Synthesis and Modifications of Phosphinic Dipeptide Analogues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
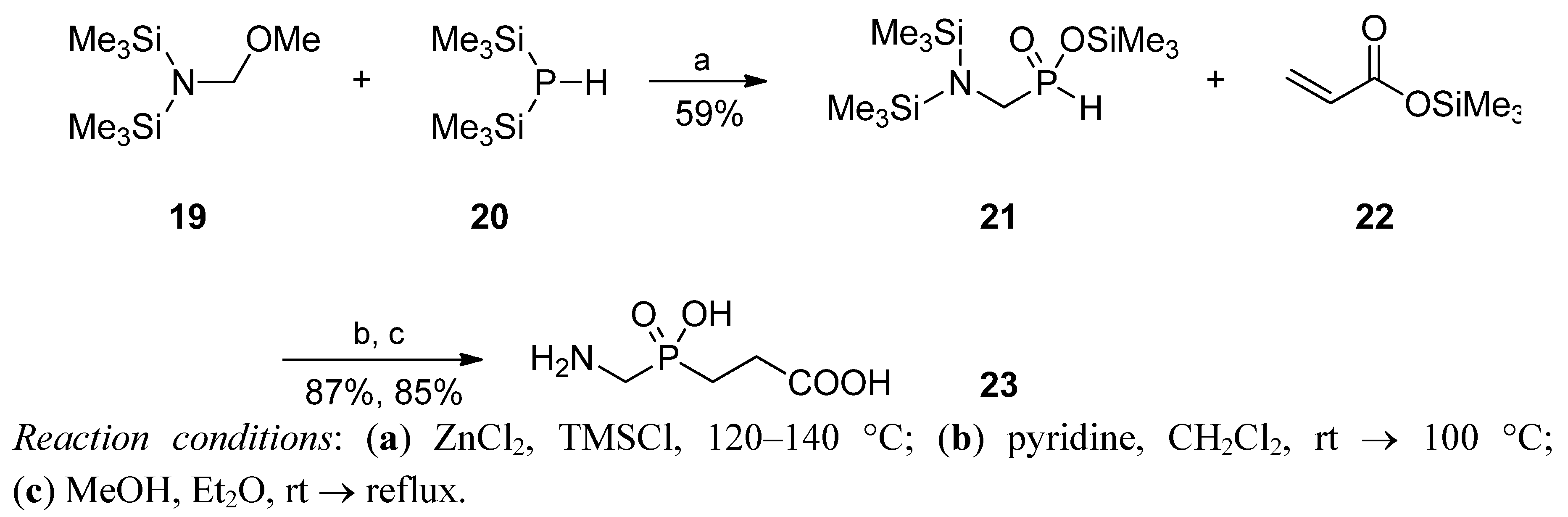{kind=link}
{kind=link}
{kind=link}
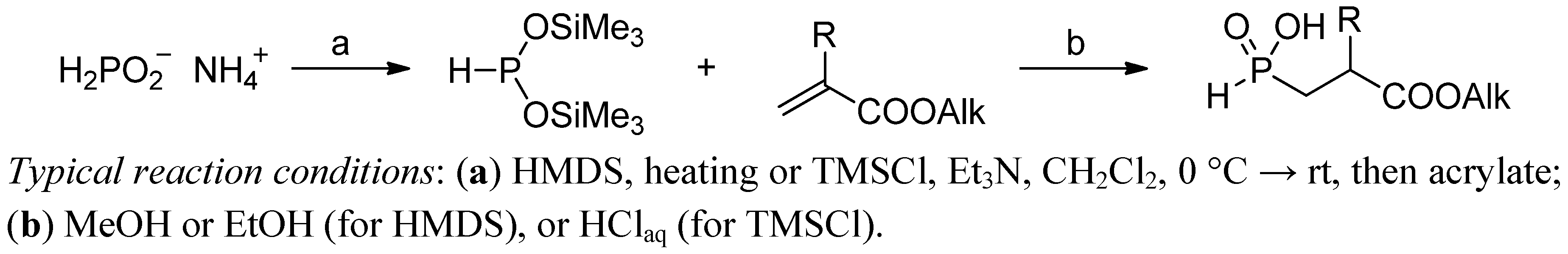{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
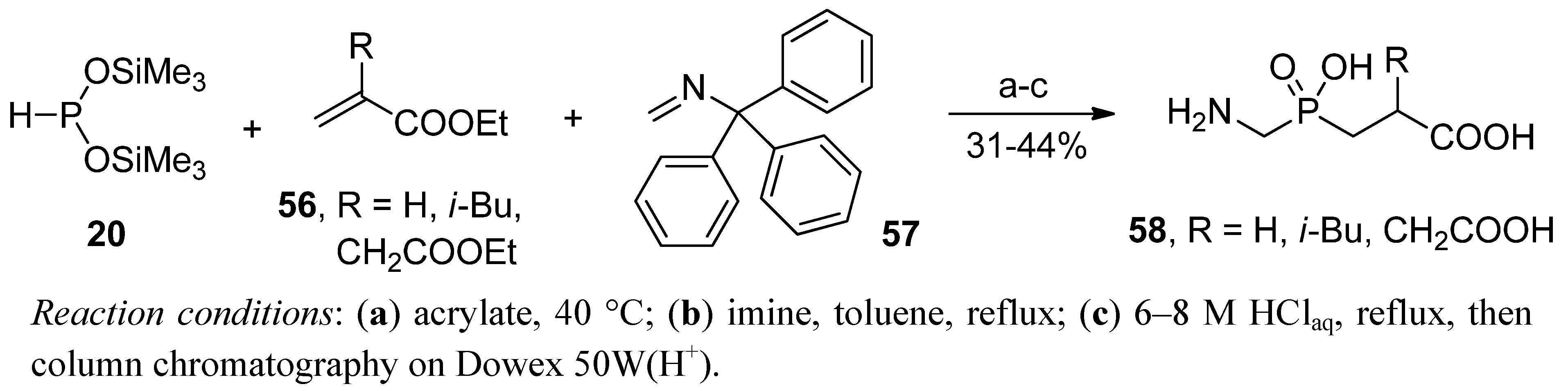{kind=link}
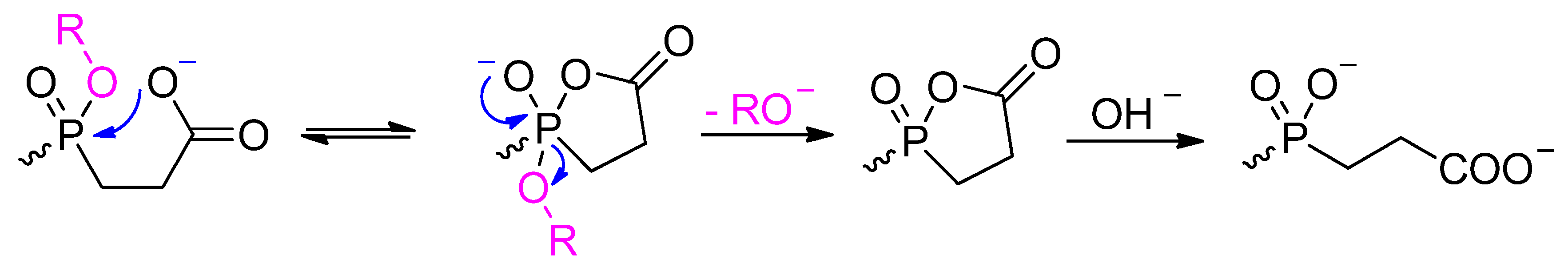{kind=link}
{kind=link}
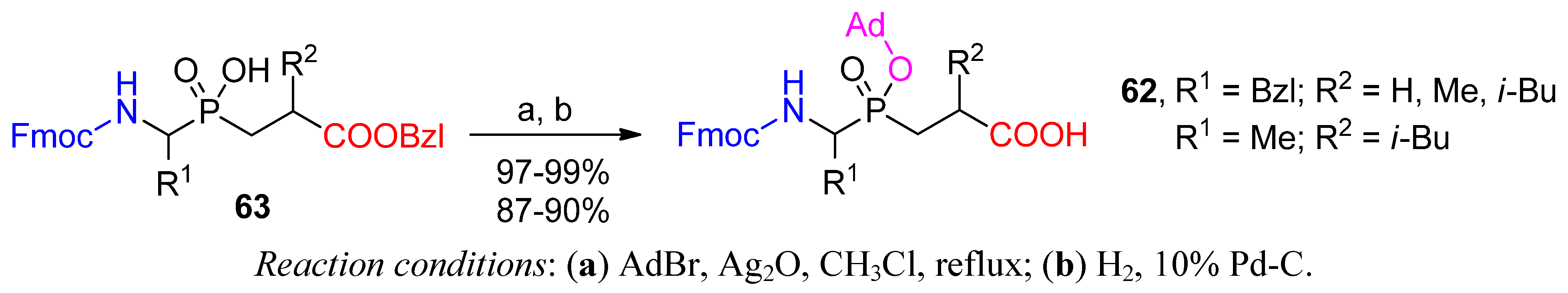{kind=link}
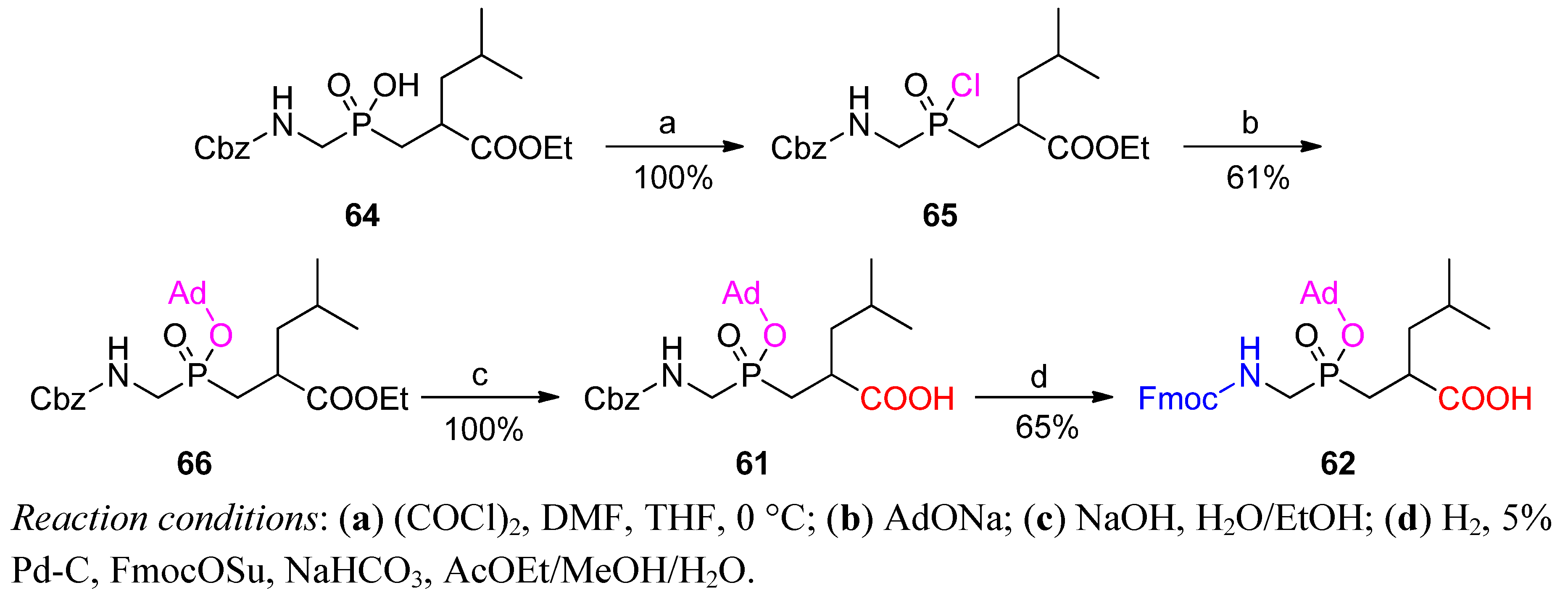{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
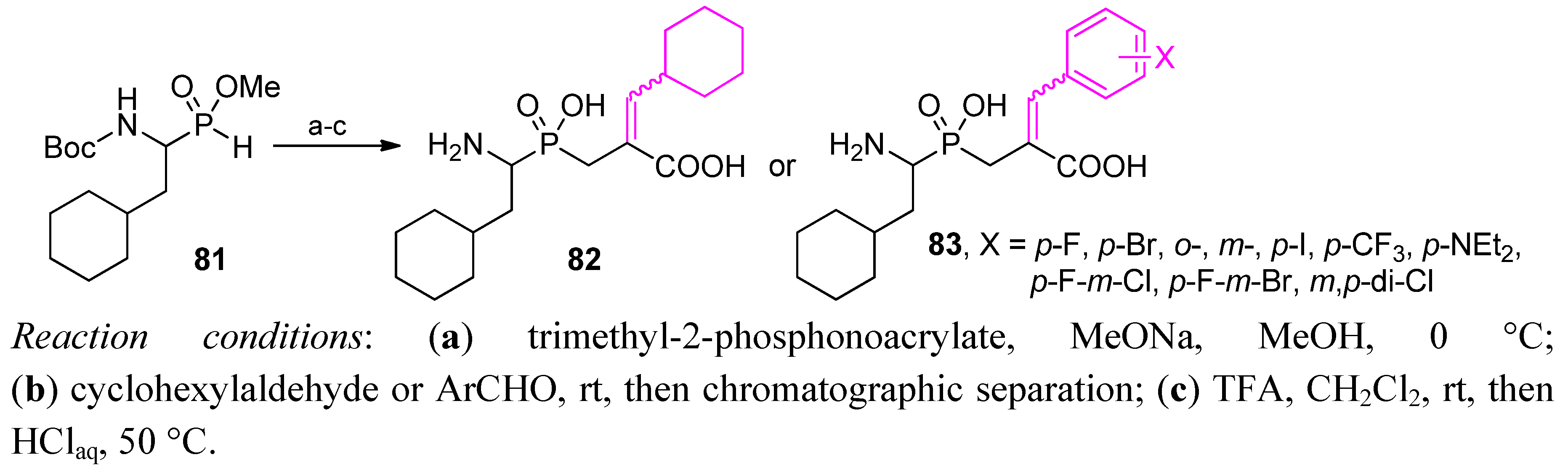{kind=link}
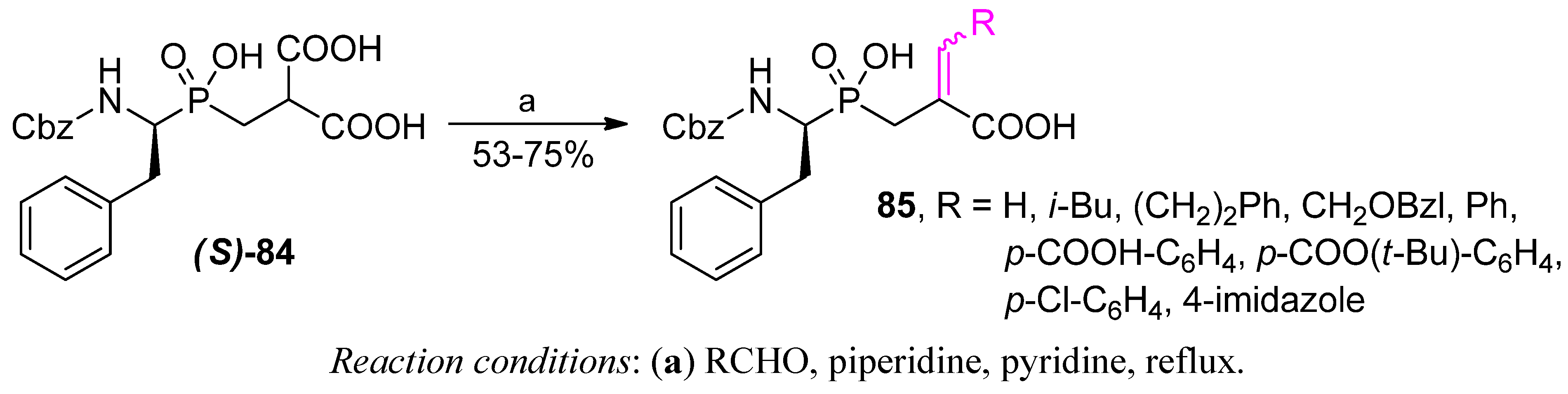{kind=link}
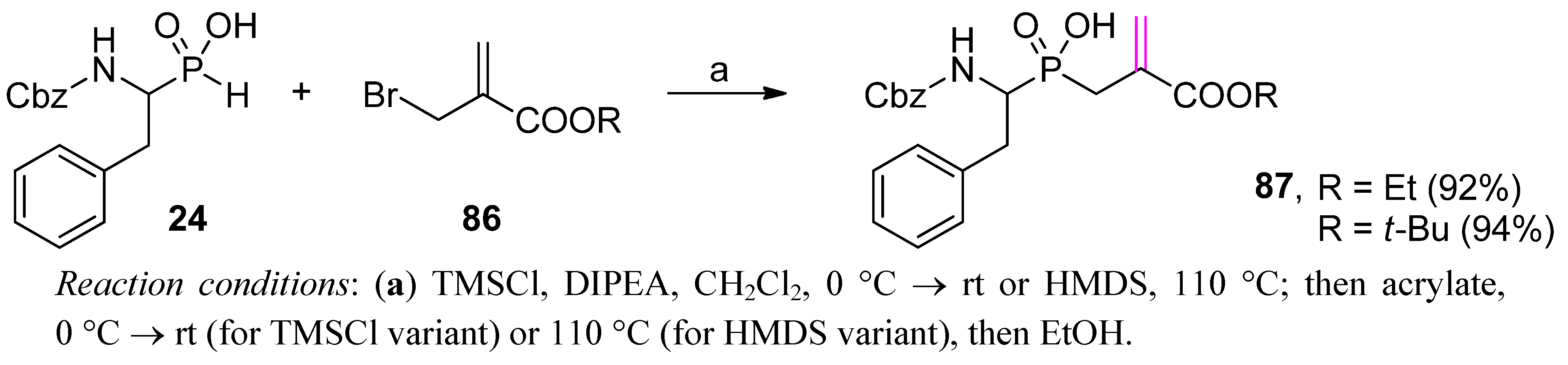{kind=link}
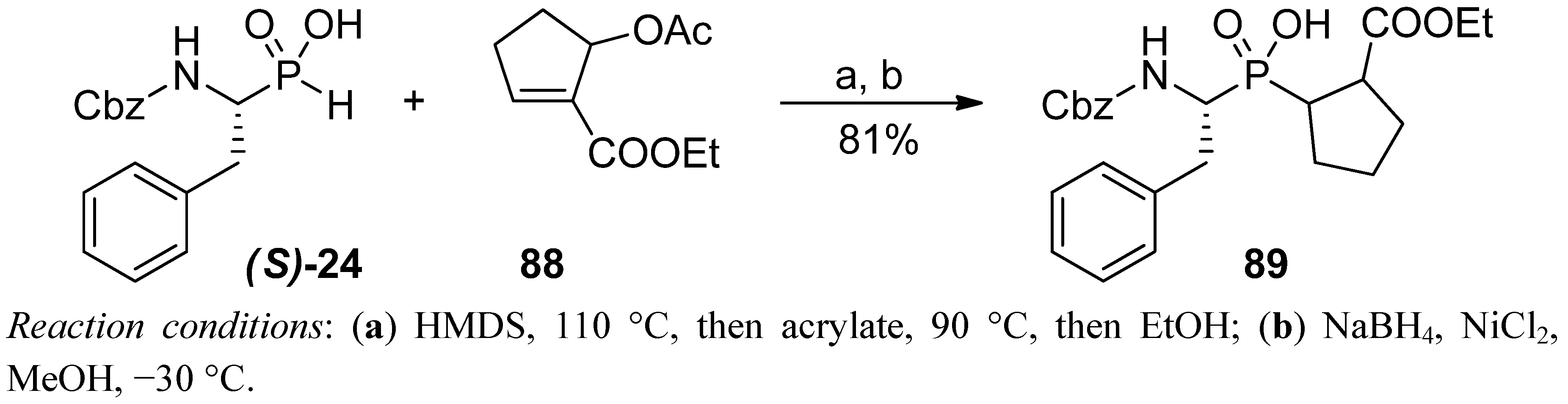{kind=link}
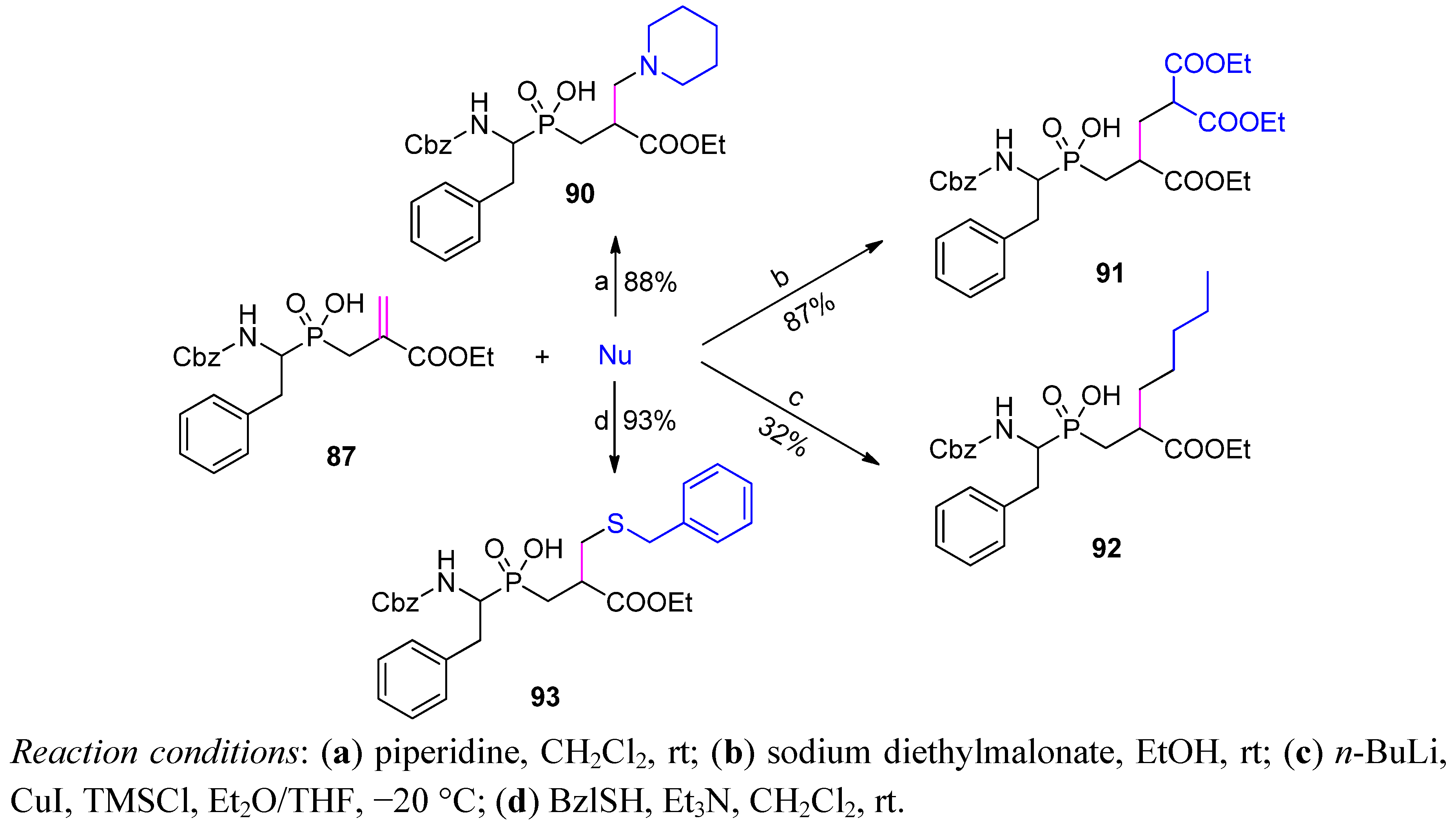{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| Aa1ψ[P(O)(OH)X]-Aa2 | phosphorus-containing dipeptide analogues (X = NH, phosphonamidates; X = O, phosphonates; X = CH2, phosphinates) |
| AaPH | α-amino-H-phosphinic (phosphonous) acid |
| Ac | acetyl |
| Ad | 1-adamantyl |
| Alk | alkyl |
| Alloc | allyloxycarbonyl |
| APN | alanyl aminopeptidase |
| Ar | aryl |
| ATP | adenosine triphosphate |
| Boc | t-butyloxycarbonyl |
| BOP | (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| BSA | N,O-bis(trimethylsilyl)-acetamide |
| i-Bu | isobutyl |
| n-Bu | n-butyl |
| sec-Bu | sec-butyl |
| t-Bu | tert-butyl |
| Bz | benzoyl |
| Bzl | benzyl |
| Cbz | benzyloxy-carbonyl |
| CPB | carboxypeptidase B |
| dba | dibenzylideneacetone |
| DEAD | diethyl azodicarboxylate |
| DCC | N,N'-dicyclohexylcarbodiimide |
| de | diastereomeric excess |
| DIPEA | diisopropylethylamine |
| DMAP | 4-dimethylaminopyridine |
| DME | 1,2-dimethoxy-ethane |
| DMF | dimethylformamide |
| EDC | 1-ethyl-3-(3'-dimethylaminopropyl)carbo-diimide |
| Et | ethyl |
| EWG | electron withdrawing group |
| Fmoc | 9-fluorenylmethoxy-carbonyl |
| HMDS | 1,1,1,3,3,3-hexamethyldisilazane |
| HOBt | 1-hydroxybenzotriazole |
| HPLC | high performance liquid chromatography |
| LAP | leucine aminopeptidase |
| Me | methyl |
| MurD | UDP-N-acetylmuramoyl-L-alanyl:D-glutamate ligase |
| Nu | nucleophile |
| Pac | phenacyl |
| PG | protecting group |
| Ph | phenyl |
| Phth | phthaloyl |
| i-Pr | isopropyl |
| PyBOP | (benzotriazol-1-yloxy)tris(pyrrolidino)phosphonium hexafluorophosphate |
| SPPS | solid-phase peptide synthesis |
| Su | succinimide |
| TAFIa | thrombin-activatable fibrinolysis inhibitor |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| Tl | triflate (trifluoromethane- sulfonate) |
| TLC | thin layer chromatography |
| TMSCl | chlorotrimethyl-silane |
| Tr | trityl (triphenylmethyl) |
| TS | transition state |
| Ts | tosyl |
| Trs | 2,4,6-triisopropylphenylsulfonyl |
1. Introduction

2. Synthesis of the Phosphinic α,α'-Dipeptide Backbone
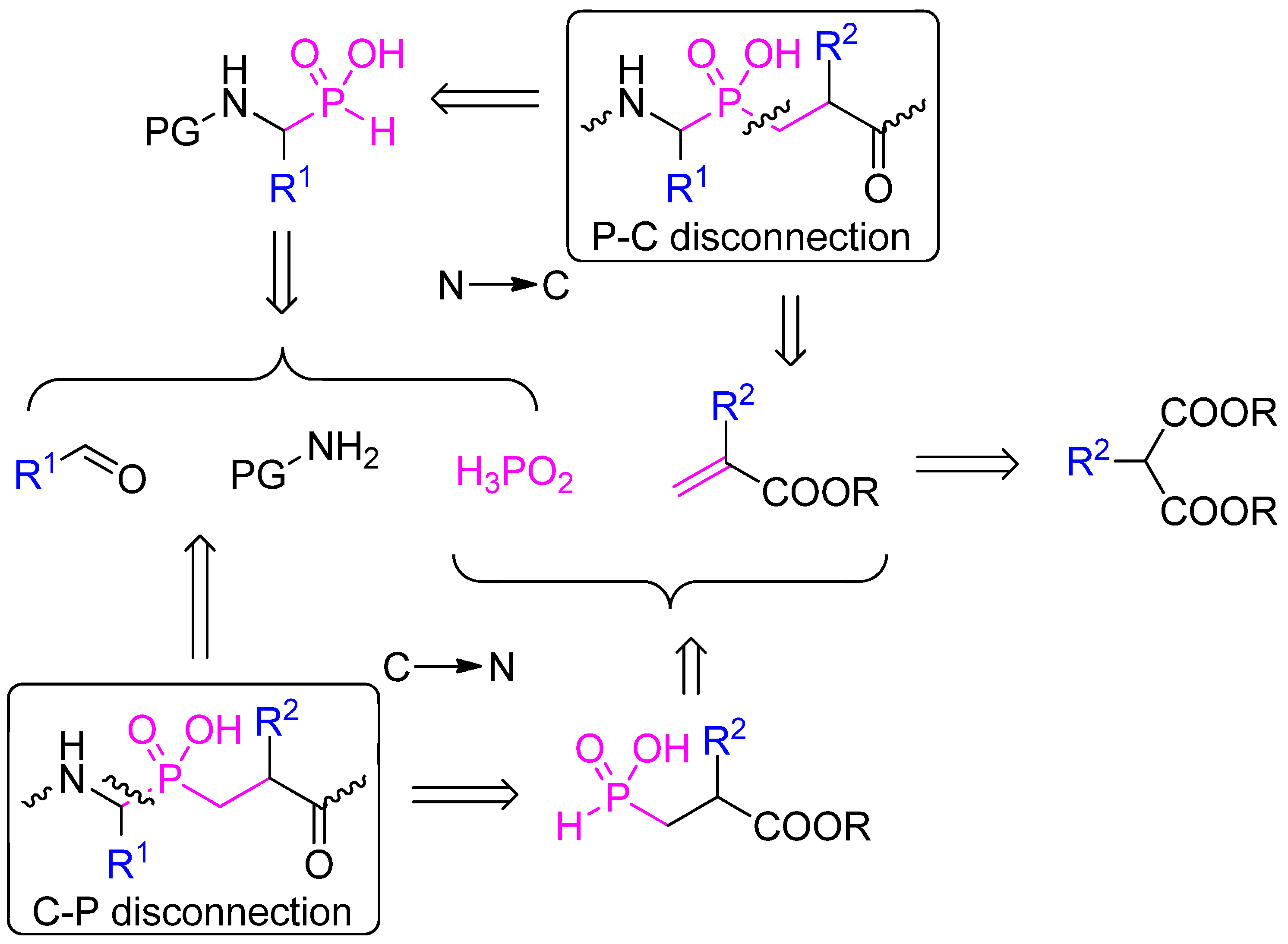
2.1. Retrosynthetic Analysis
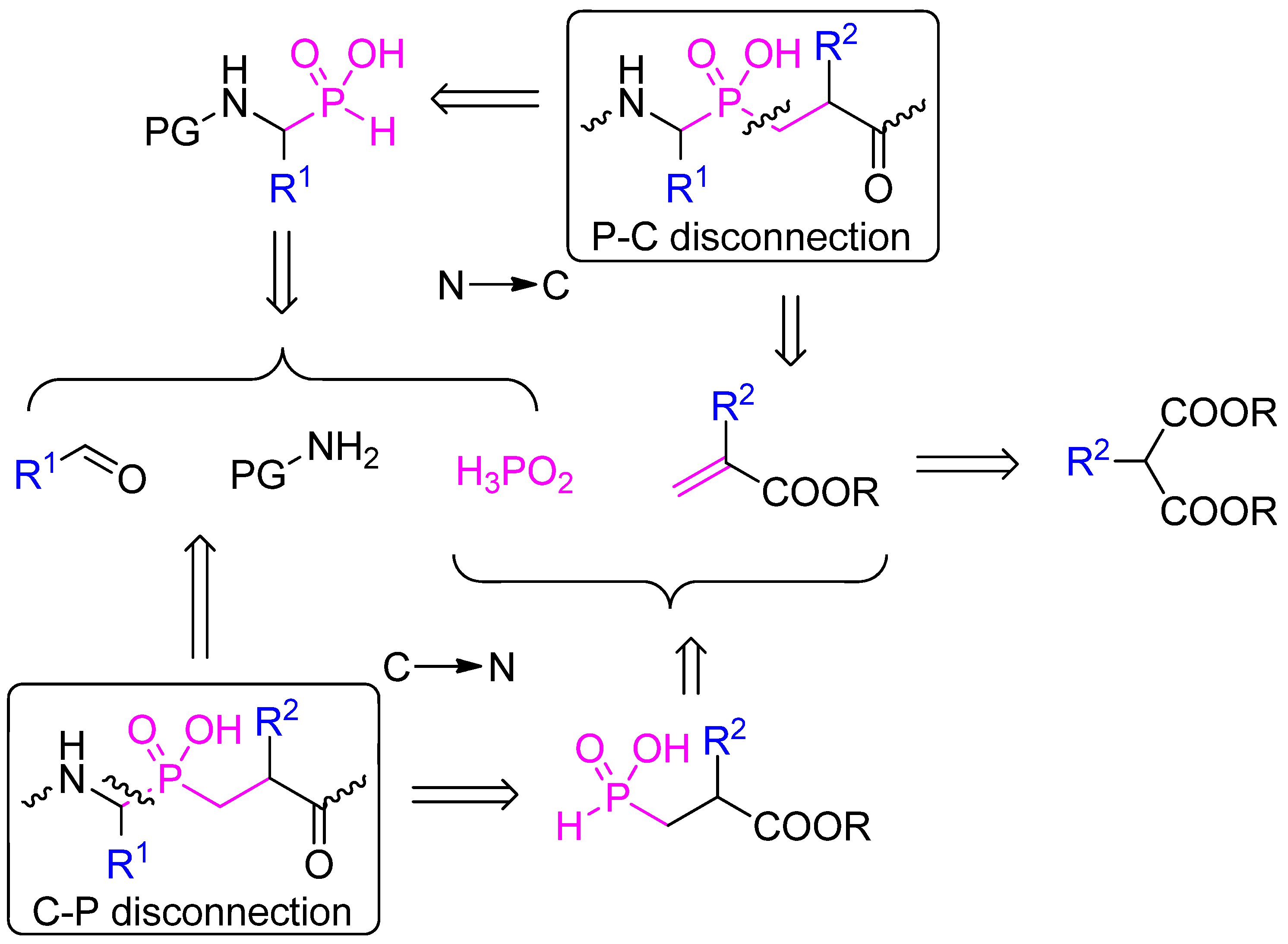
2.2. N → C Strategy
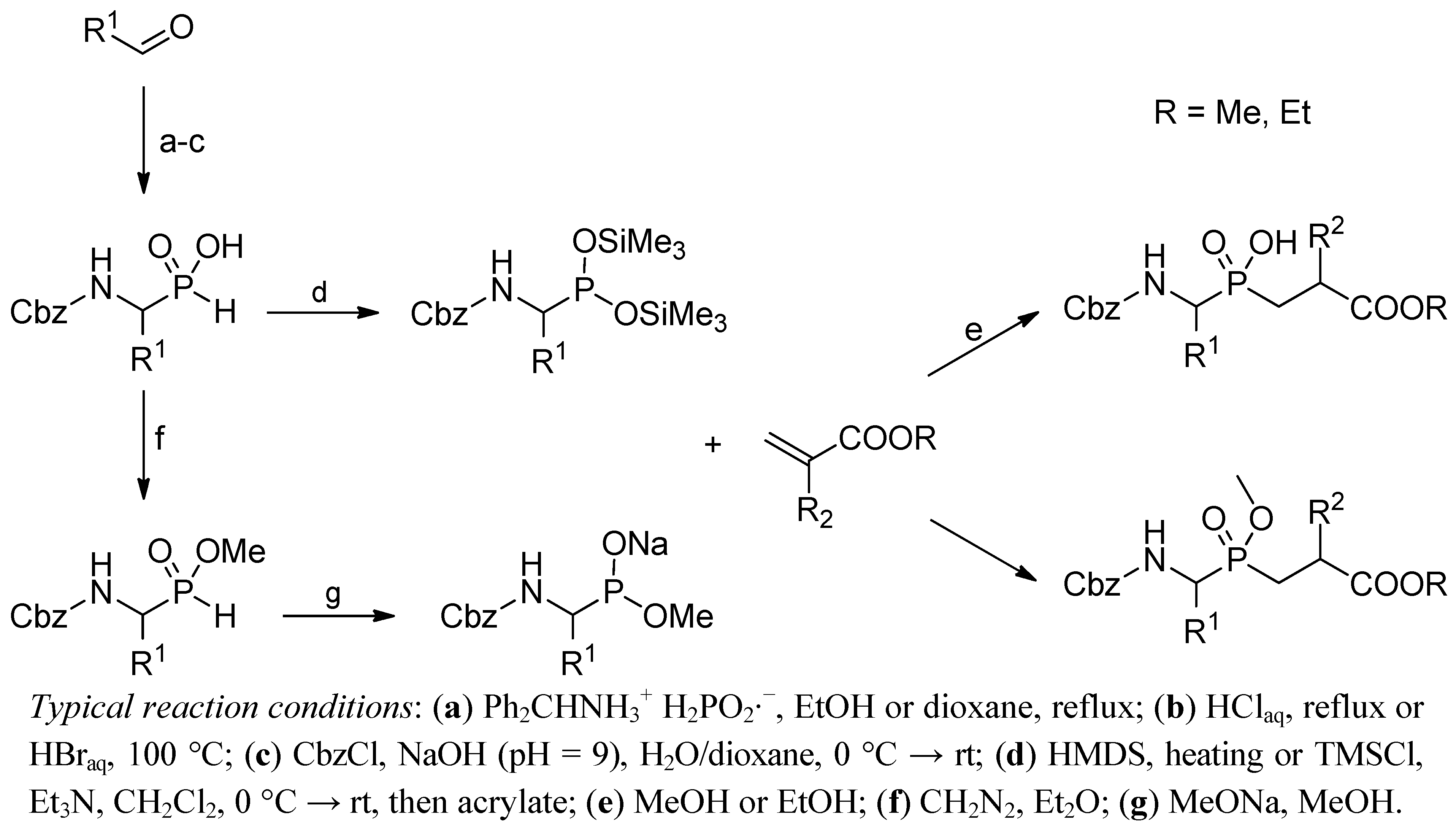

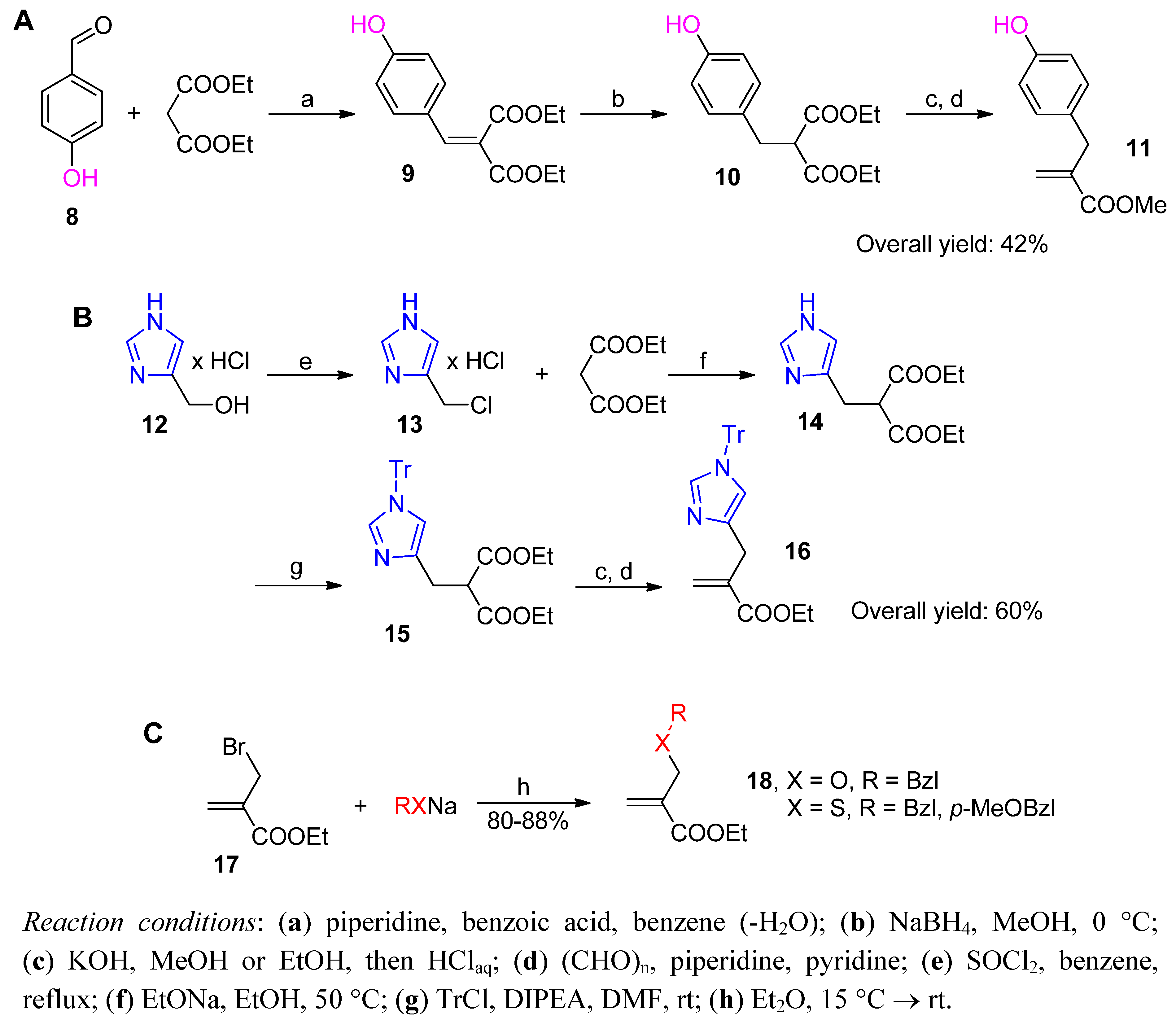
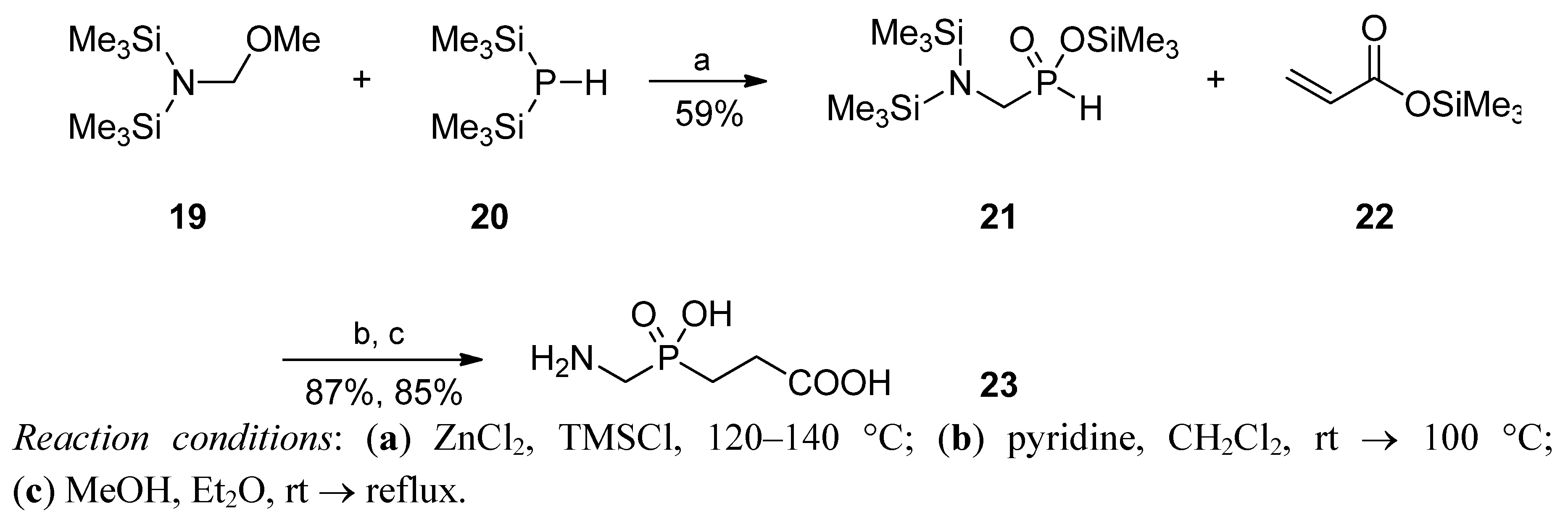
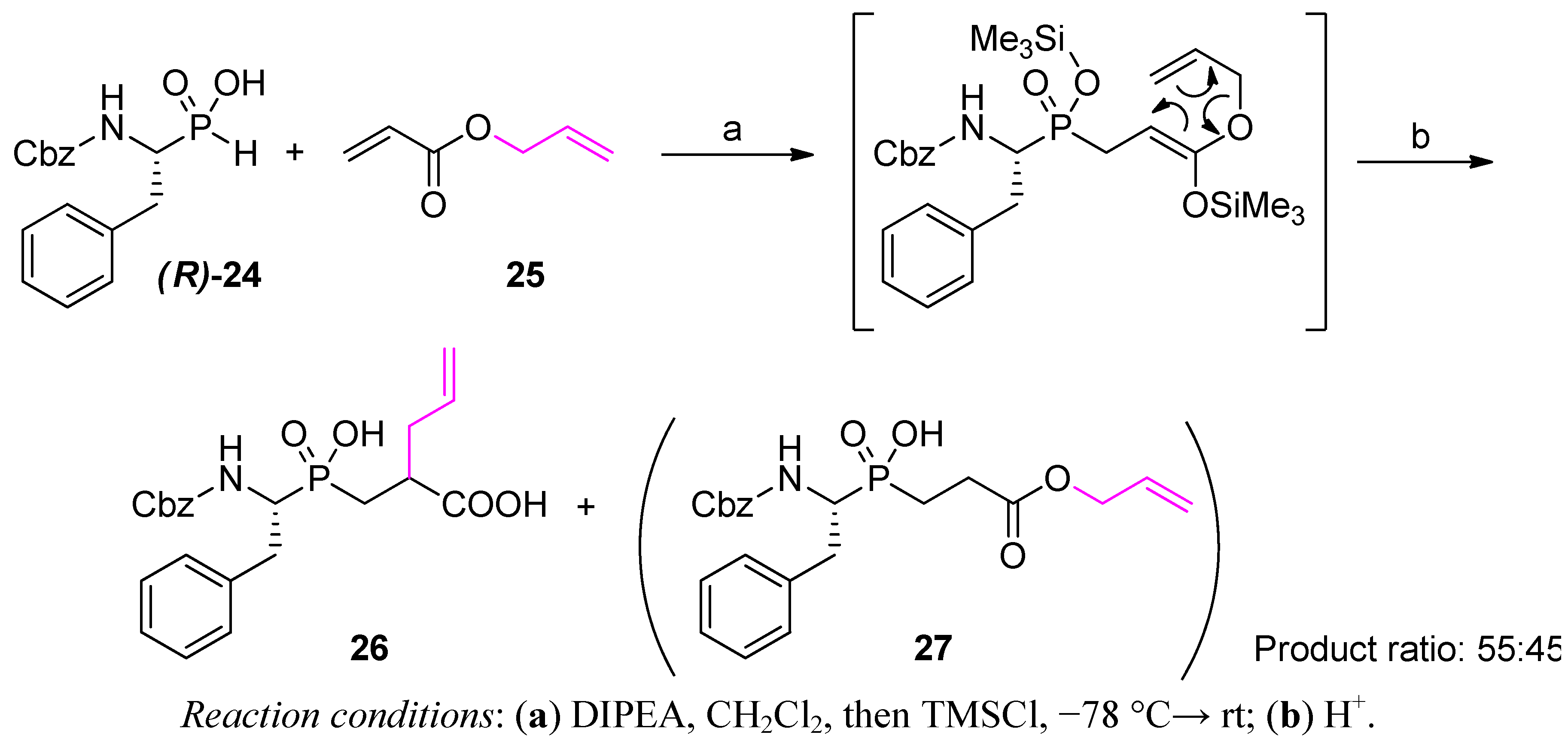
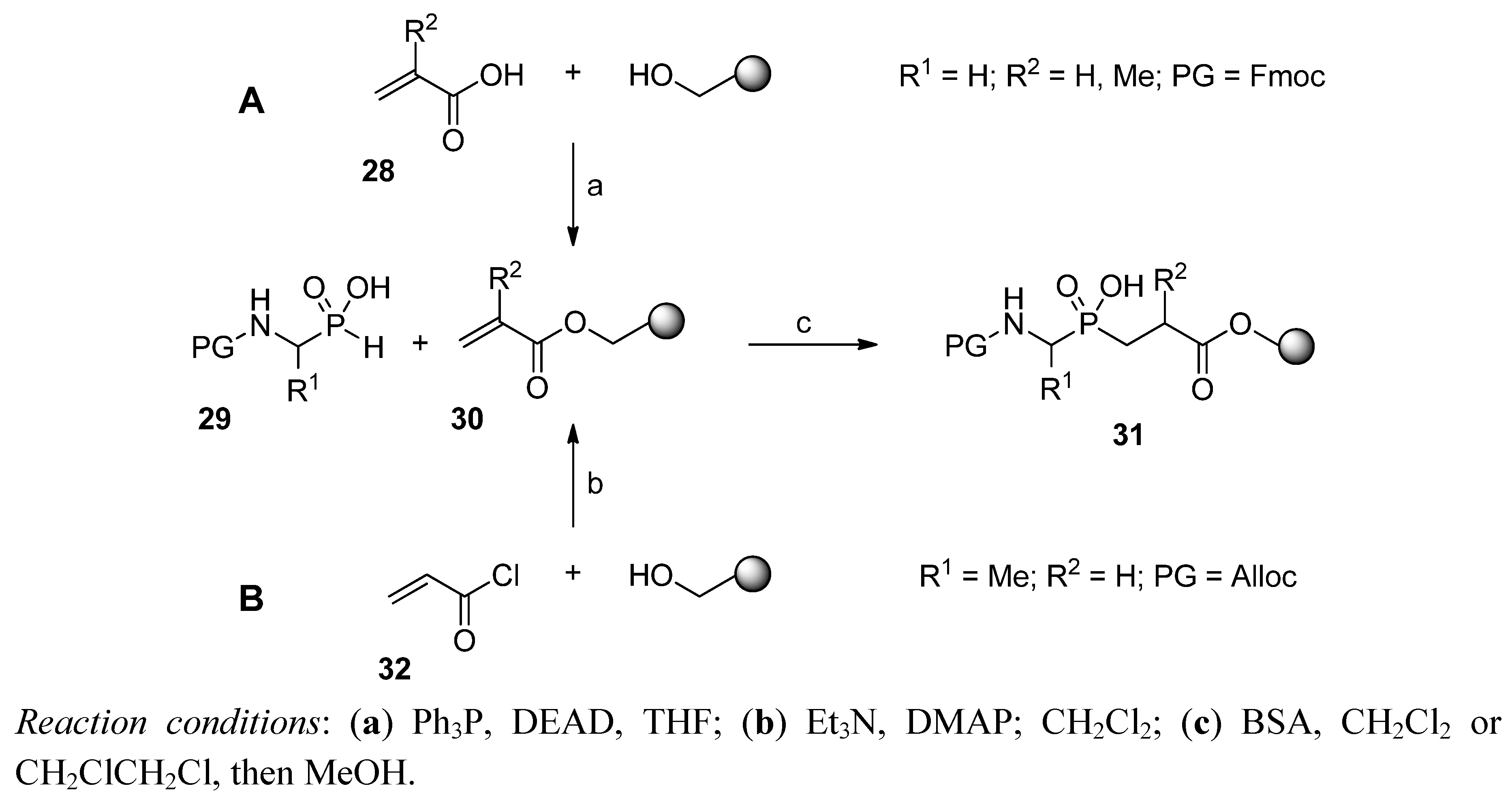
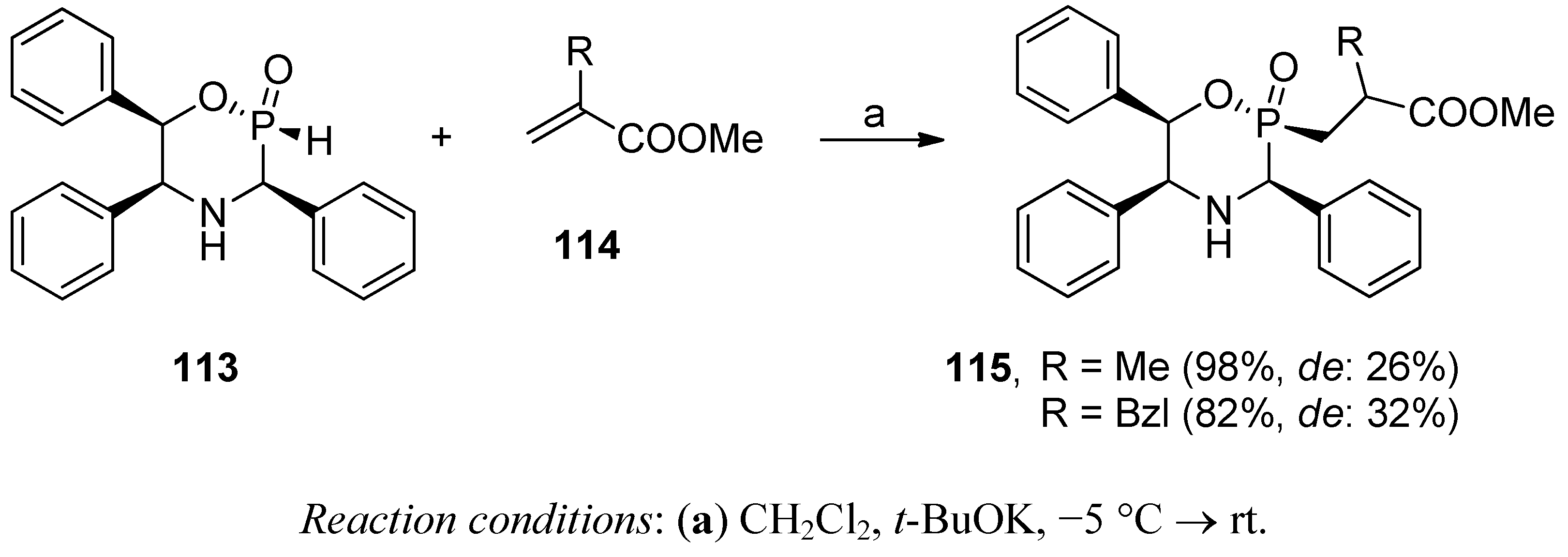
Phospha-Michael Addition on Solid Phase
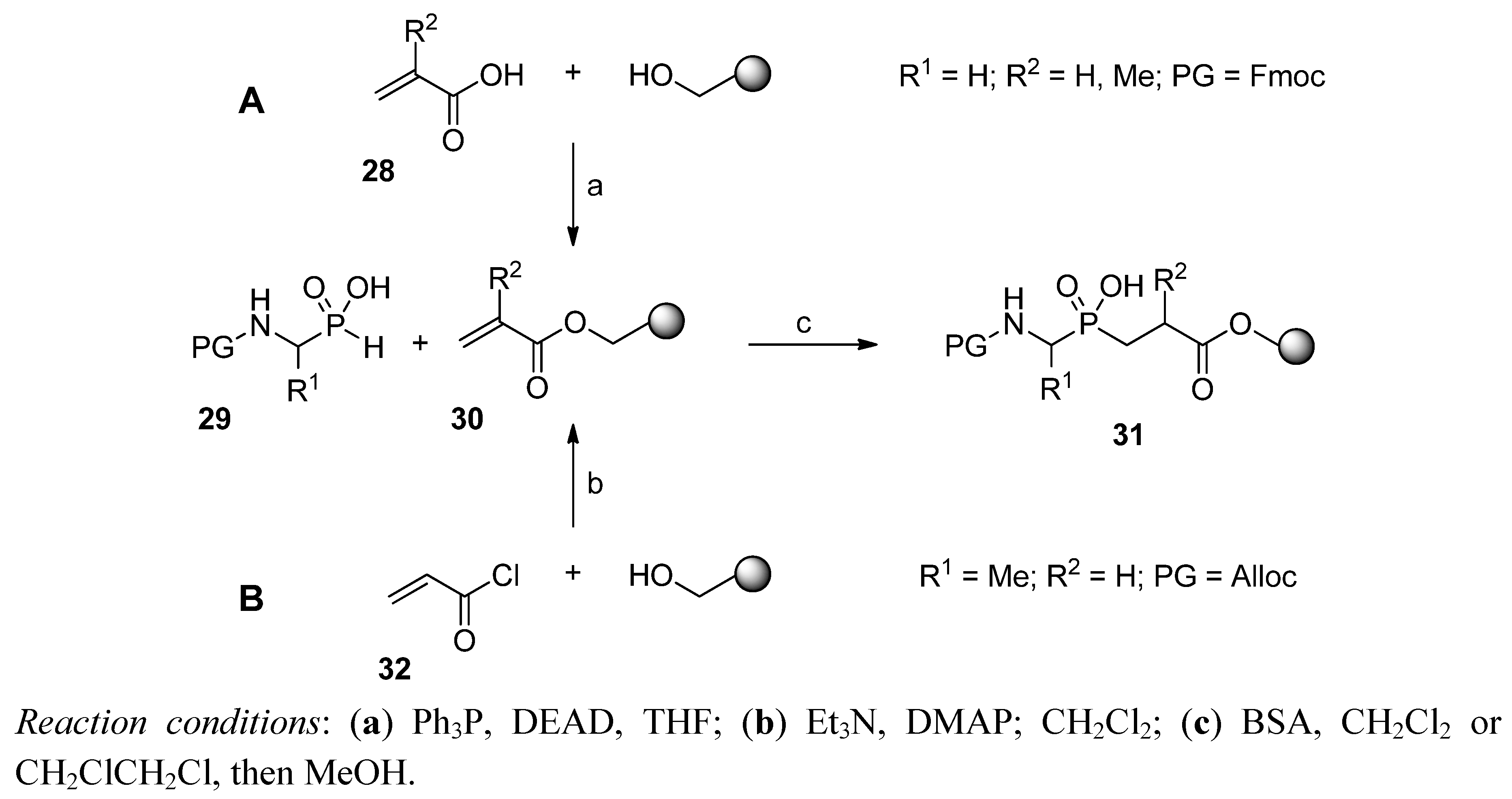
2.3. C → N Strategy
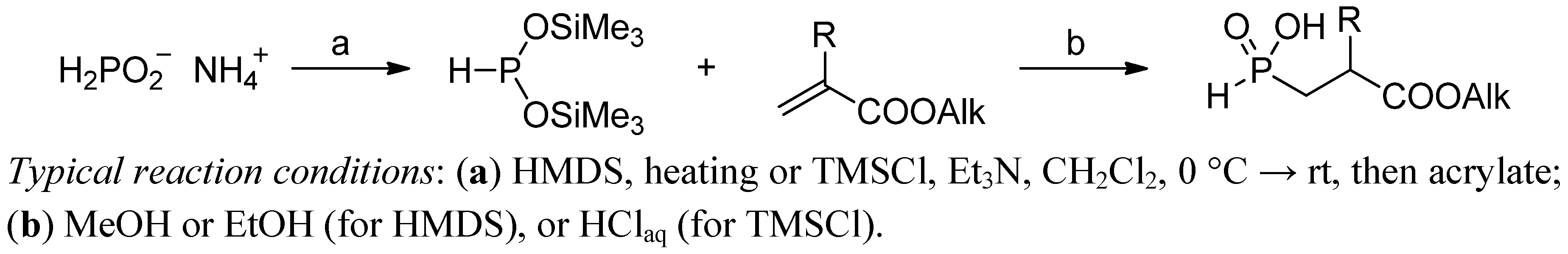
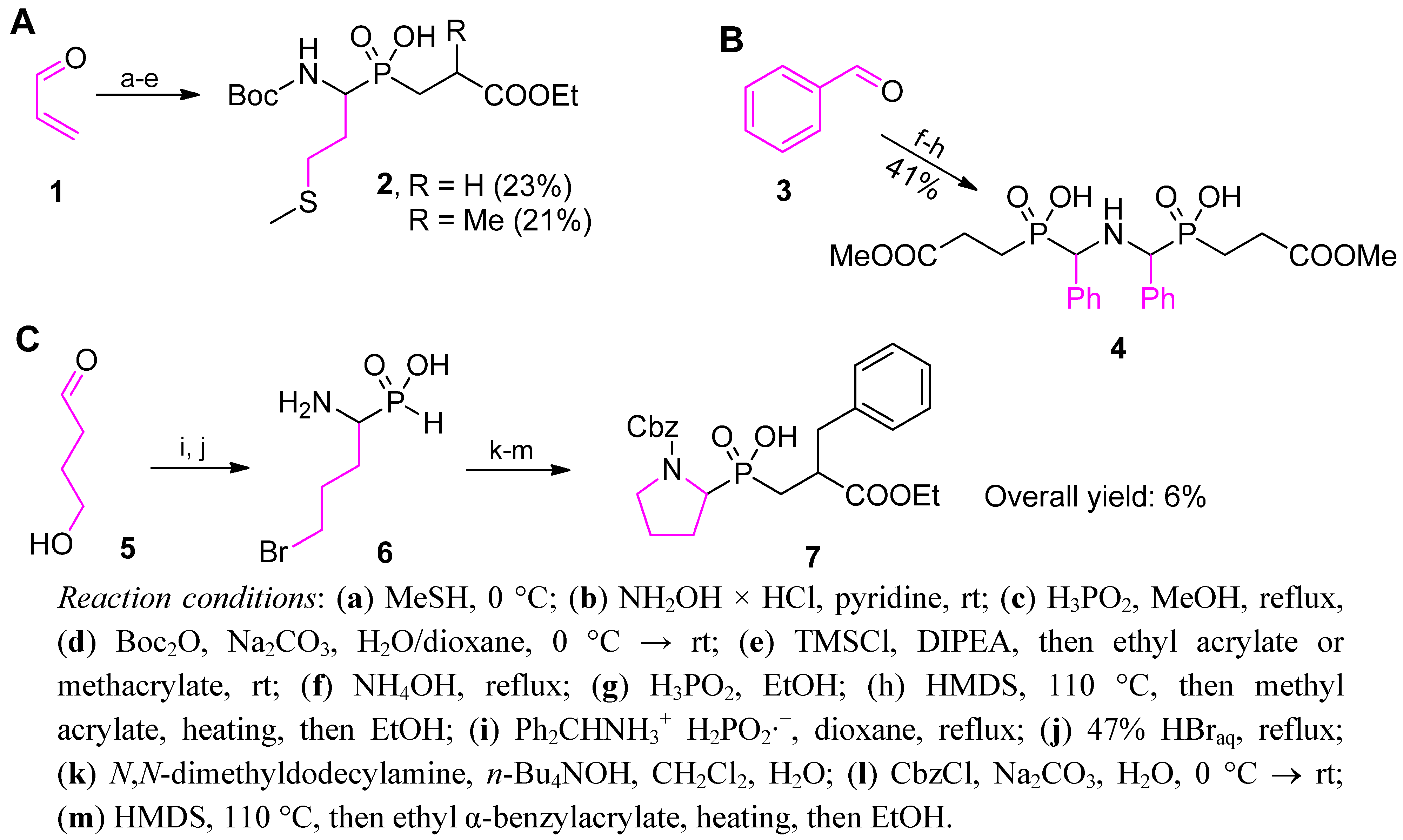
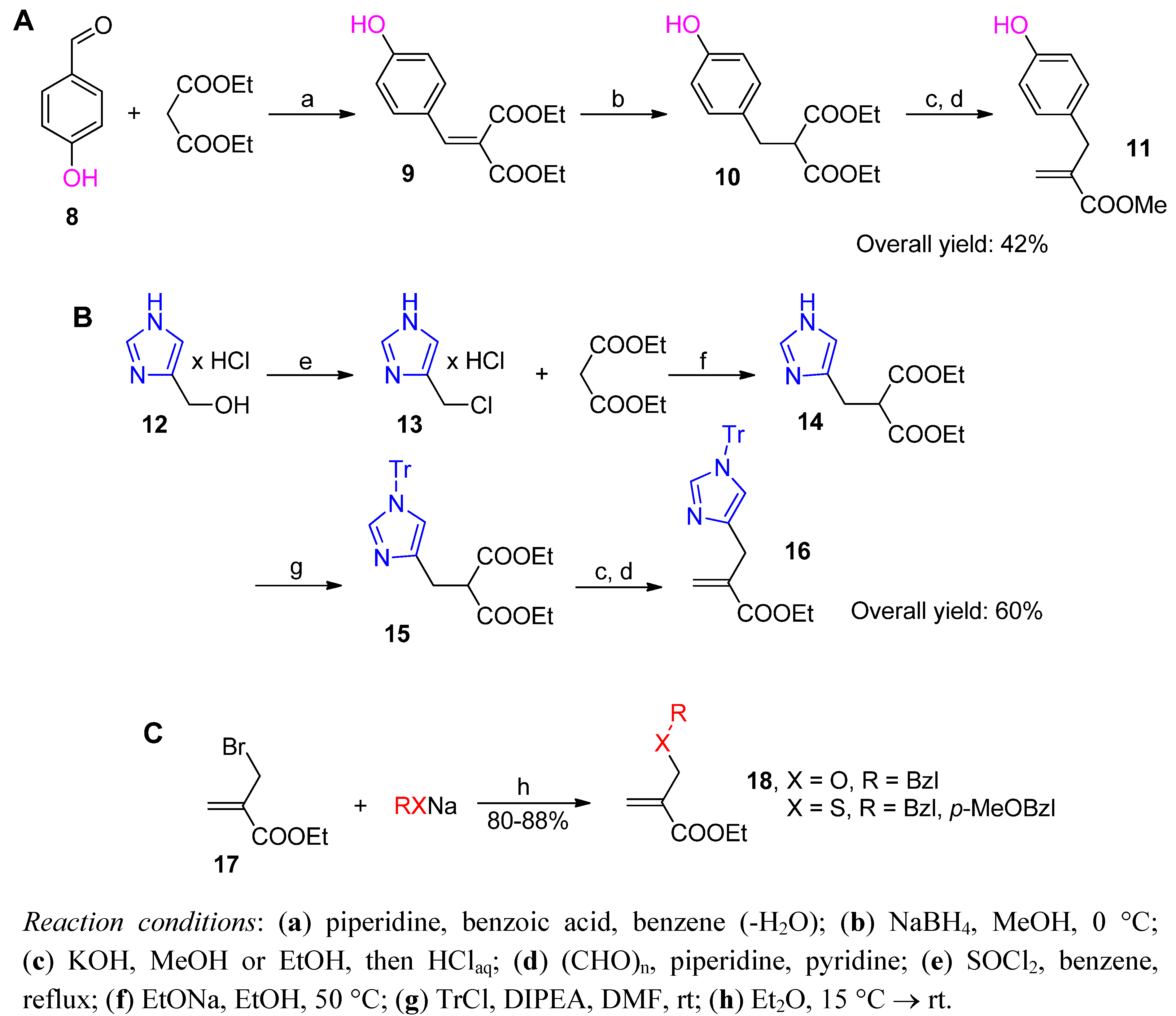
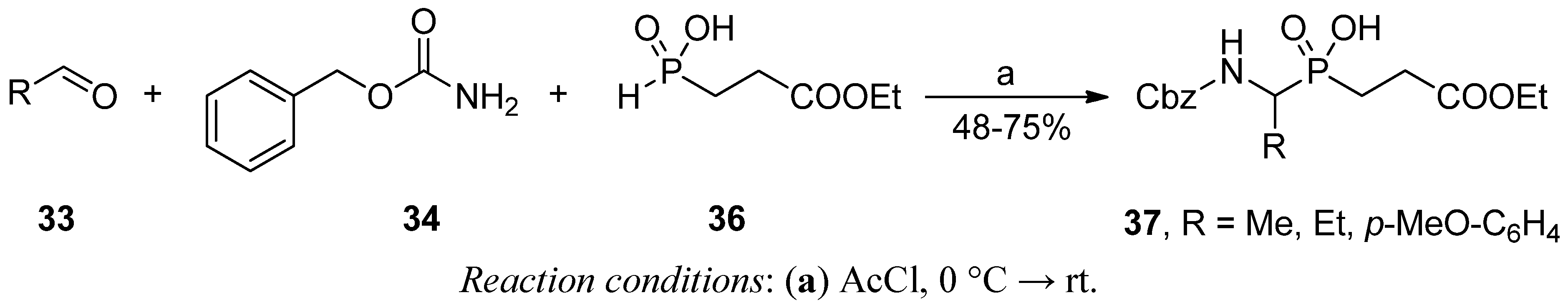
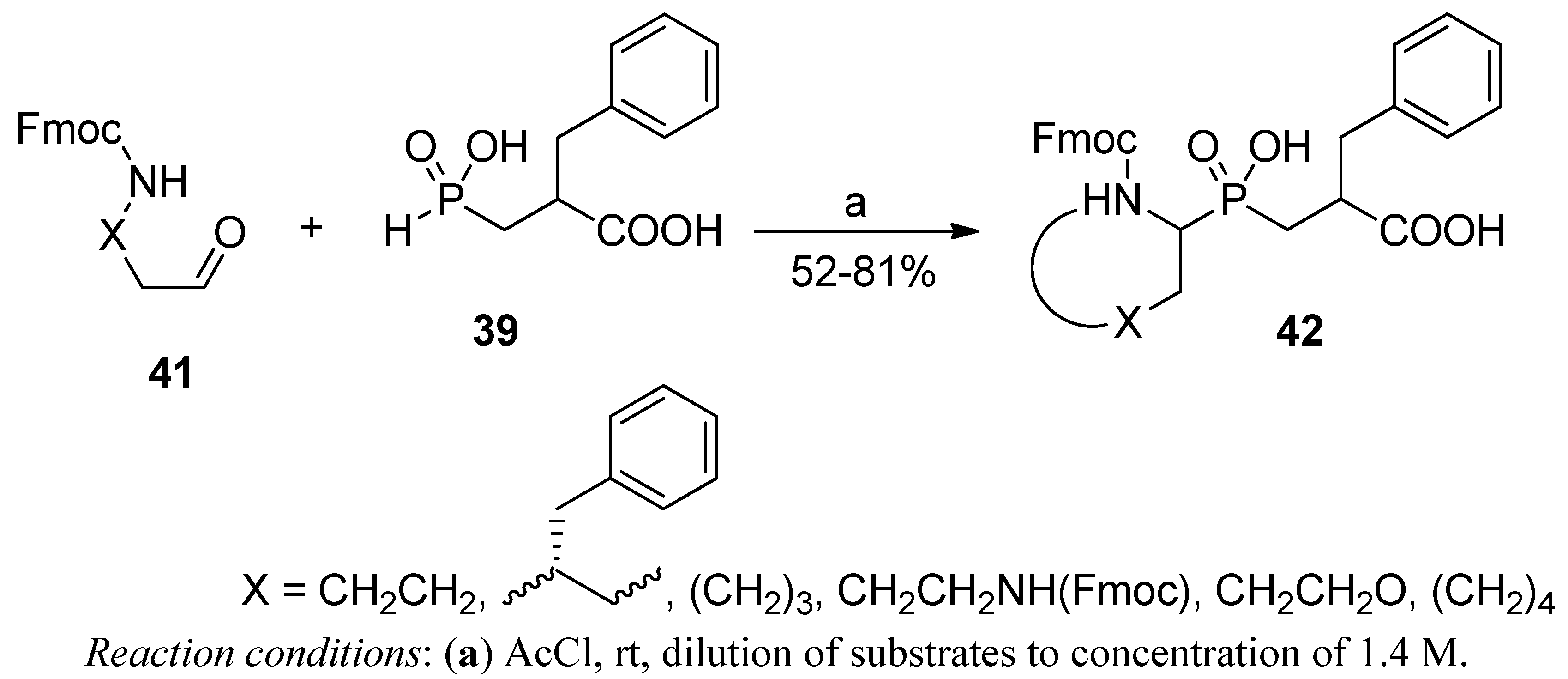
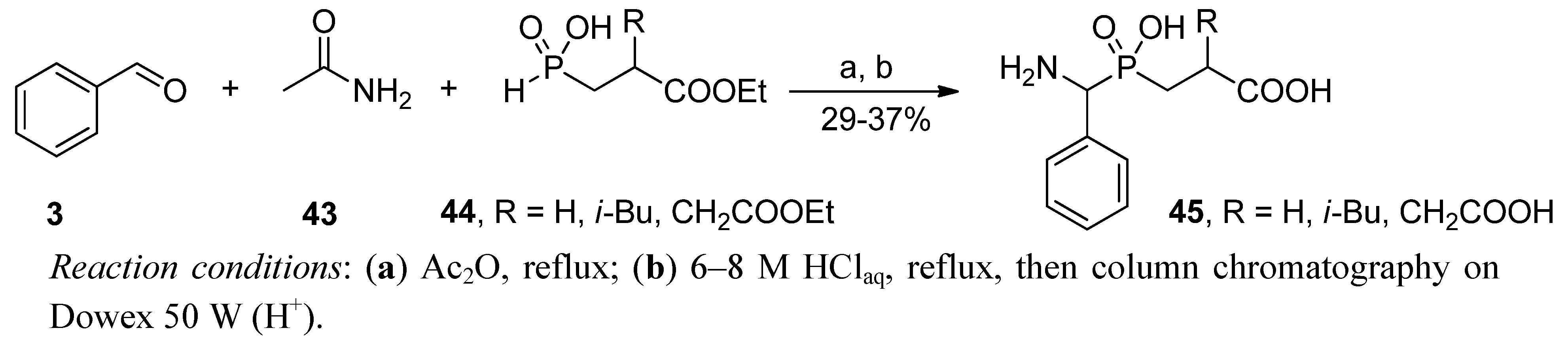
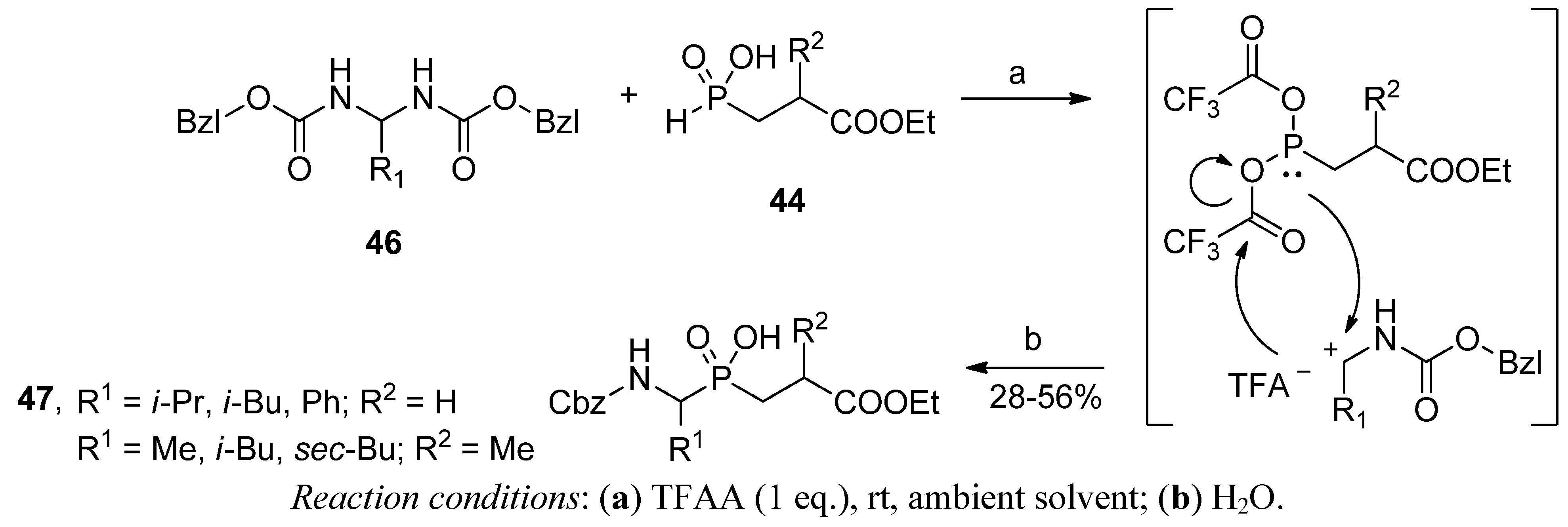
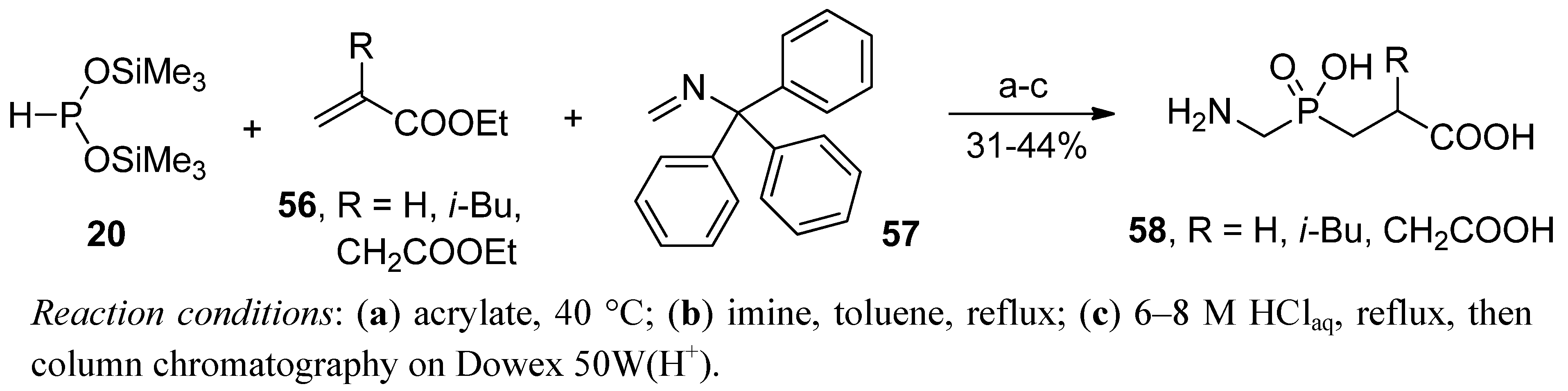
2.3.1. Amidoalkylation
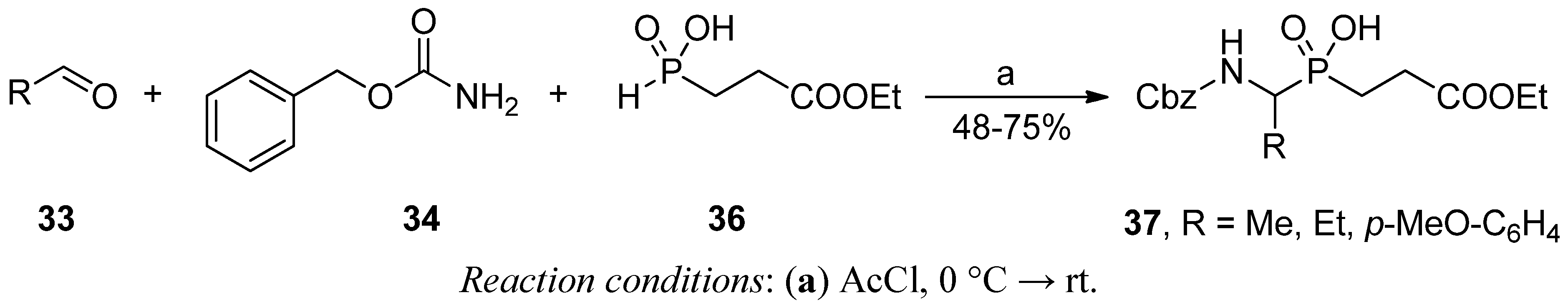

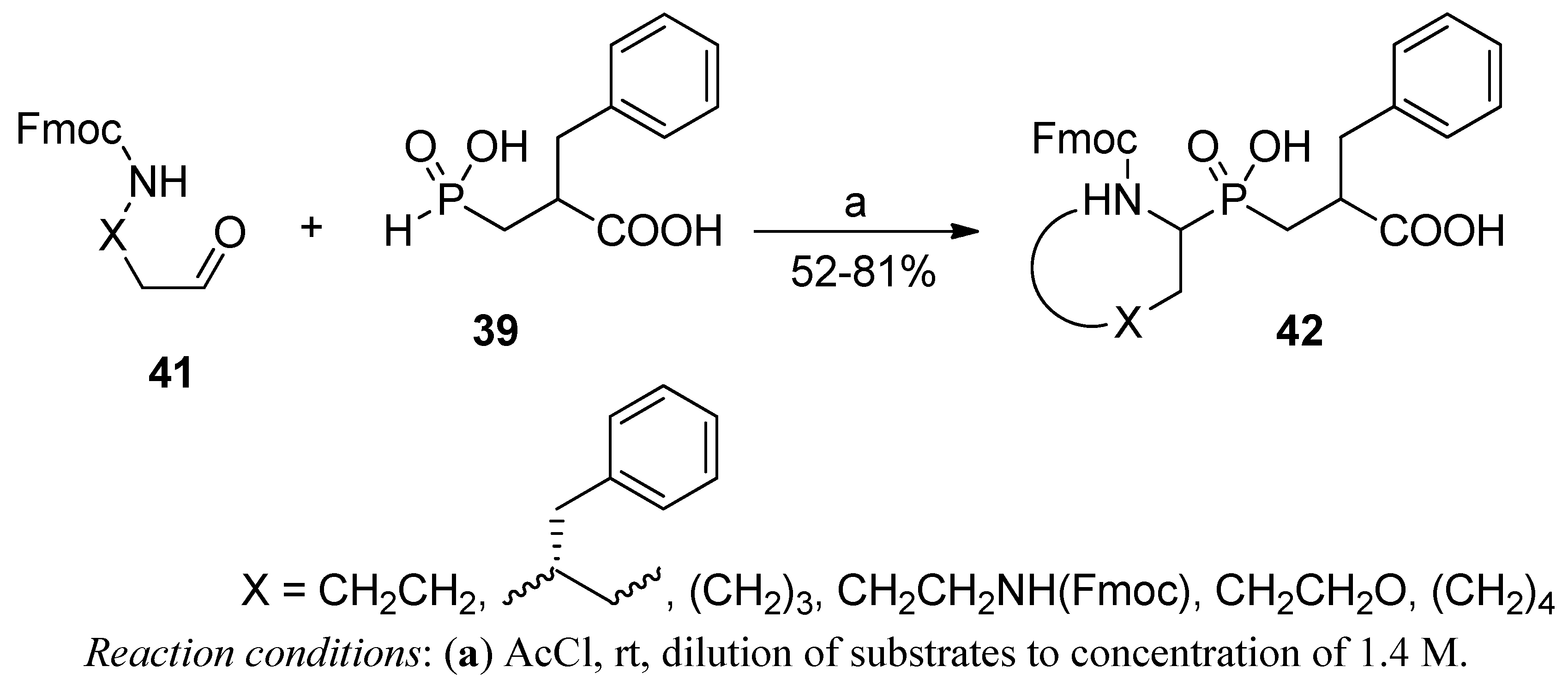
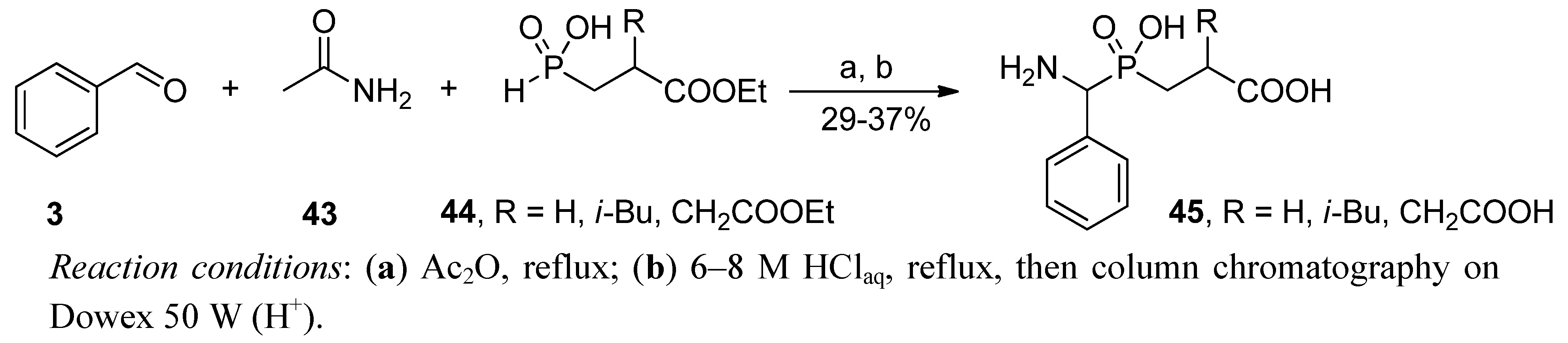
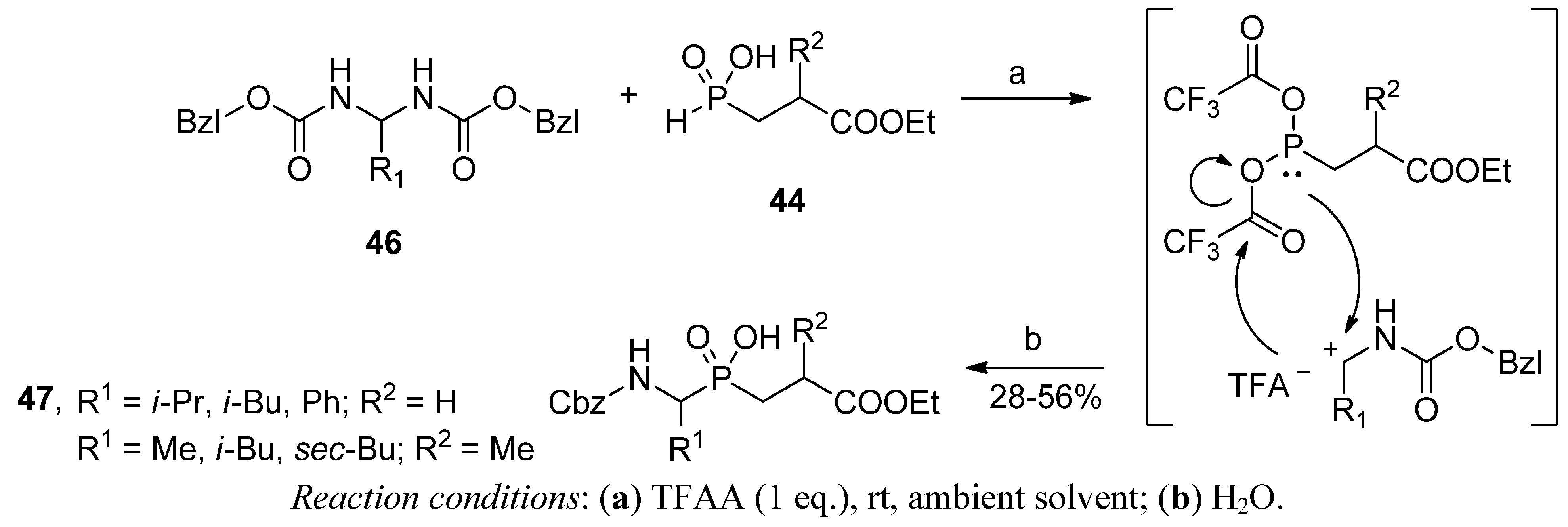
2.3.2. Phospha-Mannich and Kabachnik-Fields Reaction
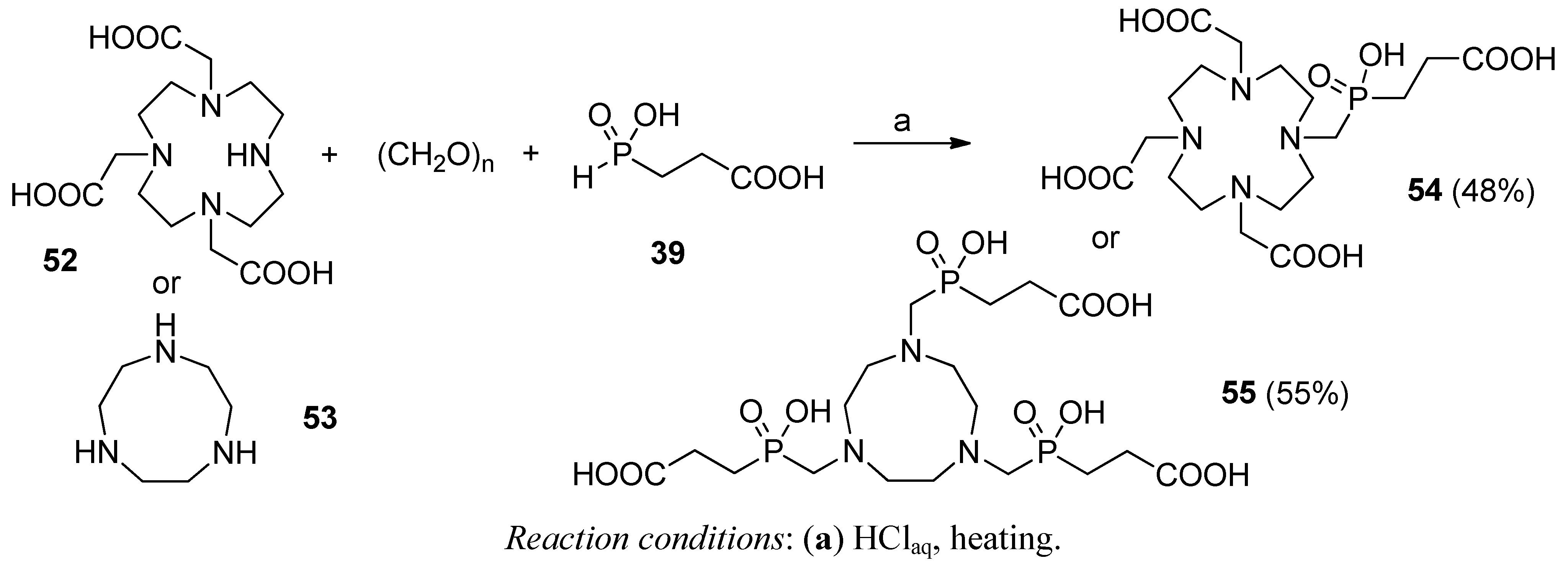
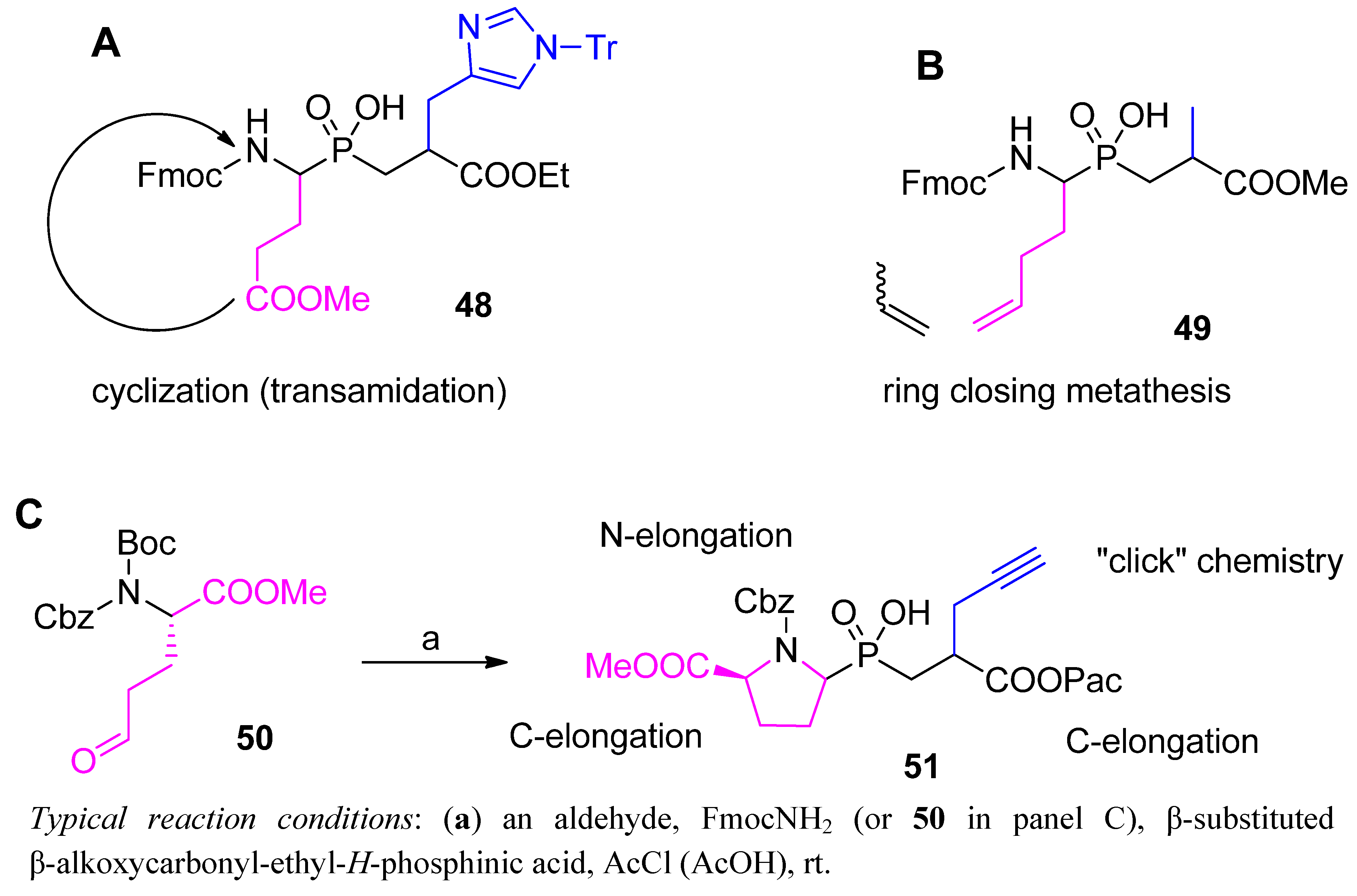
3. Modifications Following the C-P-C Formation
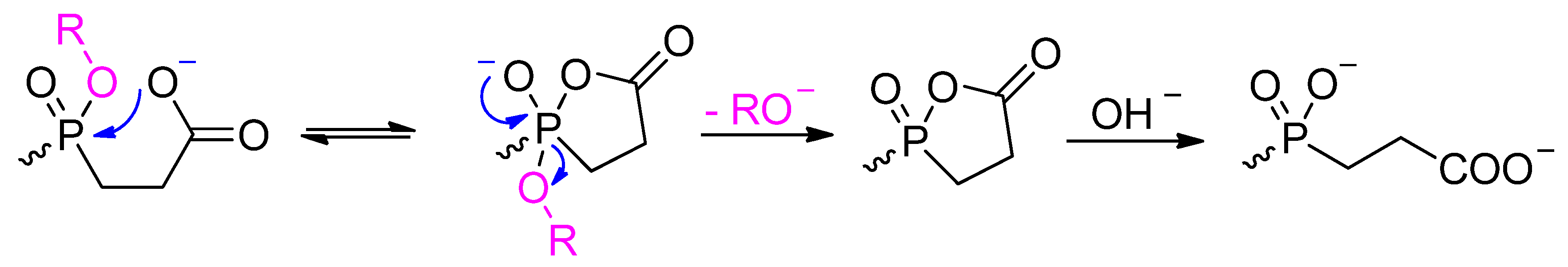
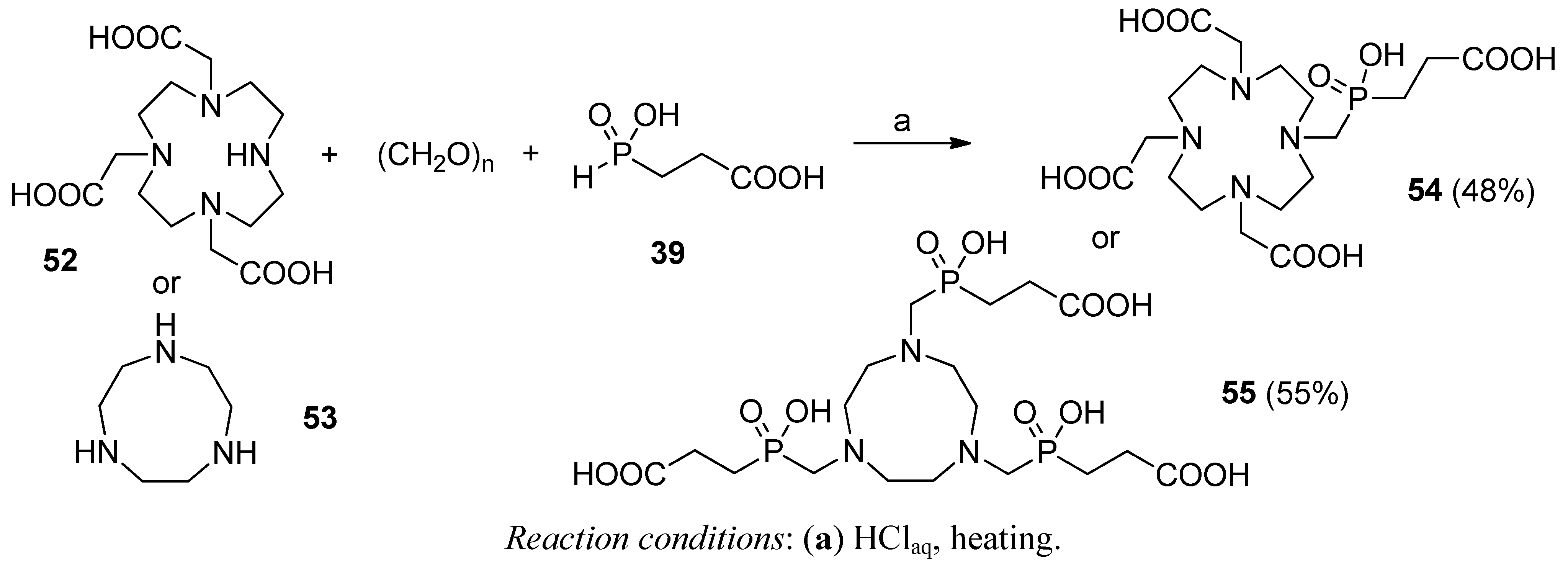
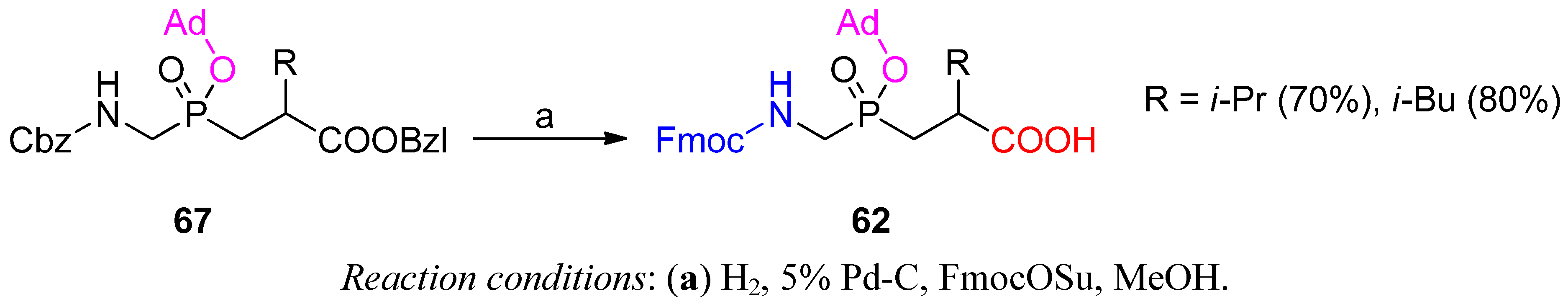
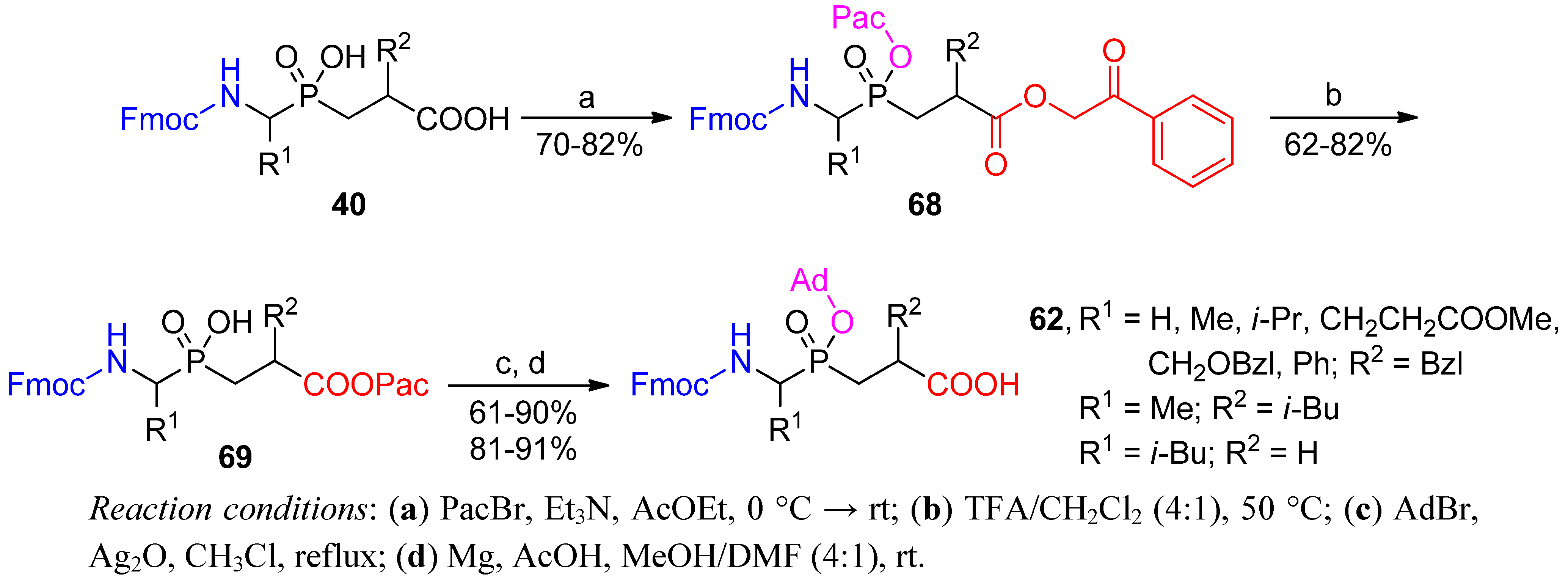
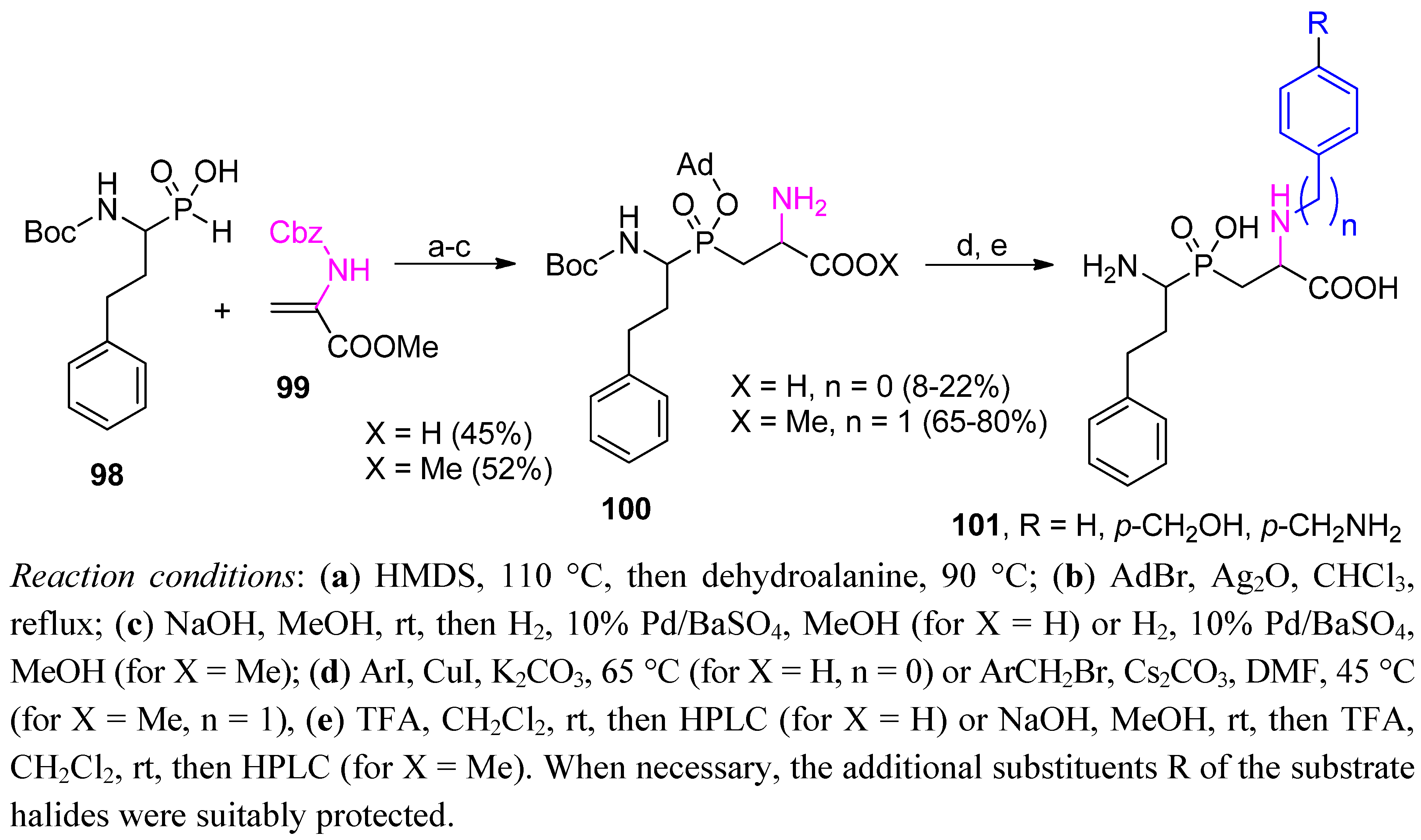
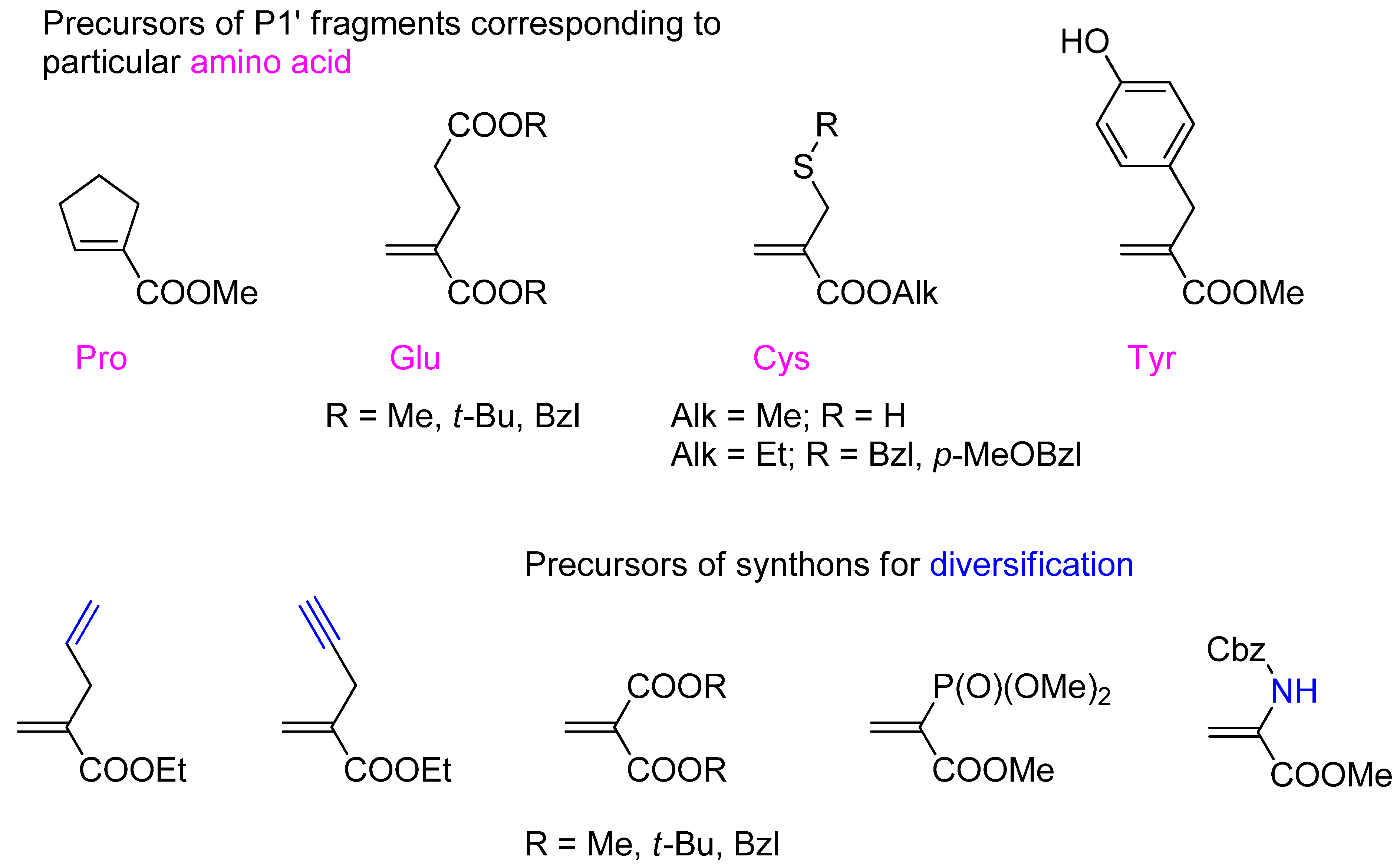
3.1. Building Blocks for Solid-Phase Peptide Synthesis
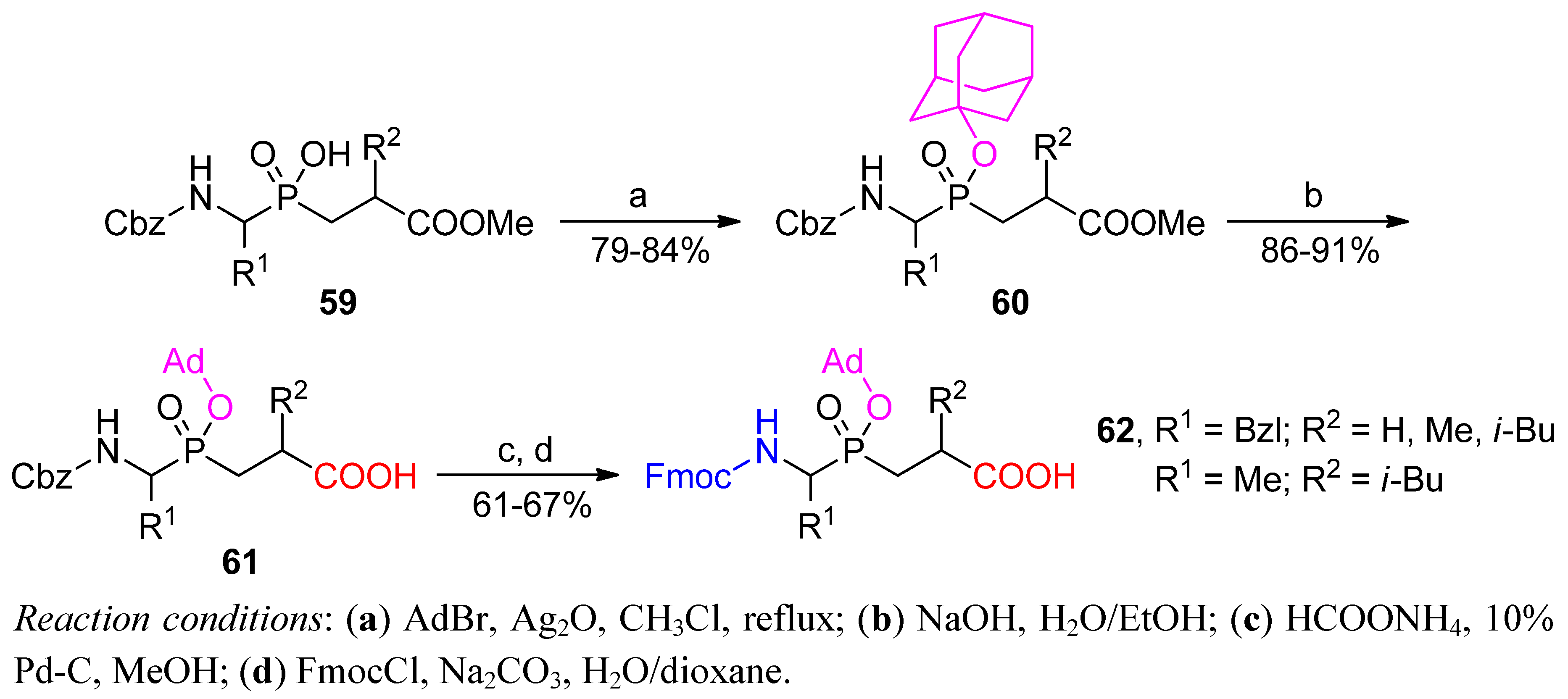
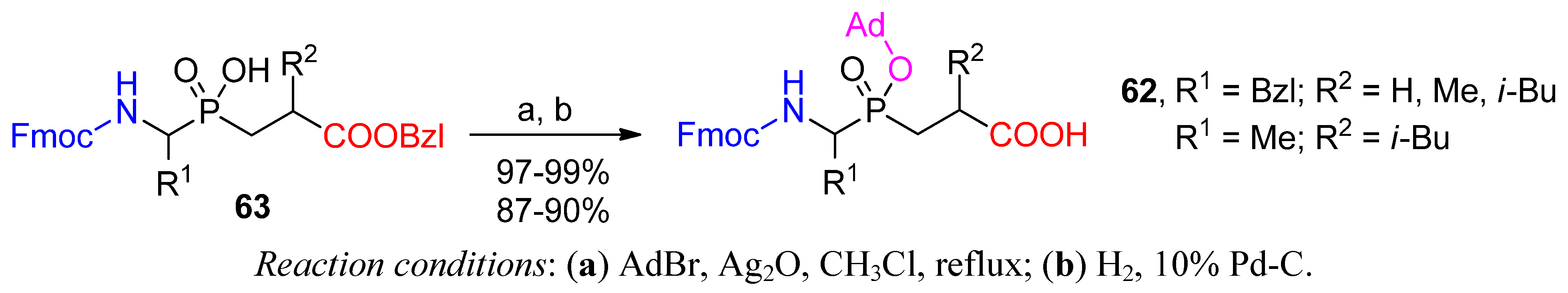
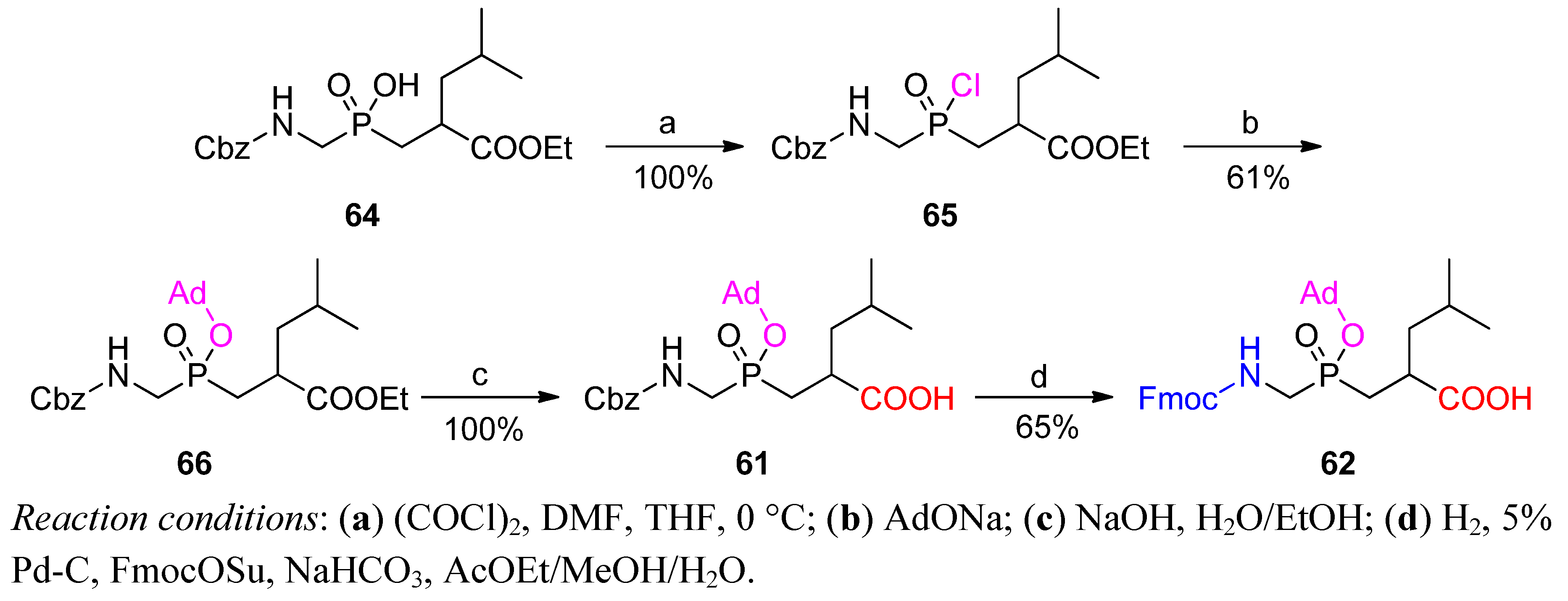
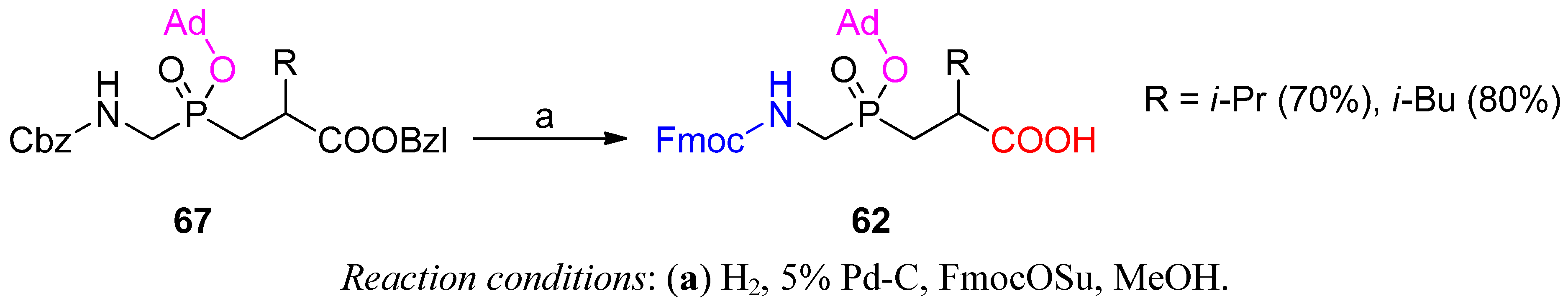
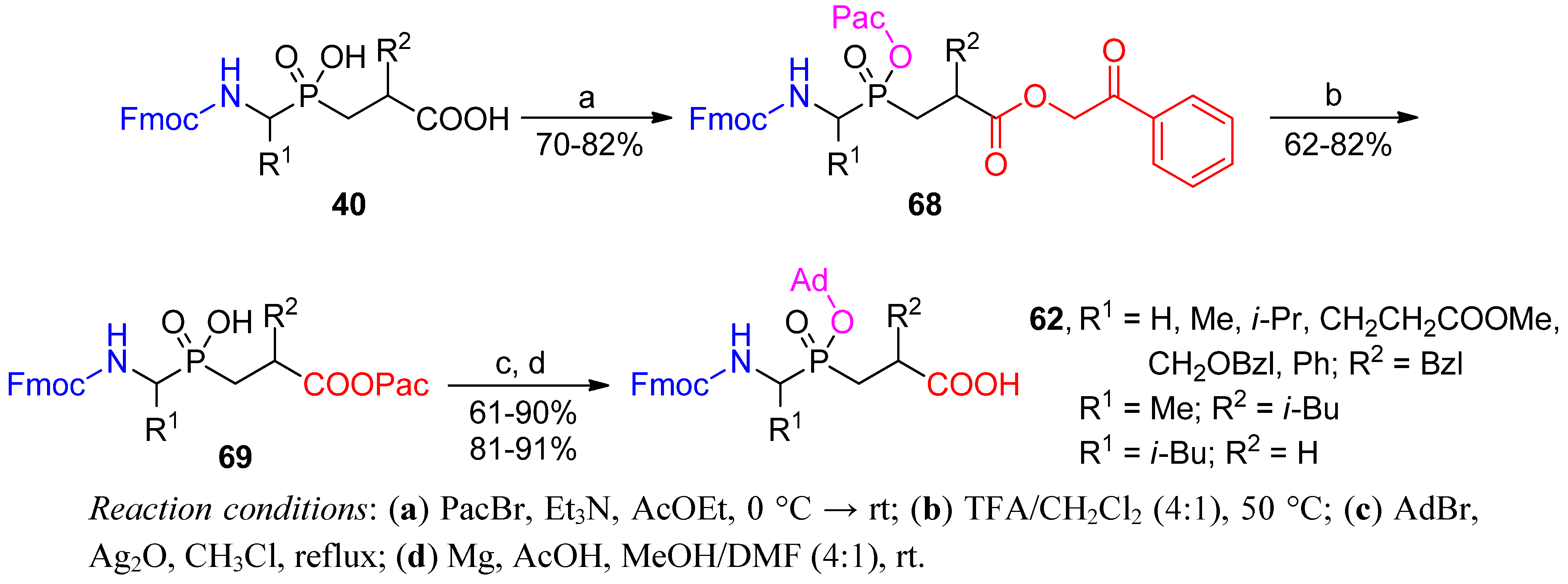
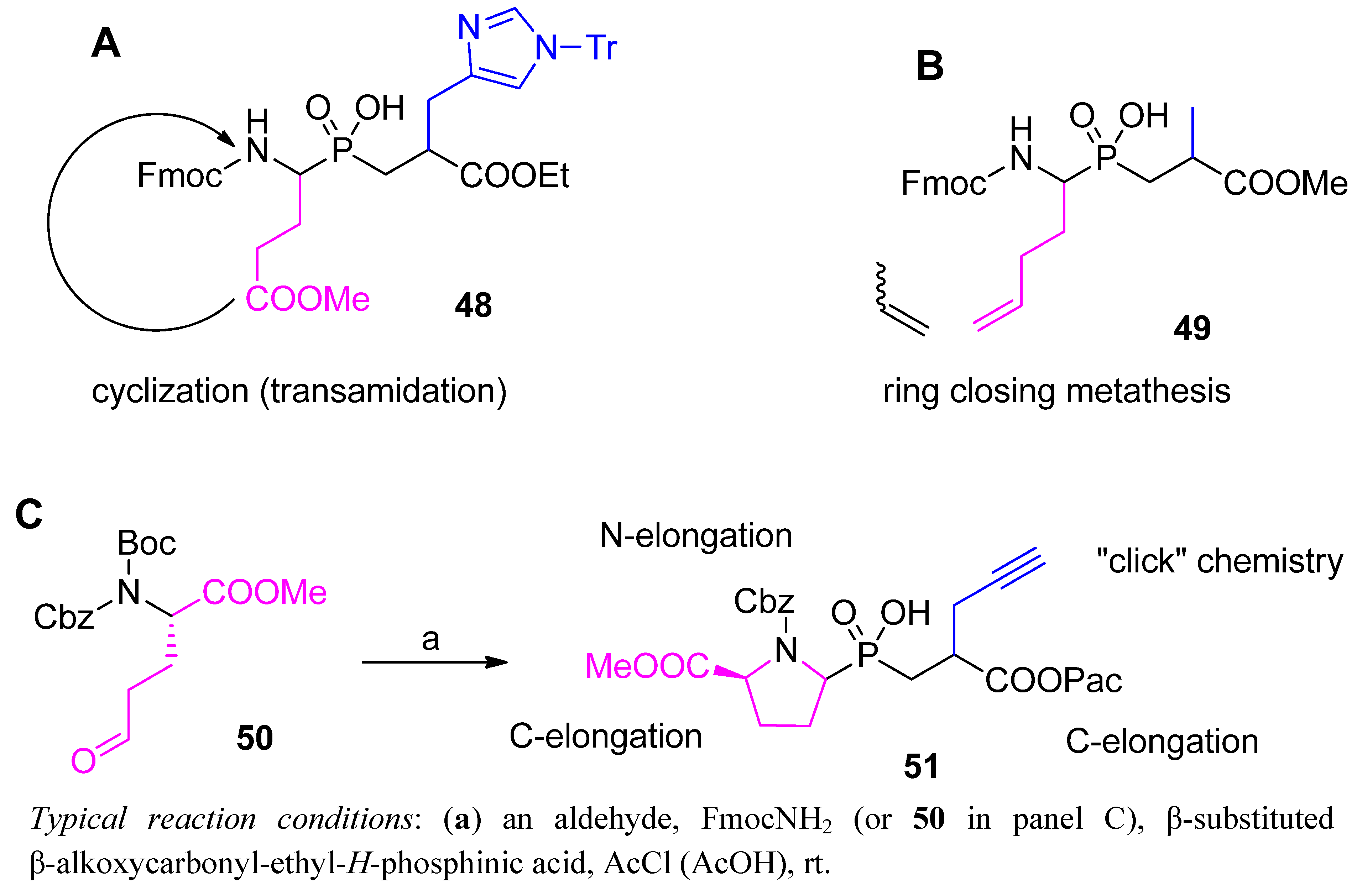
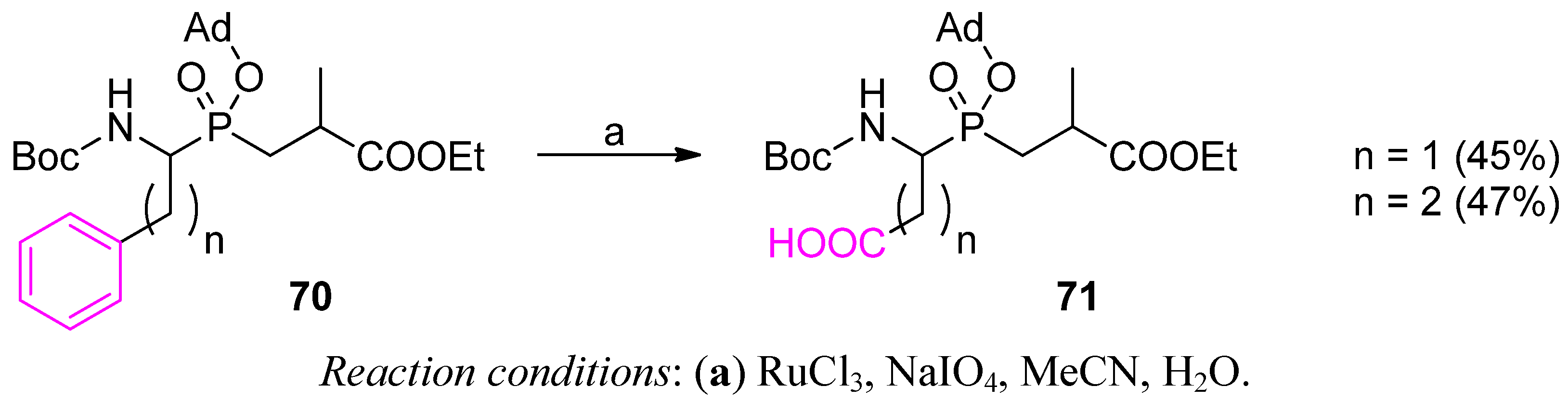
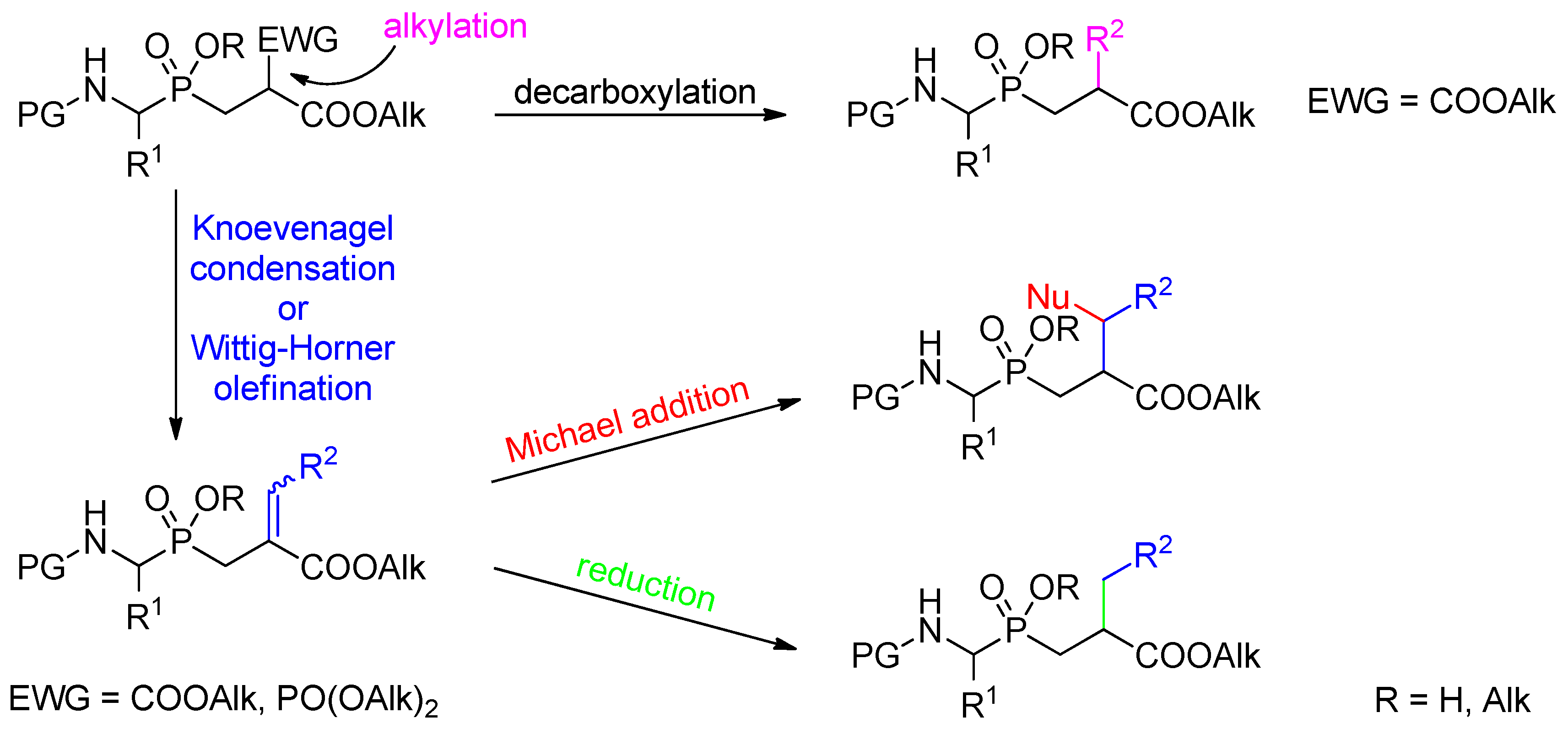
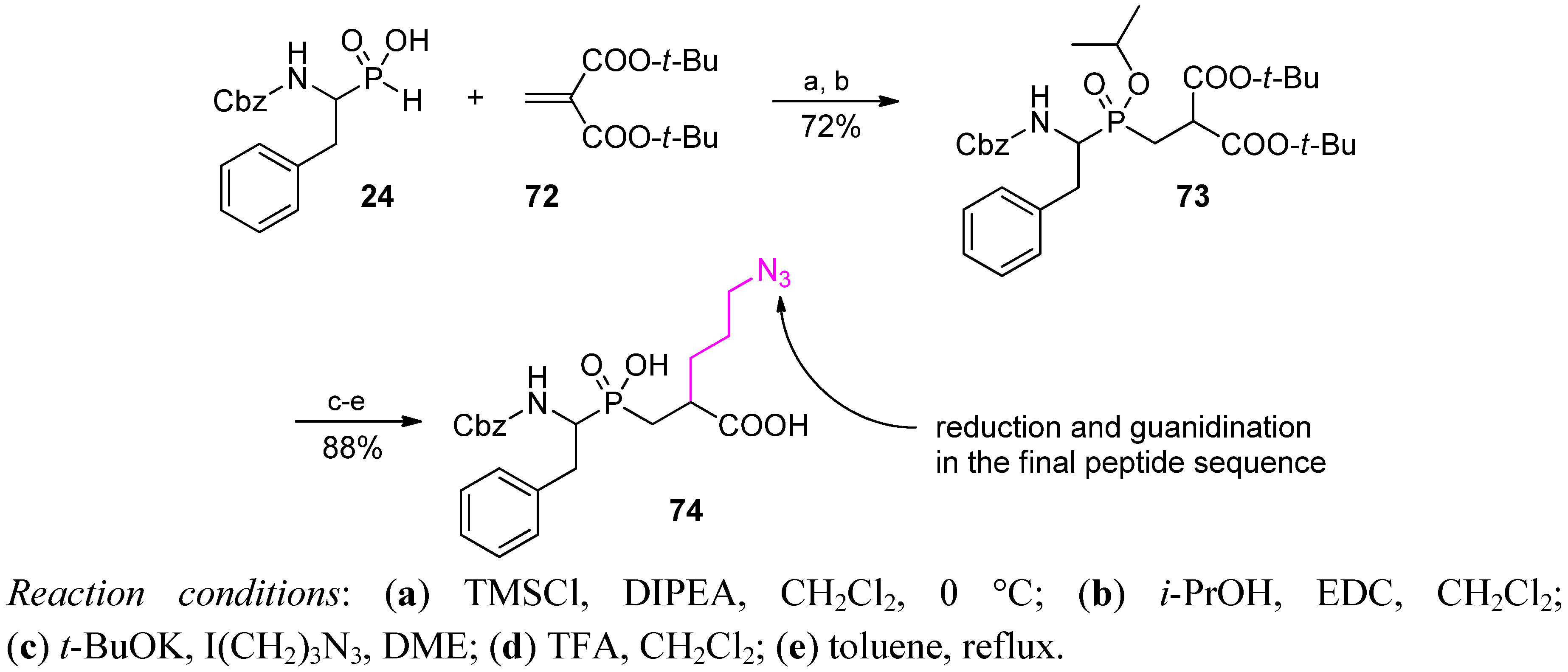
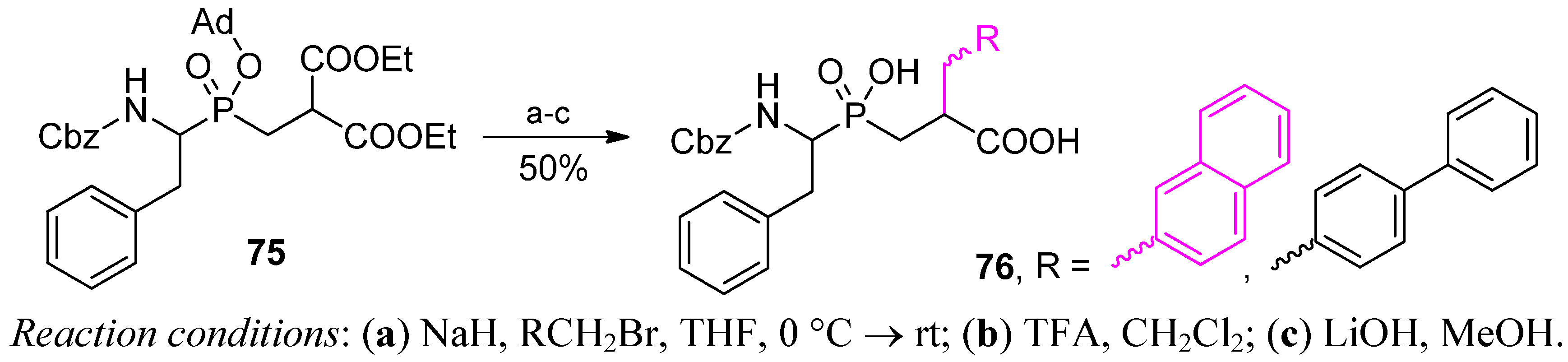
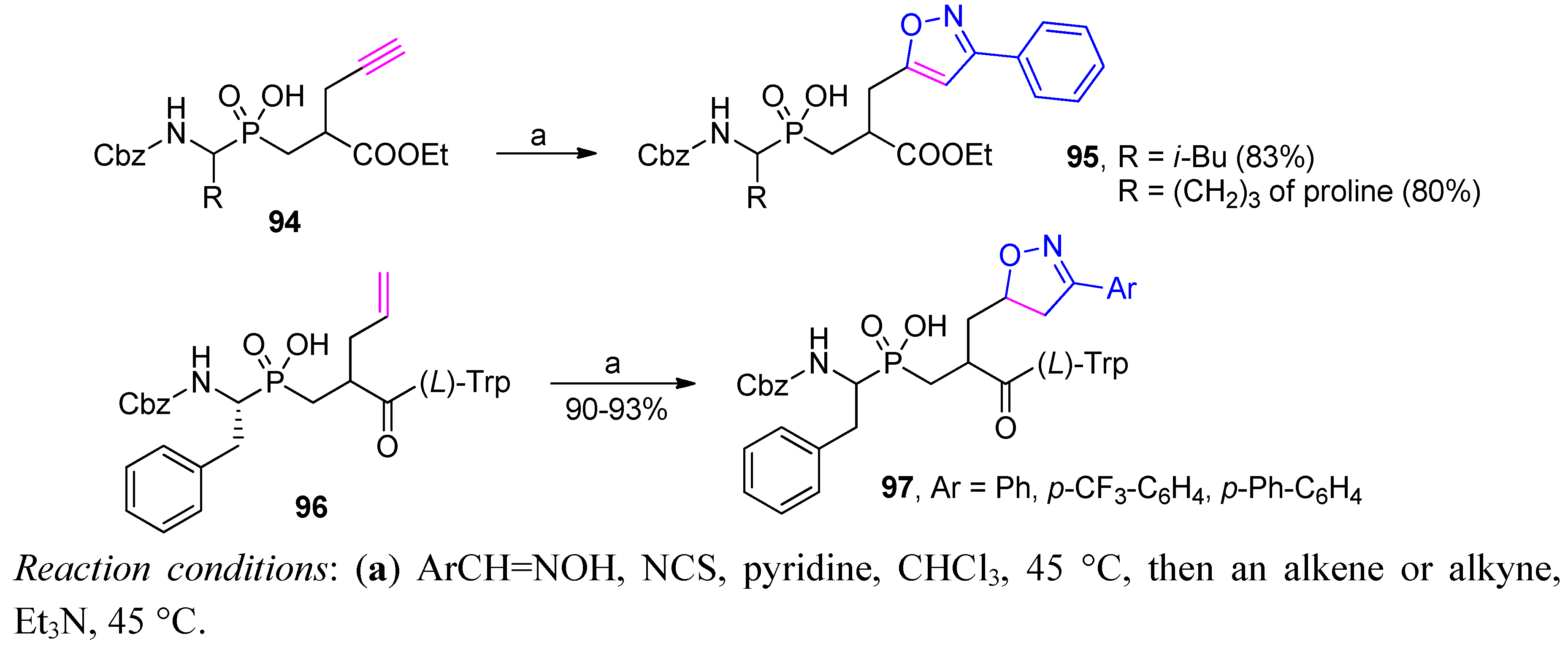
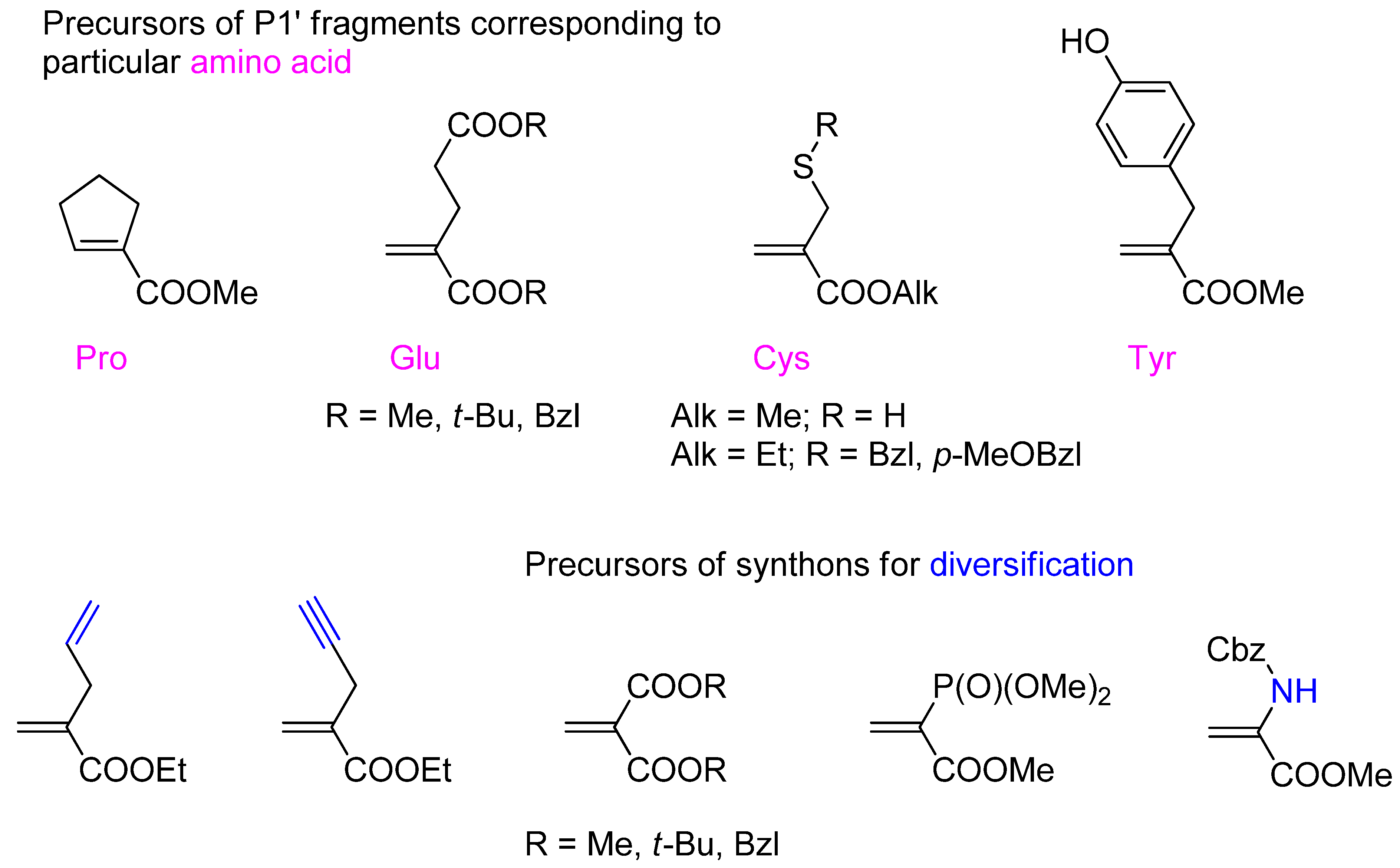
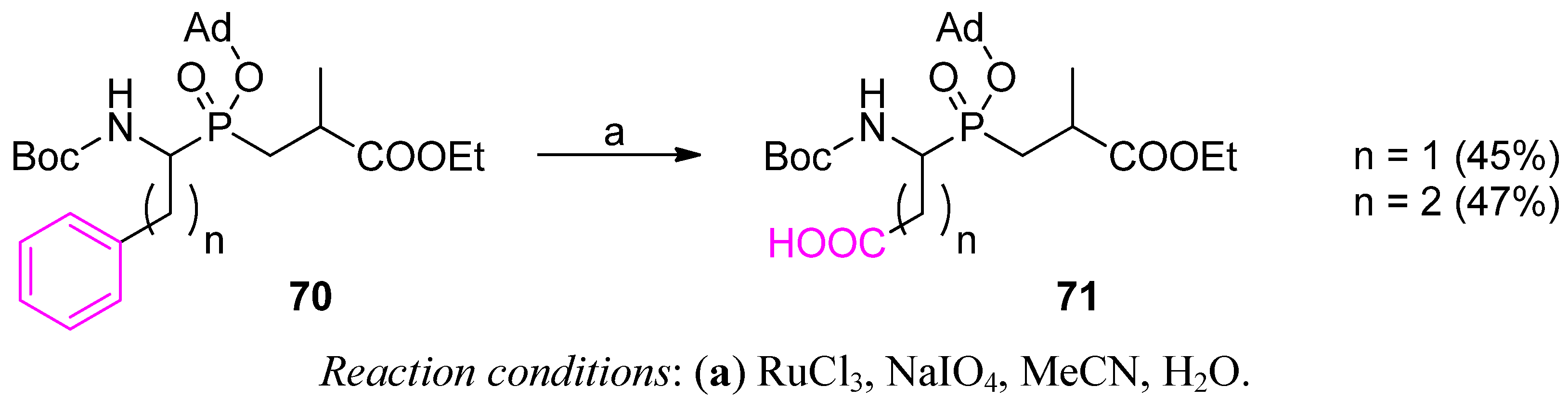
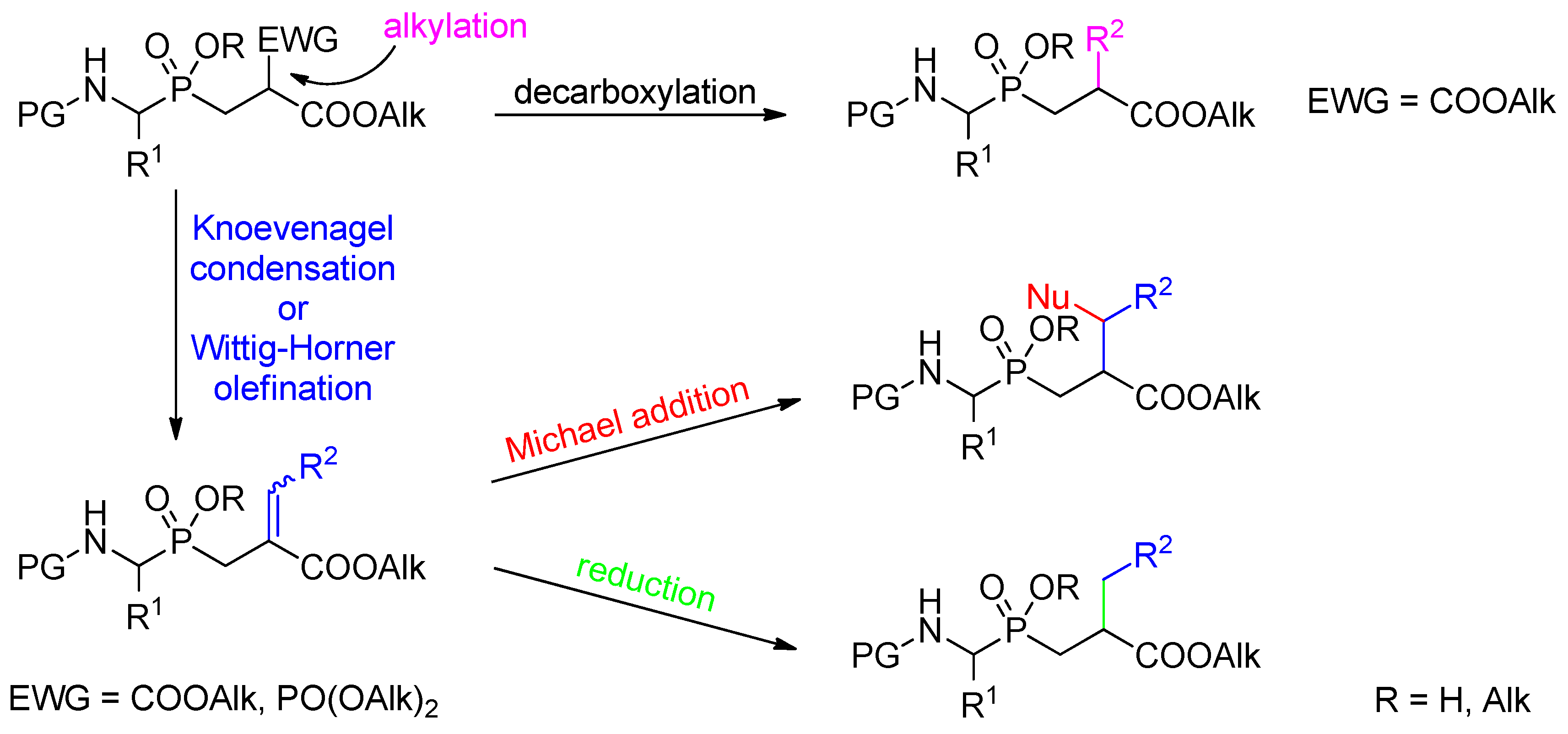
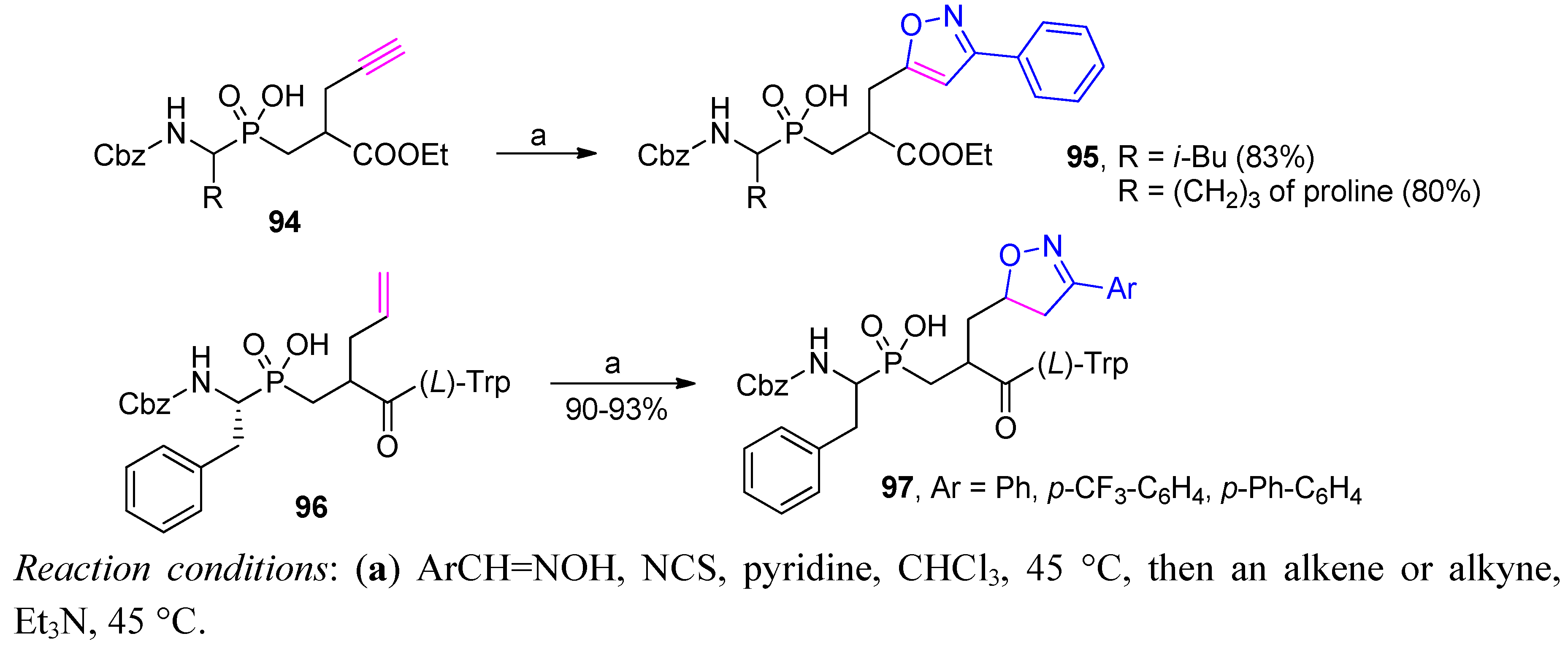
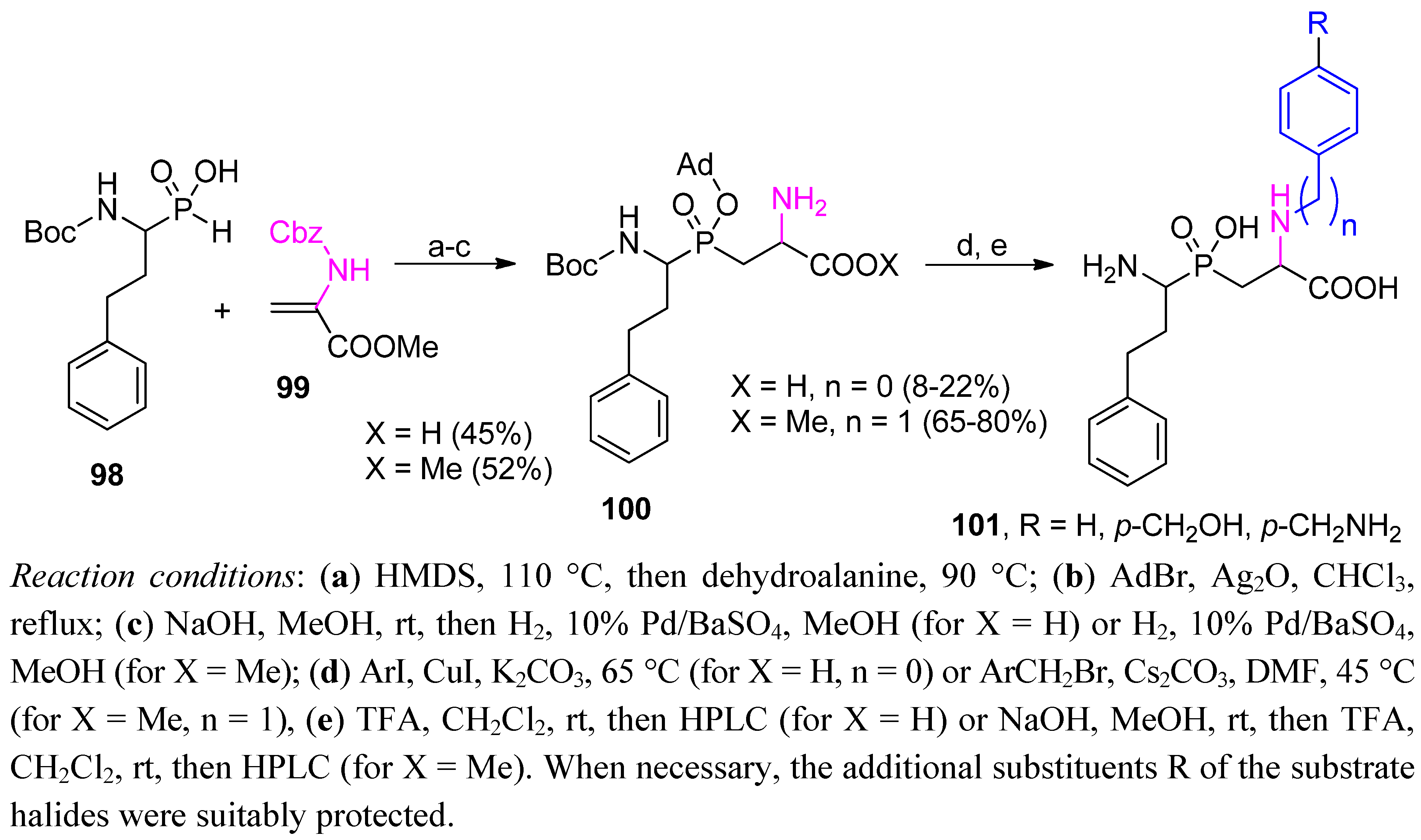
3.2. Side-Chain Substituents Modifications and Parallel Diversification
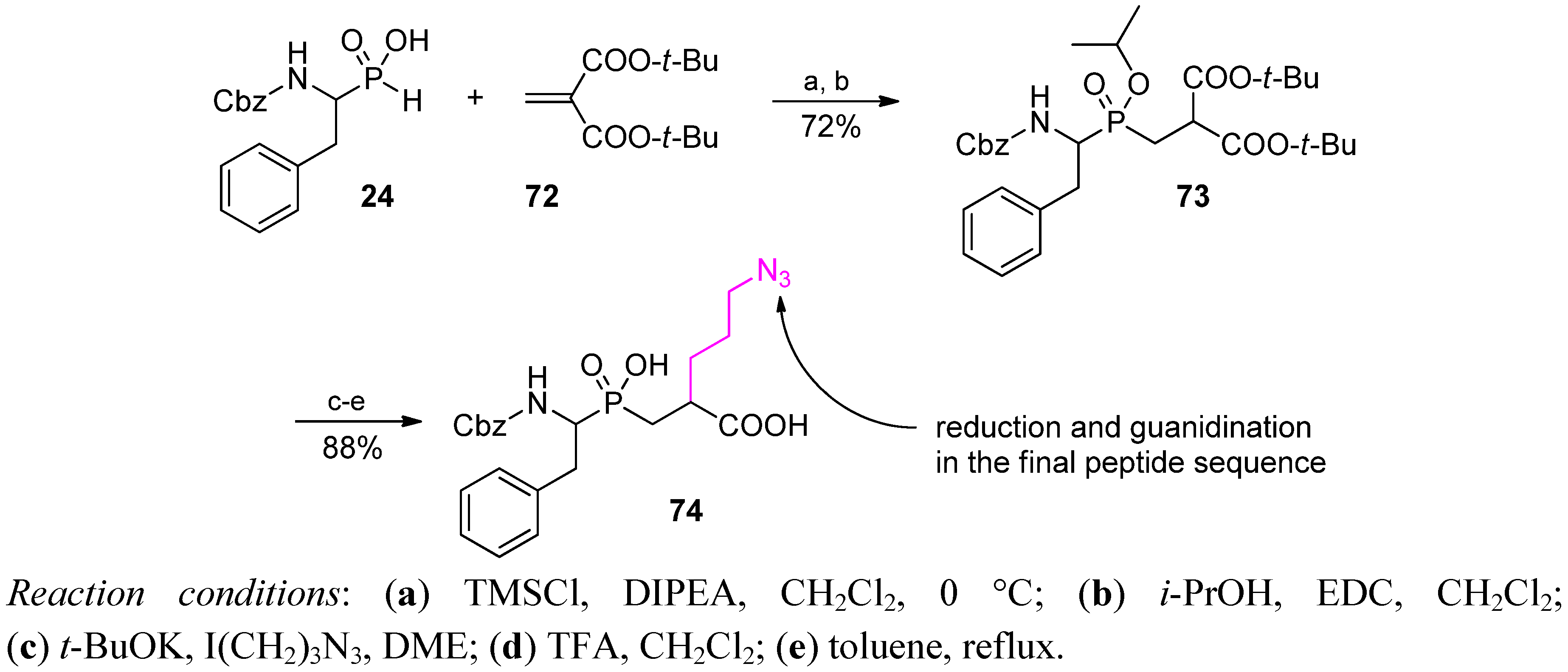
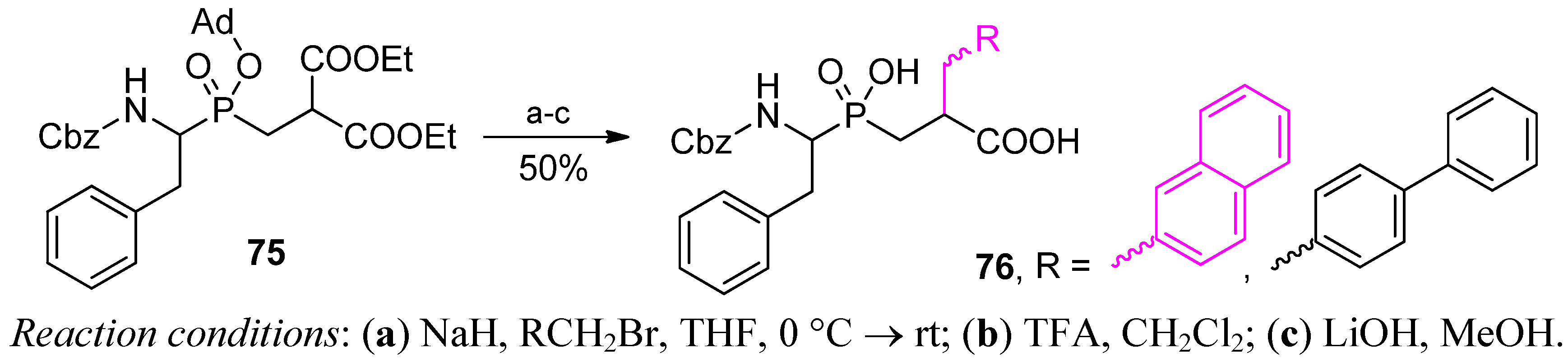
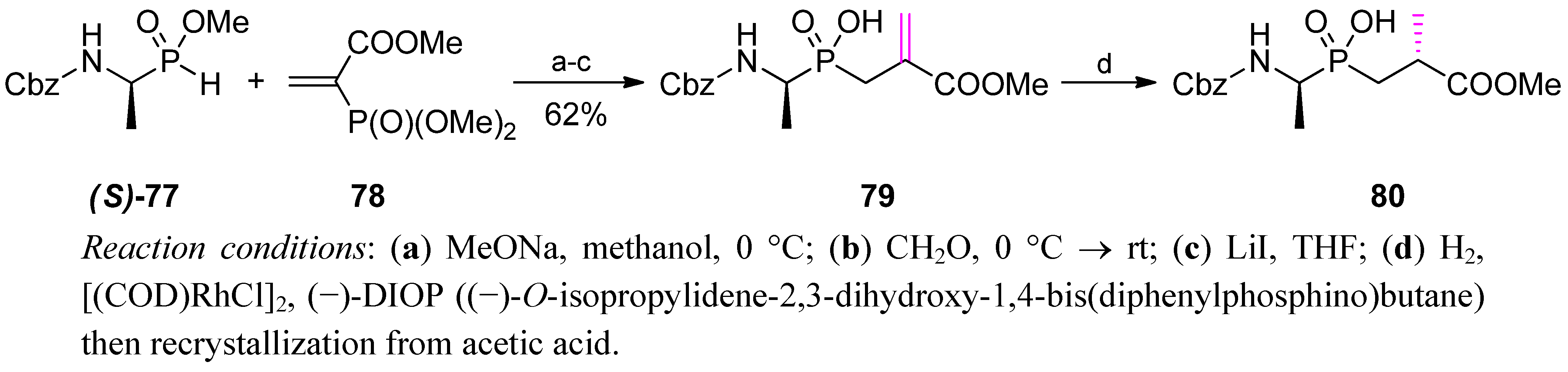
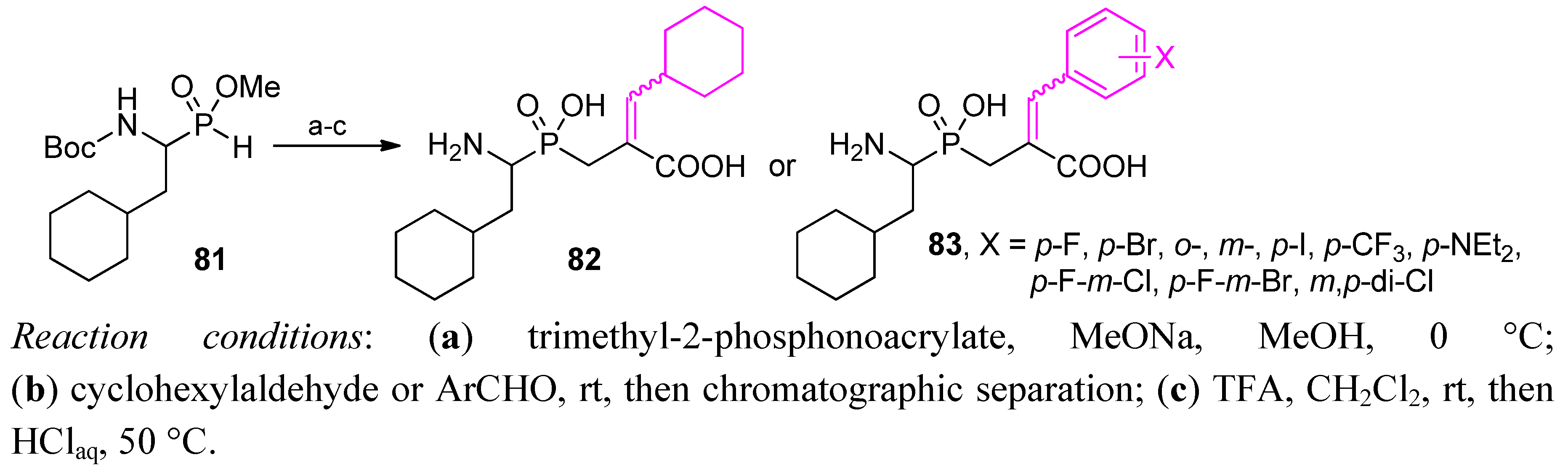
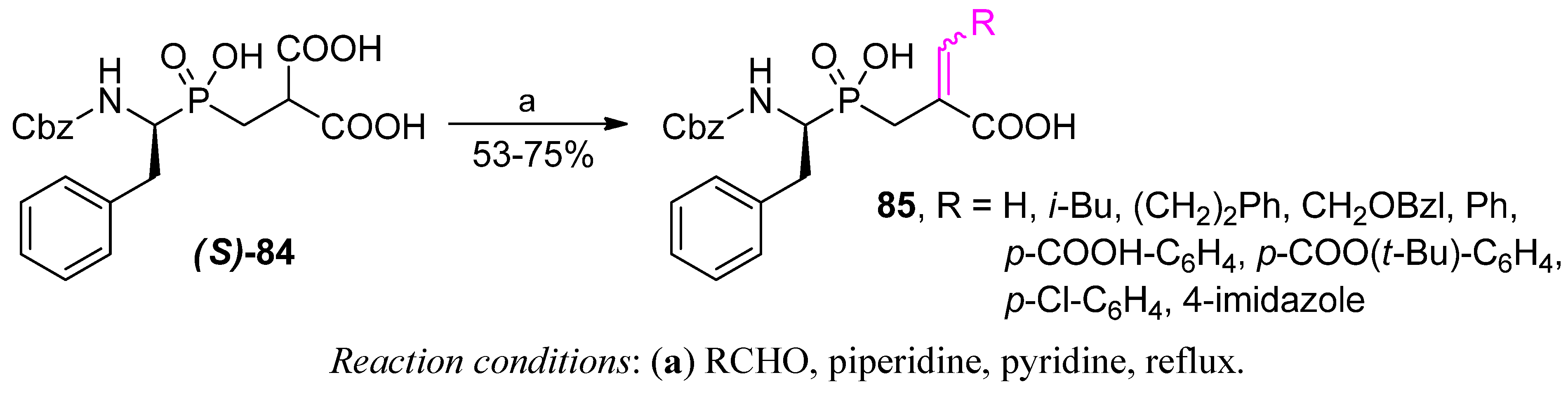
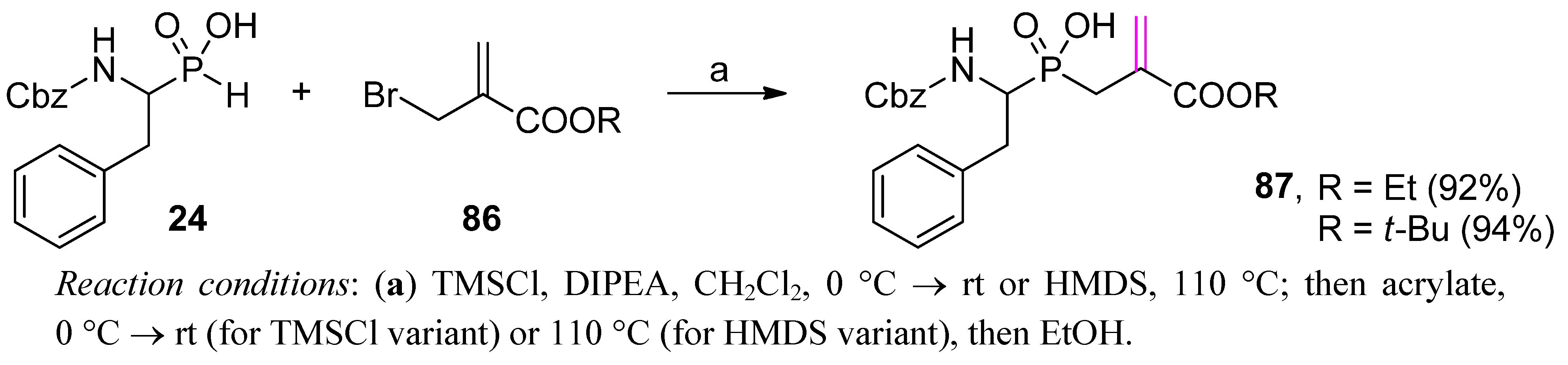
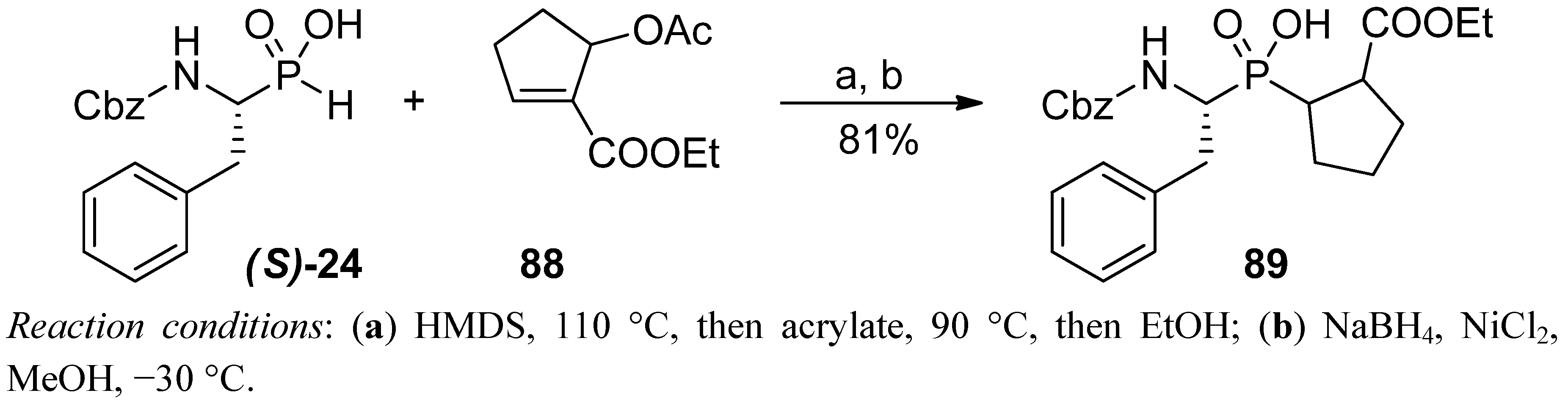
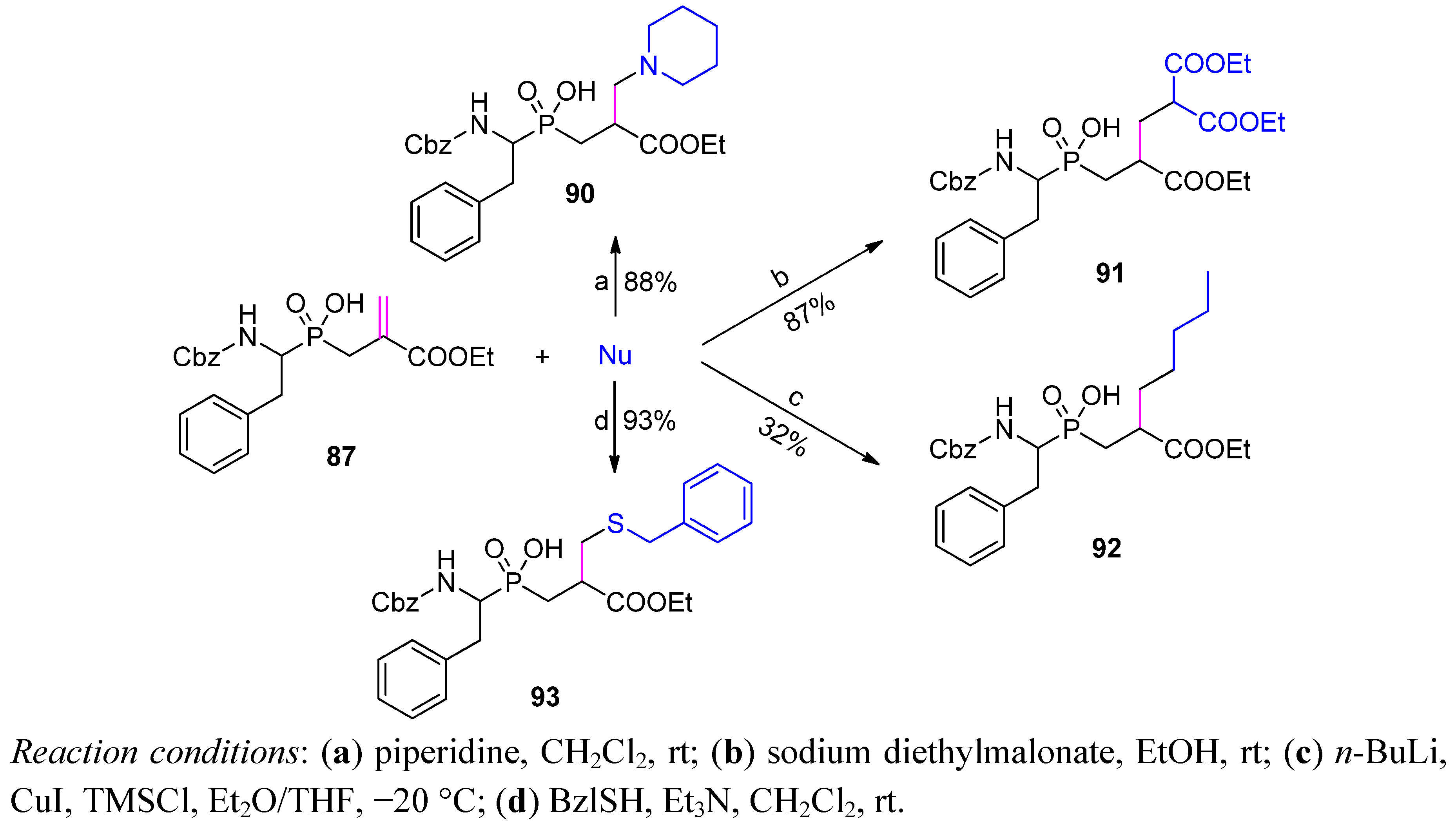
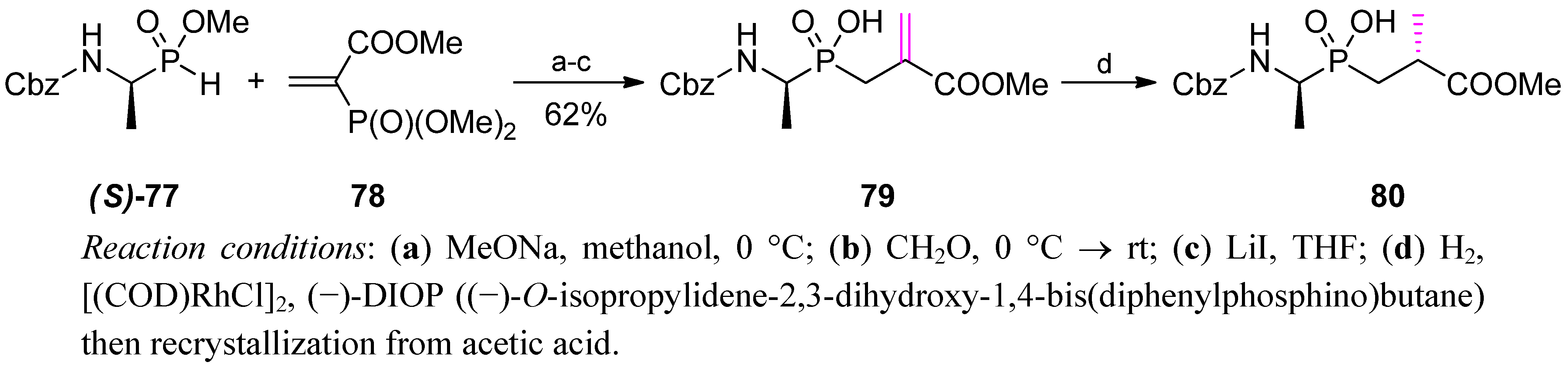
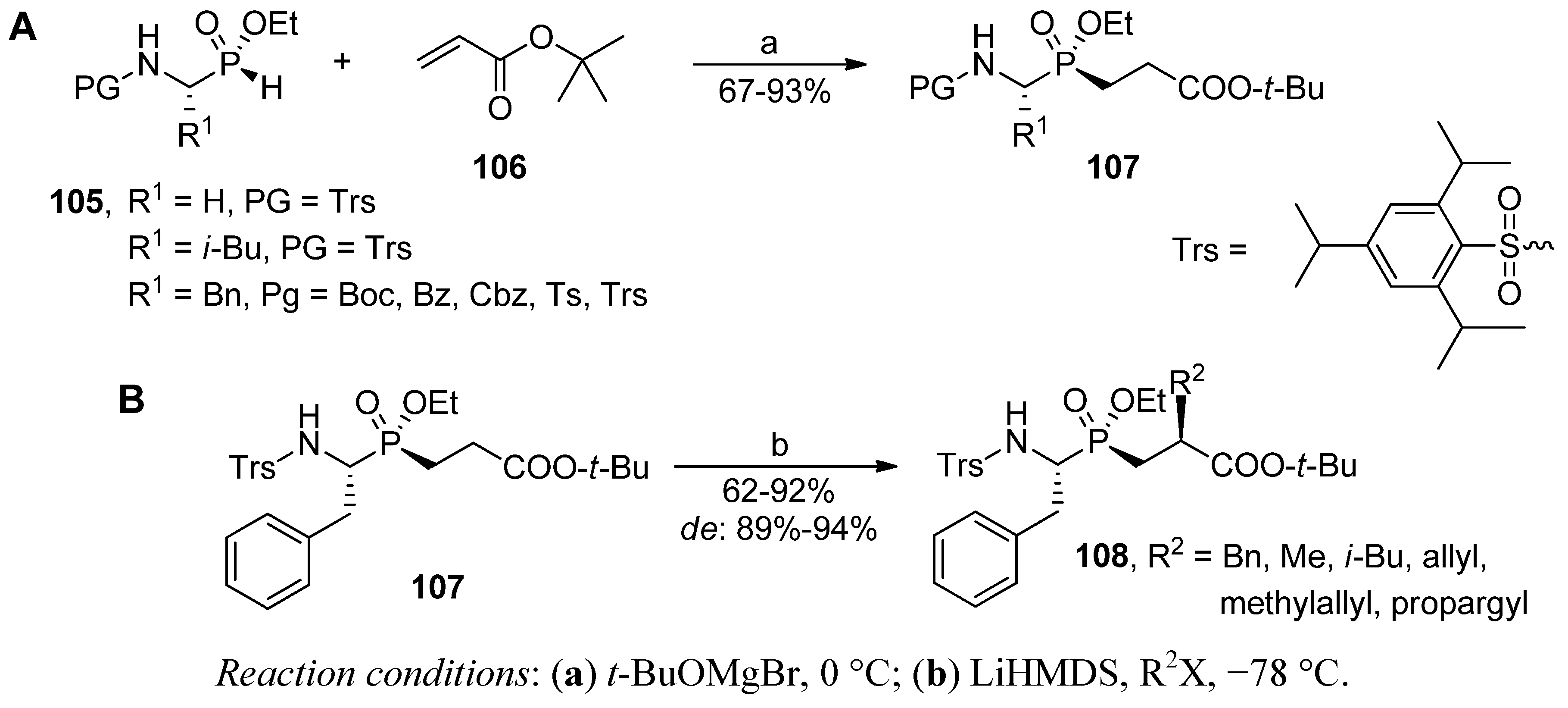
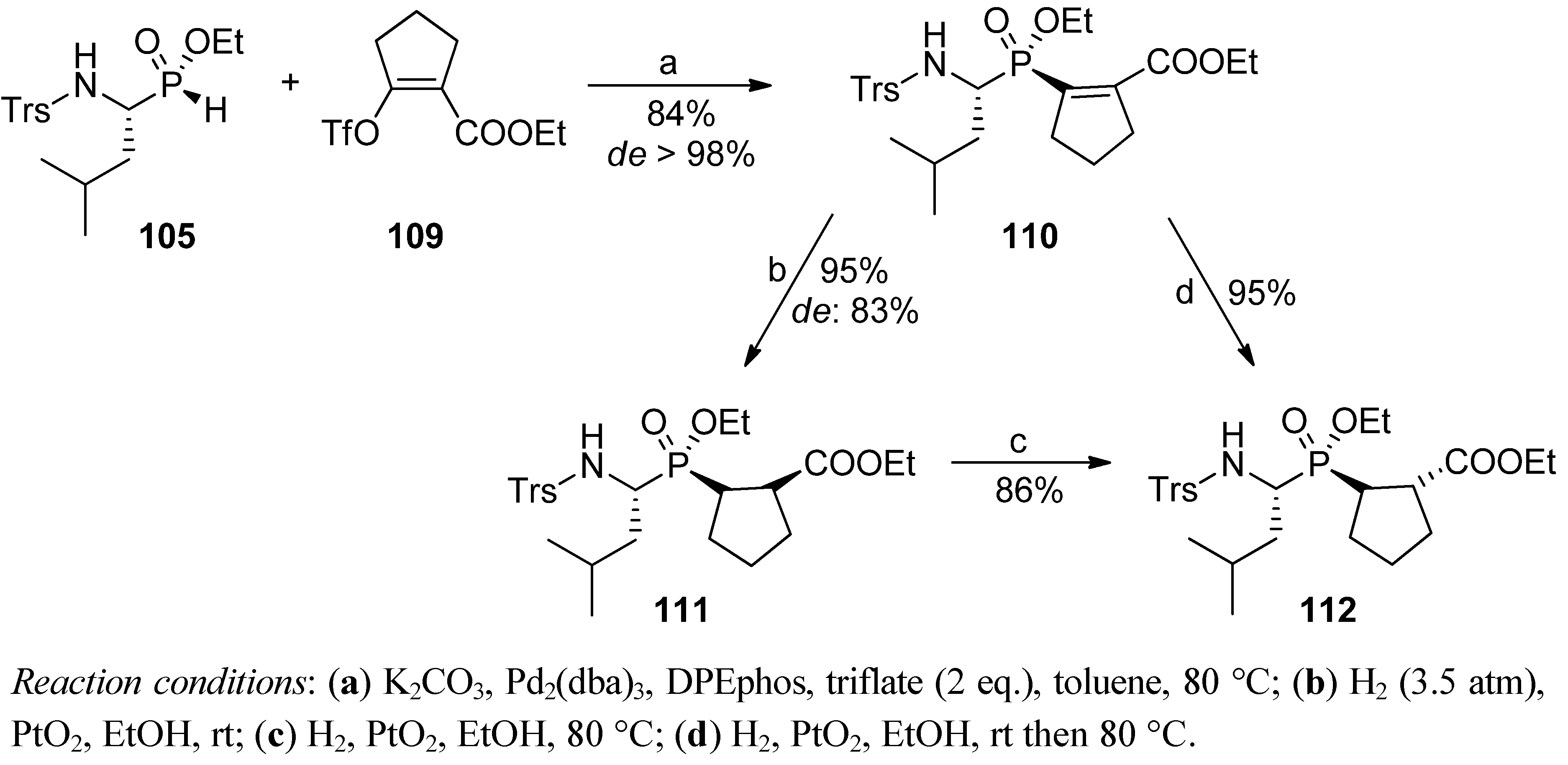
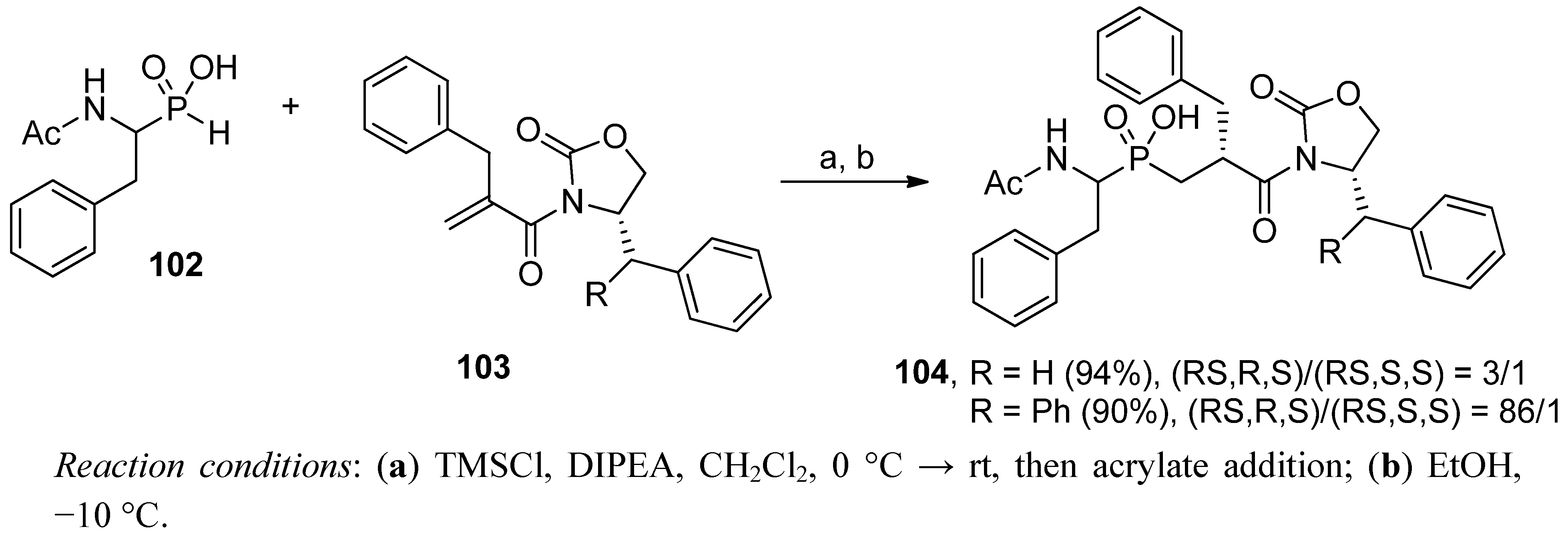
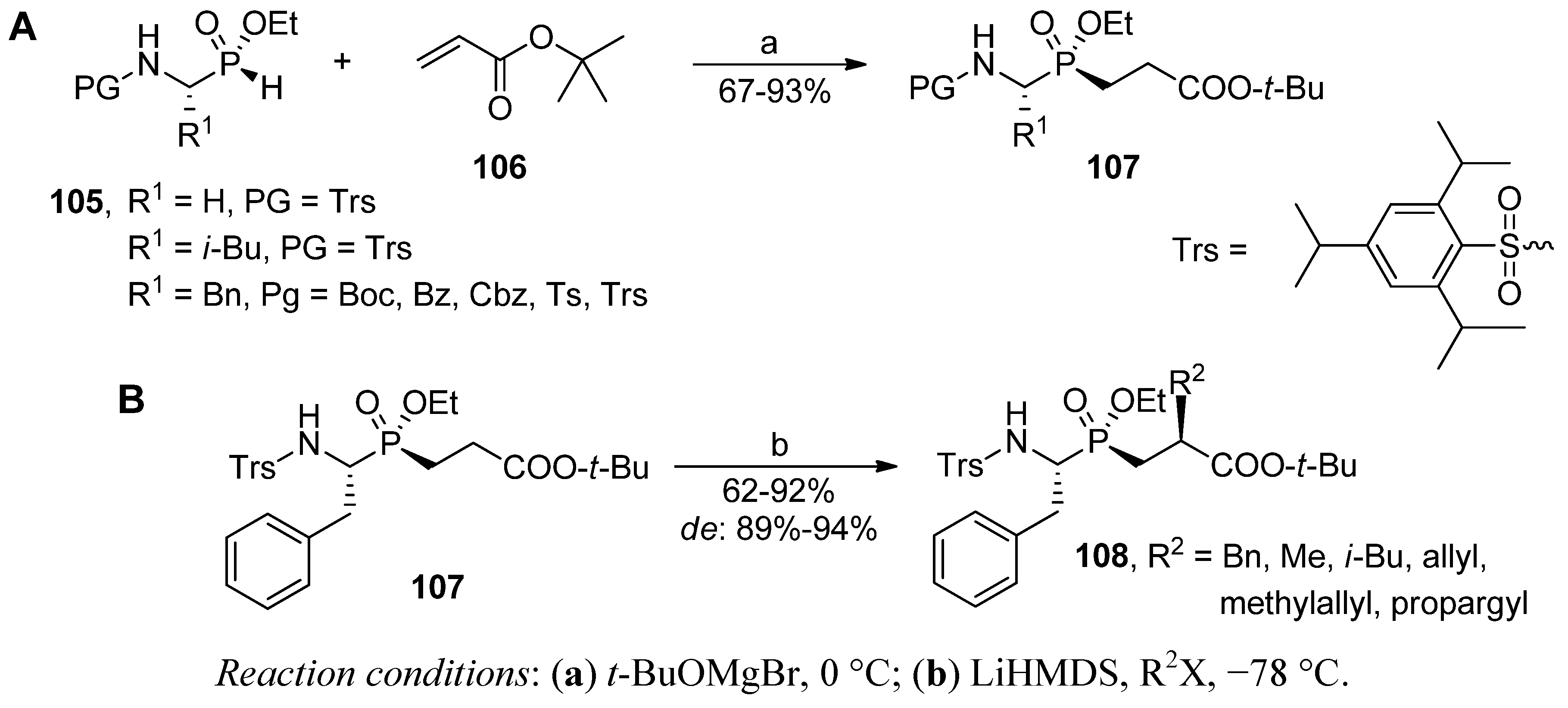
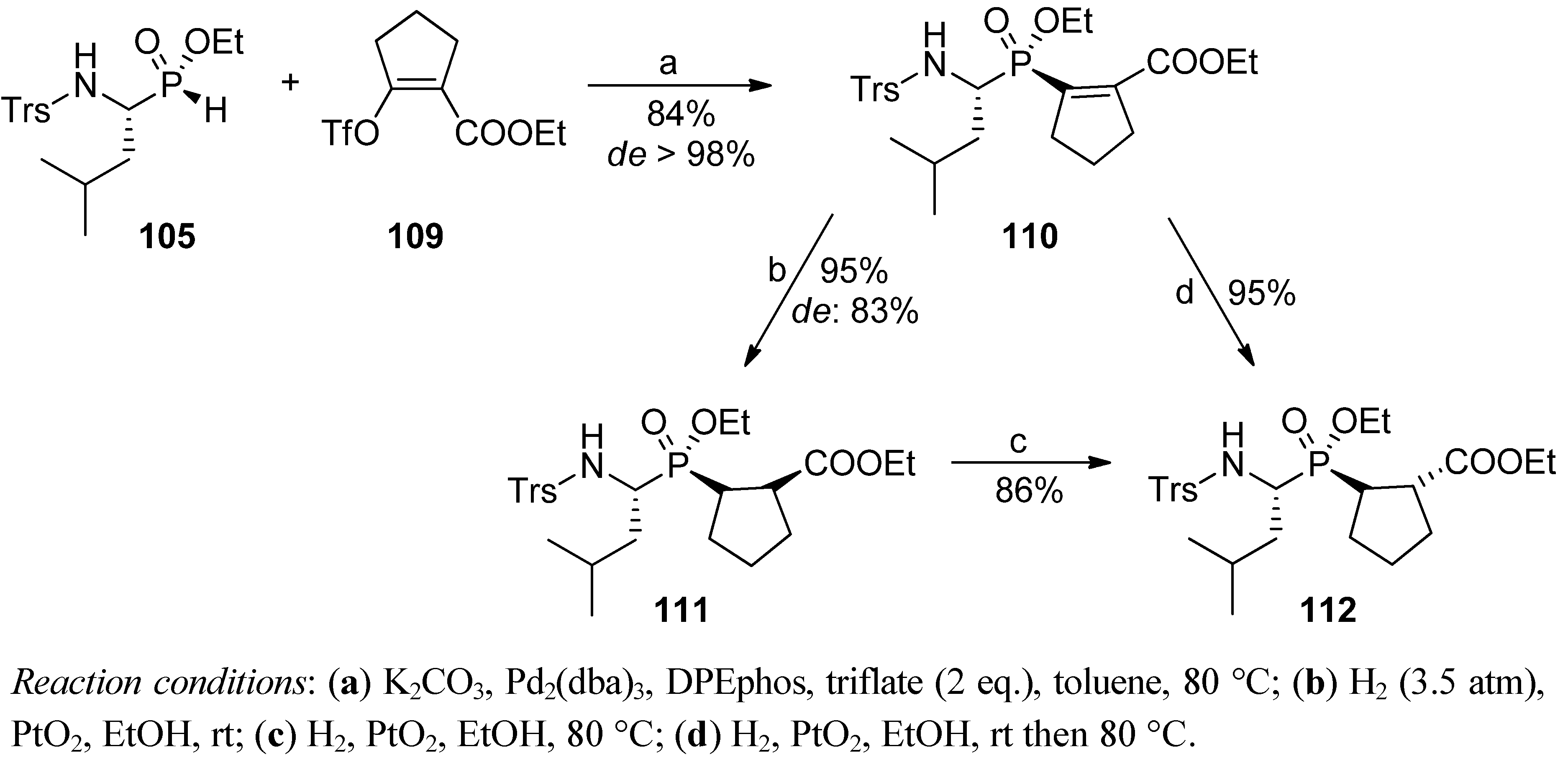
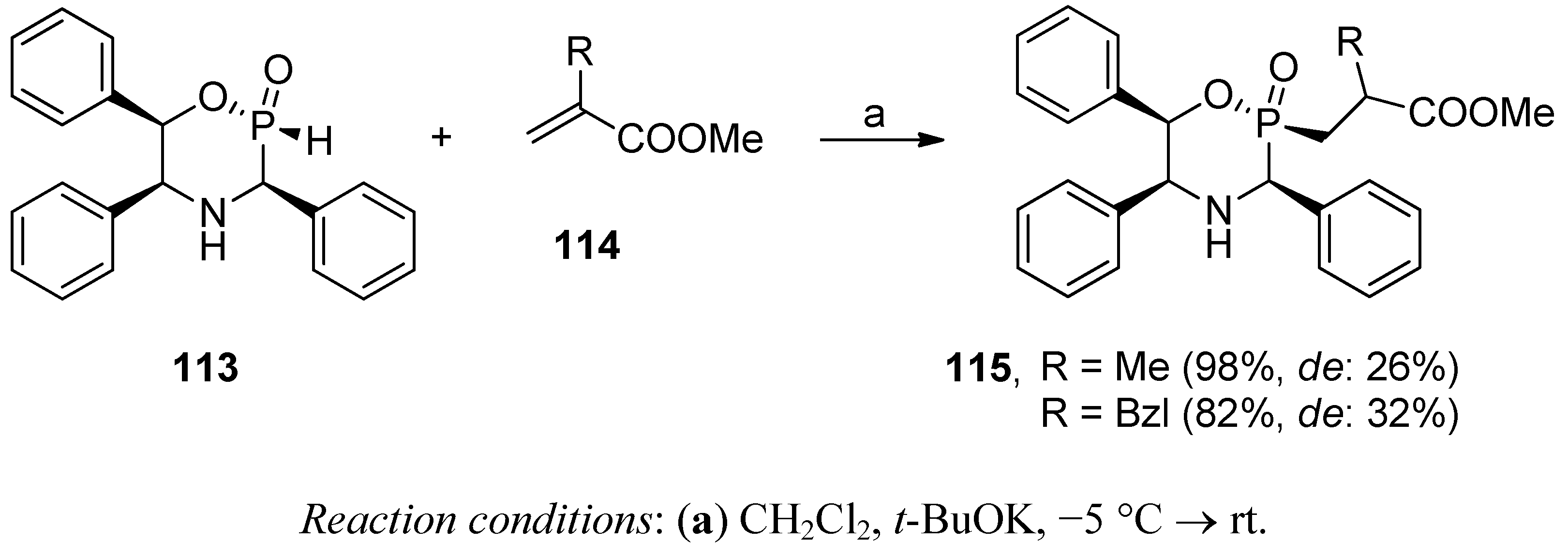
4. Stereoselective Approaches
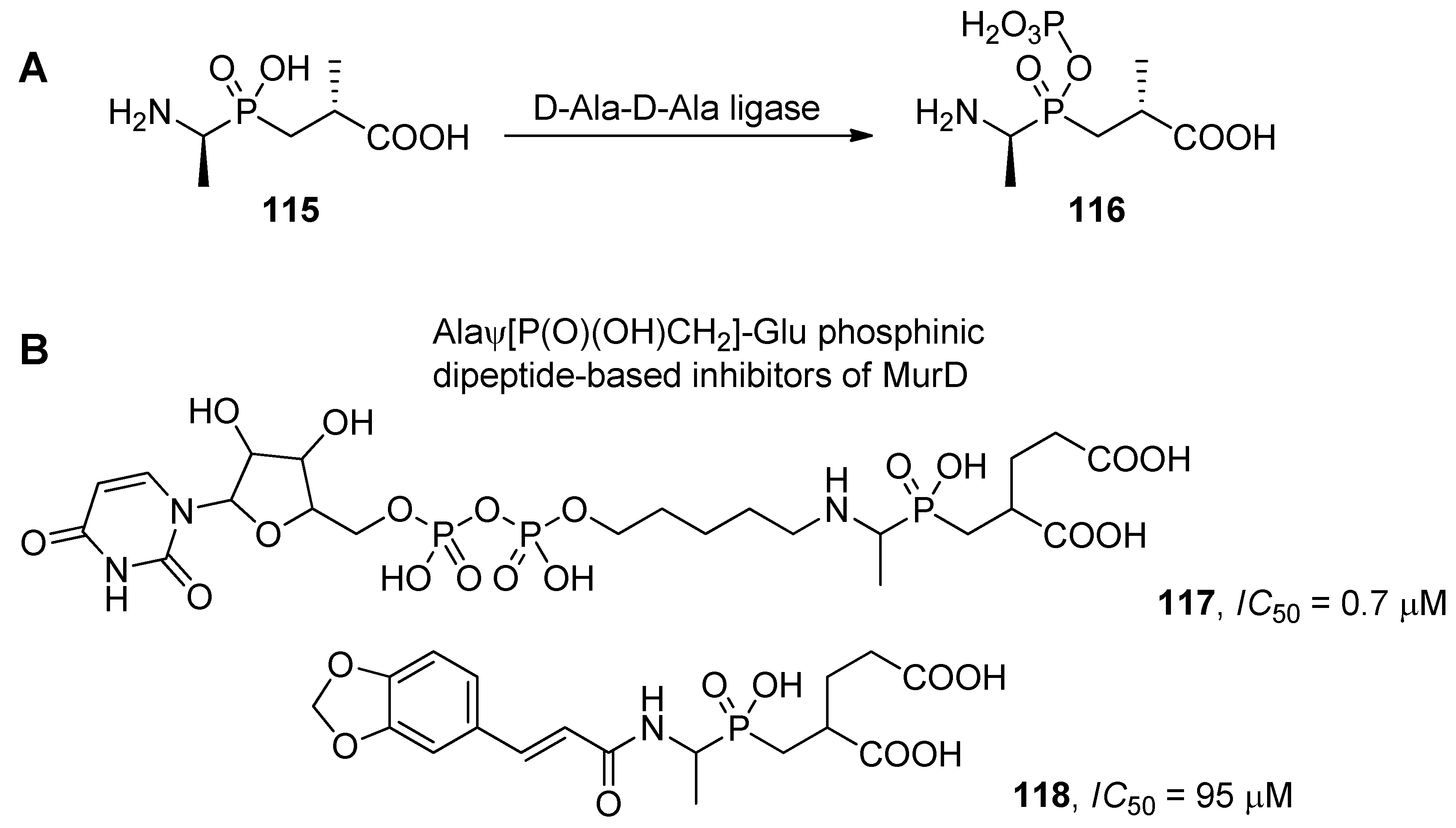
5. Applications and Conclusions

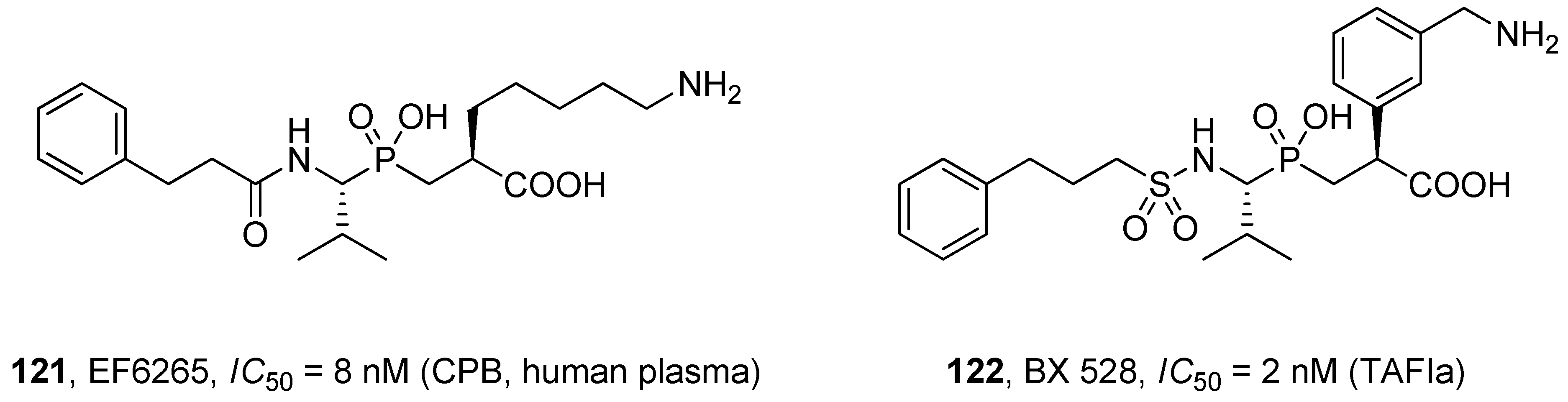
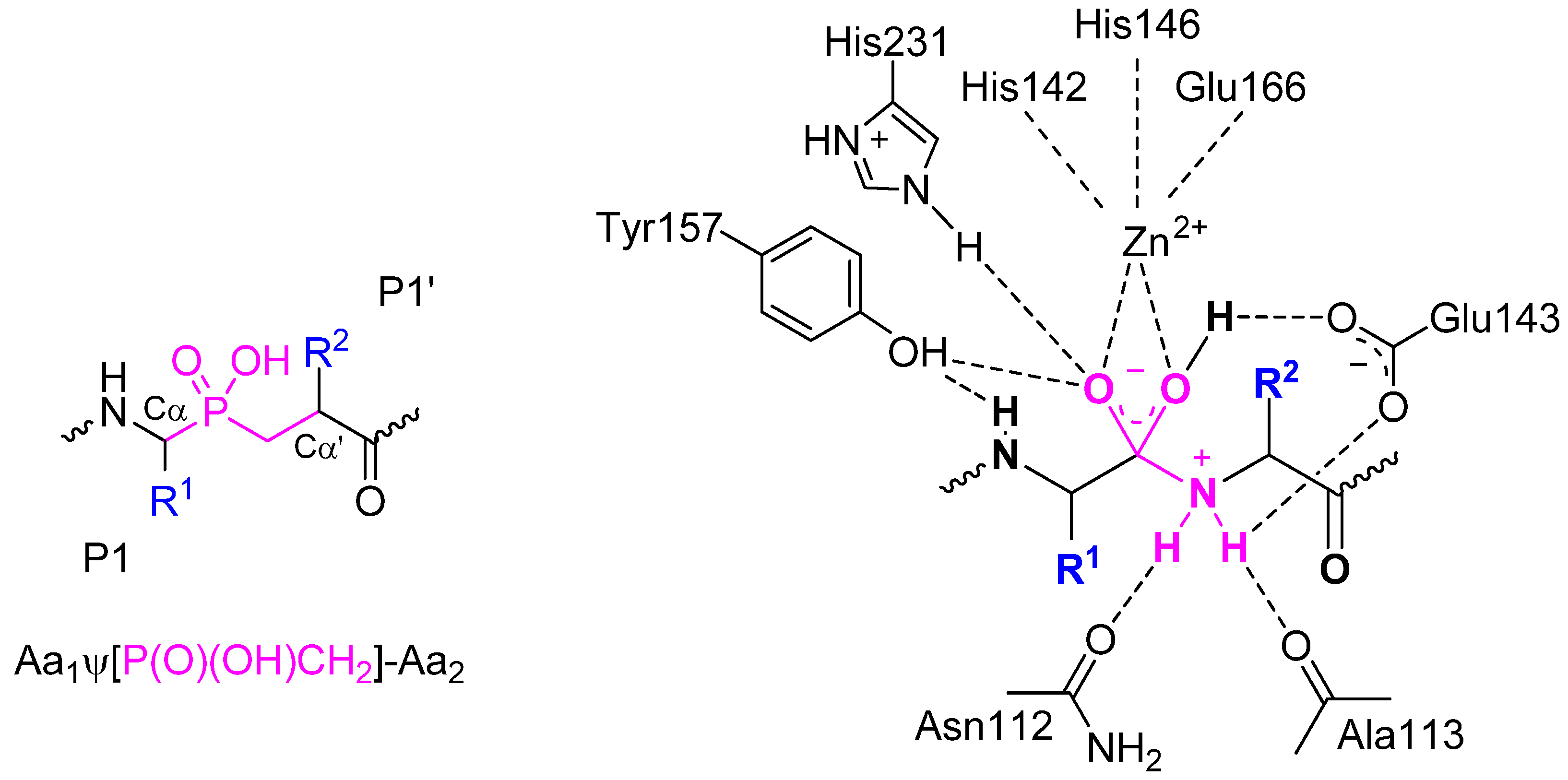
- angiotensin-converting enzyme 2, a mono-zinc carboxypeptidase, the first known human homologue of ACE [40];
- aminopeptidase A (glutamyl aminopeptidase) [122];
- NAALADase (N-acetylated-α-linked acidic dipeptidase), a metallo-dependent neuropeptidase [43];
- M18 aspartyl aminopeptidase of Plasmodium falciparum [124];
- dinuclear zinc aminopeptidase PepV from Lactobacillus delbrueckii [125];
- D-Ala-D-Ala dipeptidase VanX, required for vancomycin resistance [126];
- chymotrypsin, cathepsin G and neutrophil elastase, serine proteases [127].
Acknowledgments
References
- Matthews, B.W. Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 1988, 21, 333–340. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Pauling, L. Chemical achievement and hope for the future. Am. Sci. 1948, 36, 51–58. [Google Scholar]
- Wolfenden, R. Transition state analogues for enzyme catalysis. Nature 1969, 223, 704–705. [Google Scholar] [CrossRef]
- Kafarski, L.; Lejczak, B. The biological activity of phosphono- and phosphinopeptides. In Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons: Chichester, UK, 2000; pp. 407–442. [Google Scholar]
- Collinsová, M.; Jiráček, J. Phosphinic acid compounds in biochemistry, Biology and medicine. Curr. Med. Chem. 2000, 7, 629–647. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable potential of the α-aminophosphonate/ phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Jacobsen, N.E.; Bartlett, P.A. A phosphorus-containing dipeptide analog as an inhibitor of carboxypeptidase A. J. Am. Chem. Soc. 1981, 103, 654–657. [Google Scholar] [CrossRef]
- Bartlett, P.A.; Marlowe, C.K. Phosphonamidates as transition state analog inhibitors of thermolysin. Biochemistry 1983, 22, 4618–4624. [Google Scholar] [CrossRef]
- Holden, H.M.; Tronrud, D.E.; Monzingo, A.F.; Weaver, L.H.; Matthews, B.W. Slow- and fast-binding inhibitors of thermolysin display different modes of binding: Crystallographic analysis of extended phosphonamidate transition-state analogues. Biochemistry 1987, 26, 8542–8553. [Google Scholar] [CrossRef]
- Bartlett, P.A.; Marlowe, C.K. A possible role for water dissociation in the slow binding of phosphorus-containing transition state analog inhibitors of thermolysin. Biochemistry 1987, 26, 8553–8561. [Google Scholar] [CrossRef]
- Christianson, D.W.; Lipscomb, W.N. Comparison of carboxypeptidase A and thermolysin: Inhibition by phosphonamidates. J. Am. Chem. Soc. 1988, 110, 5560–5565. [Google Scholar] [CrossRef]
- Hanson, J.E.; Kaplan, A.P.; Bartlett, P.A. Phosphonate analogs of carboxypeptidase A are potent transition state analog inhibitors. Biochemistry 1989, 28, 6294–6305. [Google Scholar] [CrossRef]
- Kaplan, A.P.; Bartlett, P.A. An inhibitor of carboxypeptidase A with a Ki value in the femtomolar range. Biochemistry 1991, 30, 8165–8170. [Google Scholar] [CrossRef]
- Morgan, B.P.; Scholtz, J.M.; Ballinger, M.D.; Zipkin, I.D.; Bartlett, P.A. Differential binding energy: A detailed evaluation of the influence of hydrogen-bonding and hydrophobic groups on the inhibition of thermolysin by phosphorus-containing inhibitors. J. Am. Chem. Soc. 1991, 113, 297–307. [Google Scholar] [CrossRef]
- Yiotakis, A.; Vassiliou, S.; Jiráček, J.; Dive, V. Protection of the hydroxyphosphinyl function of phosphinic dipeptides by adamantyl. Application to the solid-phase synthesis of phosphinic peptides. J. Org. Chem. 1996, 61, 6601–6605. [Google Scholar] [CrossRef]
- Dive, V.; Lucet-Levannier, K.; Georgiadis, D.; Cotton, J.; Vassiliou, S.; Cuniasse, P.; Yiotakis, A. Phosphinic peptide inhibitors as tools in the study of the function of zinc metallopeptidases. Biochem. Soc. Trans. 2000, 28, 455–460. [Google Scholar] [CrossRef]
- Dive, V.; Georgiadis, D.; Matziari, M.; Makaritis, A.; Beau, F.; Cuniasse, P.; Yiotakis, A. Phosphinic peptides as zinc metalloproteinase inhibitors. Cell. Mol. Life Sci. 2004, 61, 2010–2019. [Google Scholar]
- Cuniasse, P.; Devel, L.; Makaritis, A.; Beau, F.; Georgiadis, D.; Matziari, M.; Yiotakis, A.; Dive, V. Future challenges facing the development of specific active-site-directed synthetic inhibitors of MMPs. Biochimie 2005, 87, 393–402. [Google Scholar] [CrossRef]
- Matziari, M.; Dive, V.; Yiotakis, A. Matrix metalloproteinase 11 (MMP-11; stromelysin-3) and synthetic inhibitors. Med. Res. Rev. 2007, 27, 528–552. [Google Scholar] [CrossRef]
- Yiotakis, A.; Georgiadis, D.; Matziari, M.; Makaritis, A.; Dive, V. Phosphinic peptides: Synthetic approaches and biochemical evaluation as Zn-metalloprotease inhibitors. Curr. Org. Chem. 2004, 8, 1135–1158. [Google Scholar] [CrossRef]
- Baylis, E.K.; Campbell, C.D.; Dingwall, J.G. 1-Aminoalkylphosphonous acids. Part 1. Isosteres of the protein amino acids. J. Chem. Soc. Perkin Trans. 1 1984, 2845–2853. [Google Scholar]
- Georgiadis, D.; Matziari, M.; Vassiliou, S.; Dive, V.; Yiotakis, A. A convenient method to synthesize phosphinic peptides containing an aspartyl or glutamyl aminophosphinic acid. Use of the phenyl group as the carboxyl synthon. Tetrahedron 1999, 55, 14635–14648. [Google Scholar] [CrossRef]
- Dumy, P.; Escale, R.; Girard, J.-P.; Parello, J.; Vidal, J.-P. A convenient synthetic approach to new α-(9-fluorenylmethoxycarbonylamino)alkylphosphonic acid derivatives. Synthesis 1992, 1226–1228. [Google Scholar]
- Thottathil, J.K.; Przybyla, C.A.; Moniot, J.L. Mild Arbuzov reactions of phosphonous acids. Tetrahedron Lett. 1984, 25, 4737–4740. [Google Scholar] [CrossRef]
- Thottathil, J.K.; Ryono, D.E.; Przybyla, C.A.; Moniot, J.L.; Neubeck, R. Preparation of phosphinic acids: Michael additions of phosphonous acids/esters to conjugated systems. Tetrahedron Lett. 1984, 25, 4741–4744. [Google Scholar]
- Giannousis, P.P.; Bartlett, P.A. Phosphorus amino acid analogs as inhibitors of leucine aminopeptidase. J. Med. Chem. 1987, 30, 1603–1609. [Google Scholar] [CrossRef]
- Sampson, N.S.; Bartlett, P.A. Synthesis of phosphonic acid derivatives by oxidative activation of phosphinate esters. J. Org. Chem. 1988, 53, 4500–4503. [Google Scholar] [CrossRef]
- Boyd, E.A.; Corless, M.; James, K.; Regan, A.C. A versatile route to substituted phosphinic acids. Tetrahedron Lett. 1990, 31, 2933–2936. [Google Scholar] [CrossRef]
- Boyd, E.A.; Reagan, A.C.; James, K. Synthesis of alkyl phosphinic acids from silyl phosphonites and alkyl halides. Tetrahedron Lett. 1994, 35, 4223–4226. [Google Scholar] [CrossRef]
- Parsons, W.H.; Patchett, A.A.; Bull, H.G.; Schoen, W.R.; Taub, D.; Davidson, J.; Combs, P.L.; Springer, J.P.; Gadebusch, H.; Weissberger, B.; et al. Phosphinic acid inhibitors of D-alanyl-D-alanine ligase. J. Med. Chem. 1988, 31, 1772–1778. [Google Scholar] [CrossRef]
- Allen, M.C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J.M. Renin inhibitors. Synthesis of transition-state analogue inhibitors containing phosphorus acid derivatives at the scissile bond. J. Med. Chem. 1989, 32, 1652–1661. [Google Scholar] [CrossRef]
- Cristau, H.-J.; Coulombeau, A.; Genevois-Borella, A.; Pirat, J.-L. A convenient one-pot synthesis of phosphino-dipeptide analogs. Tetrahedron Lett. 2001, 42, 4491–4494. [Google Scholar]
- Cristau, H.-J.; Coulombeau, A.; Genevois-Borella, A.; Sanchez, F.; Pirat, J.-L. Preparation of phosphinodipeptide analogs as building blocks for pseudopeptides synthesis. J. Organomet. Chem. 2002, 643/644, 381–391. [Google Scholar]
- Vassiliou, S.; Mucha, A.; Cuniasse, P.; Georgiadis, D.; Lucet-Levannier, K.; Beau, F.; Kannan, R.; Murphy, G.; Knäuper, V.; Rio, M.-C.; et al. Phosphinic pseudo-tripeptides as potent inhibitors of matrix metalloproteinases: a structure-activity study. J. Med. Chem. 1999, 42, 2610–2620. [Google Scholar] [CrossRef]
- Chen, H.; Noble, F.; Mothé, A.; Meudal, H.; Coric, P.; Danascimento, S.; Roques, B.P.; George, P.; Fournié-Zaluski, M.-C. Phosphinic derivatives as new dual enkephalin-degrading enzyme inhibitors: Synthesis, biological properties, and antinociceptive activities. J. Med. Chem. 2000, 43, 1398–1408. [Google Scholar] [CrossRef]
- Miller, D.J.; Hammond, S.M.; Anderluzzi, D.; Bugg, T.D.H. Aminoalkylphosphinate inhibitors of D-Ala-D-Ala adding enzyme. J. Chem. Soc. Perkin Trans. 1 1998, 131–142. [Google Scholar]
- Liboska, R.; Pícha, J.; Hančlová, I.; Buděšínský, M.; Šanda, M.; Jiráček, J. Synthesis of methionine- and norleucine-derived phosphinopeptides. Tetrahedron Lett. 2008, 49, 5629–5631. [Google Scholar] [CrossRef]
- Kaboudin, B.; Saadati, F. A simple, novel and convenient method for the synthesis of 1-aminophosphinic acids: synthesis of a novel C2-symmetric phosphinic acid pseudodipeptide. Tetrahedron Lett. 2009, 50, 1450–1452. [Google Scholar] [CrossRef]
- Mores, A.; Matziari, M.; Beau, F.; Cuniasse, P.; Yiotakis, A.; Dive, V. Development of potent and selective phosphinic peptide inhibitors of angiotensin-converting enzyme 2. J. Med. Chem. 2008, 51, 2216–2226. [Google Scholar] [CrossRef]
- Demange, L.; Dugave, C. Synthesis of phosphinic alanyl-proline surrogates Alaψ(PO2R-CH)Pro as potential inhibitors of the human cyclophilin hCyp-18. Tetrahedron Lett. 2001, 42, 6295–6297. [Google Scholar] [CrossRef]
- Tanner, M.E.; Vaganay, S.; van Heijenoort, J.; Blanot, D. Phosphinate inhibitors of the D-glutamic acid-adding enzyme of peptidoglycan biosynthesis. J. Org. Chem. 1996, 61, 1756–1760. [Google Scholar] [CrossRef]
- Jackson, P.F.; Tays, K.L.; Maclin, K.M.; Ko, Y.-S.; Li, W.; Vitharana, D.; Tsukamoto, T.; Stoermer, D.; Lu, X.-C.M.; Wozniak, K.; Slusher, B.S. Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J. Med. Chem. 2001, 44, 4170–4175. [Google Scholar] [CrossRef]
- Kende, A.S.; Dong, H.-Q.; Liu, X.; Ebetino, F.H. A useful synthesis of the Phe-Arg phosphinic dipeptide isostere. Tetrahedron Lett. 2002, 43, 4973–4976. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef]
- Vassiliou, S.; Xeilari, M.; Yiotakis, A.; Grembecka, J.; Pawełczak, M.; Kafarski, P.; Mucha, A. A synthetic method for diversification of the P1' substituent in phosphinic dipeptides as a tool for exploration of the specificity of the S1' binding pockets of leucine aminopeptidases. Bioorg. Med. Chem. 2007, 15, 3187–3200. [Google Scholar] [CrossRef]
- Makaritis, A.; Georgiadis, D.; Dive, V.; Yiotakis, A. Diastereoselective solution and multipin-based combinatorial array synthesis of a novel class of potent phosphinic metalloprotease inhibitors. Chem. Eur. J. 2003, 9, 2079–2094. [Google Scholar] [CrossRef]
- Jullien, N.; Makritis, A.; Georgiadis, D.; Beau, F.; Yiotakis, A.; Dive, V. Phosphinic tripeptides as dual angiotensin-converting enzyme C-domain and endothelin-converting enzyme-1 inhibitors. J. Med. Chem. 2010, 53, 208–220. [Google Scholar] [CrossRef]
- Stetter, H.; Kuhlmann, H. Eine einfache Herstellung von α-Alkylacrylsäure-estern. Synthesis 1979, 29–30. [Google Scholar] [CrossRef]
- Matziari, M.; Bauer, K.; Dive, V.; Yiotakis, A. Synthesis of the phosphinic analogue of thyrotropin releasing hormone. J. Org. Chem. 2008, 73, 8591–8593. [Google Scholar] [CrossRef]
- Borloo, M.; Jiao, X.-Y.; Wójtowicz, H.; Rajan, P.; Verbruggen, C.; Augustyns, K.; Haemers, A. A convenient one-pot preparation of disubstituted phosphinic acids derived from simple amino acids and proline. Synthesis 1995, 1074–1076. [Google Scholar]
- Fougère, C.; Guénin, E.; Hardouin, J.; Lecouvey, M. Rapid and efficient synthesis of unsymmetrical phosphinic acids R'P(O)OHR''. Eur. J. Org. Chem. 2009, 6048–6054. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis of new organophosphorus-substituted mono- and bis(trimethylsilyl)amines with PCH2N fragments and their derivatives. Heteroatom Chem. 2010, 21, 71–77. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis of new organophosphorus-substituted derivatives of functionalized propionates and their analogues. Heteroatom Chem. 2008, 19, 418–428. [Google Scholar] [CrossRef]
- Livantsov, M.V.; Prishchenko, A.A.; Novikova, O.P.; Livantsova, L.I.; Grigor'ev, E.V. Synthesis of phosphorus-substituted derivatives of methylsuccinic acid. Russ. J. Gen. Chem. 2003, 73, 659–660. [Google Scholar] [CrossRef]
- Rogakos, V.; Georgiadis, D.; Dive, V.; Yiotakis, A. A modular rearrangement approach toward medicinally relevant phosphinic structures. Org. Lett. 2009, 11, 4696–4699. [Google Scholar] [CrossRef]
- Dorff, P.H.; Chiu, G.; Goldstein, S.W.; Morgan, B.P. Solid phase synthesis of phosphinopeptoids as transition state analog inhibitors. Tetrahedron Lett. 1998, 39, 3375–3378. [Google Scholar] [CrossRef]
- Buchardt, J.; Meldal, M. Novel methodology for the solid-phase synthesis of phosphinic peptides. J. Chem. Soc. Perkin Trans. 1 2000, 3306–3310. [Google Scholar] [CrossRef]
- Chen, S.; Coward, J.K. A general method for the synthesis of N-protected α-aminophosphinic acids. Tetrahedron Lett. 1996, 37, 4335–4338. [Google Scholar] [CrossRef]
- Yuan, C.; Wang, G.; Chen, S. Studies on organophosphorus compounds; XLVI. A facile and direct route to dialkyl 1-(benzyloxycarbonylamino)alkylphosphonates and dialkyl or diphenyl α-(benzyloxycarbonylamino)benzylphosphonates. Synthesis 1990, 522–524. [Google Scholar]
- Matziari, M.; Yiotakis, A. Shortcut to Fmoc-protected phosphinic pseudodipeptidic blocks. Org. Lett. 2005, 7, 4049–4052. [Google Scholar] [CrossRef]
- Nasopoulou, M.; Georgiadis, D.; Matziari, M.; Dive, V.; Yiotakis, A. A versatile annulation protocol toward novel constrained phosphinic peptidomimetics. J. Org. Chem. 2007, 72, 7222–7228. [Google Scholar] [CrossRef]
- Rozhko, L.F.; Ragulin, V.V. Method for the synthesis of phosphinic acids from hypophosphites V. The synthesis of pseudo-α,α-dipeptides. Amino Acids 2005, 29, 139–143. [Google Scholar] [CrossRef]
- Dmitriev, M.E.; Ragulin, V.V. New opinions on the amidoalkylation of hydrophosphorylic compounds. Tetrahedron Lett. 2010, 51, 2613–2616. [Google Scholar] [CrossRef]
- Dmitriev, M.E.; Rossinets, E.A.; Ragulin, V.V. Amidoalkylation of hydrophosphoryl compounds. Russ. J. Gen. Chem. 2011, 81, 1092–1104. [Google Scholar] [CrossRef]
- Dmitriev, M.E.; Ragulin, V.V. Arbuzov-type reaction of acylphosphonites and N-alkoxycarbonylimine cations generated in situ with trifluoroacetic anhydride. Tetrahedron Lett. 2012, 53, 1634–1636. [Google Scholar] [CrossRef]
- Huber, T.; Manzenrieder, F.; Kuttruff, C.A.; Dorner-Ciossek, C.; Kessler, H. Prolonged stability by cyclization: Macrocyclic phosphino dipeptide isostere inhibitors of β-secretase (BACE1). Bioorg. Med. Chem. Lett. 2009, 19, 4427–4431. [Google Scholar] [CrossRef]
- Foersterova, M.; Svobodova, I.; Lubal, P.; Taborsky, P.; Kotek, J.; Hermann, P.; Lukes, I. Thermodynamic study of lanthanide(III) complexes with bifunctional monophosphinic acid analogues of H4dota and comparative kinetic study of yttrium(III) complexes. J. Chem. Soc. Dalton Trans. 2007, 535–549. [Google Scholar]
- Notni, J.; Hermann, P.; Havlickova, J.; Kotek, J.; Kubicek, V.; Plutnar, J.; Loktionova, N.; Riss, P.J.; Roesch, F.; Lukes, I. A triazacyclononane-based bifunctional phosphinate ligand for the preparation of multimeric 68Ga tracers for positron emission tomography. Chem. Eur. J. 2010, 16, 7174–7185. [Google Scholar]
- Foersterova, M.; Petrik, M.; Laznickova, A.; Laznicek, M.; Hermann, P.; Lukes, I.; Melichar, F. Complexation and biodistribution study of 111In and 90Y complexes of bifunctional phosphinic acid analogs of H4dota. Appl. Radiat. Isotopes 2009, 67, 21–29. [Google Scholar] [CrossRef]
- Lacerda, S.; Marques, F.; Campello, P.; Gano, L.; Kubicek, V.; Hermann, P.; Santos, I. Chemical, radiochemical and biological studies of Sm and Ho complexes of H4dota analogues containing one methylphosphonic/phosphinic acid pendant arm. J. Labelled Comp. Radiopharm. 2010, 53, 36–43. [Google Scholar]
- Notni, J.; Simecek, J.; Hermann, P.; Wester, H.-J. TRAP, a powerful and versatile framework for gallium-68 radiopharmaceuticals. Chem. Eur. J. 2011, 17, 14718–14722. [Google Scholar] [CrossRef]
- Ragulin, V.V. One-pot synthesis of α-amino phosphinic acids. Russ. J. Gen. Chem. 2004, 74, 142–143. [Google Scholar] [CrossRef]
- Georgiadis, D.; Dive, V.; Yiotakis, A. Synthesis and comparative study on the reactivity of peptidyl-type phosphinic esters. Intramolecular effects in the alkaline and acidic cleavage of methyl β-carboxyphosphinates. J. Org. Chem. 2001, 66, 6604–6610. [Google Scholar] [CrossRef]
- Reiter, L.A.; Jones, B.P. Amide-assisted hydrolysis of β-carboxamido-substituted phosphinic acid esters. J. Org. Chem. 1997, 62, 2808–2812. [Google Scholar] [CrossRef]
- Nasopoulou, M.; Matziari, M.; Dive, V.; Yiotakis, A. Chemoselective protection of solid-phase compatible Fmoc-phosphinic building blocks. J. Org. Chem. 2006, 71, 9525–9527. [Google Scholar] [CrossRef]
- Georgiadis, D.; Matziari, M.; Yiotakis, A. A highly efficient method for the preparation of phosphinic pseudodipeptidic blocks suitably protected for solid-phase peptide synthesis. Tetrahedron 2001, 57, 3471–3478. [Google Scholar] [CrossRef]
- Buchardt, J.; Ferreras, M.; Krog-Jensen, C.; Delaissé, J.-M.; Foged, N.T.; Meldal, M. Phosphinic peptide matrix metalloproteinase-9 inhibitors by solid-phase synthesis using a building block approach. Chem. Eur. J. 1999, 5, 2877–2884. [Google Scholar] [CrossRef]
- Bhowmick, M.; Sappidi, R.R.; Fields, G.B.; Lepore, S.D. Efficient synthesis of Fmoc-protected phosphinic pseudodipeptides: Building blocks for the synthesis of matrix metalloproteinase inhibitors. Biopolymers 2011, 96, 1–3. [Google Scholar] [CrossRef]
- Jiráček, J.; Yiotakis, A.; Vincent, B.; Lecoq, A.; Nicolau, A.; Checler, F.; Dive, V. Development of highly potent and selective phosphinic peptide inhibitors of zinc endopeptidase 24-15 using combinatorial chemistry. J. Biol. Chem. 1995, 270, 21701–21706. [Google Scholar]
- Jiráček, J.; Yiotakis, A.; Vincent, B.; Checler, F.; Dive, V. Development of the first potent and selective inhibitor of the zinc endopeptidase neurolysin using a systematic approach based on combinatorial chemistry of phosphinic peptides. J. Biol. Chem. 1996, 271, 19606–19611. [Google Scholar] [CrossRef]
- Dive, V.; Cotton, J.; Yiotakis, A.; Michaud, A.; Vassiliou, S.; Jiráček, J.; Vazeux, G.; Chauvet, M.-T.; Cuniasse, P.; Corvol, P. RXP 407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc. Natl. Acad. Sci. USA 1999, 96, 4330–4335. [Google Scholar]
- Buchardt, J.; Schiødt, C.B.; Krog-Jensen, C.; Delaissé, J.-M.; Foged, N.T.; Meldal, M. Solid phase combinatorial library of phosphinic peptides for discovery of matrix metalloproteinase inhibitors. J. Comb. Chem. 2000, 2, 624–638. [Google Scholar] [CrossRef]
- Campagne, J.-M.; Coste, J.; Guillou, L.; Heitz, A.; Jouin, P. Solid phase synthesis of phosphinic peptides. Tetrahedron Lett. 1993, 34, 4181–4184. [Google Scholar] [CrossRef]
- Mucha, A.; Pawełczak, M.; Hurek, J.; Kafarski, P. Synthesis and activity of phosphinic tripeptide inhibitors of cathepsin C. Bioorg. Med. Chem. Lett. 2004, 14, 3113–3116. [Google Scholar] [CrossRef]
- Matziari, M.; Nasopoulou, M.; Yiotakis, A. Active methylene phosphinic peptides: A new diversification approach. Org. Lett. 2006, 8, 2317–2319. [Google Scholar] [CrossRef]
- Gurulingappa, H.; Buckhaults, P.; Kumar, S.K.; Kinzler, K.W.; Vogelstein, B.; Khan, S.R. Design, synthesis and evaluation of new RDP inhibitors. Tetrahedron Lett. 2003, 44, 1871–1873. [Google Scholar] [CrossRef]
- Gurulingappa, H.; Buckhalts, P.; Kinzler, K.W.; Vogelstein, B.; Khan, S.R. Synthesis and evaluation of aminophosphinic acid derivatives as inhibitors of renal dipeptidase. Bioorg. Med. Chem. Lett. 2004, 14, 3531–3533. [Google Scholar] [CrossRef]
- Matziari, M.; Georgiadis, D.; Dive, V.; Yiotakis, A. Convenient synthesis and diversification of dehydroalaninyl phosphinic peptide analogues. Org. Lett. 2001, 3, 659–662. [Google Scholar] [CrossRef]
- Georgiadis, D.; Cuniasse, P.; Cotton, J.; Yiotakis, A.; Dive, V. Structural determinants of RXPA380, a potent and highly selective inhibitor of the angiotensin-converting enzyme C-domain. Biochemistry 2004, 43, 8048–8054. [Google Scholar] [CrossRef]
- Matziari, M.; Beau, F.; Cuniasse, P.; Dive, V.; Yiotakis, A. Evaluation of P1'-diversified phosphinic peptides leads to the development of highly selective inhibitors of MMP-11. J. Med. Chem. 2004, 47, 325–336. [Google Scholar] [CrossRef]
- Matziari, M.; Dellis, D.; Dive, V.; Yiotakis, A.; Samios, J. Conformational and solvation studies via computer simulation of the novel large scale diastereoselectively synthesized phosphinic MMP Inhibitor RXP03 diluted in selected solvents. J. Phys. Chem. B 2010, 114, 421–428. [Google Scholar] [CrossRef]
- Liu, X.; Hu, X.E.; Tian, X.; Mazur, A.; Ebetino, F.H. Enantioselective synthesis of phosphinyl peptidomimetics via an asymmetric Michael reaction of phosphinic acids with acrylate derivatives. J. Organomet. Chem. 2002, 646, 212–222. [Google Scholar] [CrossRef]
- Yamagishi, T.; Ichikawa, H.; Haruki, T.; Yokomatsu, T. Diastereoselective synthesis of α,β'-disubstituted aminomethyl(2-carboxyethyl)phosphinates as phosphinyl dipeptide isosteres. Org. Lett. 2008, 10, 4347–4350. [Google Scholar] [CrossRef]
- Yamagishi, T.; Tashiro, N.; Yokomatsu, T. Diastereoselective synthesis of the Leu-Pro type phosphinyl dipeptide isostere. J. Org. Chem. 2011, 76, 5472–5476. [Google Scholar] [CrossRef]
- Monbrun, J.; Dayde, B.; Cristau, H.-J.; Volle, J.-N.; Virieux, D.; Pirat, J.-L. Diastereoselective Michael addition of 2H-2-oxo-1,4,2-oxaza phosphinanes to olefins. Tetrahedron 2011, 67, 540–545. [Google Scholar] [CrossRef]
- Lämmerhofer, M.; Hebenstreit, D.; Gavioli, E.; Lindner, W.; Mucha, A.; Kafarski, P.; Wieczorek, P. High-performance liquid chromatographic enantiomer separation and determination of absolute configurations of phosphinic acid analogues of dipeptides and their α-aminophosphinic acid precursors. Tetrahedron: Asymmetry 2003, 14, 2557–2565. [Google Scholar] [CrossRef]
- Preinerstorfer, B.; Lubda, D.; Mucha, A.; Kafarski, P.; Lindner, W.; Lämmerhofer, M. Stereoselective separations of chiral phosphinic acid pseudodipeptides by CEC using silica monoliths modified with an anion-exchange-type chiral selector. Electrophoresis 2006, 27, 4312–4320. [Google Scholar] [CrossRef]
- Mucha, A.; Lämmerhofer, M.; Lindner, W.; Pawełczak, M.; Kafarski, P. Individual stereoisomers of phosphinic dipeptide inhibitor of leucine aminopeptidase. Bioorg. Med. Chem. Lett. 2008, 18, 1550–1554. [Google Scholar] [CrossRef]
- Ellsworth, B.A.; Tom, N.J.; Bartlett, P.A. Synthesis and evaluation of inhibitors of bacterial D-alanine: D-alanine ligases. Chem. Biol. 1996, 3, 37–44. [Google Scholar] [CrossRef]
- Fan, C.; Park, I.-S.; Walsh, C.T.; Knox, J.R. D-Alanine:D-alanine ligase: Phosphonate and phosphinate intermediates with wild type and the Y216F mutant. Biochemistry 1997, 36, 2531–2538. [Google Scholar] [CrossRef]
- Kuzin, A.P.; Sun, T.; Jorczak-Baillass, J.; Healy, V.L.; Walsh, C.T.; Knox, J.R. Enzymes of vancomycin resistance: The structure of D-alanine-D-lactate ligase of naturally resistant Leuconostoc mesenteroides. Structure 2000, 8, 463–470. [Google Scholar] [CrossRef]
- Roper, D.I.; Huyton, T.; Vagin, A.; Dodson, G. The molecular basis of vancomycin resistance in clinically relevant Enterococci: Crystal structure of D-alanyl-D-lactate ligase (VanA). Proc. Natl. Acad. Sci. USA 2000, 97, 8921–8925. [Google Scholar] [CrossRef]
- Gegnas, L.D.; Waddell, S.T.; Chabin, R.M.; Reddy, S.; Wong, K.K. Inhibitors of the bacterial cell wall biosynthesis enzyme Mur D. Bioorg. Med. Chem. Lett. 1998, 8, 1643–1648. [Google Scholar] [CrossRef]
- trancar, K.; Blanot, D.; Gobec, S. Design, synthesis and structure-activity relationships of new phosphinate inhibitors of MurD. Bioorg. Med. Chem. Lett. 2006, 16, 343–348. [Google Scholar] [CrossRef]
- trancar, K.; Boniface, A.; Blanot, D.; Gobec, S. Phosphinate inhibitors of UDP-N-acetylmuramoyl-L-alanyl-D-glutamate: L-Lysine ligase (MurE). Arch. Pharm. Chem. Life Sci. 2007, 340, 127–134. [Google Scholar] [CrossRef]
- Shomura, Y.; Hinokuchi, E.; Ikeda, H.; Senoo, A.; Takahashi, Y.; Saito, J.; Komori, H.; Shibata, N.; Yonetani, Y.; Higuchi, Y. Structural and enzymatic characterization of BacD, an L-amino acid dipeptide ligase from Bacillus subtilis. Protein Sci. 2012, 21, 707–716. [Google Scholar] [CrossRef]
- Chen, H.; Roques, B.P.; Fournié-Zaluski, M.-C. Design of the first highly potent and selective aminopeptidase N (EC 3.4.11.2) inhibitor. Bioorg. Med. Chem. Lett. 1999, 9, 1511–1516. [Google Scholar] [CrossRef]
- Yang, K.-W.; Golich, F.C.; Sigdel, T.K.; Crowder, M.W. Phosphinate, sulfonate, and sulfonamidate dipeptides as potential inhibitors of Escherichia coli aminopeptidase N. Bioorg. Med. Chem. Lett. 2005, 15, 5150–5153. [Google Scholar] [CrossRef]
- Pícha, J.; Liboska, R.; Buděšínský, M.; Jiráček, J.; Pawełczak, M.; Mucha, A. Unusual activity pattern of leucine aminopeptidase inhibitors based on phosphorus containing derivatives of methionine and norleucine. J. Enz. Inhib. Med. Chem. 2011, 26, 155–161. [Google Scholar] [CrossRef]
- Mucha, A.; Drąg, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef]
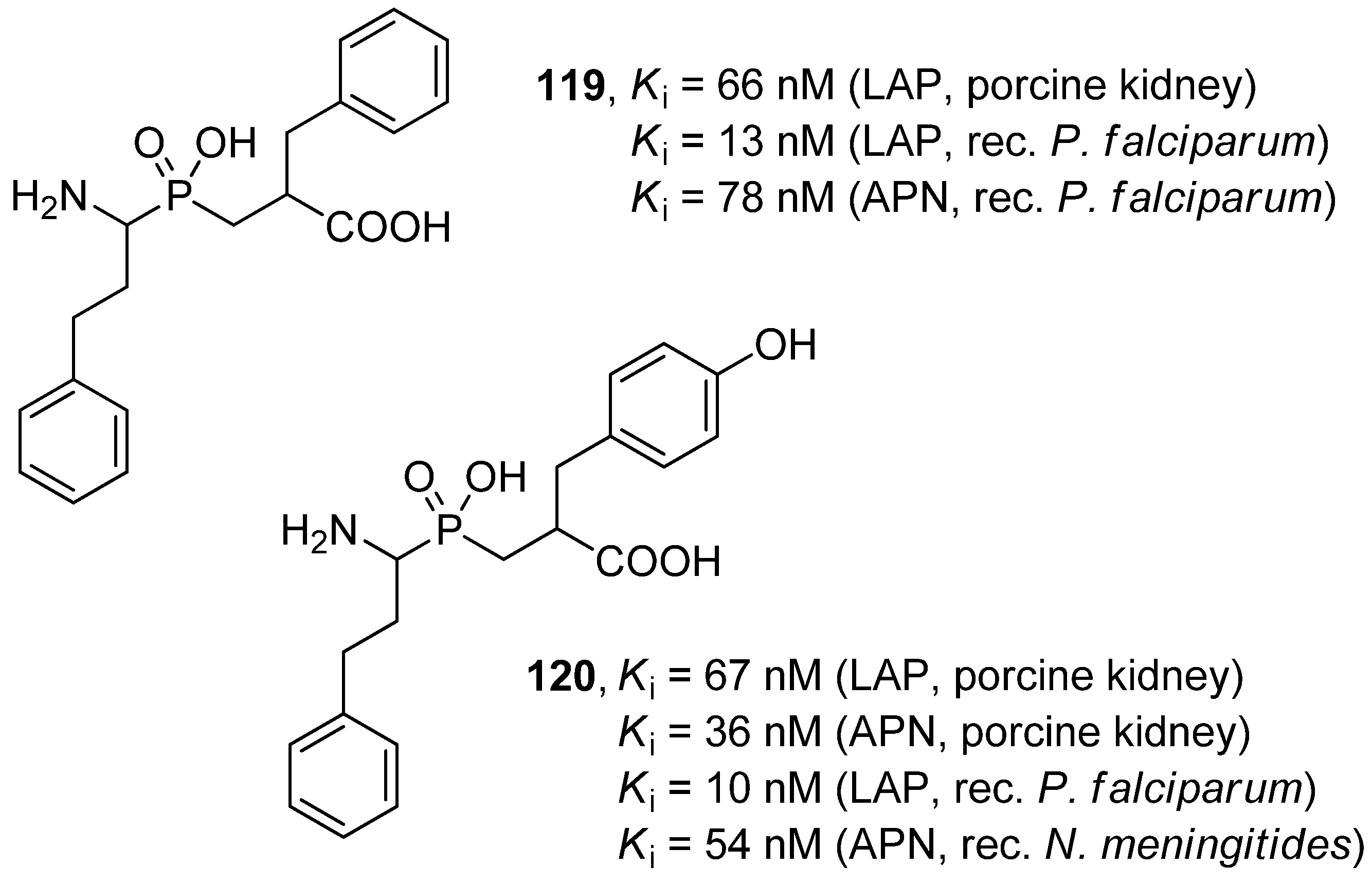
- Skinner-Adams, T.S.; Lowther, J.; Teuscher, F.; Stack, C.M.; Grembecka, J.; Mucha, A.; Kafarski, P.; Trenholme, K.R.; Dalton, J.P.; Gardiner, D.L. Identification of phosphinate dipeptide analog inhibitors directed against the Plasmodium falciparum M17 leucine aminopeptidase as lead antimalarial compounds. J. Med. Chem. 2007, 50, 6024–6031. [Google Scholar]
- Skinner-Adams, T.S.; Stack, C.M.; Trenholme, K.R.; Brown, C.L.; Grembecka, J.; Lowther, J.; Mucha, A.; Drąg, M.; Kafarski, P.; McGowan, S.; et al. Plasmodium falciparum neutral aminopeptidases: New targets for anti-malarials. Trends Biochem. Sci. 2010, 35, 53–61. [Google Scholar] [CrossRef]
- Węglarz-Tomczak, E.; Poręba, M.; Byzia, A.; Berlicki, Ł.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Drąg, M.; Mucha, A. An integrated approach to the ligand binding specificity of Neisseria meningitidis M1 alanine aminopeptidase by fluorogenic substrate profiling, inhibitory studies and molecular modeling. Biochimie 2012. [Google Scholar] [CrossRef]
- McGowan, S.; Porter, C.J.; Lowther, J.; Stack, C.M.; Golding, S.J.; Skinner-Adams, T.S.; Trenholme, K.R.; Teuscher, F.; Donnelly, S.M.; Grembecka, J.; et al. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc. Natl. Acad. Sci. USA 2009, 106, 2537–2542. [Google Scholar]
- McGowan, S.; Oellig, C.A.; Birru, W.A.; Caradoc-Davies, T.T.; Stack, C.M.; Lowther, J.; Skinner-Adams, T.; Mucha, A.; Kafarski, P.; Grembecka, J.; et al. Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases. Proc. Natl. Acad. Sci. USA 2010, 107, 2449–2454. [Google Scholar]
- Willemse, J.L.; Heylen, E.; Nesheim, M.E.; Hendriks, D.F. Carboxypeptidase U (TAFIa): A new drug target for fibrinolytic therapy? J. Thromb. Haemost. 2009, 7, 1962–1971. [Google Scholar] [CrossRef]
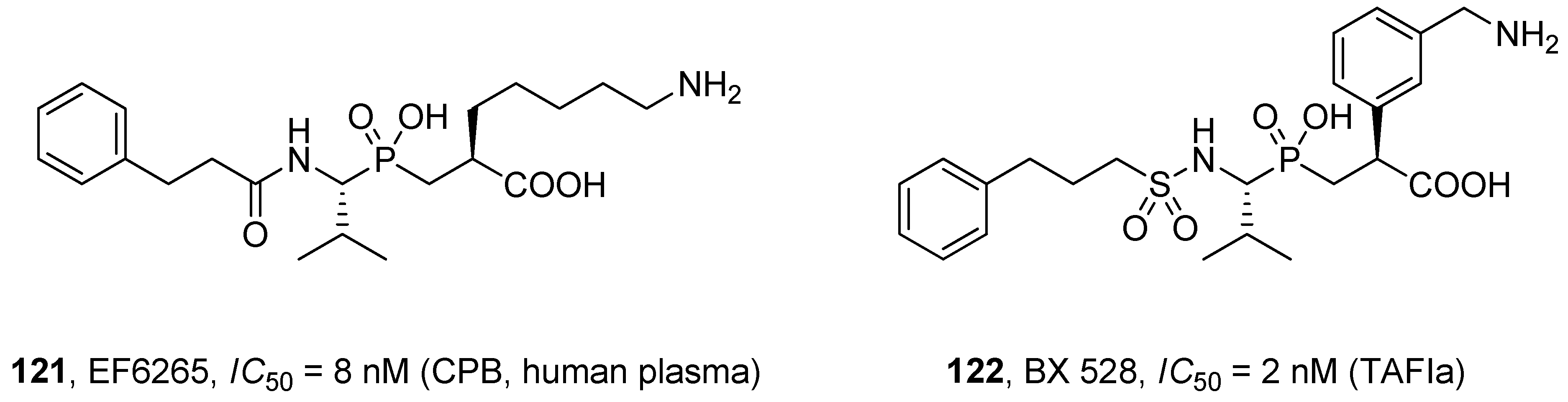
- Suzuki, K.; Muto, Y.; Fushihara, K.; Kanemoto, K.; Iida, H.; Sato, E.; Kikuchi, C.; Matsushima, T.; Kato, E.; Nomoto, M.; et al. Enhancement of fibrinolysis by EF6265 [(S)-7-amino-2-[[[(R)-2-methyl-1-(3-phenylpropanoylamino)propyl]hydroxyphosphinoyl]methyl] hepta-noic acid], a specific inhibitor of plasma carboxypeptidase B. J. Pharmacol. Exp. Ther. 2004, 309, 607–615. [Google Scholar] [CrossRef]
- Wang, Y.X.; Zhao, L.; Nagashima, M.; Vincelette, J.; Sukovich, D.; Li, W.; Subramanyam, B.; Yuan, S.; Emayan, K.; Islam, I.; et al. A novel inhibitor of activated thrombin-activatable fibrinolysis inhibitor (TAFIa)-Part I: Pharmacological characterization. Thromb. Haemost. 2007, 97, 45–53. [Google Scholar]
- Wang, Y.X.; da Cunha, V.; Vincelette, J.; Zhao, L.; Nagashima, M.; Kawai, K.; Yuan, S.; Emayan, K.; Islam, I.; Hosoya, J.; et al. A novel inhibitor of activated thrombin activatable fibrinolysis inhibitor (TAFIa)-part II: Enhancement of both exogenous and endogenous fibrinolysis in animal models of thrombosis. Thromb. Haemost. 2007, 97, 54–61. [Google Scholar]
- Adler, M.; Buckman, B.; Bryant, J.; Chang, Z.; Chu, K.; Emayan, K.; Hrvatin, P.; Islam, I.; Morser, J.; Sukovich, D.; et al. Structures of potent selective peptide mimetics bound to carboxypeptidase B. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 149–157. [Google Scholar] [CrossRef]
- Georgiadis, D.; Vazeux, G.; Llorens-Cortes, C.; Yiotakis, A.; Dive, V. Potent and selective inhibition of zinc aminopeptidase A (EC 3.4.11.7, APA) by glutamyl aminophosphinic peptides: Importance of glutamyl aminophosphinic residue in the P1 position. Biochemistry 2000, 39, 1152–1155. [Google Scholar]
- Cummings, J.A.; Nguyen, T.T.; Fedorov, A.A.; Kolb, P.; Xu, C.; Fedorov, E.V.; Shoichet, B.K.; Barondeau, D.P.; Almo, S.C.; Raushel, F.M. Structure, Mechanism, And substrate profile for Sco3058: The closest bacterial homologue to human renal dipeptidase. Biochemistry 2010, 49, 611–622. [Google Scholar]
- Teuscher, F.; Lowther, J.; Skinner-Adams, T.S.; Spielmann, T.; Dixon, M.W.A.; Stack, C.M.; Donnelly, S.; Mucha, A.; Kafarski, P.; Vassiliou, S.; et al. The M18 aspartyl aminopeptidase of the human malaria parasite Plasmodium falciparum. J. Biol. Chem. 2007, 282, 30817–30826. [Google Scholar]
- Jozic, D.; Bourenkow, G.; Bartunik, H.; Scholze, H.; Dive, V.; Henrich, B.; Huber, R.; Bode, W.; Maskos, K. Crystal structure of the dinuclear zinc aminopeptidase PepV from Lactobacillus delbrueckii unravels its preference for dipeptides. Structure 2002, 10, 1097–1106. [Google Scholar] [CrossRef]
- Wu, Z.; Walsh, C.T. Phosphinate analogs of D-, D-dipeptides: Slow-binding inhibition and proteolysis protection of VanX, a D-, D-dipeptidase required for vancomycin resistance in Enterococcus faecium. Proc. Natl. Acad. Sci. USA 1995, 92, 11603–11607. [Google Scholar] [CrossRef]
- Walker, B.; Wharry, S.; Hamilton, R.J.; Martin, S.L.; Healy, A.; Walker, B.J. Asymmetric preference of serine proteases toward phosphonate and phosphinate esters. Biochem. Biophys. Res. Commun. 2000, 276, 1235–1239. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mucha, A. Synthesis and Modifications of Phosphinic Dipeptide Analogues. Molecules 2012, 17, 13530-13568. https://doi.org/10.3390/molecules171113530
Mucha A. Synthesis and Modifications of Phosphinic Dipeptide Analogues. Molecules. 2012; 17(11):13530-13568. https://doi.org/10.3390/molecules171113530
Chicago/Turabian StyleMucha, Artur. 2012. "Synthesis and Modifications of Phosphinic Dipeptide Analogues" Molecules 17, no. 11: 13530-13568. https://doi.org/10.3390/molecules171113530
APA StyleMucha, A. (2012). Synthesis and Modifications of Phosphinic Dipeptide Analogues. Molecules, 17(11), 13530-13568. https://doi.org/10.3390/molecules171113530