Org. Lett. 2008, 10, 2019–2022. [Google Scholar] [CrossRef]
- Tiseni, P.S.; Peters, R. Catalytic Asymmetric Formation of d-Lactones by [4+2]-Cycloaddition of Zwitterionic Dienolates Generated fromα,β-Unsaturated Acid Chlorides. Angew. Chem. Int. Ed. 2007, 46, 5325–5328. [Google Scholar] [CrossRef]
- Koch, F.M.; Peters, R. Lewis-Acid/Lewis-Base Catalyzed [2+2]-Cycloaddition of Sulfenes and Aldehydes: A Versatile Entry to Chiral Sulfonyl and Sulfinyl Derivatives. Chem. Eur. J. 2011, 17, 3679–3692. [Google Scholar]
- Zajac, M.; Peters, R. Catalytic Asymmetric Synthesis of β-Sultams as Precursors for Taurine Derivatives. Chem. Eur. J. 2009, 15, 8204–8222. [Google Scholar] [CrossRef]
- Peters, R.; Fischer, D.F.; Jautze, S.; Koch, F.M.; Kull, T.; Tiseni, P.S.; Xin, Z.-Q.; Zajac, M. Catalytic Methods for Direct Access to Chiral High-Added-Value Products. Chimia 2008, 62, 497–505. [Google Scholar]
- Koch, F.M.; Peters, R. Rapid Asymmetric Access to β-Hydroxysulfinic Acids and Allylsulfonic Acids by Chemoselective Reduction of β-Sultones. Synlett 2008, 1505–1509. [Google Scholar]
- Zajac, M.; Peters, R. Catalytic Asymmetric Formation of β-Sultams. Org. Lett. 2007, 9, 2007–2010. [Google Scholar] [CrossRef]
- Koch, F.M.; Peters, R. First Catalytic Enantio- and Diastereoselective Formation of β-Sultones: Ring-Strained Precursors for Enantioenriched β-Hydroxysulfonyl Derivatives. Angew. Chem. Int. Ed. 2007, 46, 2685–2689. [Google Scholar]
- Bhatt, S.; Nayak, S.K. Reductive deoxygenation of ortho-hydroxyaromatic aldehydes to 1,2-bis(hydroxyaryl)ethanes: Application to the synthesis of ethylene bridged calixarene-analogous metacyclophanes. Tetrahedron Lett. 2009, 50, 5823. [Google Scholar] [CrossRef]
- Hofsløkken, N.U.; Skattebøl, L. Convenient Method for the ortho-Formylation of Phenols. Acta Chem. Scand. 1999, 53, 258. [Google Scholar] [CrossRef]
- Wang, Q.; Wilson, C.; Blake, A.J.; Collinson, S.R.; Tasker, P.A.; Schröder, M. The one-pot halomethylation of 5-substituted salicylaldehydes as convenient precursors for the preparation of heteroditopic ligands for the binding of metal salts. Tetrahedron Lett. 2006, 47, 8983–8986. [Google Scholar] [CrossRef]
- Precipitation of catalyst 14aB is for instance visible at −70 °C prior to addition of the reagents.
- Larrow, J.F.; Jacobsen, E.N. Asymmetric processes catalyzed by chiral (salen)metal complexes. Top. Organomet. Chem. 2004, 6, 123–152. [Google Scholar]
- Pommier, A.; Pons, J.M. Recent Advances in β-Lactone Chemistry. Synthesis 1993, 441–459. [Google Scholar] [CrossRef]
- Konsler, R.G.; Karl, J.; Jacobsen, E.N. Cooperative Asymmetric Catalysis with Dimeric Salen Complexes. J. Am. Chem. Soc. 1998, 120, 10780–10781. [Google Scholar] [CrossRef]
- Weber, M.; Jautze, S.; Frey, W.; Peters, R. Bis-Palladacycle Catalyzed Brønsted-Acid/-Base Promoted Asymmetric Tandem Azlactone Formation-Michael Addition. J. Am. Chem. Soc. 2010, 132, 12222–12225. [Google Scholar] [CrossRef]
- Jautze, S.; Peters, R. Bimetallic Enantioselective Catalysis of Michael Additions Forming Quaternary Stereocenters. Angew. Chem. Int. Ed. Engl. 2008, 47, 9284–9288. [Google Scholar] [CrossRef]
- Jautze, S.; Seiler, P.; Peters, R. Synthesis of nearly Enantiopure Allylic Amines by Aza-Claisen Rearrangement of Z-Configured Allylic Trifluoroacetimidates Catalyzed by Highly Active Ferrocenylbispalladacycles. Chem. Eur. J. 2008, 14, 1430–1444. [Google Scholar] [CrossRef]
- Jautze, S.; Seiler, P.; Peters, R. Angew. Chem. Int. Ed. Engl. 2007, 46, 1260–1264. [CrossRef]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear Effects in Asymmetric Catalysis. Angew. Chem. Int. Ed. Engl. 2009, 48, 456–494. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Manby, F.R.; Schütz, M.; Celani, P.; Knizia, G.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, 2010; 1, a package of ab initio programs .
- Wang, X.-L.; Wan, H.; Guan, G.-F. Structure and Interaction of Ion-Pairs of [EPy]Cl and [EPy]Br in Gas and Liquid Phases. Acta Phys.-Chim. Sin. 2008, 24, 2077–2082. [Google Scholar]
- 86. This might also explain why the standard salen ligand with tBu moieties showed no reactivity at all for the title reaction.
- Katsuki, T. Some recent advances in metallosalen chemistry. Synlett 2003, 281–297. [Google Scholar] [CrossRef]
- Katsuki, T. Unique asymmetric catalysis of cis-βmetal complexes of salen and its related Schiff-base ligands. Chem. Soc. Rev. 2004, 33, 437–444. [Google Scholar] [CrossRef]
- Jacobsen, H. Computational Studies of the Hydrolytic Kinetic Resolution of Terminal Epoxides Catalysed by Co(salen) Complexes: Evaluation of Suitable Model Systems and Appropriate Density Functional Approaches. In Frontiers in Organometallic Chemistry; Cato, M.A., Ed.; Nova Science Publishers: New York, NY, USA, 2006; pp. 1–16. [Google Scholar]
- Darensbourg, D.J.; Billodeaux, D.R. Aluminum Salen Complexes and Tetrabutylammonium Salts: A Binary Catalytic System for Production of Polycarbonates from CO2 and Cyclohexene Oxide. Inorg. Chem. 2005, 44, 1433–1442. [Google Scholar] [CrossRef]
- Yu, X.; Scheller, D.; Rademacher, O.; Wolff, T. Selectivity in the Photodimerization of 6-Alkylcoumarins. J. Org. Chem. 2003, 68, 7386–7399. [Google Scholar] [CrossRef]
- Aldoshin, S.M.; Sanina, N.A.; Minkin, V.I.; Voloshin, N.A.; Ikorskii, V.N.; Oveharenko, V.I.; Smirnov, V.A.; Nagaeva, N.K. Molecular photochromic ferromagnetic based on the layered polymeric tris-oxalate of Cr(III), Mn(II) and 1-[(1′,3′,3′-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2′-indoline]-8-yl)methyl]pyridinium. J. Mol. Struct. 2007, 826, 69–74. [Google Scholar] [CrossRef]
- Samples Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meier, P.; Broghammer, F.; Latendorf, K.; Rauhut, G.; Peters, R. Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects. Molecules 2012, 17, 7121-7150. https://doi.org/10.3390/molecules17067121
Meier P, Broghammer F, Latendorf K, Rauhut G, Peters R. Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects. Molecules. 2012; 17(6):7121-7150. https://doi.org/10.3390/molecules17067121
Chicago/Turabian StyleMeier, Patrick, Florian Broghammer, Katja Latendorf, Guntram Rauhut, and René Peters. 2012. "Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects" Molecules 17, no. 6: 7121-7150. https://doi.org/10.3390/molecules17067121
APA StyleMeier, P., Broghammer, F., Latendorf, K., Rauhut, G., & Peters, R. (2012). Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects. Molecules, 17(6), 7121-7150. https://doi.org/10.3390/molecules17067121