Synthesis and Characterization of Some New C2 Symmetric Chiral Bisamide Ligands Derived from Chiral Feist’s Acid

 ,
, 
Abstract
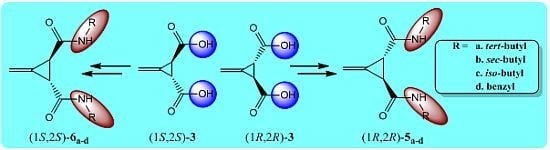
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
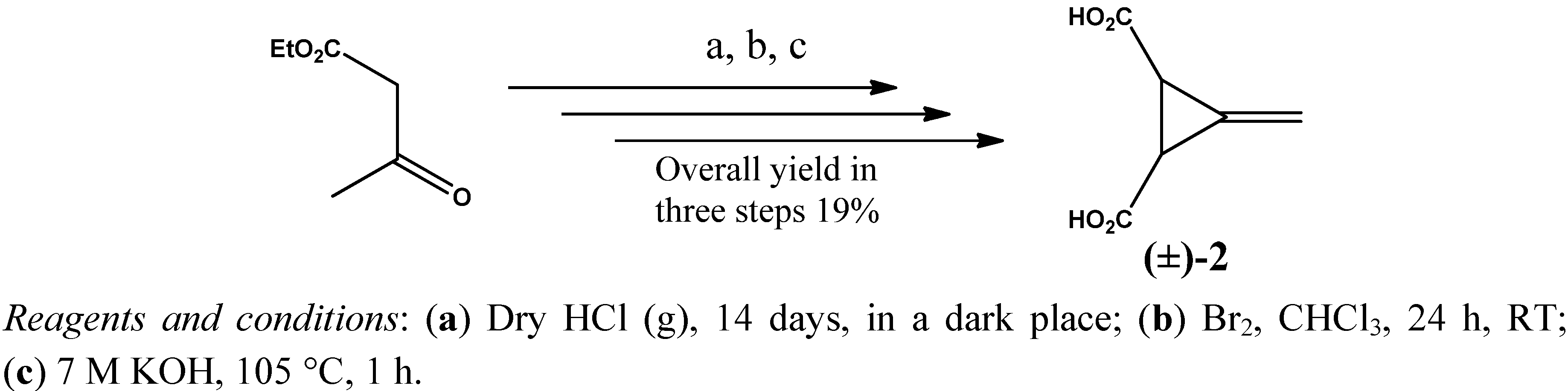
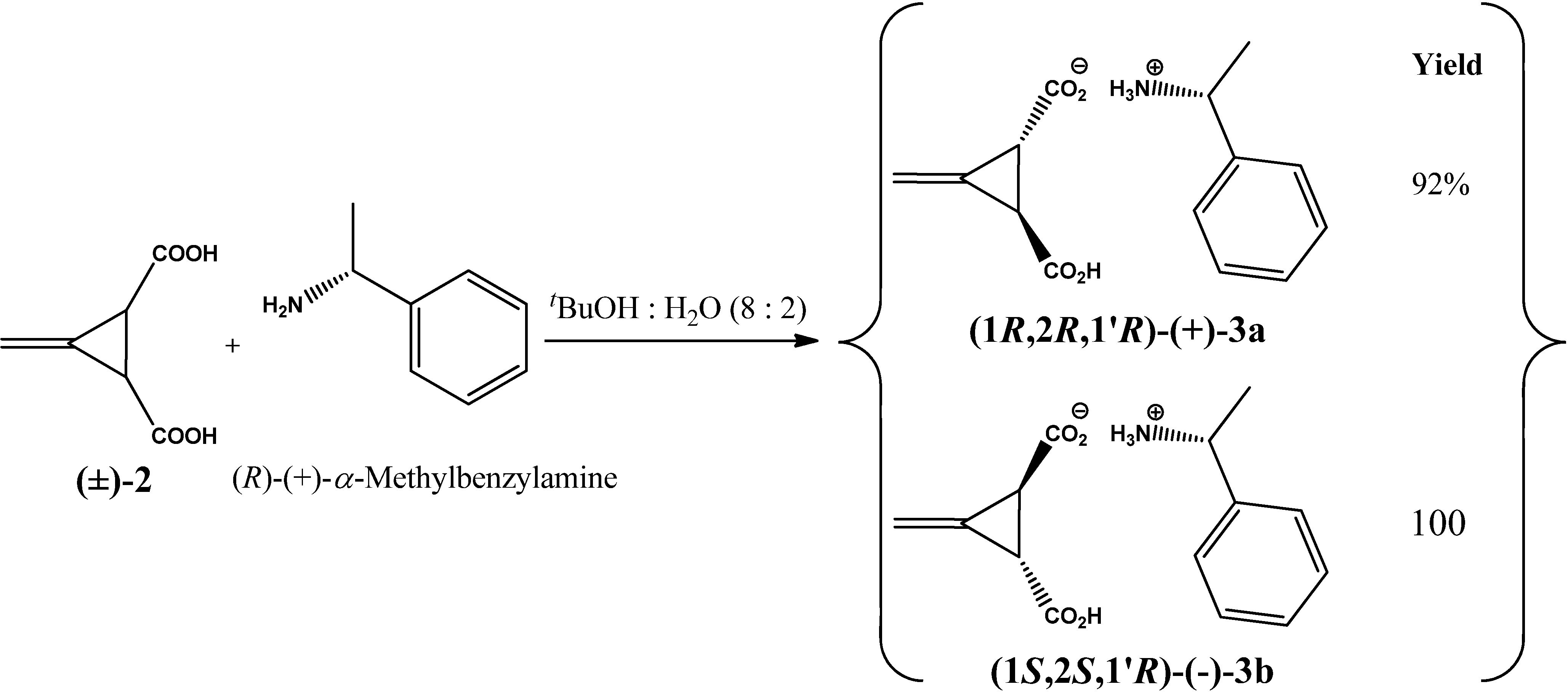
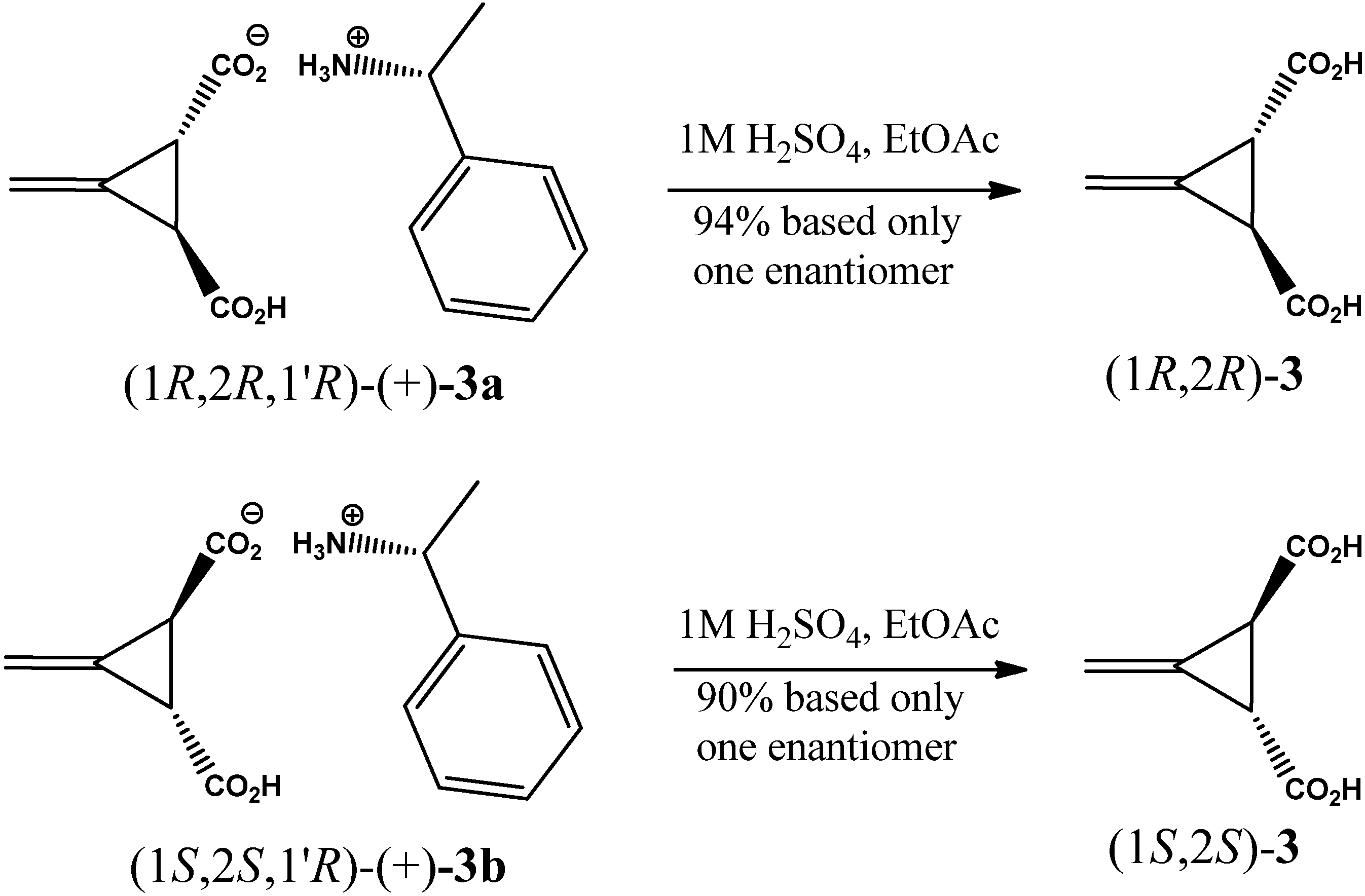
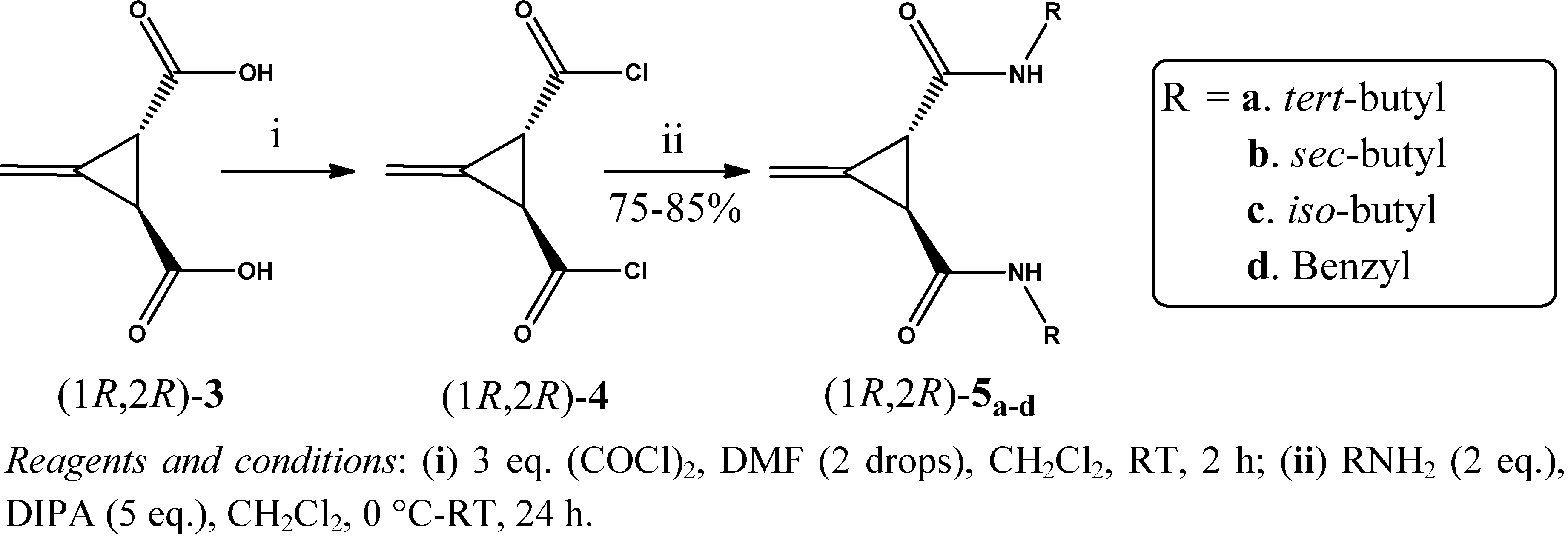
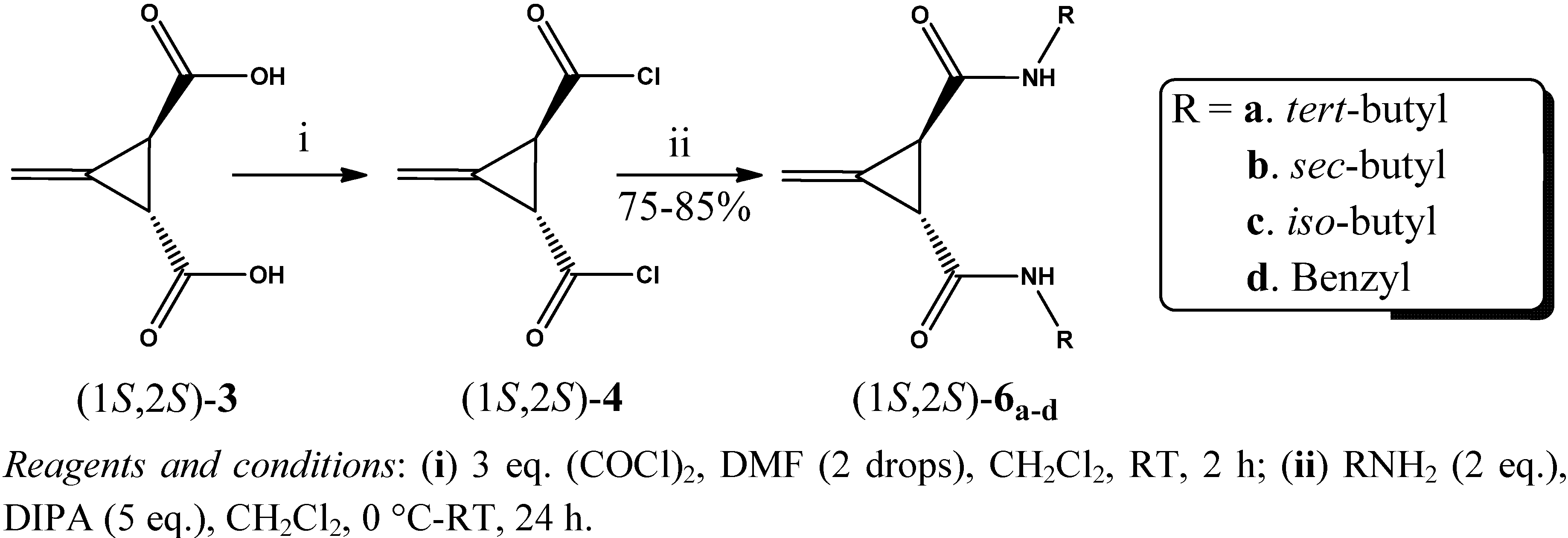
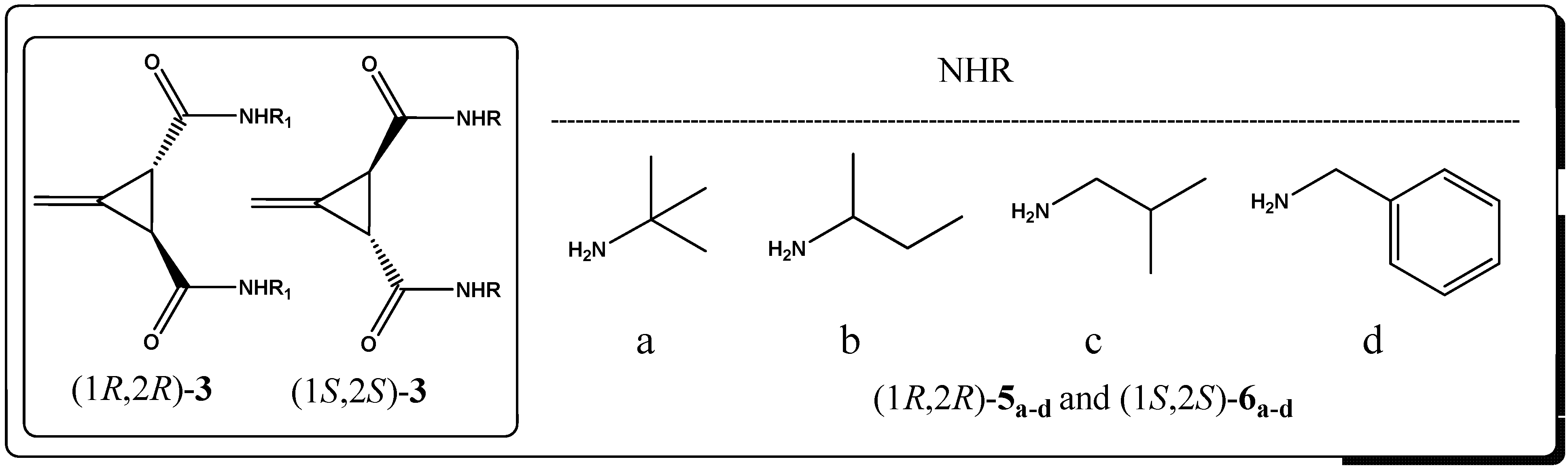
2. Results and Discussion
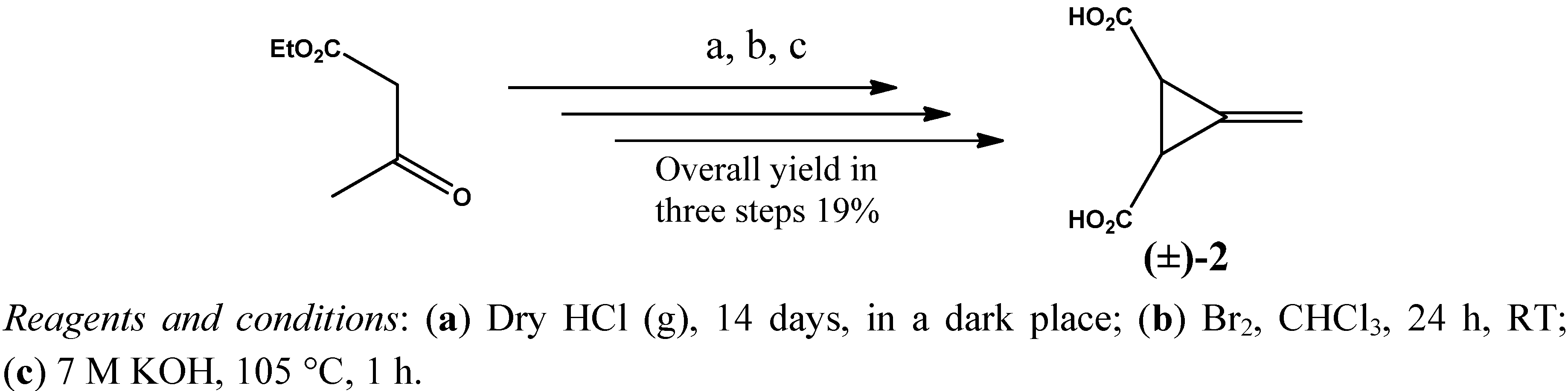
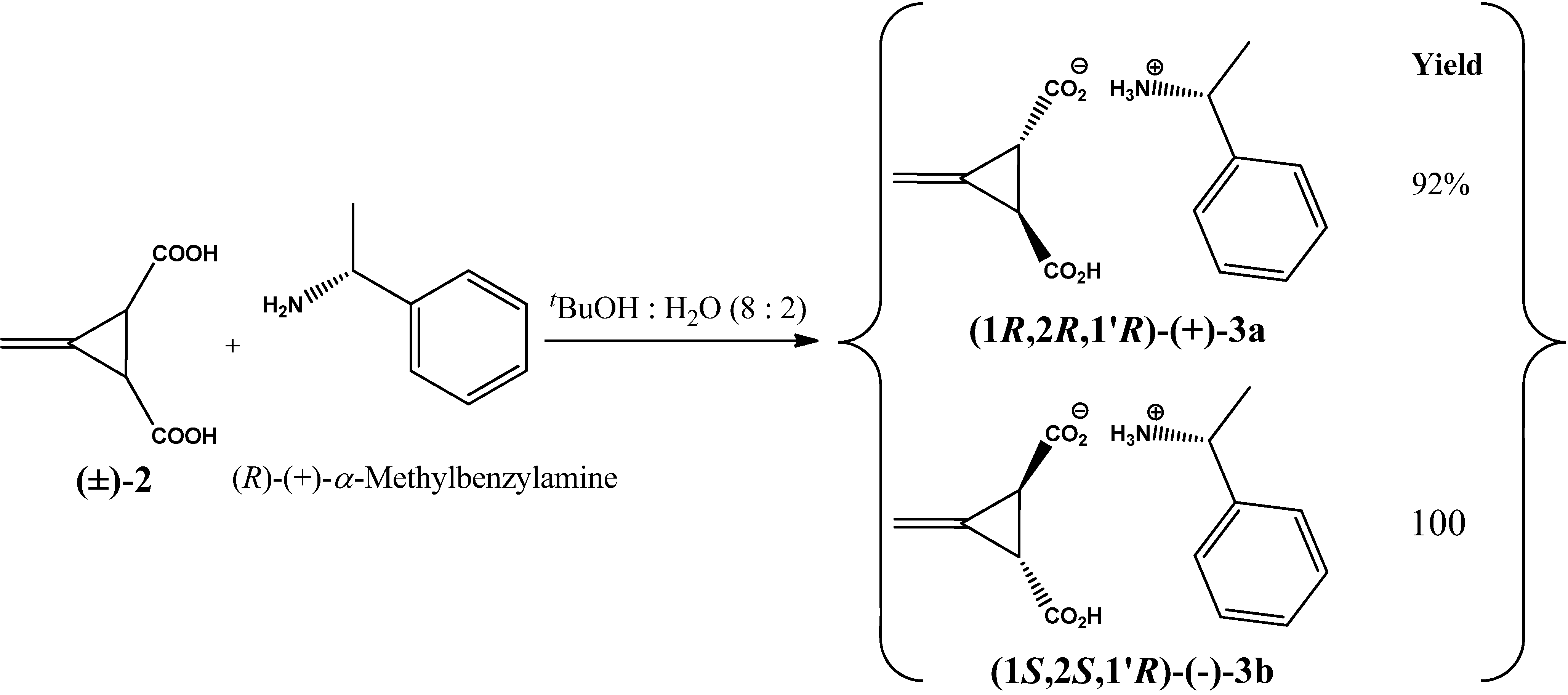
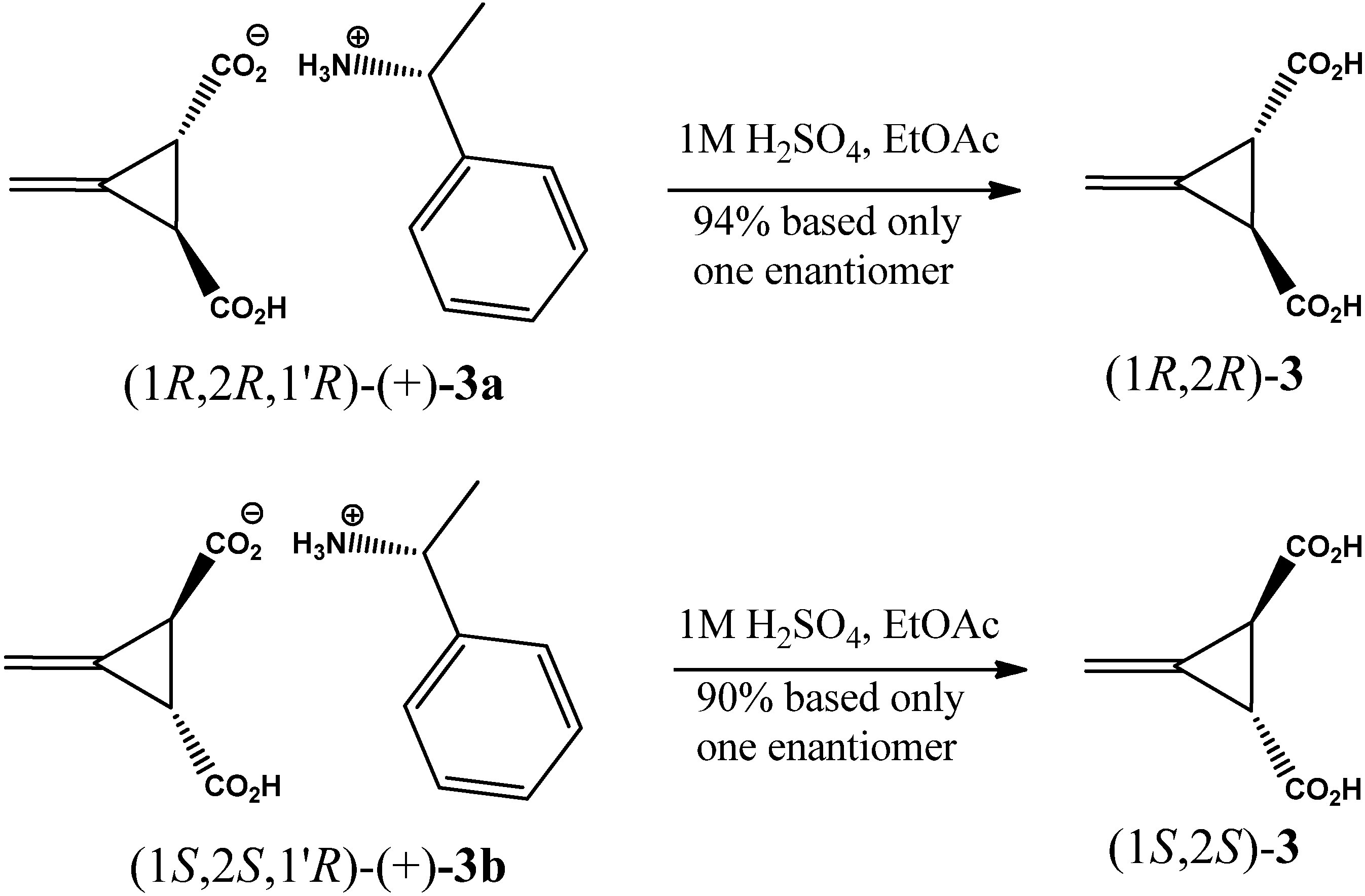
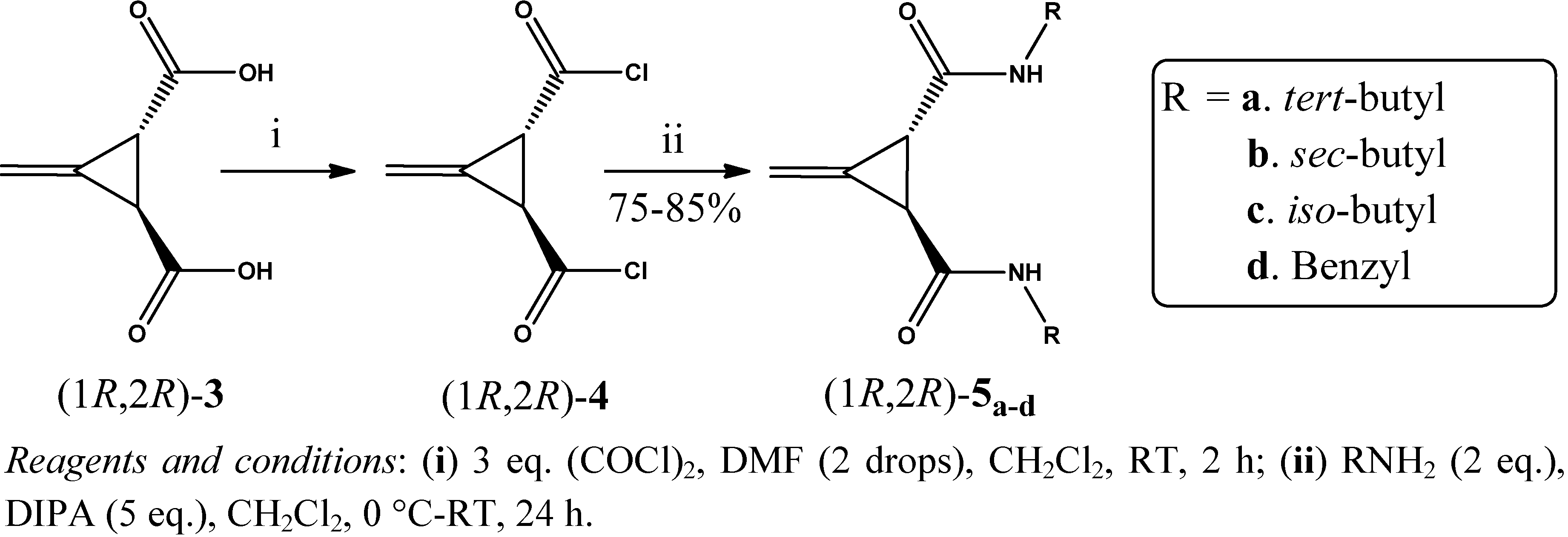
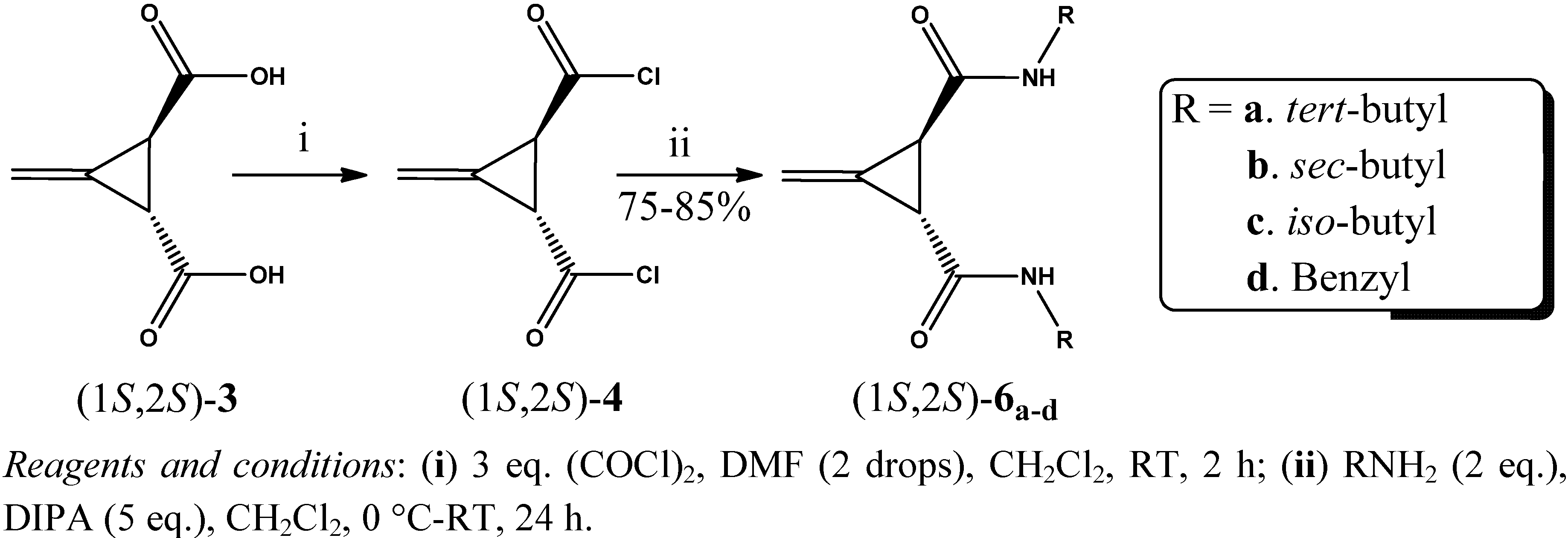
3. Experimental
3.1. General
3.2. Resolution of trans-(±)-3-Methylenecyclopropane-1,2-dicarboxylic Acid [(±)-2]
3.3. General Procedure for the Preparation of Acid Chlorides (1R,2R)-4 and (1S,2S)-4 (GP1)
3.4. General Procedure for the Synthesis of Amides (1R,2R)-5a–d and (1S,2S)-6a–d (GP2)
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Larcheveque, M.; Ignatova, E.; Cuvigny, T. Asymmetric alkylation of chiral N,N-disubstituted amide. J. Organomet. Chem. 1979, 177, 5–15. [Google Scholar]
- Altava, B.; Burguete, M.I.; Carbó, N.; Escorihuela, J.; Luis, S.V. Chiral bis(amino amides) as chiral solvating agents for enantiomeric excess determination of α-hydroxy and arylpropionic acids. Tetrahedron Asymmetry 2010, 21, 982–989. [Google Scholar] [CrossRef]
- Honda, A.; Waltz, K.M.; Carroll, P.J.; Wals, P.J. Atropisomeric amides: Achiral ligands with chiral conformations. Chirality 2003, 15, 615–621. [Google Scholar] [CrossRef]
- Al Majid, A.M.A.; Al-Othman, Z.A.; Islam, M.S. Synthesis of some C2 symmetric bidentate ligands, and their complexes derived from Feist’s acid. Helv. Chim. Acta 2012, 92, 268–277. [Google Scholar]
- Verbroom, R.C.; Plietker, B.J.; Bäckvall, J.E. New chiral diamide ligands containing redox-active hydroquinone groups. Synthesis and results in the palladium(II)-catalyzed 1,4-diacetoxylation of 1,3-dienes. J. Organomet. Chem. 2003, 687, 508–517. [Google Scholar]
- Bateman, L.; Breeden, S.W.; O’Leary, P. New chiral diamide ligands: Synthesis and application in allylic alkylation. Tetrahedron Asymmetry 2008, 19, 391–396. [Google Scholar] [CrossRef]
- Grasa, G.A.; Gerosa, A.Z.; Medlock, J.A.; Hems, W.P. Asymmetric hydrogenation of isobutyrophenone using a [(diphosphine) rucl2 (1,4-diamine)] catalyst. Org. Lett. 2005, 7, 1449–1451. [Google Scholar] [CrossRef]
- Pritchett, S.; Gantgel, P.; Wals, P.J. Synthesis and crystal structures of chiral titanium bis(sulfonamido) bis(amide) complexes: Differences in ligand hapticity caused by crystal packing forces. Organometallics 1997, 16, 5130–5132. [Google Scholar] [CrossRef]
- Noyori, R.; Kitamura, M.; Lee, D.; Hayashi, S.; Tanaka, S.; Yoshimura, M. Catalytic Leuckart-Wallach-type reductive amination of ketones. Angew. Chem. Int. Ed. Engl. 1991, 30, 49–69. [Google Scholar] [CrossRef]
- Soai, K.; Niwa, S. Enantioselective addition of organozinc reagent to aldehydes. Chem. Rev. 1992, 92, 833–856. [Google Scholar] [CrossRef]
- Erdik, E. Organozinc Reagents in Organic Synthesis; CRC Press: New York, NY, USA, 1996. [Google Scholar]
- Soai, K.; Shibata, T. Alkylation of Carbonyl Groups. In Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999; Volume II, pp. 911–923, Chapter 26. [Google Scholar]
- Pu, L.; Yu, H.B. Catalytic asymmetric organozinc addition to carbonyl compound. Chem. Rev. 2001, 101, 757–824. [Google Scholar] [CrossRef]
- Oguni, N.; Omi, T. Enantioselective addition of diethylzinc to benzaldehyde catalyzed by a small amount of chiral 2-amino-1-alcohols. Tetrahedron Lett. 1984, 25, 2823–2824. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef]
- Soai, K.; Ookawa, A.; Kaba, T.; Ogawa, K. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes using chiral pyrrolidinylmethanols and their metal salts. J. Am. Chem. Soc. 1987, 109, 7111–7115. [Google Scholar] [CrossRef]
- Oguni, N.; Matsuda, Y.; Kaneko, T. Asymmetric amplifying phenomena in enantioselective addition of diethylzinc to benzaldehyde. J. Am. Chem. Soc. 1988, 110, 7877–7878. [Google Scholar] [CrossRef]
- Soai, K.; Yokoyama, S.; Hayasaka, T. Chiral N,N-dialkylnorephedrines as catalysts of the highly enantioselective addition of dialkylzincs to aliphatic and aromatic aldehydes. The asymmetric synthesis of secondary aliphatic and aromatic alcohols of high optical purity. J. Org. Chem. 1991, 56, 4264–4268. [Google Scholar] [CrossRef]
- Zhao, G.; Li, X.G.; Wang, X.R. Enantioselective additions of diphenylzinc to aldehydes using chiral pyrrolidinylmethanol derivatives as catalysts. Tetrahedron Asymmetry 2001, 12, 399–403. [Google Scholar] [CrossRef]
- DiMauro, F.F.; Kozlowski, M.C. Salen-derived catalysts containing secondary basic groups in the addition of diethylzinc to aldehydes. Org. Lett. 2001, 3, 3053–3059. [Google Scholar] [CrossRef]
- Wipf, P.; Wang, X. A new ligand scaffold for catalytic asymmetric alkylzinc additions to aldehydes. Org. Lett. 2002, 4, 1197–1200. [Google Scholar] [CrossRef]
- Nugent, W.A. An amino alcohol ligand for highly enantioselective addition of organozinc reagents to aldehydes: Serendipity rules. Org. Lett. 2002, 4, 2133–2136. [Google Scholar] [CrossRef]
- Atodiresei, I.; Schiffers, I.; Bolm, C. Asymmetric synthesis of chiral bisoxazolines and their use as ligands in metal catalysis. Tetrahedron Asymmetry 2006, 17, 620–633. [Google Scholar] [CrossRef]
- Nahar, K.R.; Solanki, A.K.; Bhandari, A.M. Imido derivatives of titanium (IV). Z. Anorg. Allg. Chem. 1979, 449, 187–192. [Google Scholar] [CrossRef]
- Imai, Y.; Zhang, W.; Kida, T.; Nakatsuji, Y.; Ikeda, I. Novel C2-symmetric chiral bisoxazoline ligands in rhodium(I)-catalyzed asymmetric hydrosilylation. Tetrahedron Asymmetry 1996, 7, 2453–2462. [Google Scholar] [CrossRef]
- Procter, G. Asymmetric Synthesis; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Kosak, R.; Johnson, T.A. Catalysis of Organic Reaction; Marcel Dekker: New York, NY, USA, 1994; Chapter 4. [Google Scholar]
- Noyori, R. Asymmetric Catalysis in Organic Synthesis; John Wiley & Sons: New York, NY, USA, 1994; Chapter 2. [Google Scholar]
- Kimura, K.; Sugiyama, E.; Ishizuki, T.; Kunieda, T. Dramatic reversal of enantioselectivity in β-aminoalcohol-catalyzed addition of diethylzinc to aldehydes. Tetrahedron Lett. 1992, 33, 3147–3150. [Google Scholar] [CrossRef]
- Doering, W.E.; Roth, H.D. Stereochemistry of the methylenecyclopropane rearrangement: Feist’s ester. Tetrahedron 1970, 26, 2825–2835. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Booth, B.L.; Gomes, J.T. C2-Symmetric ligands for asymmetric catalysis based on Feist’s acid. J. Chem. Res. 1998, 78–79. [Google Scholar]
- Godfrey, J.D.Jr.; Muller, R.H.; Kissick, T.P.; Sing, J. Process for the Preparation of an Antiviral Agent. U.S. Patent 5185463, 1993. [Google Scholar]
- Goss, F.R.; Ingold, C.K.; Thorpe, J.F. The chemistry of the glutaconic acids. Part XIV. Three-carbon tautomerism in the cyclopropane series. J. Chem. Soc. 1923, 123, 327–361. [Google Scholar]
- Sample Availability: Samples of the compounds (1R,2R)-3, (1S,2S)-3, 5a–d, 6a–d are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al Majid, A.M.A.; Islam, M.S.; Al-Othman, Z.A.; Al-Salhoob, A.F.; Barakat, A. Synthesis and Characterization of Some New C2 Symmetric Chiral Bisamide Ligands Derived from Chiral Feist’s Acid. Molecules 2012, 17, 5550-5563. https://doi.org/10.3390/molecules17055550
Al Majid AMA, Islam MS, Al-Othman ZA, Al-Salhoob AF, Barakat A. Synthesis and Characterization of Some New C2 Symmetric Chiral Bisamide Ligands Derived from Chiral Feist’s Acid. Molecules. 2012; 17(5):5550-5563. https://doi.org/10.3390/molecules17055550
Chicago/Turabian StyleAl Majid, Abdullah M. A., Mohammad Shahidul Islam, Zeid A. Al-Othman, Ahlam F. Al-Salhoob, and Assem Barakat. 2012. "Synthesis and Characterization of Some New C2 Symmetric Chiral Bisamide Ligands Derived from Chiral Feist’s Acid" Molecules 17, no. 5: 5550-5563. https://doi.org/10.3390/molecules17055550
APA StyleAl Majid, A. M. A., Islam, M. S., Al-Othman, Z. A., Al-Salhoob, A. F., & Barakat, A. (2012). Synthesis and Characterization of Some New C2 Symmetric Chiral Bisamide Ligands Derived from Chiral Feist’s Acid. Molecules, 17(5), 5550-5563. https://doi.org/10.3390/molecules17055550