Chemical Architecture and Applications of Nucleic Acid Derivatives Containing 1,2,3-Triazole Functionalities Synthesized via Click Chemistry

{kind=link}
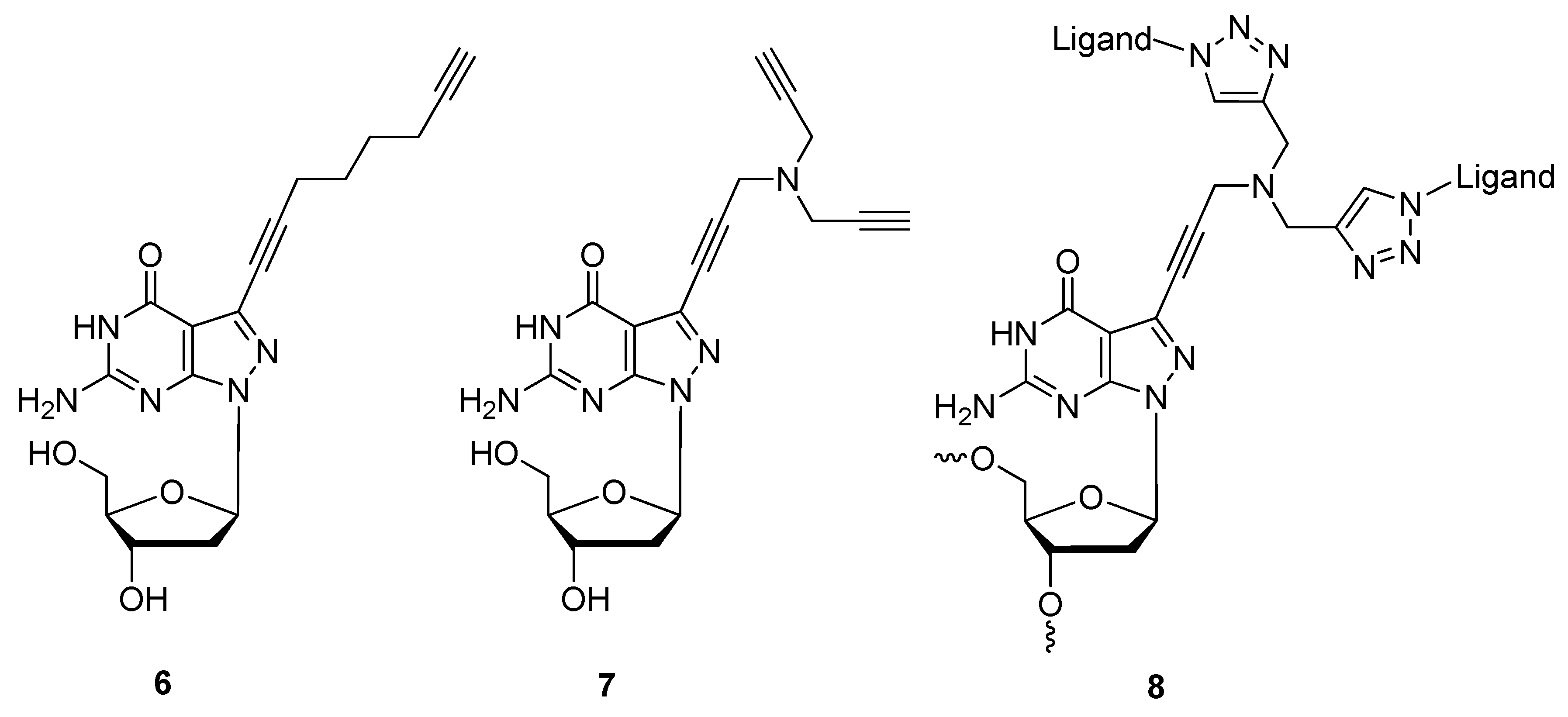{kind=link}
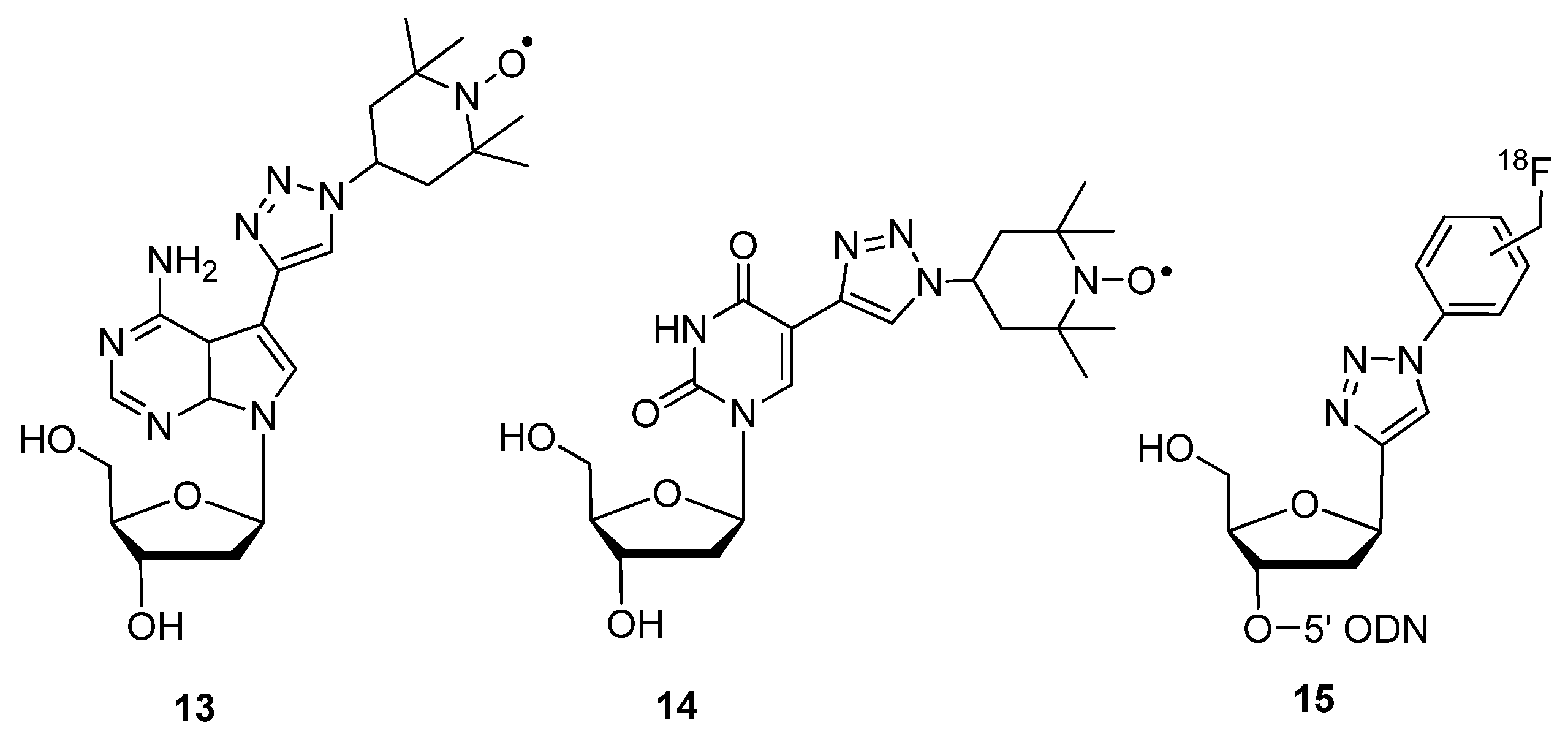{kind=link}
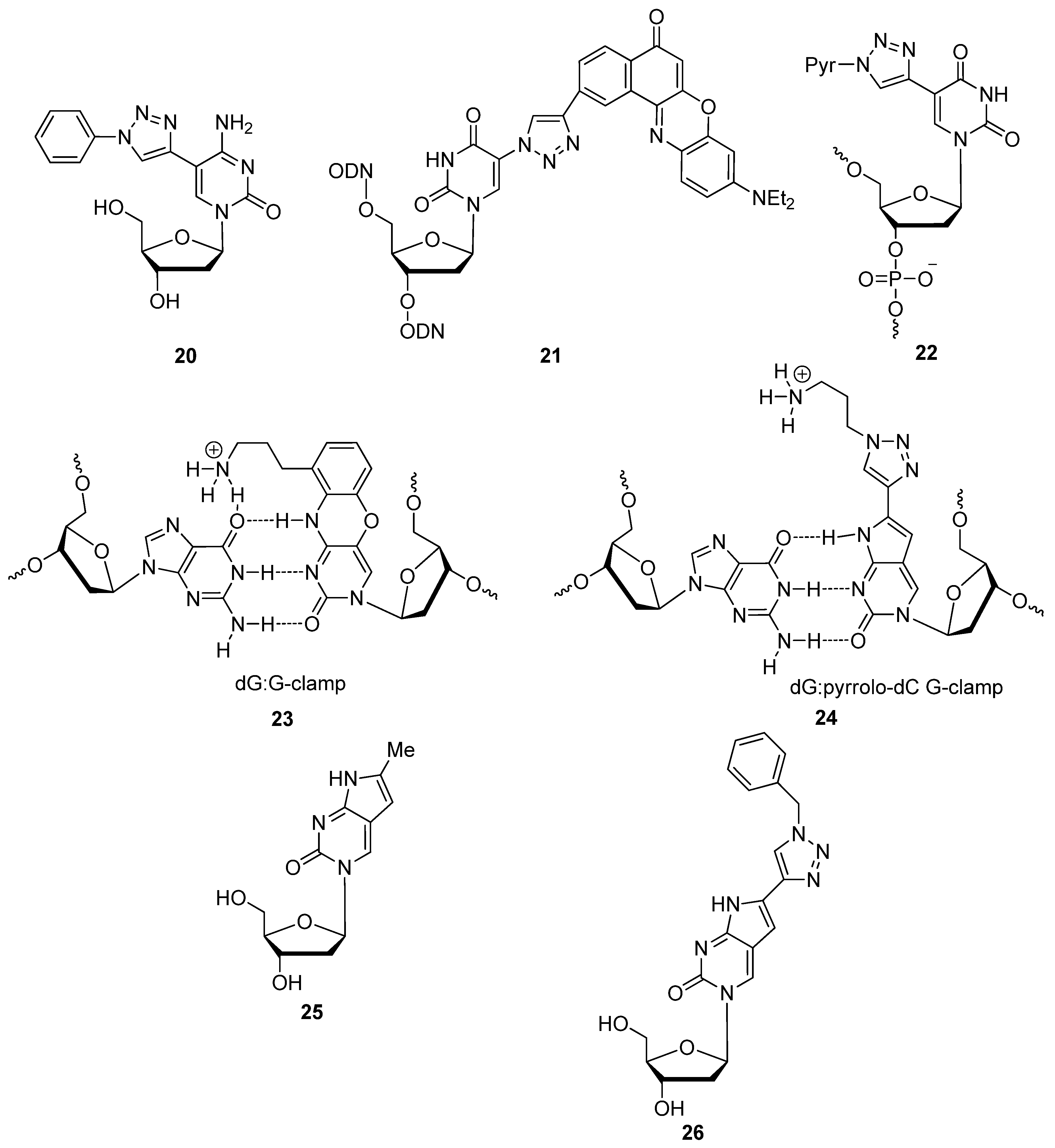{kind=link}
{kind=link}
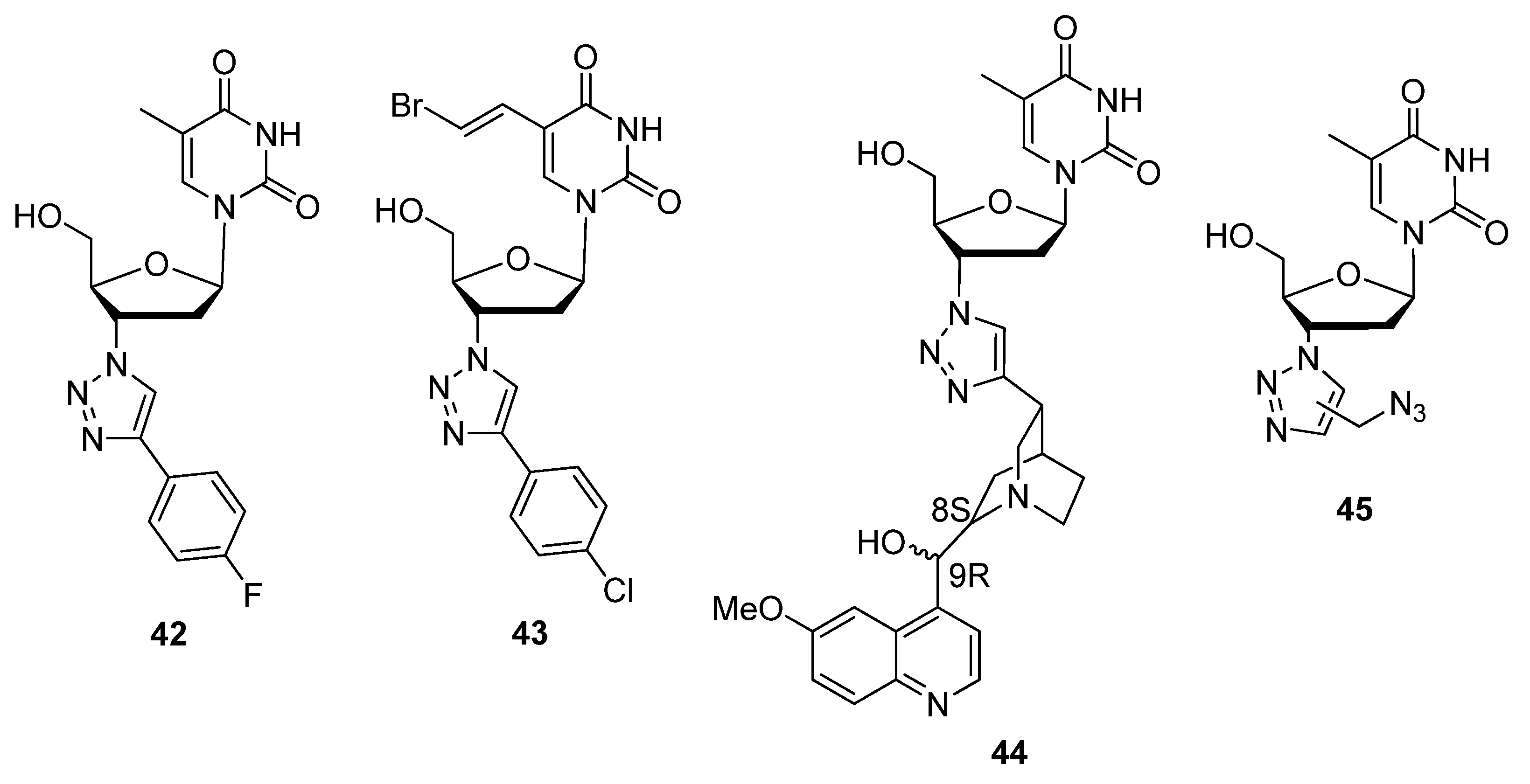{kind=link}
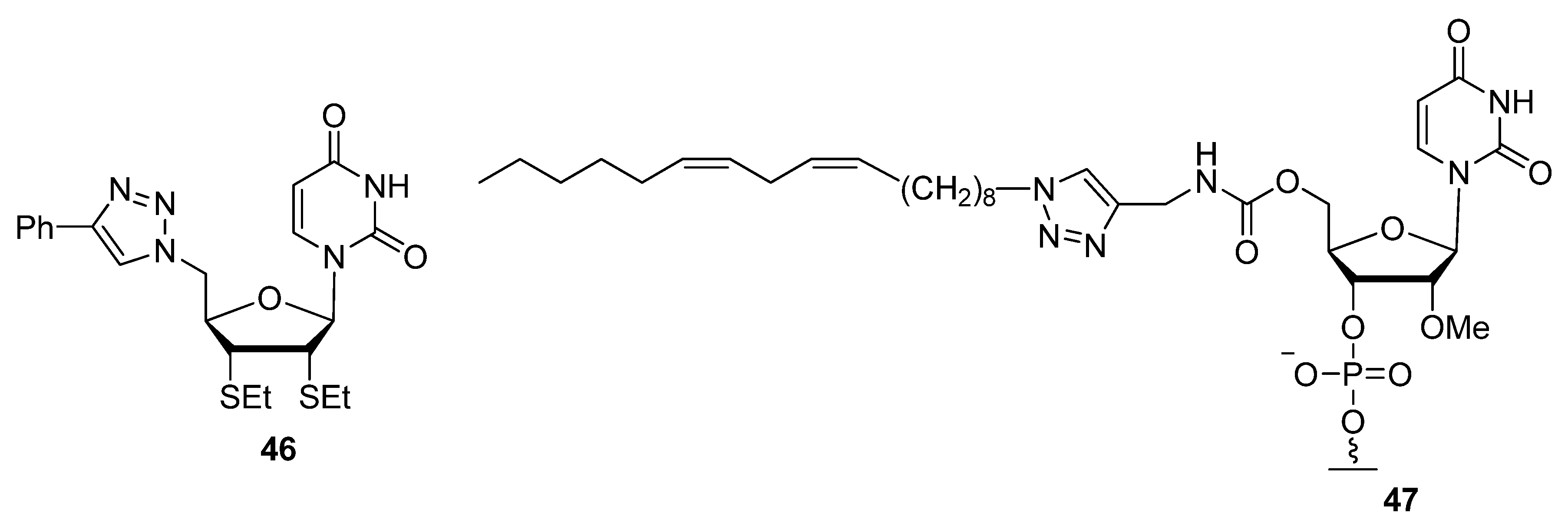{kind=link}
{kind=link}
{kind=link}
{kind=link}
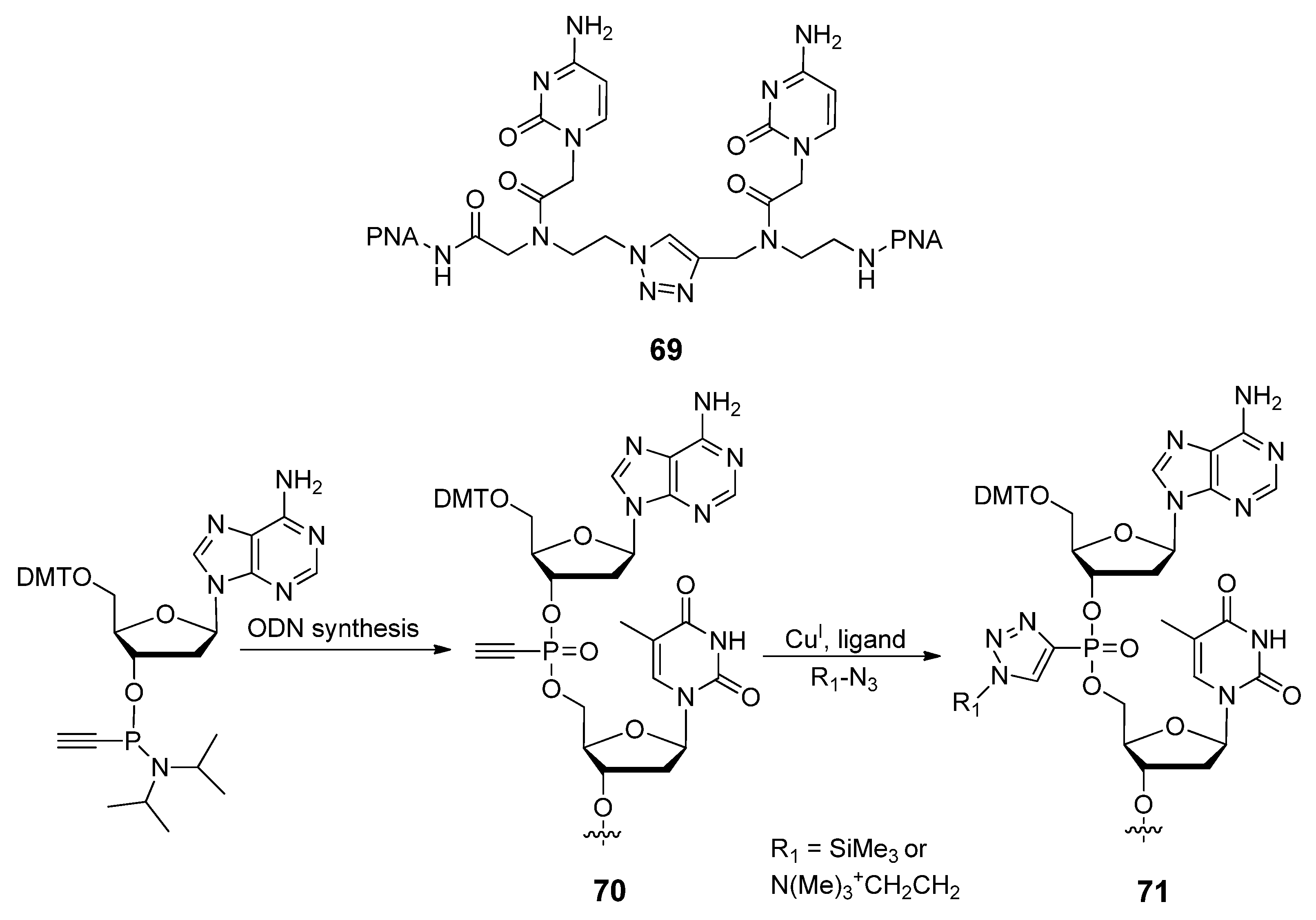{kind=link}
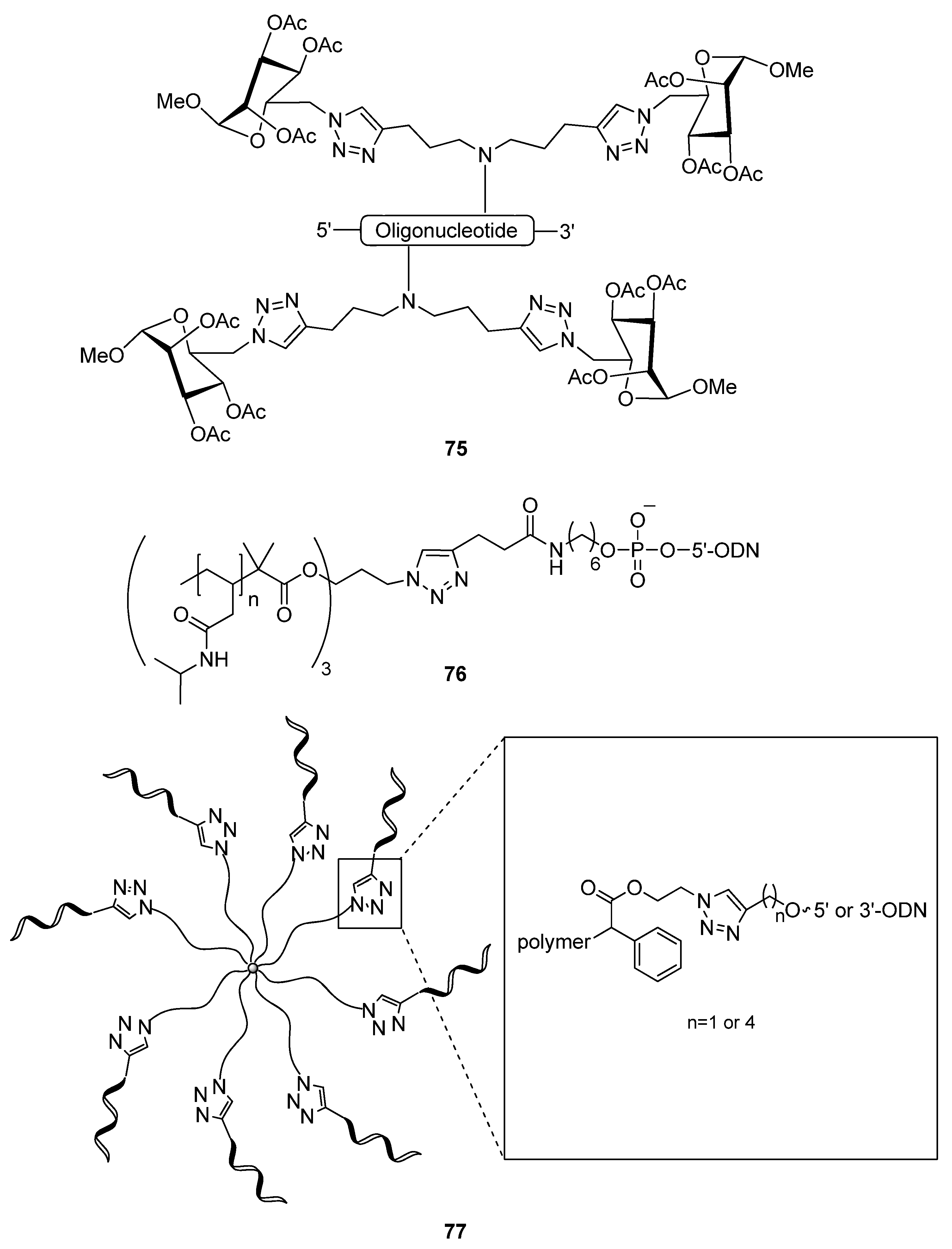{kind=link}
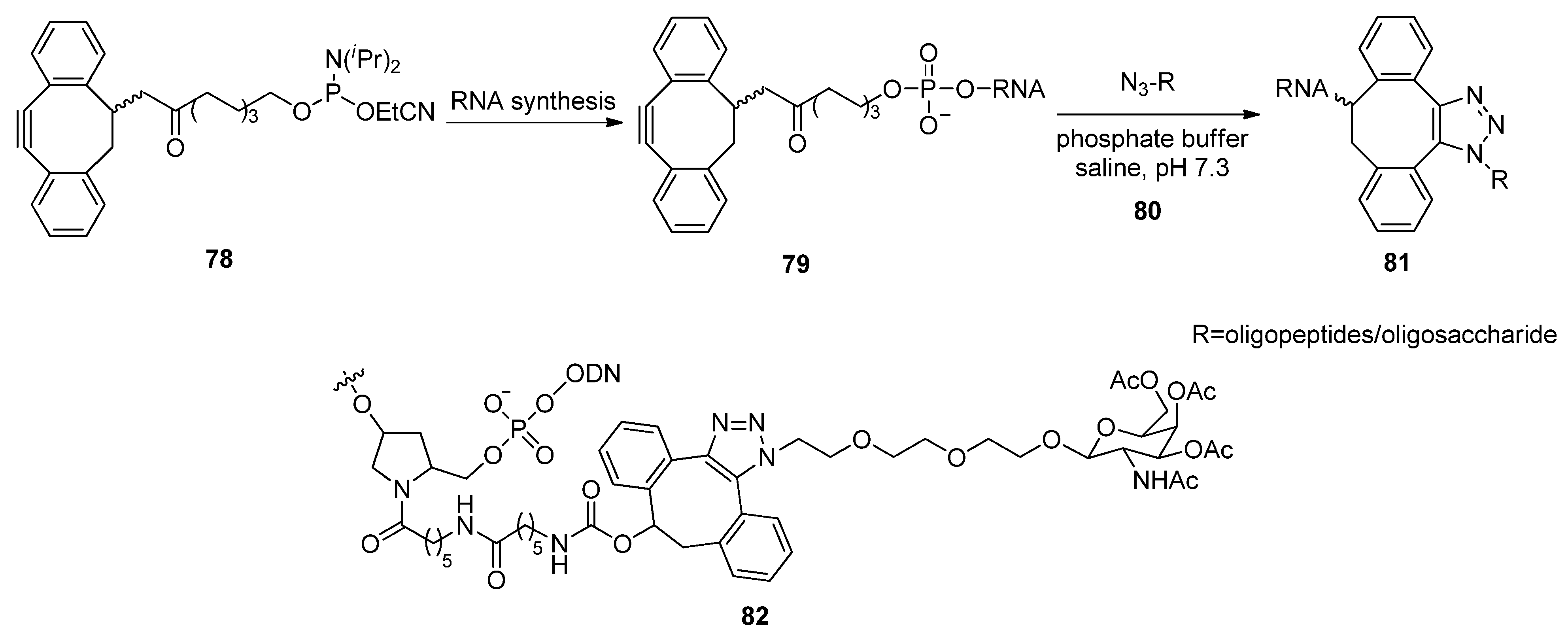{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
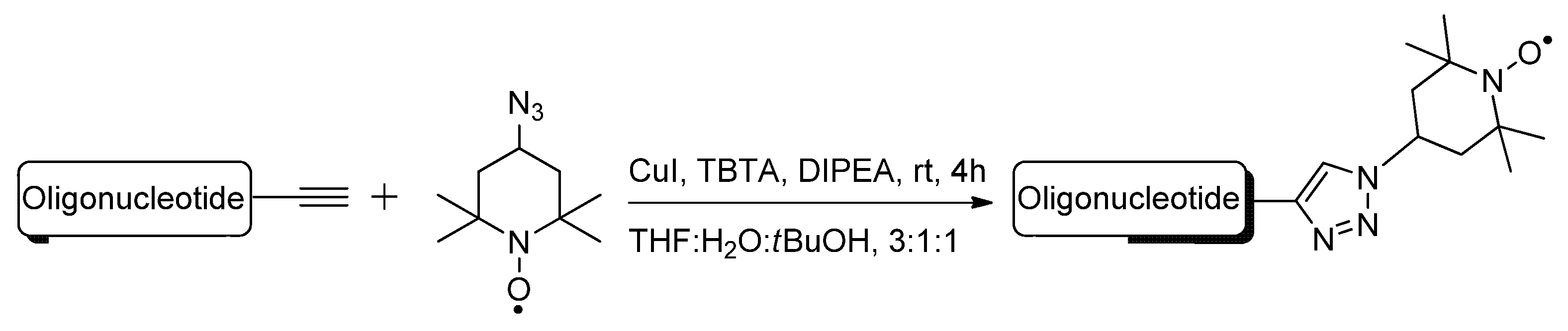{kind=link}
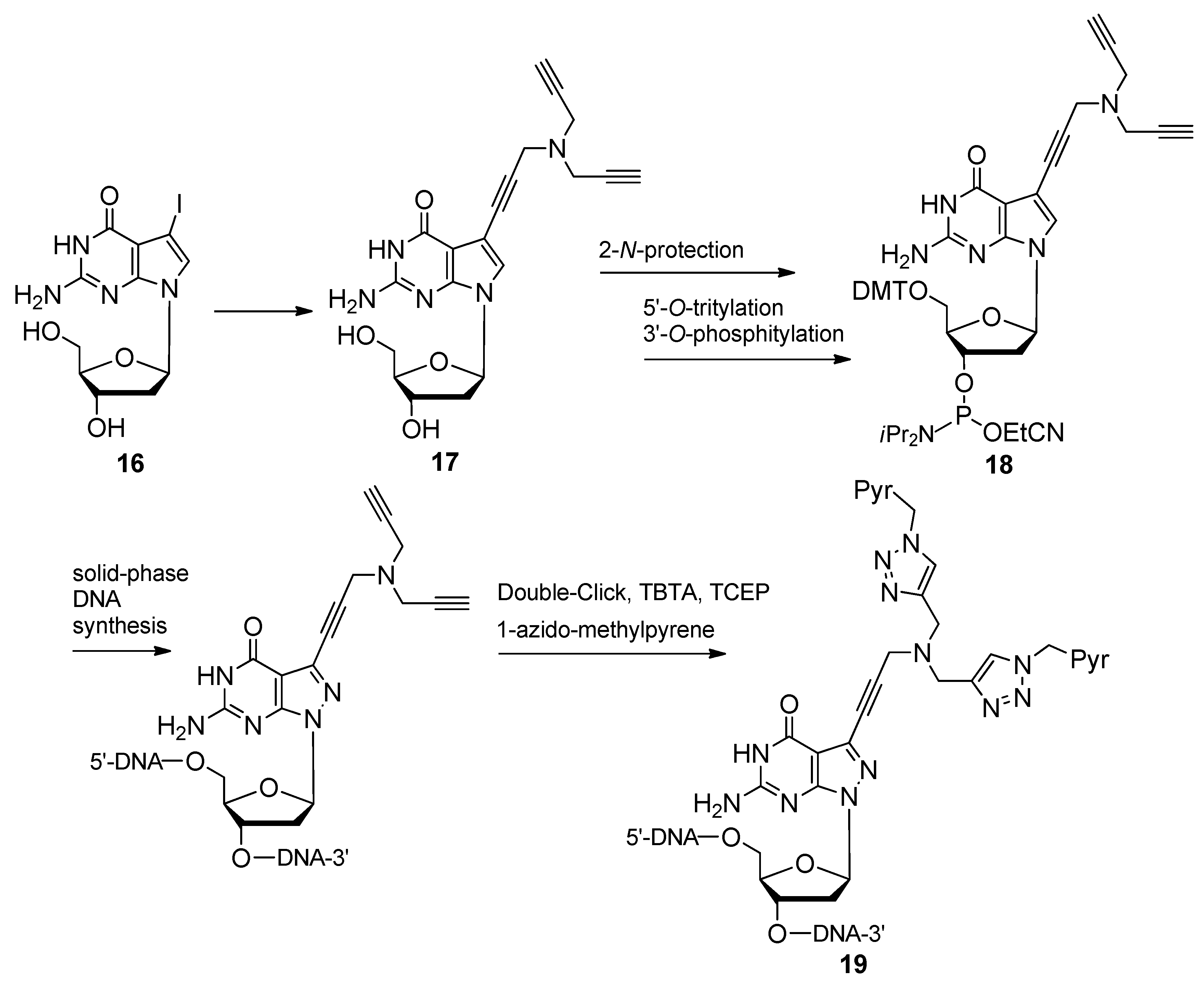{kind=link}
{kind=link}
{kind=link}
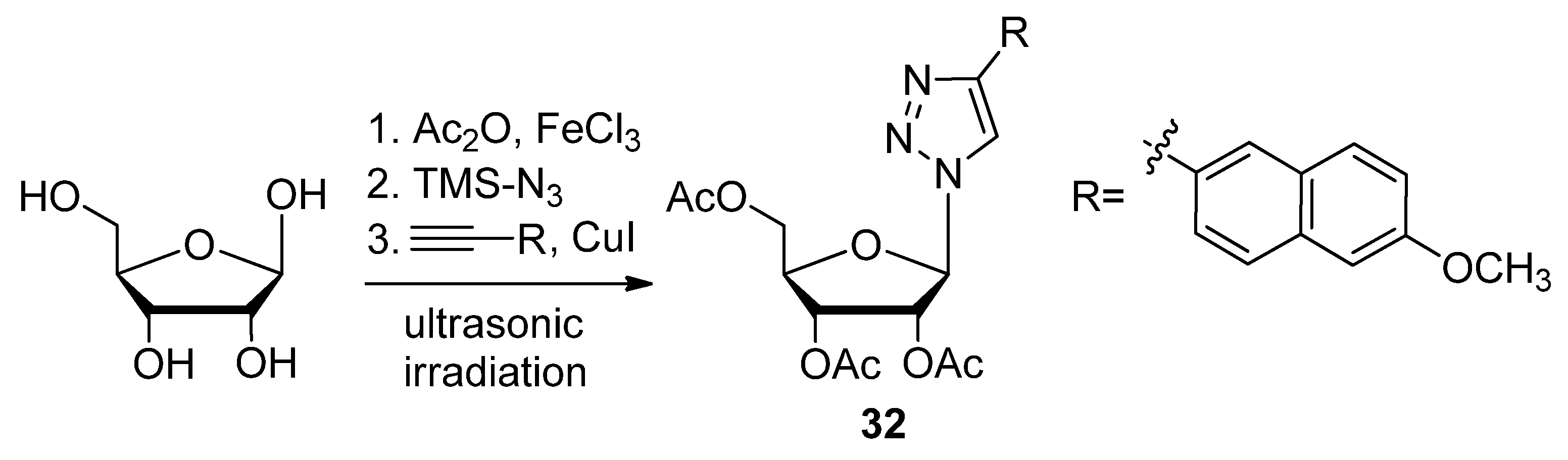{kind=link}
{kind=link}
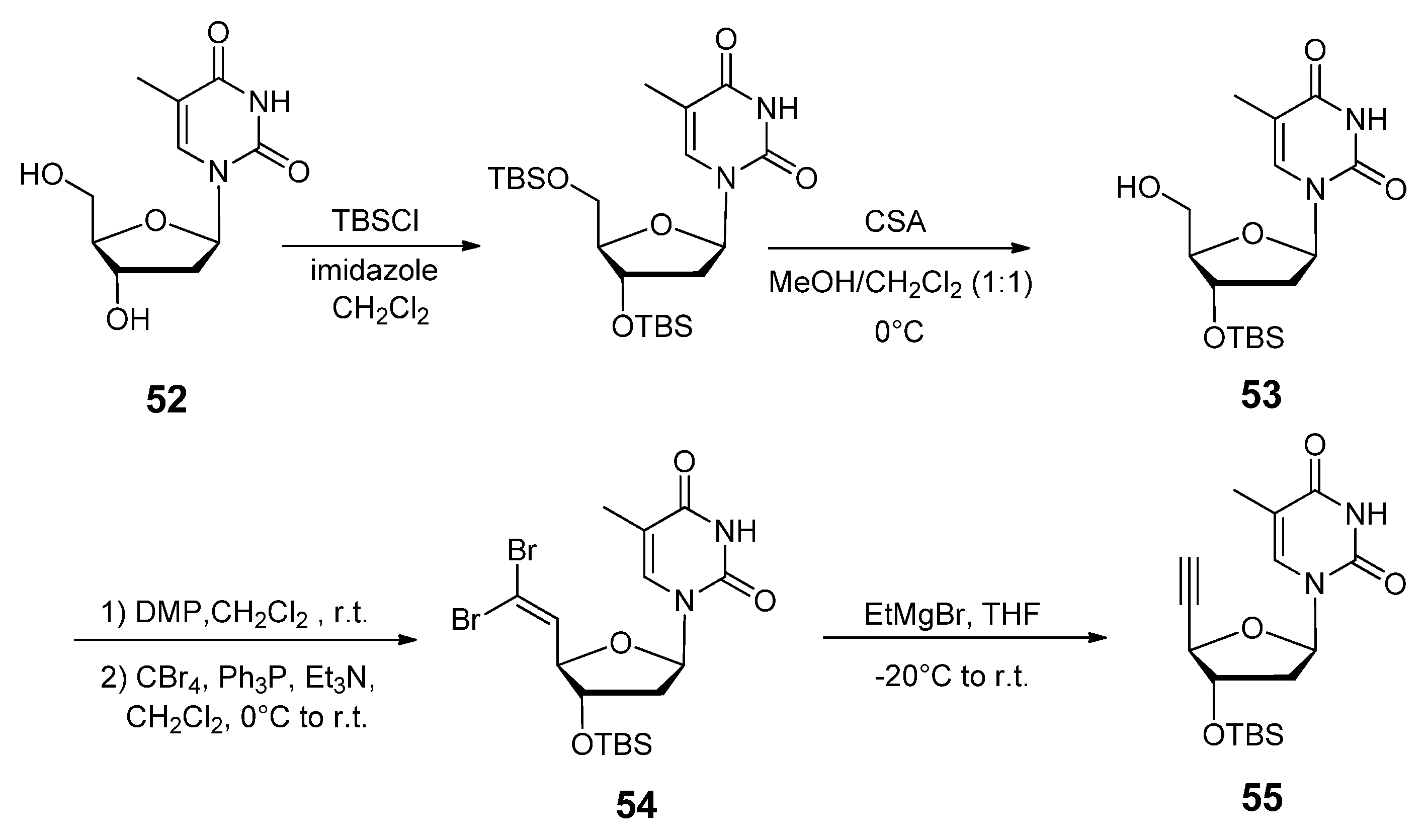{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Click Modification of Nucleic Acids
2.1. Nitrogenous Bases
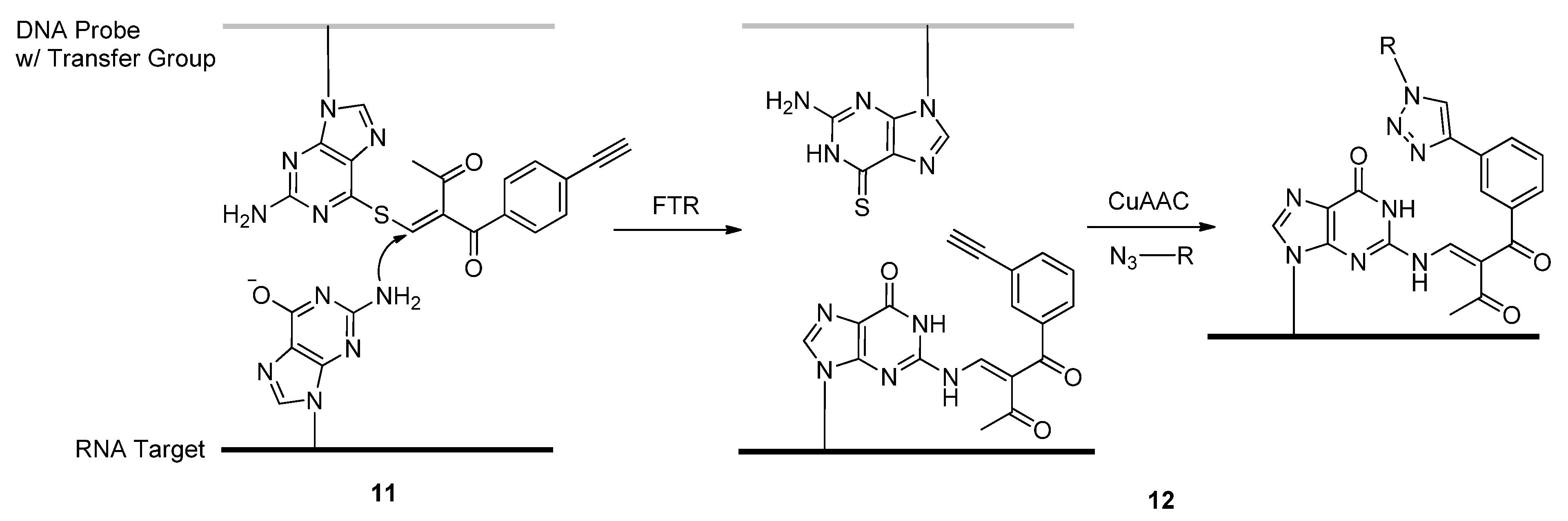
2.1.1. ASOs and siRNAs
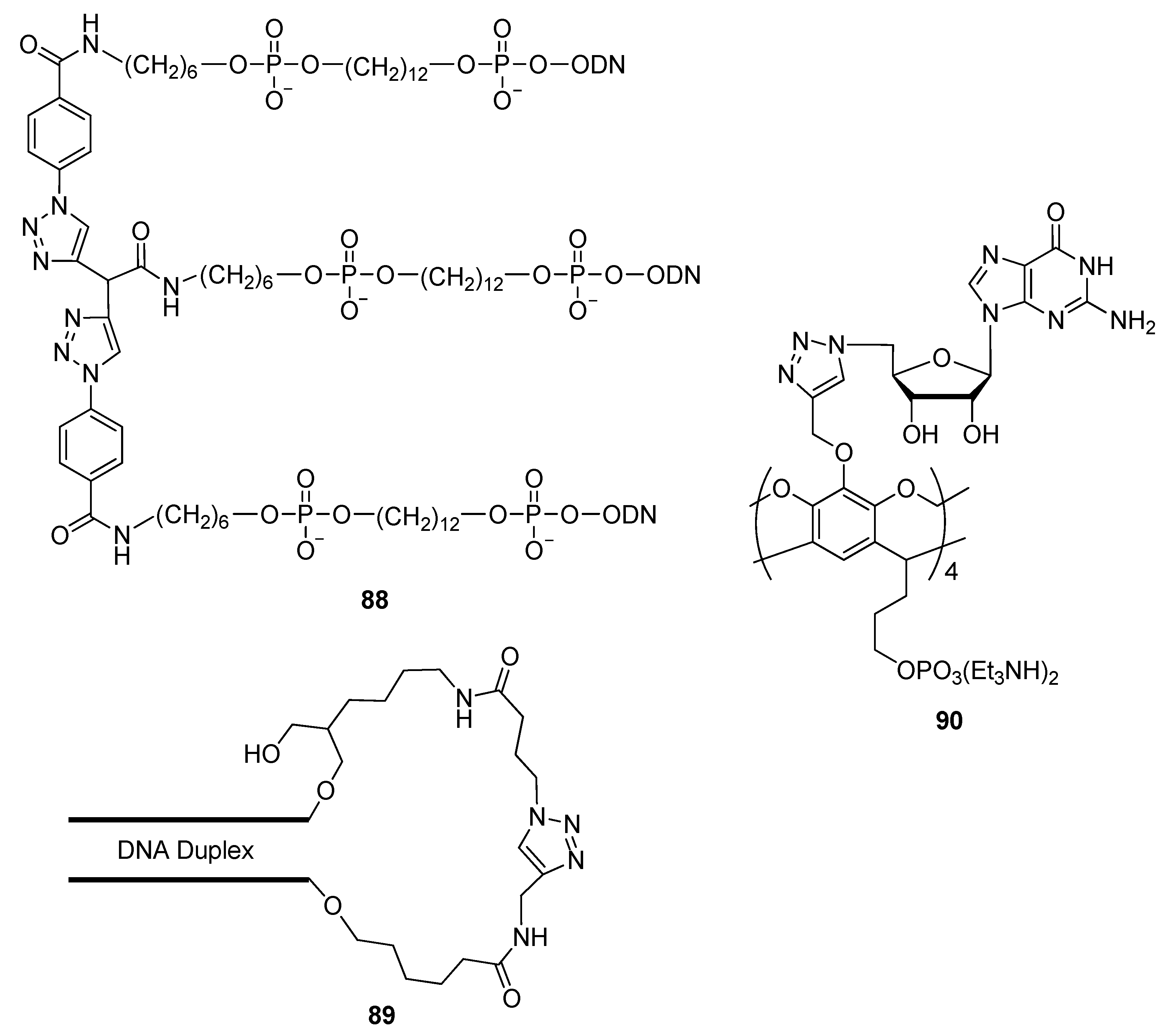
2.1.2. Oligonucleotide Based Nano-Materials
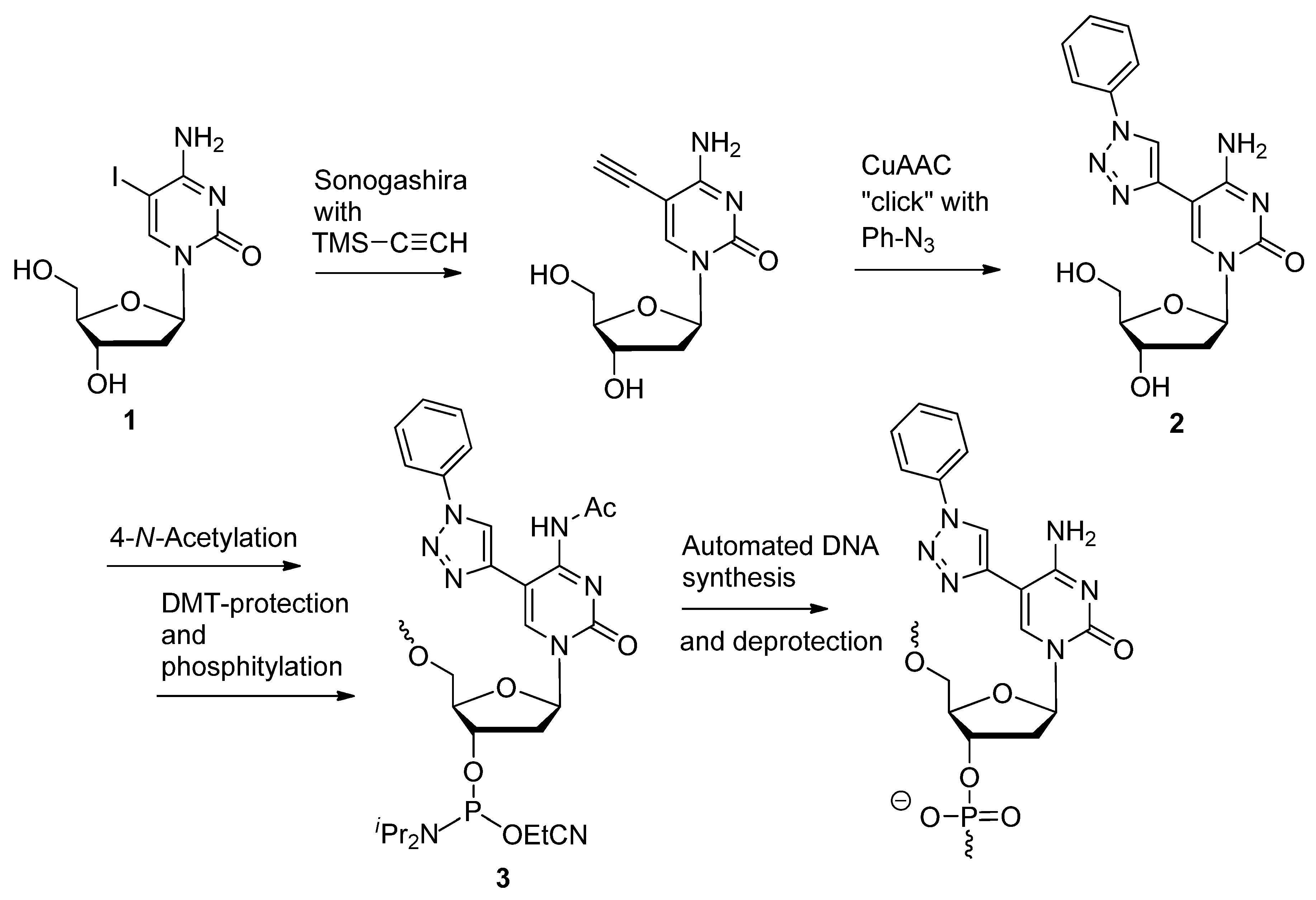


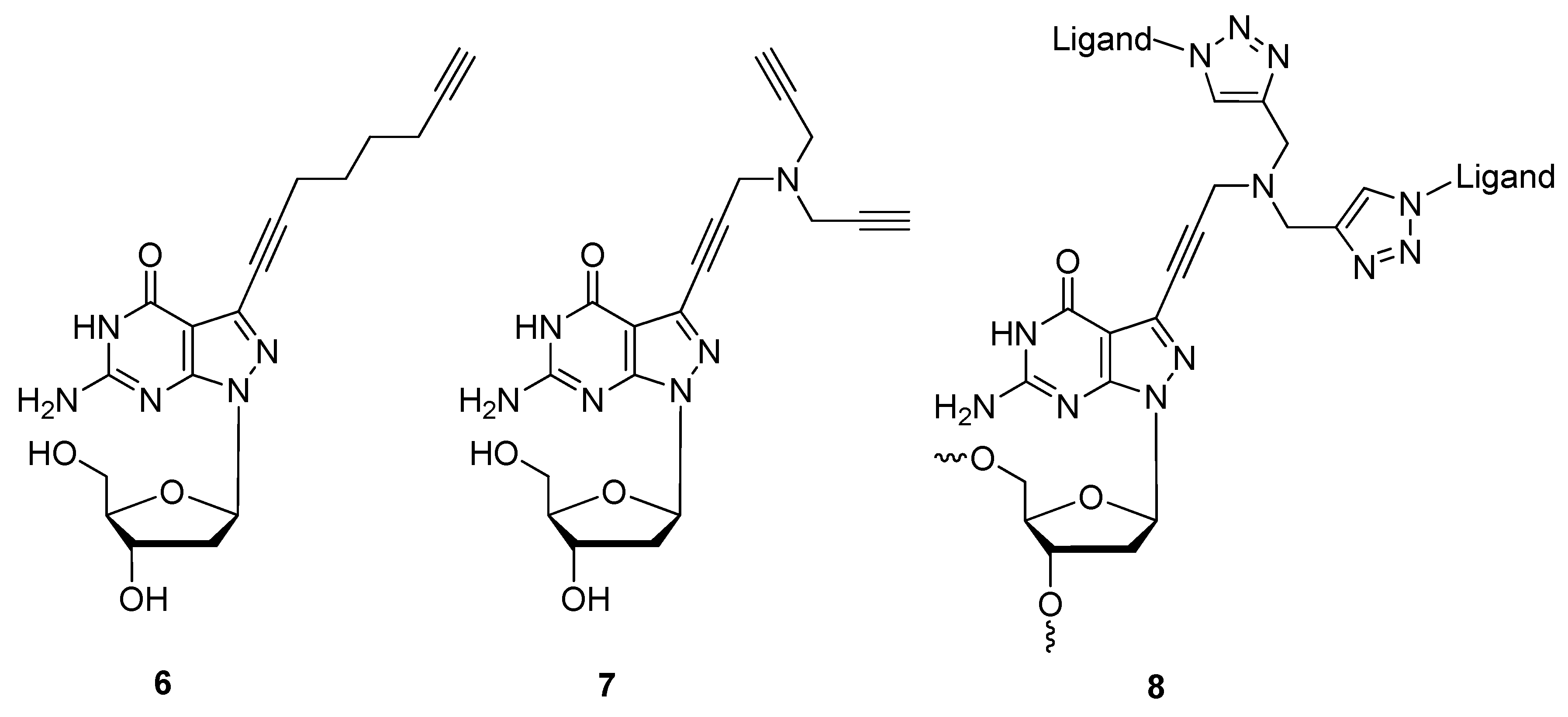
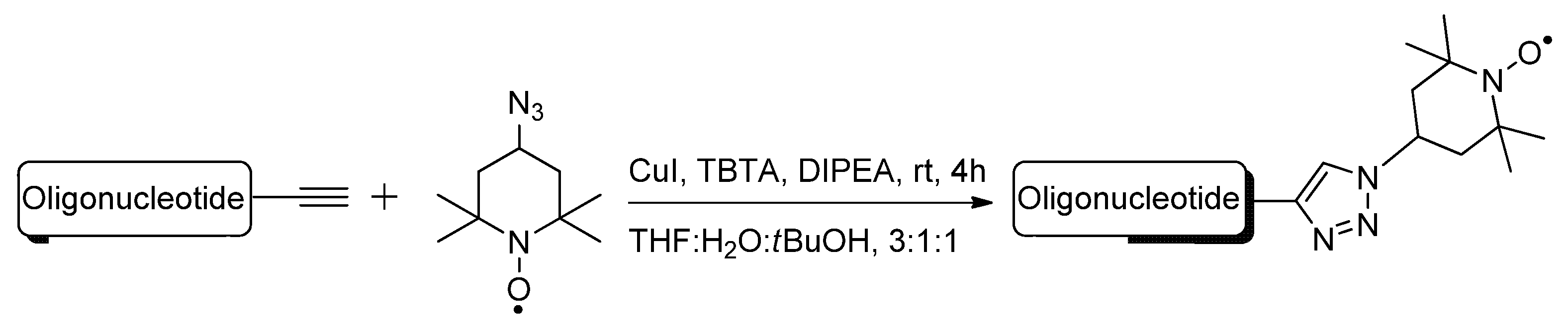
2.1.3. Structural Determination
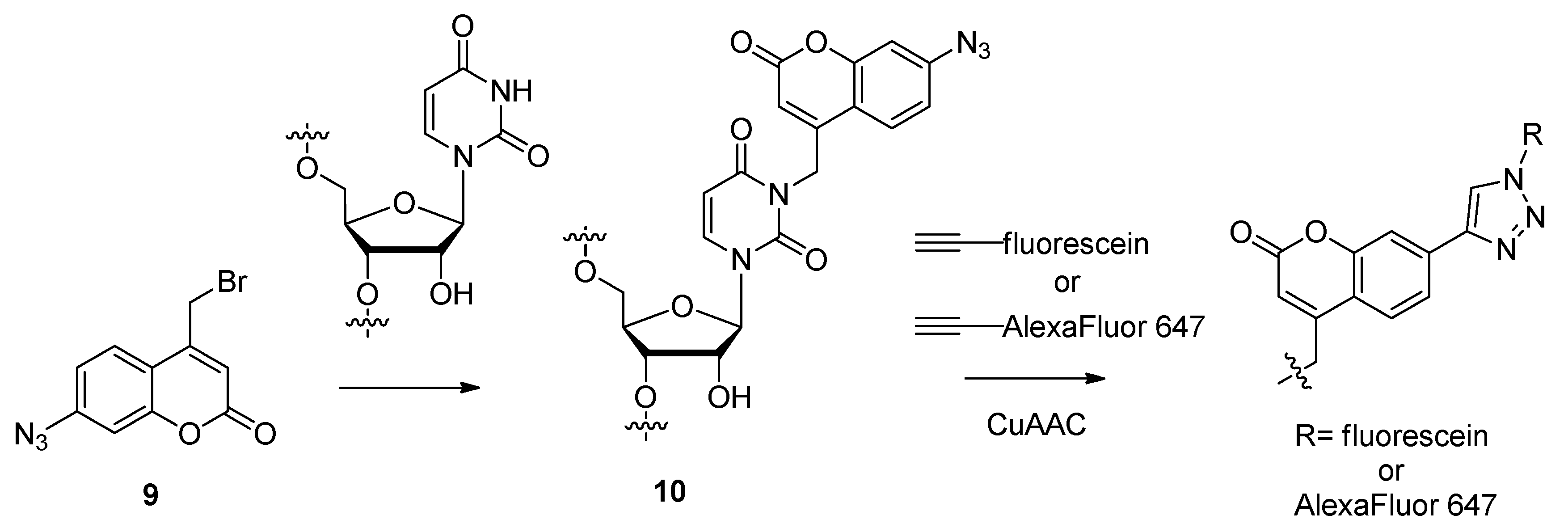
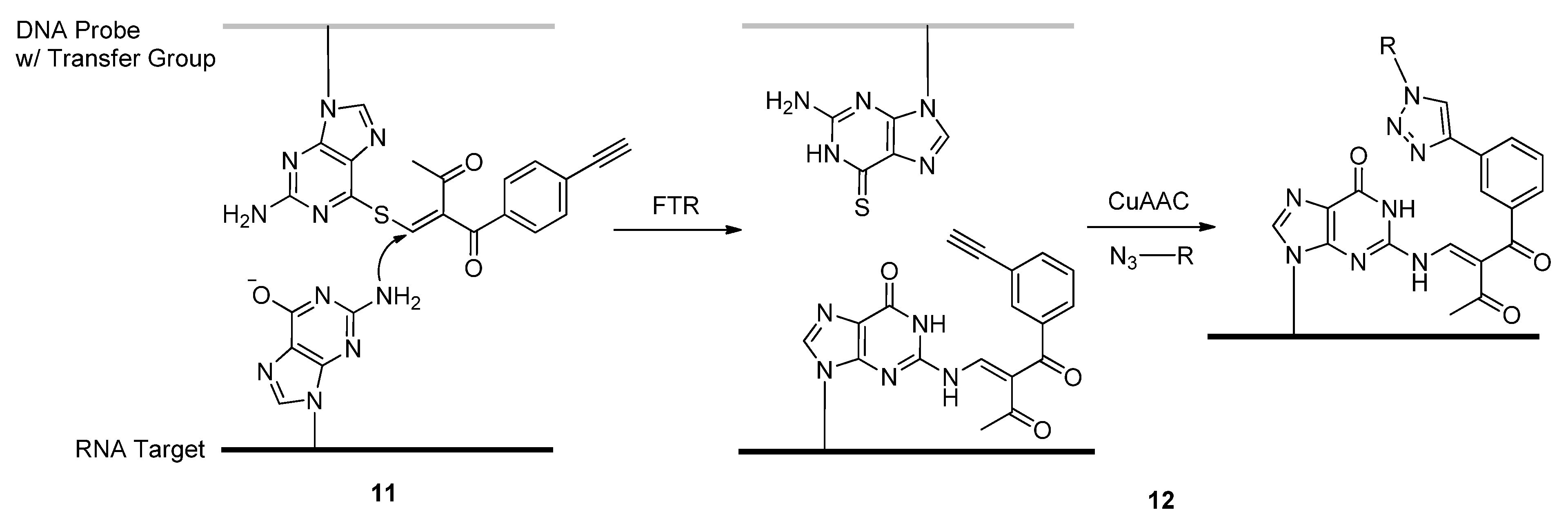
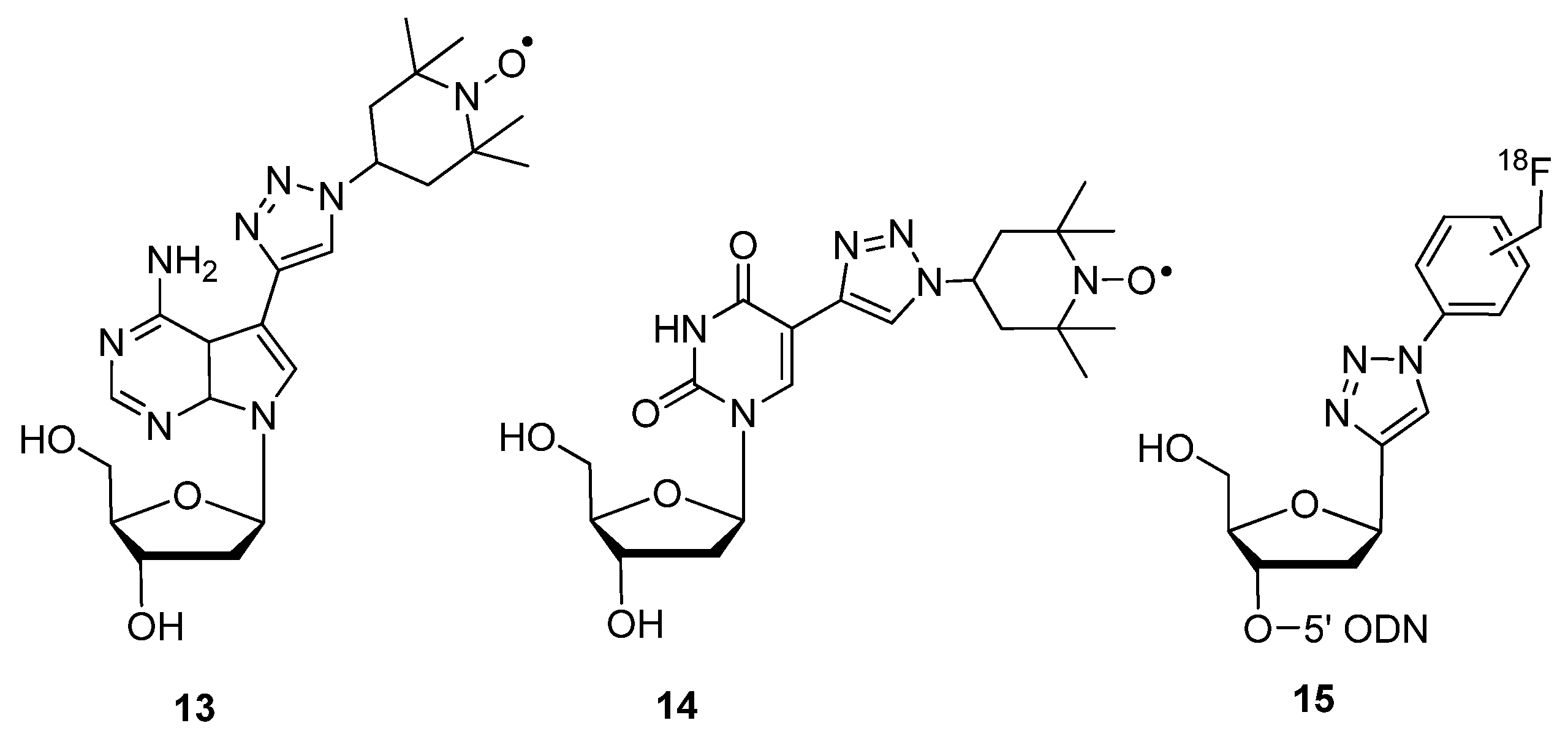
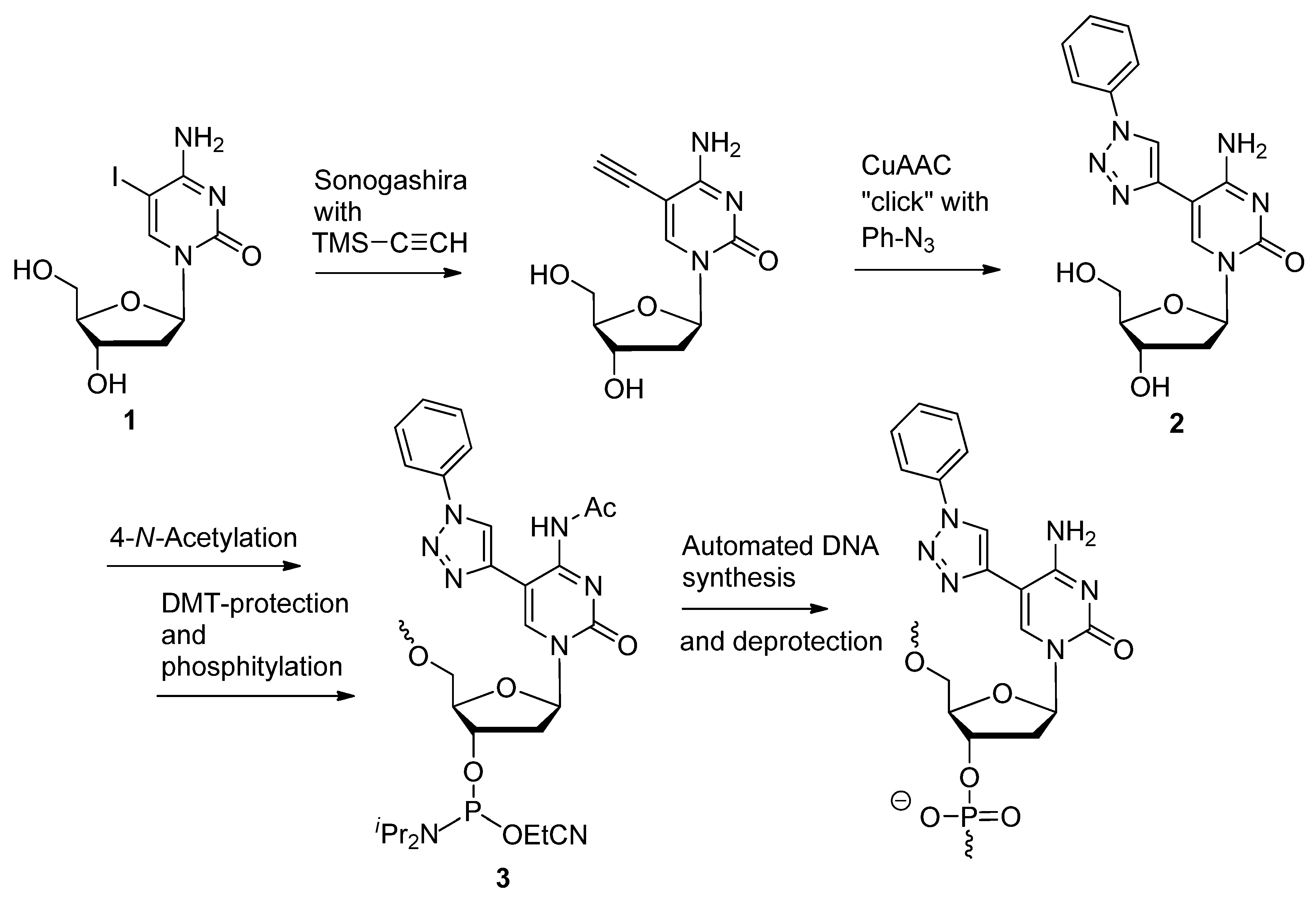
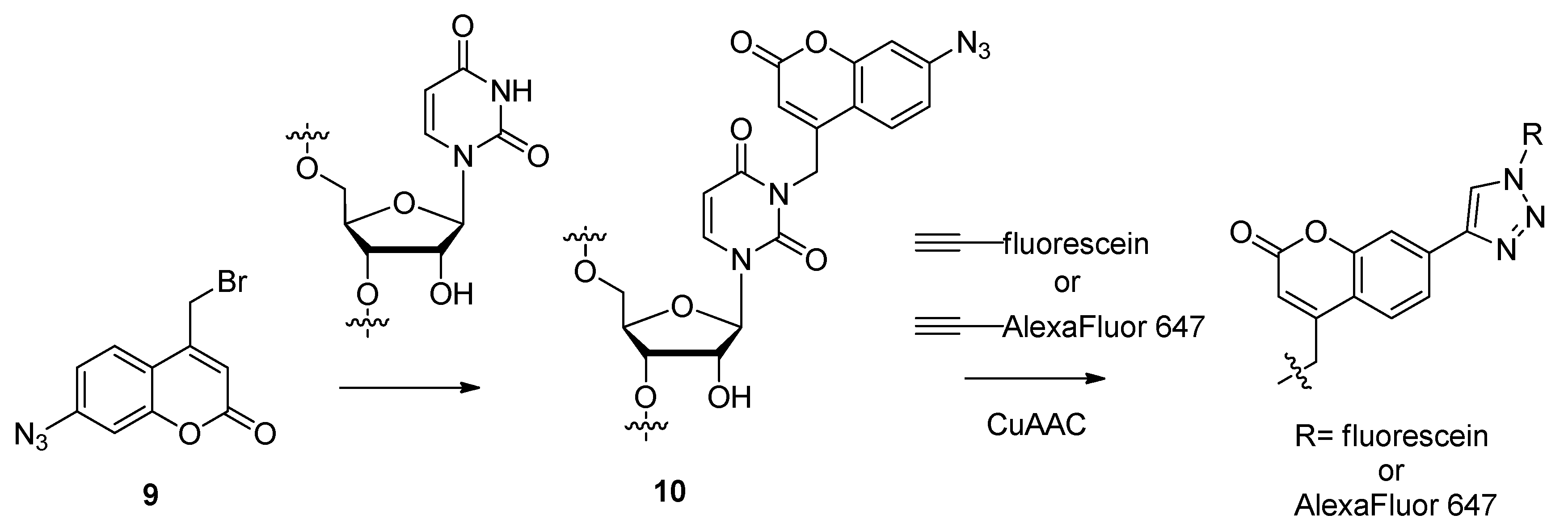
2.1.4. Fluorescent Labeling
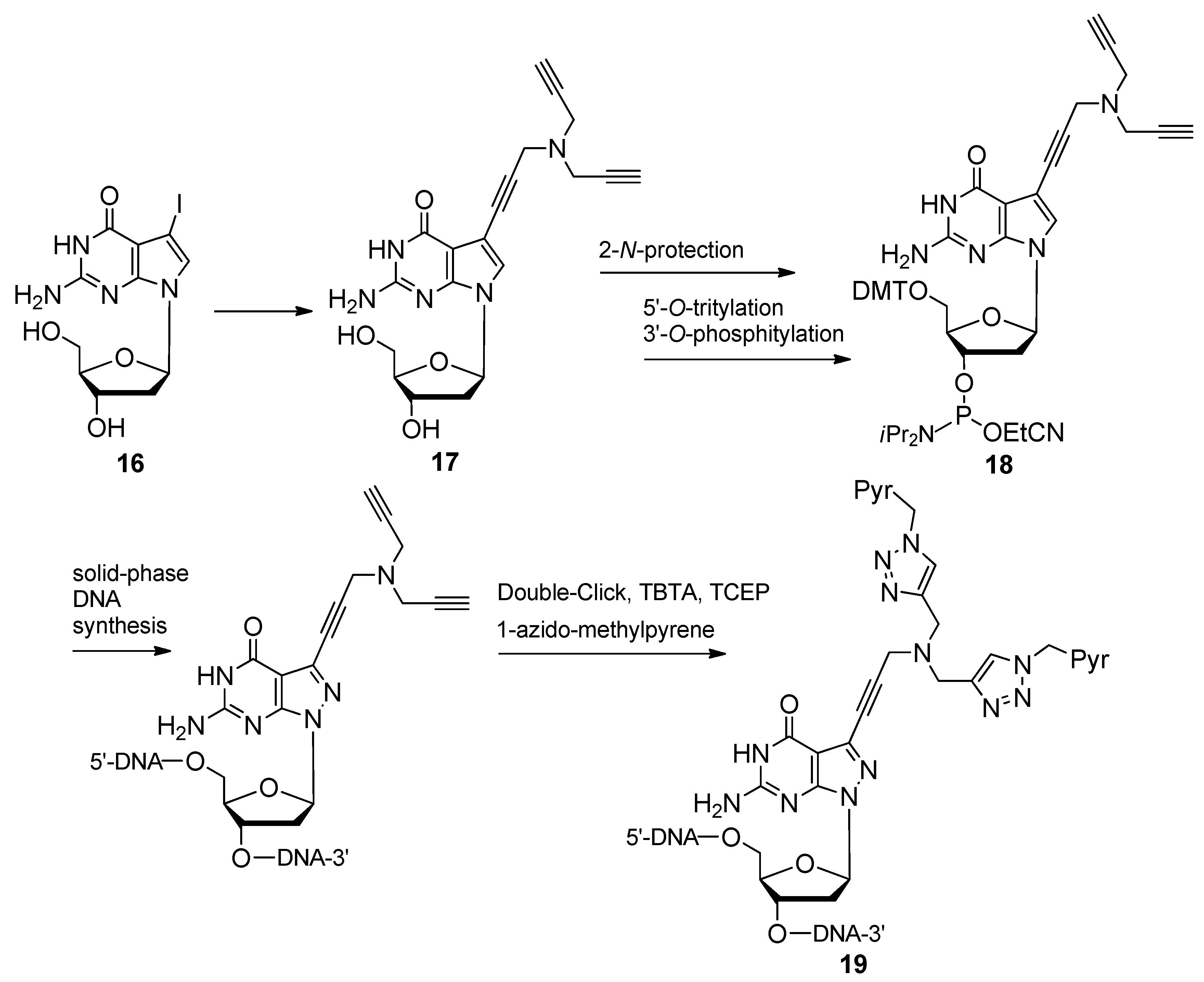
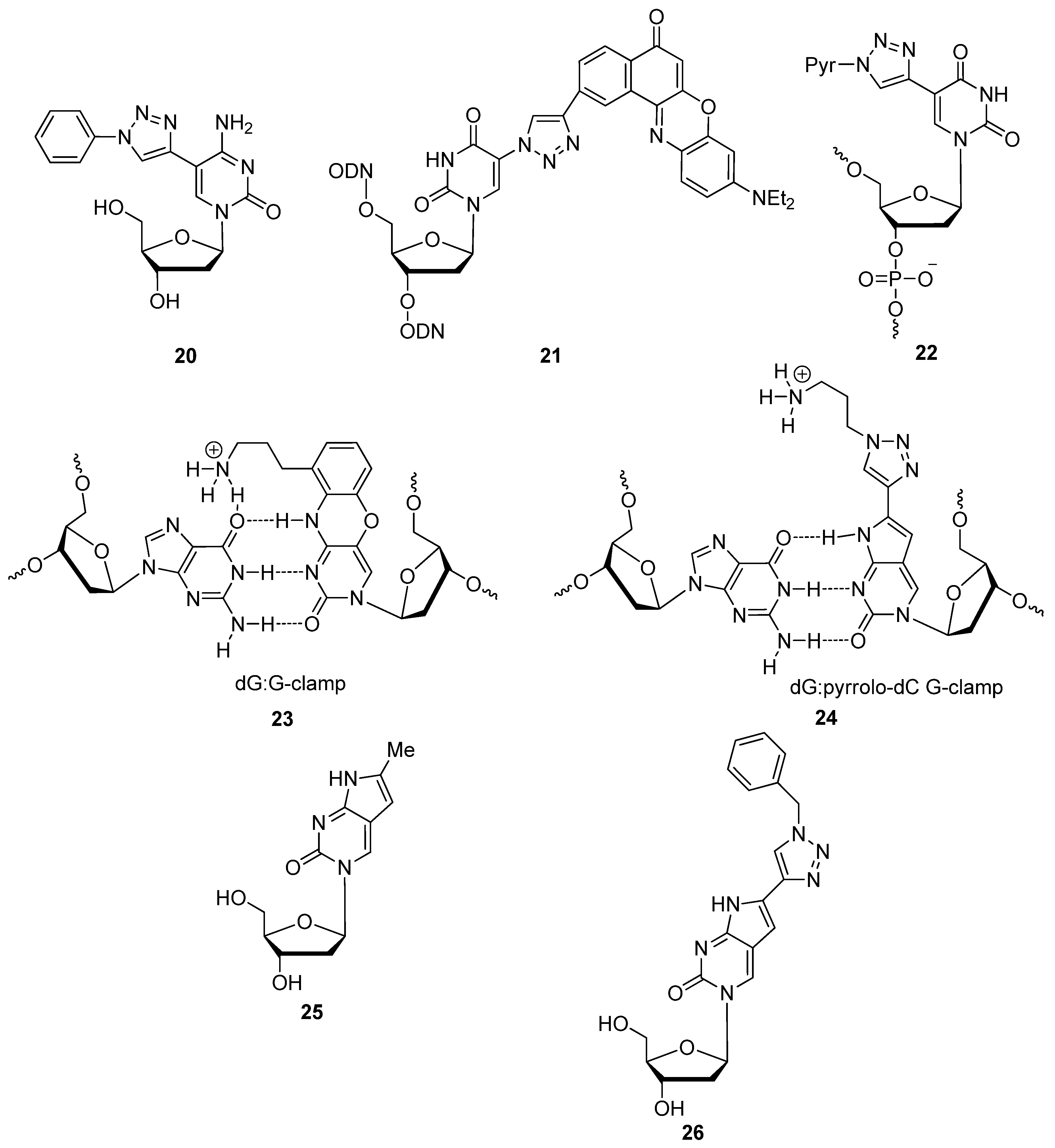
2.1.5. Anticancer and Antiviral Development
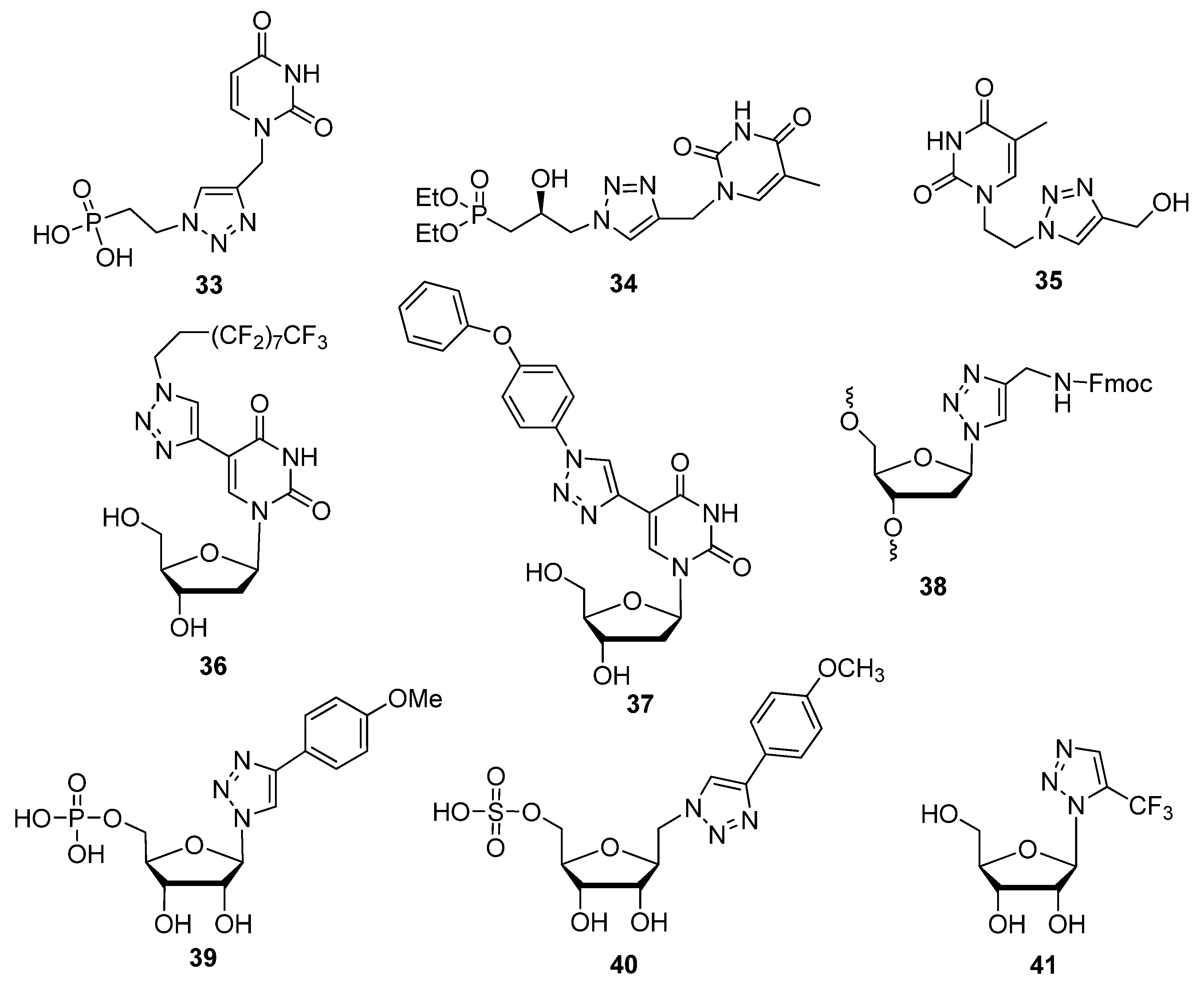
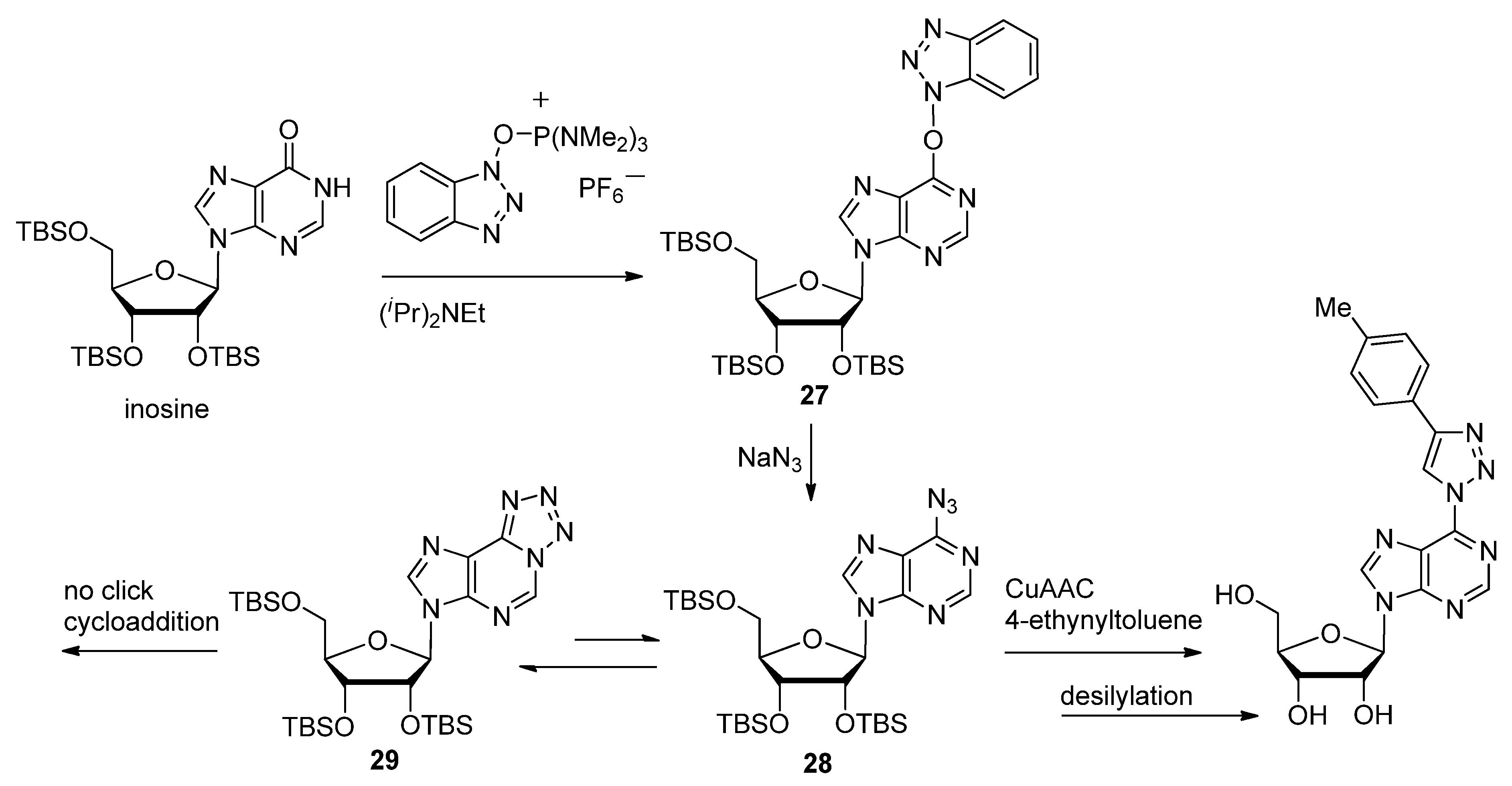
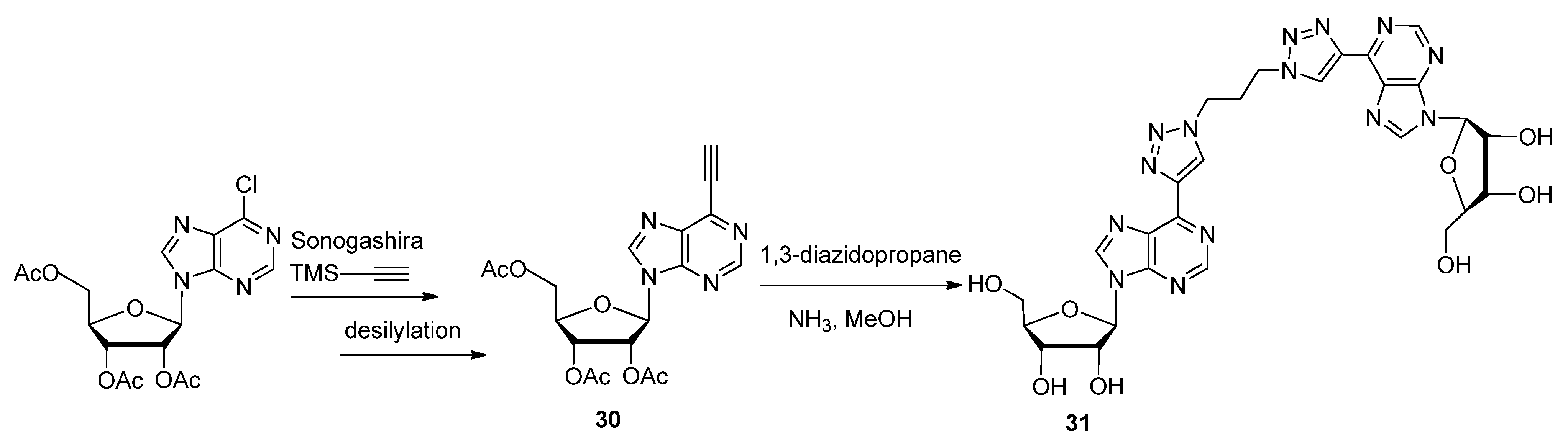
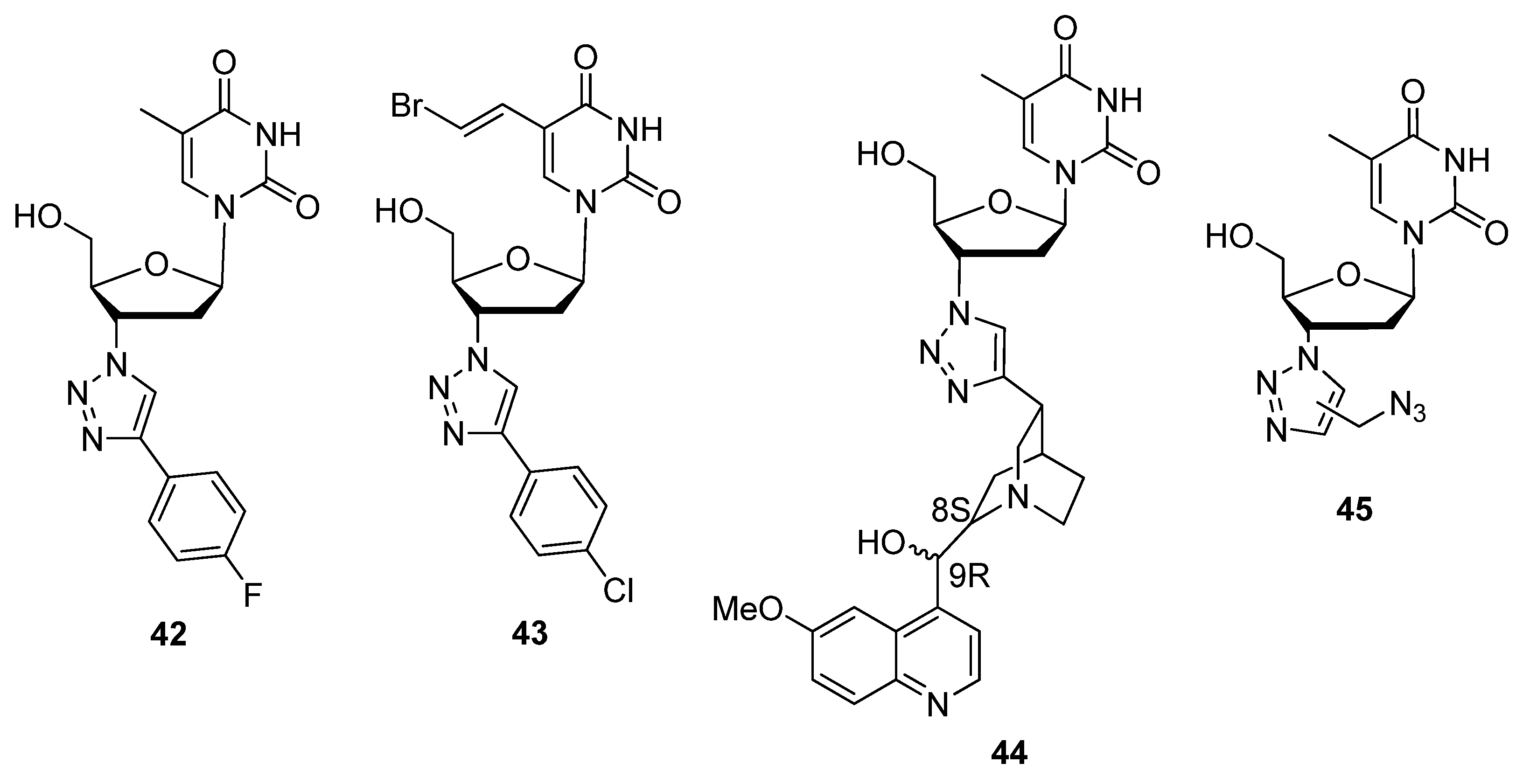
2.2. Sugar Modifications
2.2.1. C3′-Modified Sugars
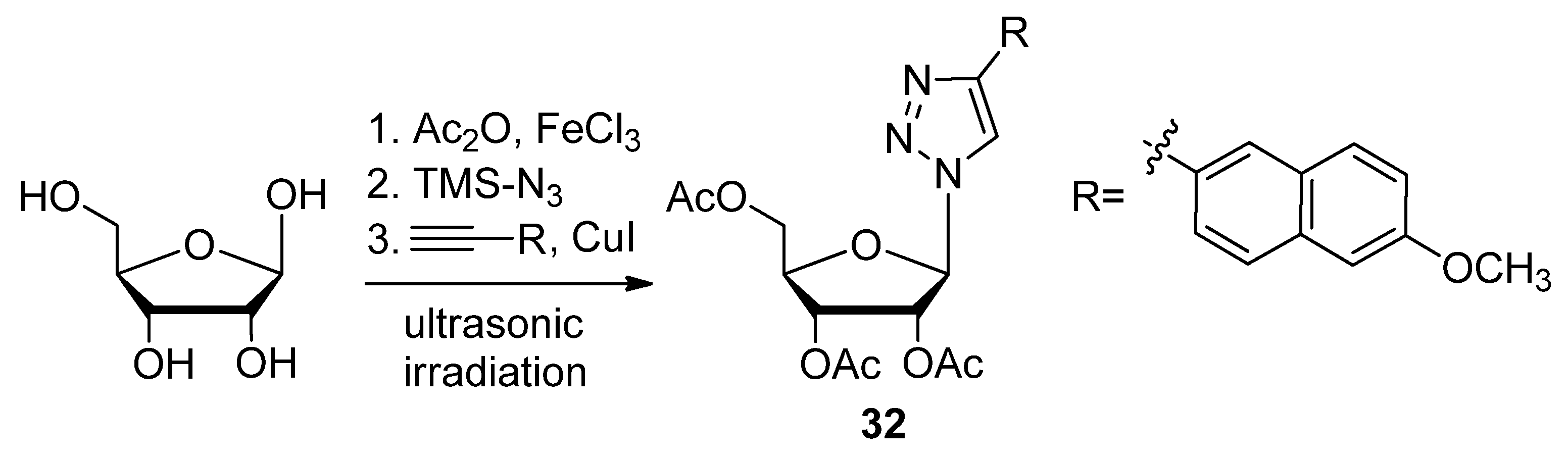
2.2.2. C5′-Modified Sugars
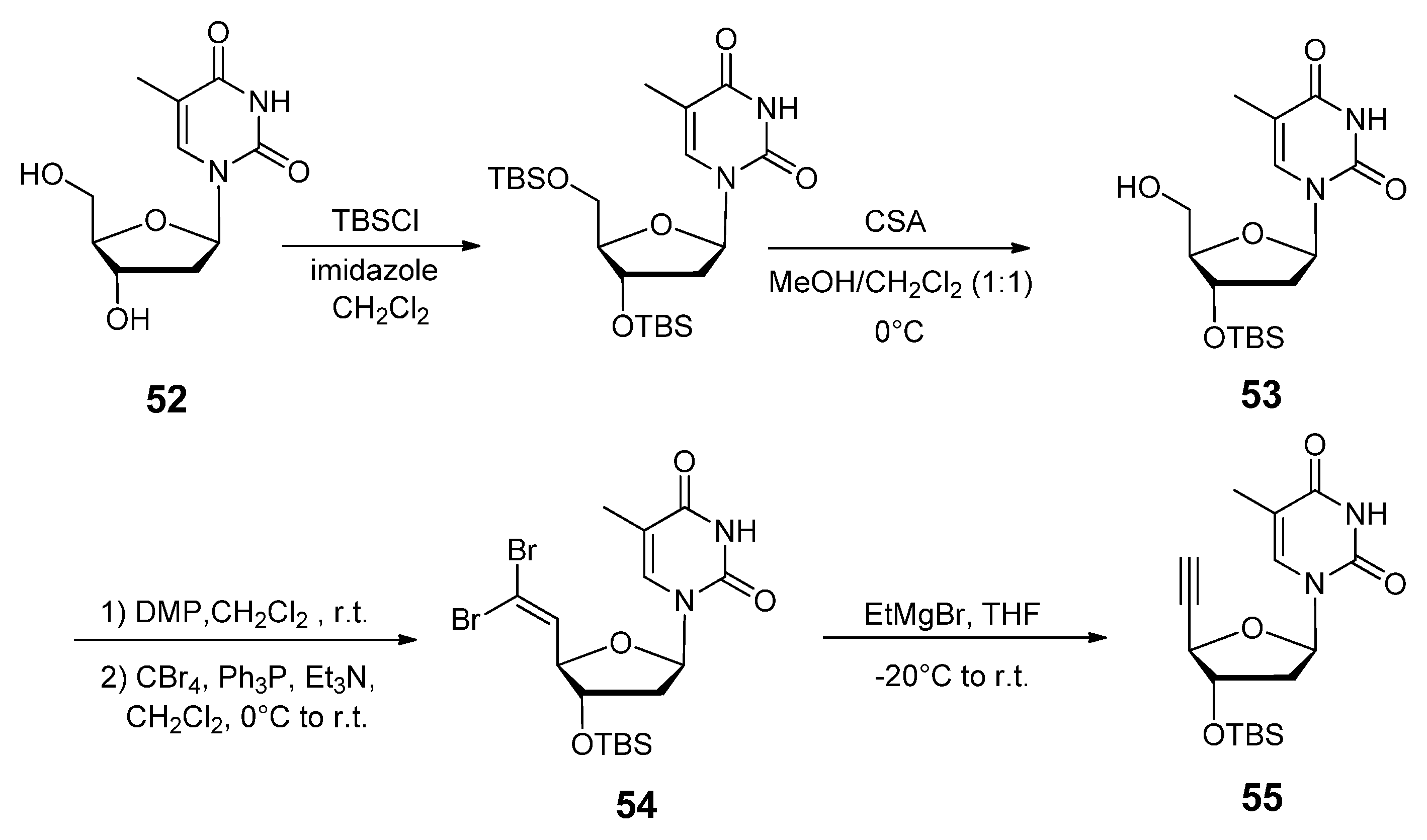
2.2.3. C2′-Modified Sugars
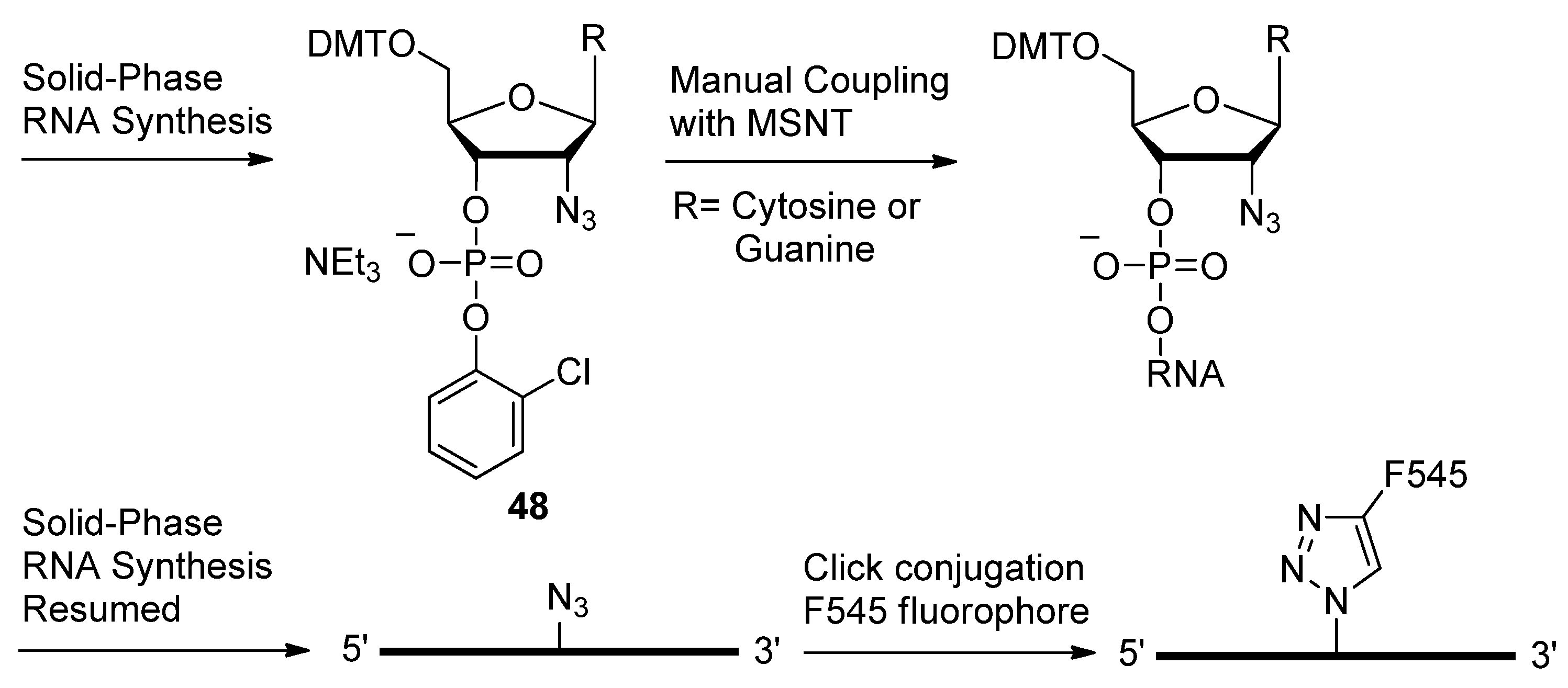
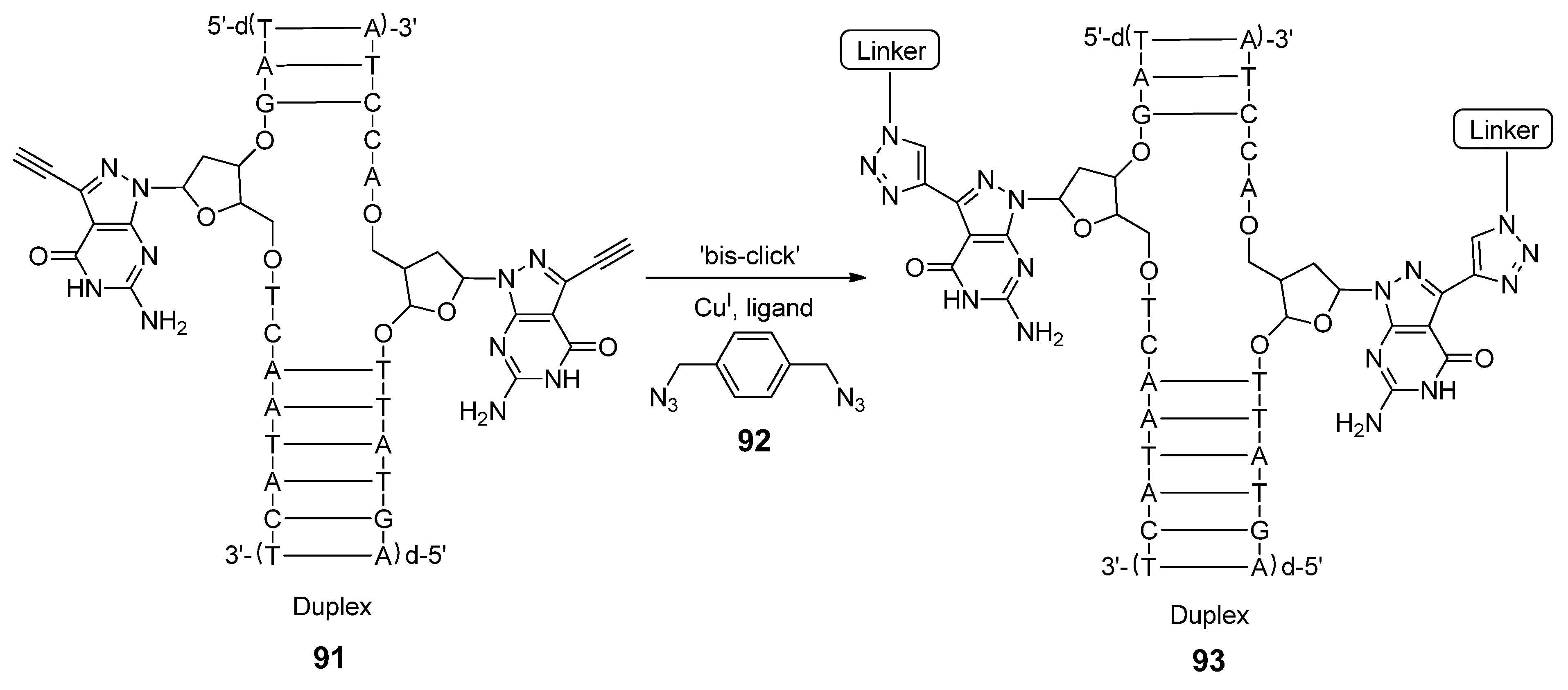
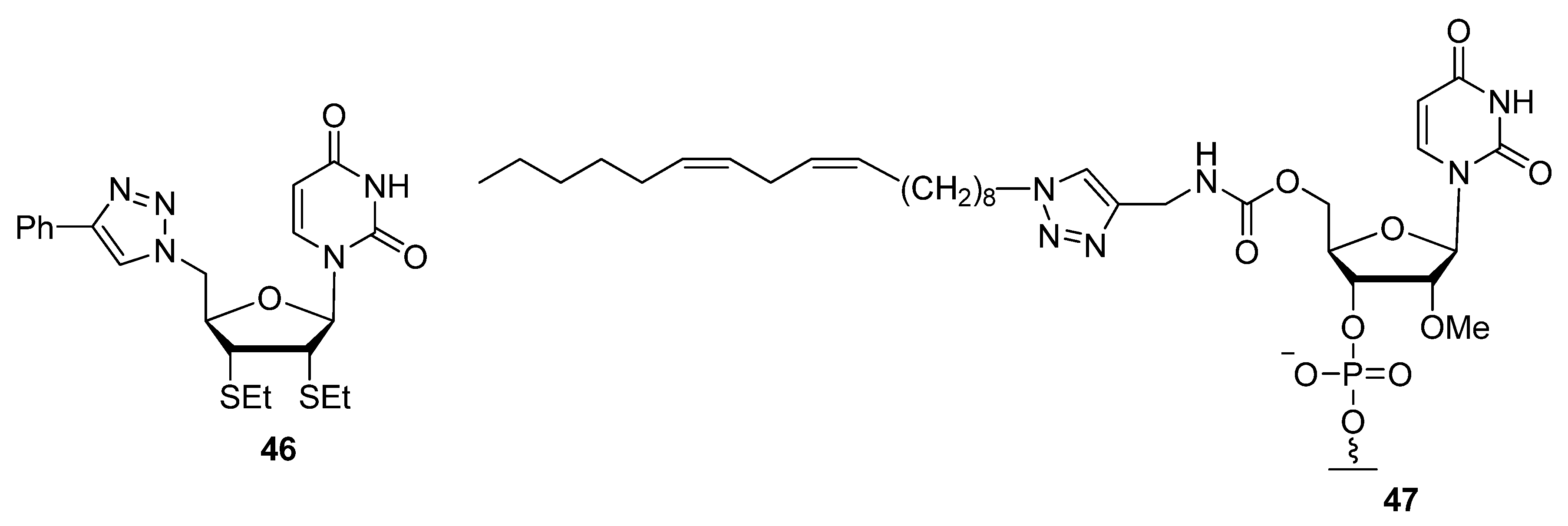
2.3. Backbone Modifications
2.3.1. DNA Backbone Modification
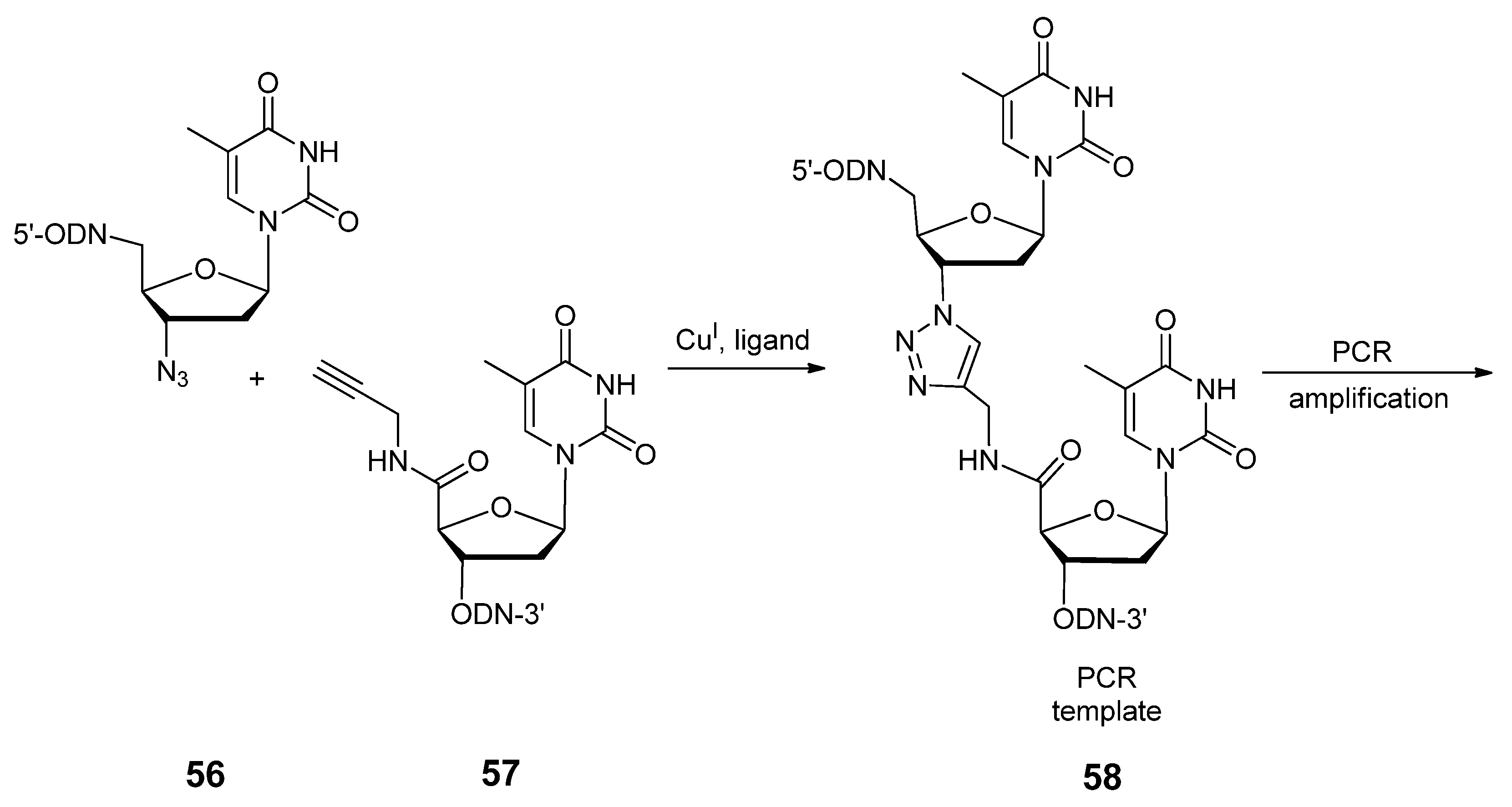
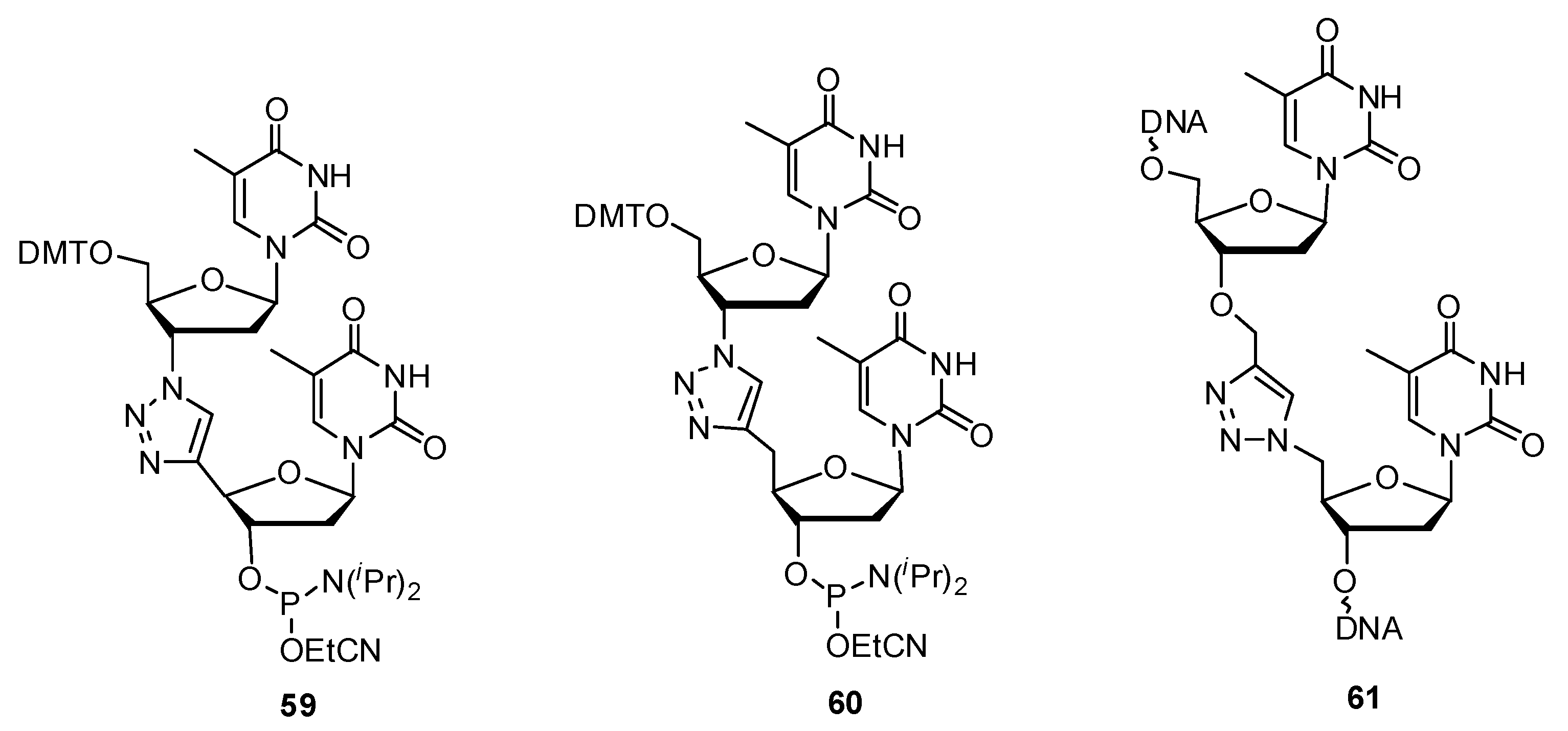
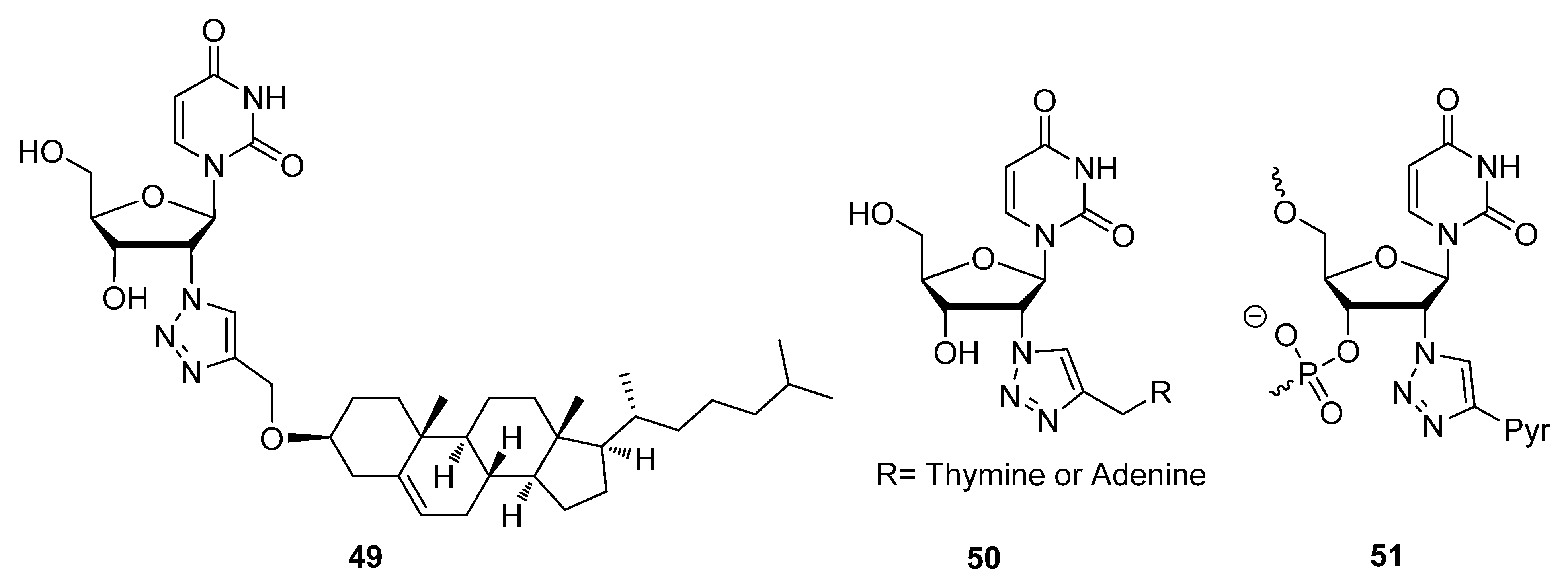
2.3.2. RNA Backbone Modifications
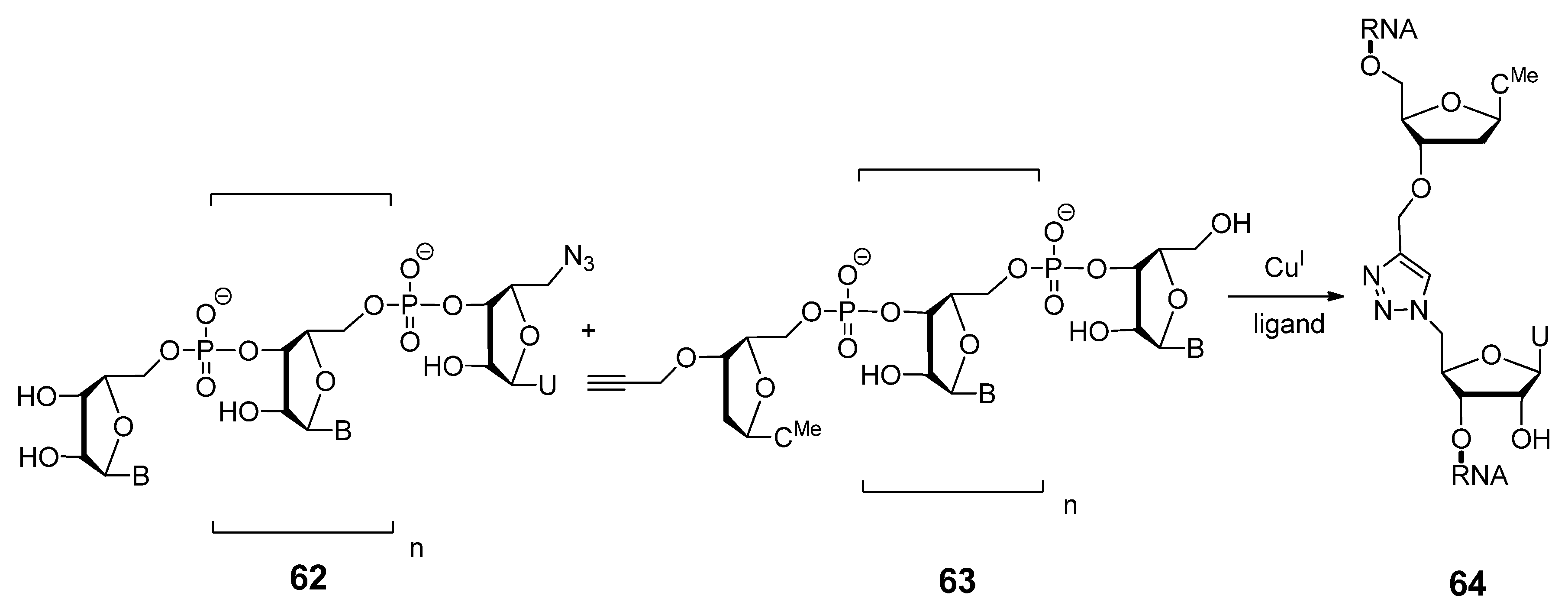
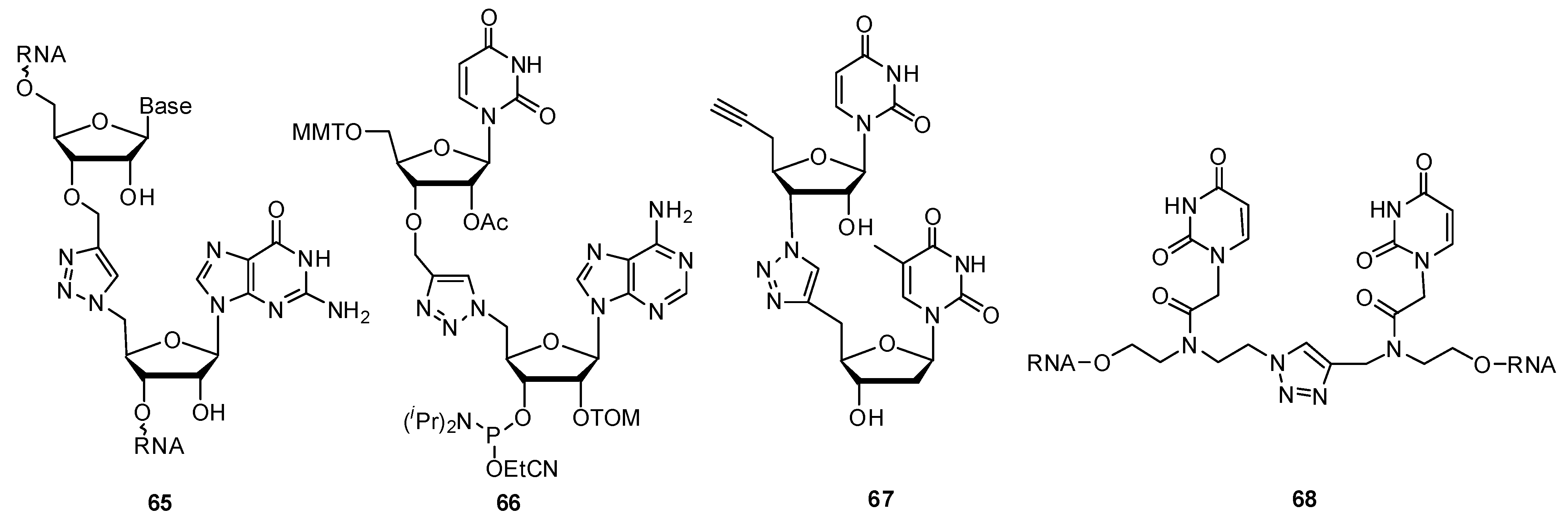
2.3.3. Other Backbone Modifications
2.4. Bio-conjugation
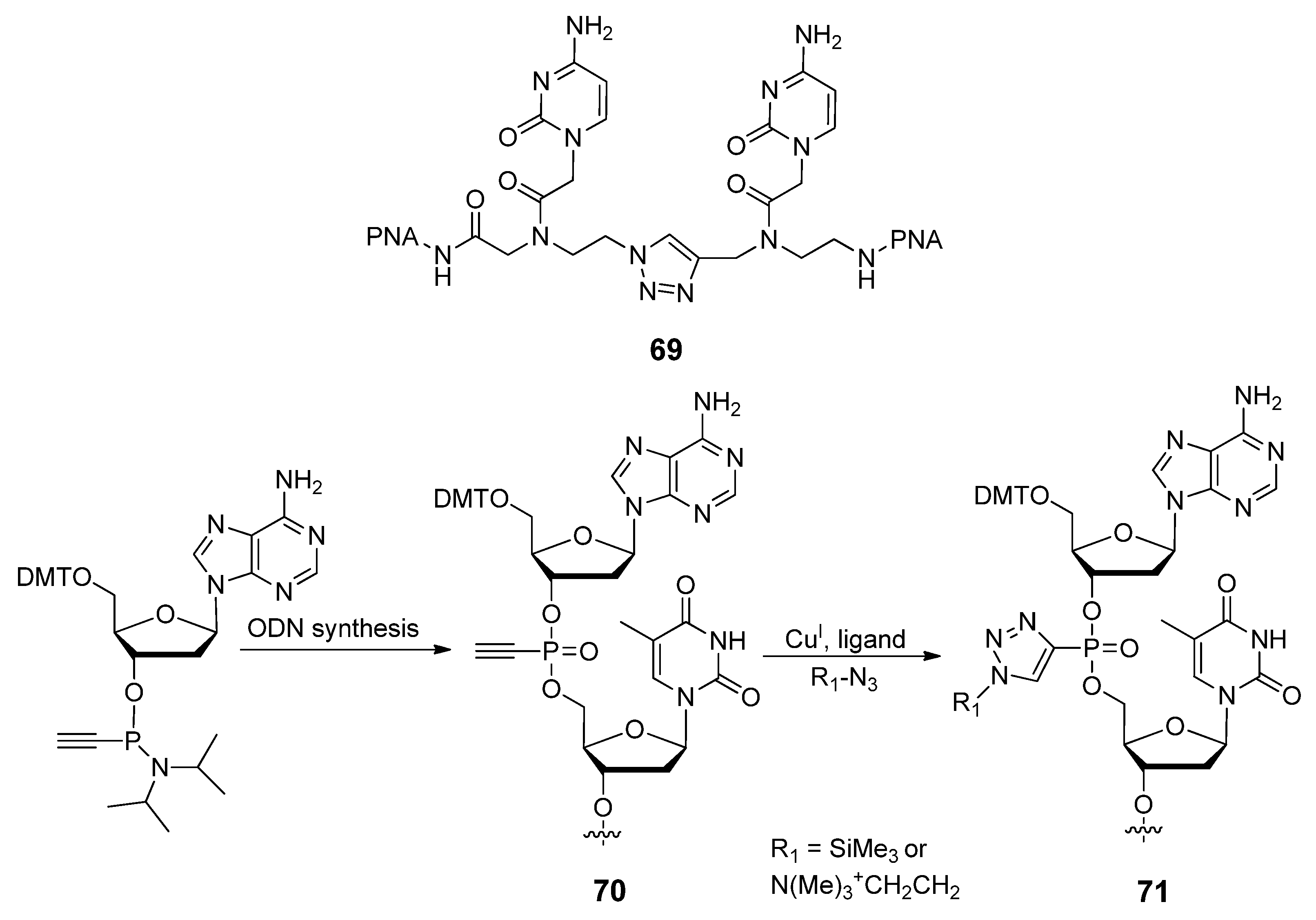
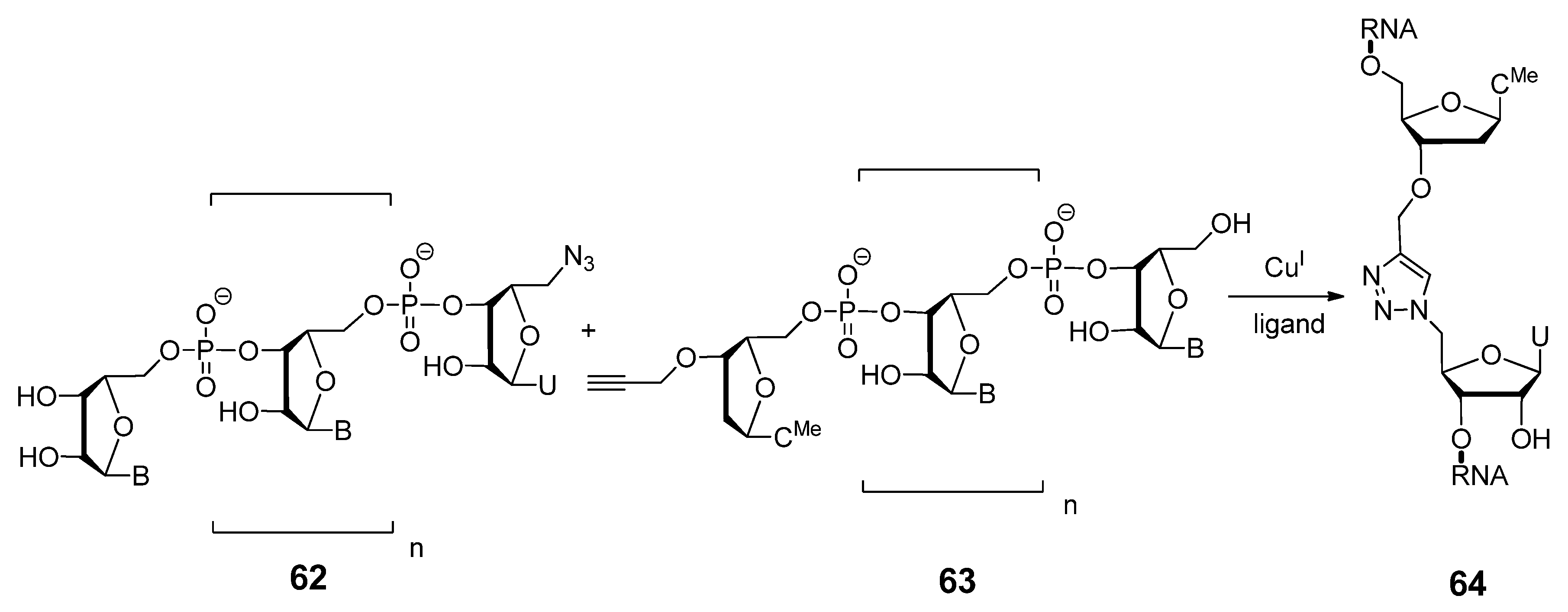
2.4.1. Nucleic Acids Delivery

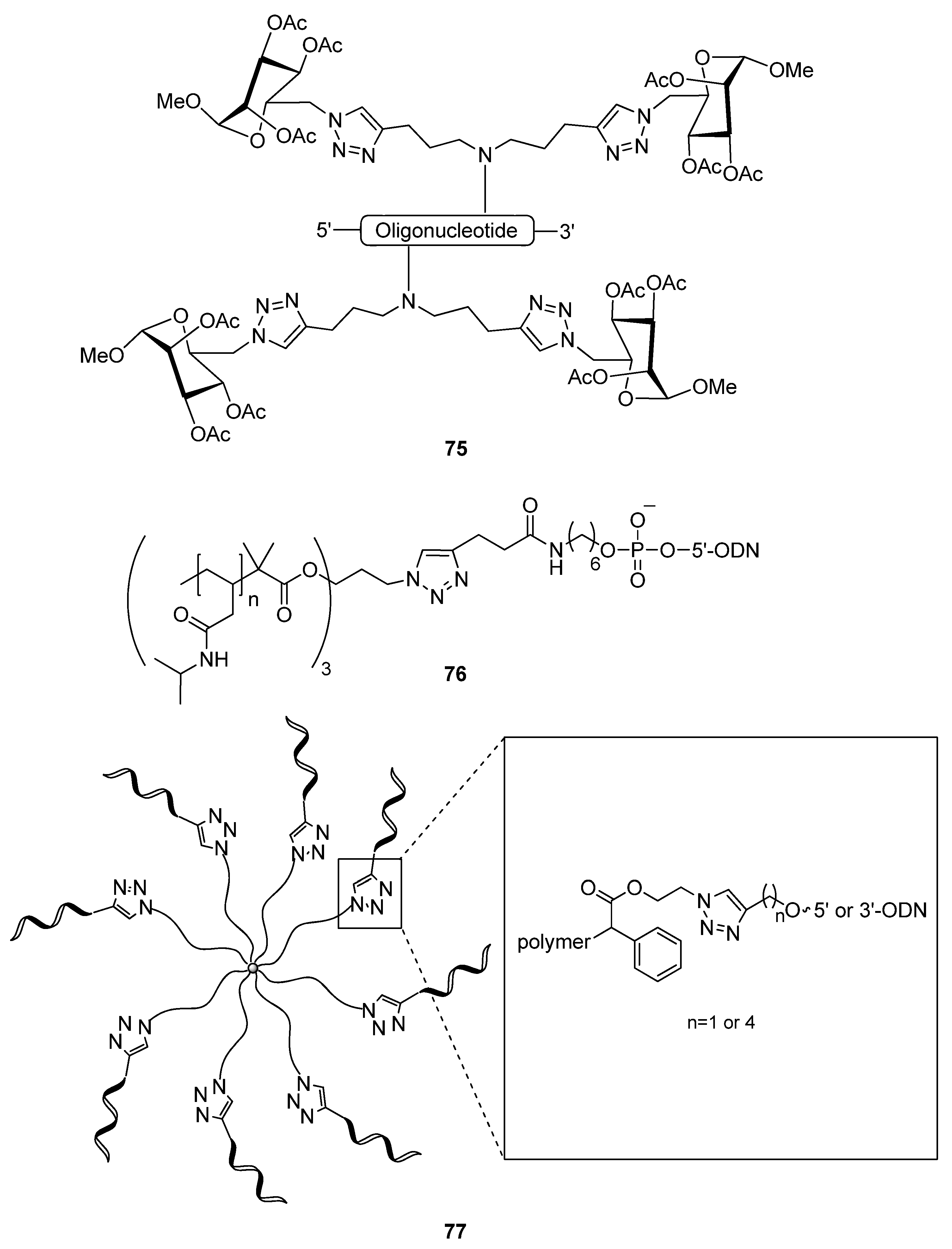
2.4.2. Copper-Free Click Ligation

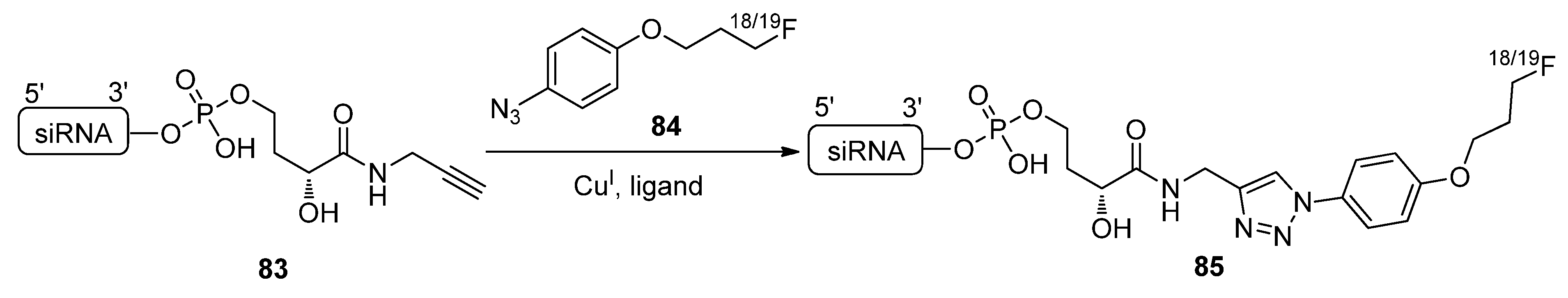
2.4.3. Oligonucleotide Labeling
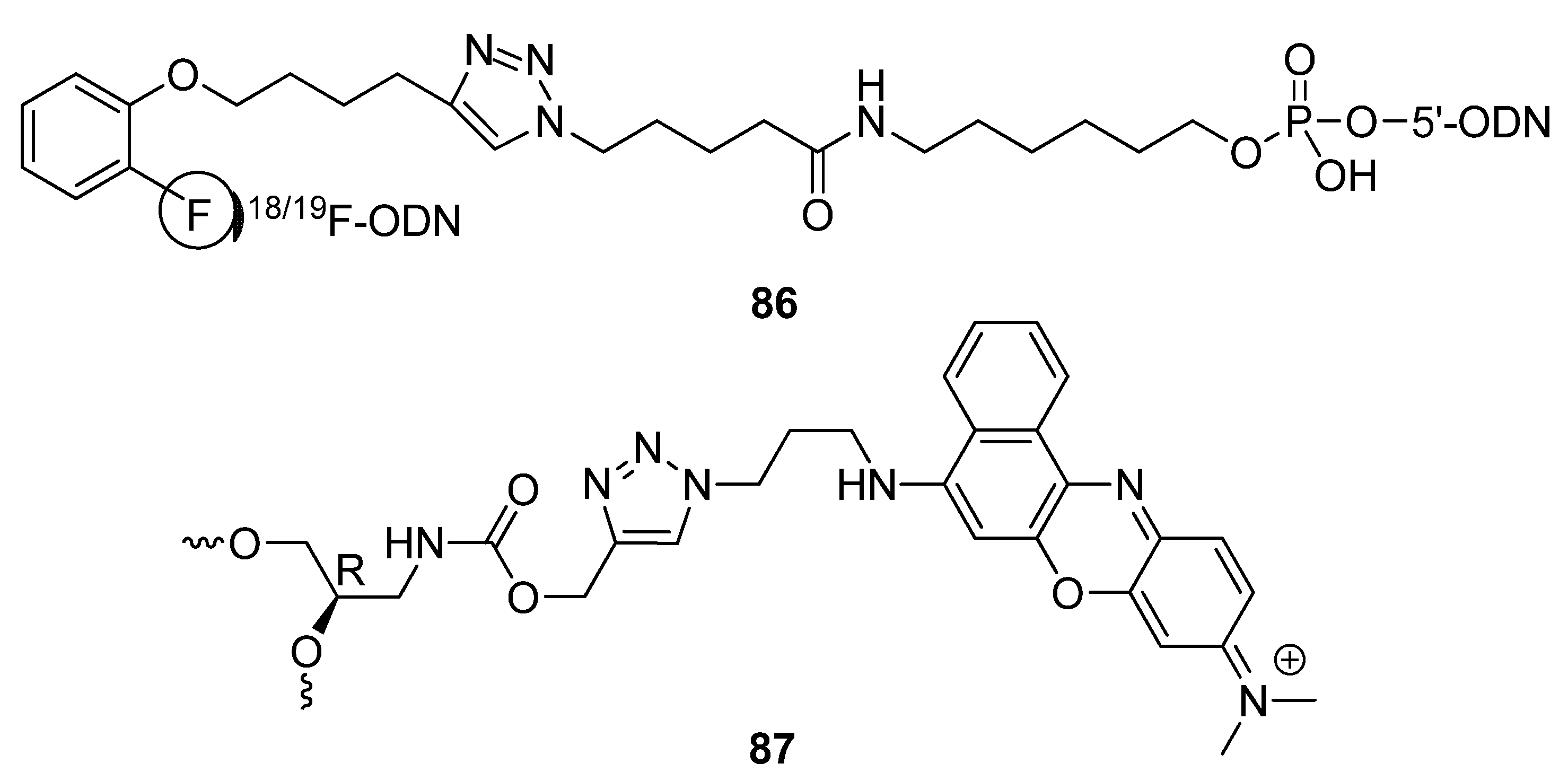
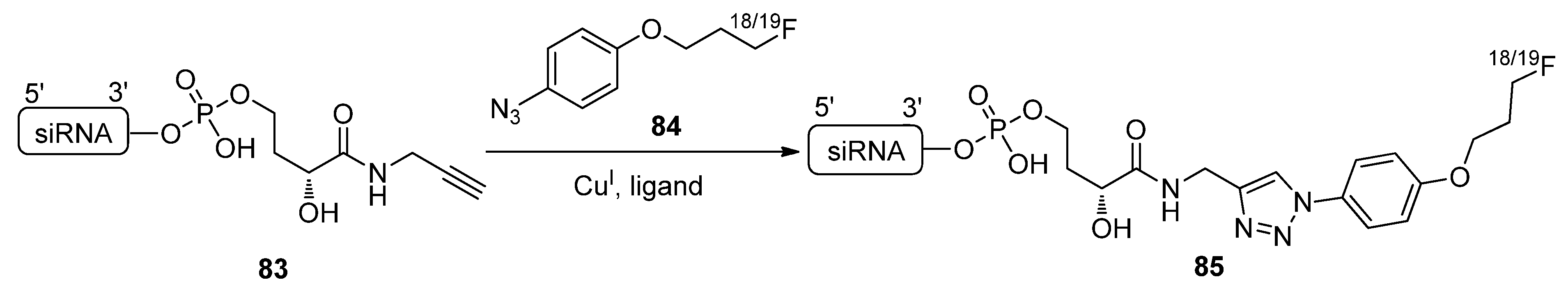
2.4.4. Other Applications of “Click Conjugation”
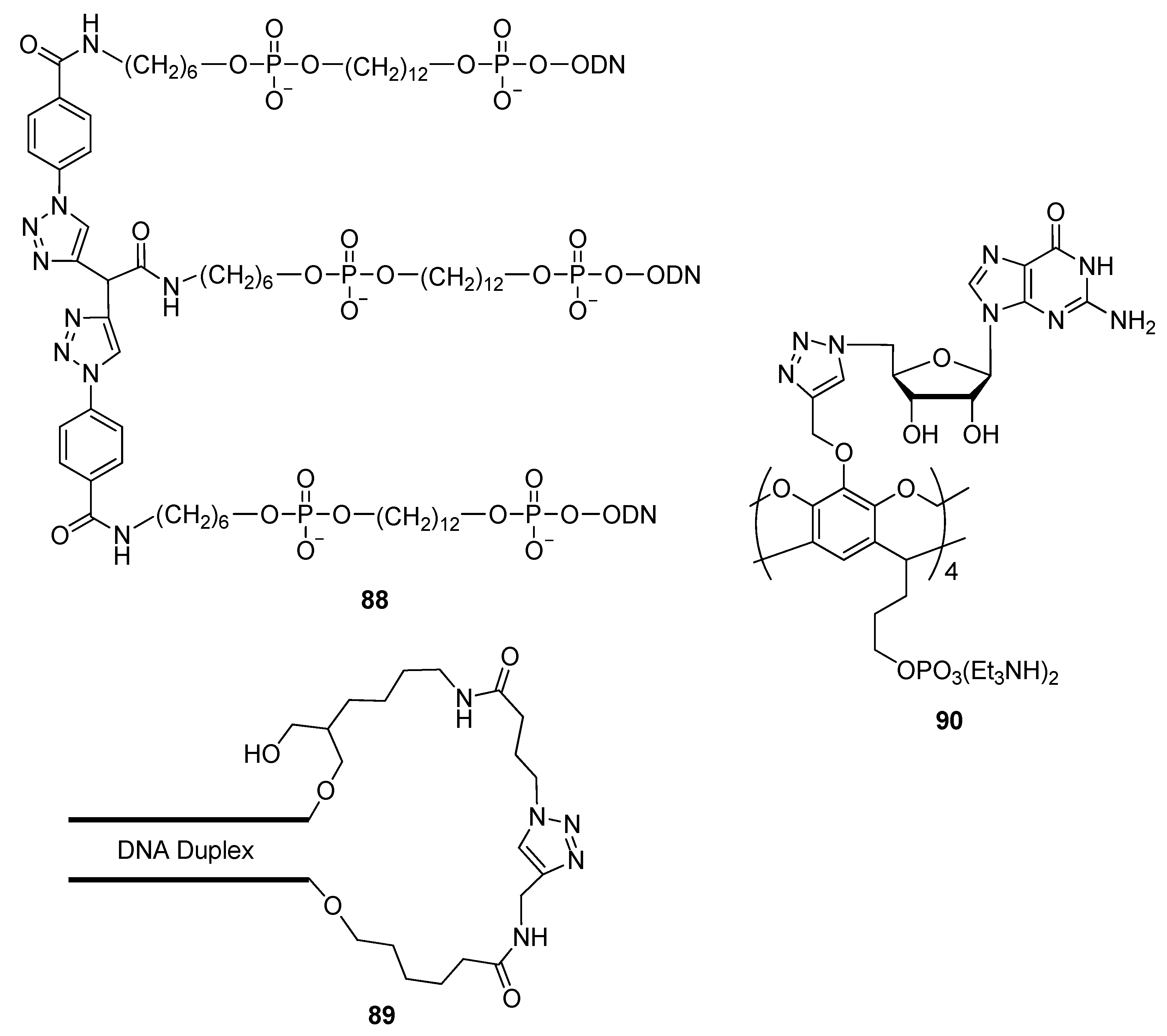
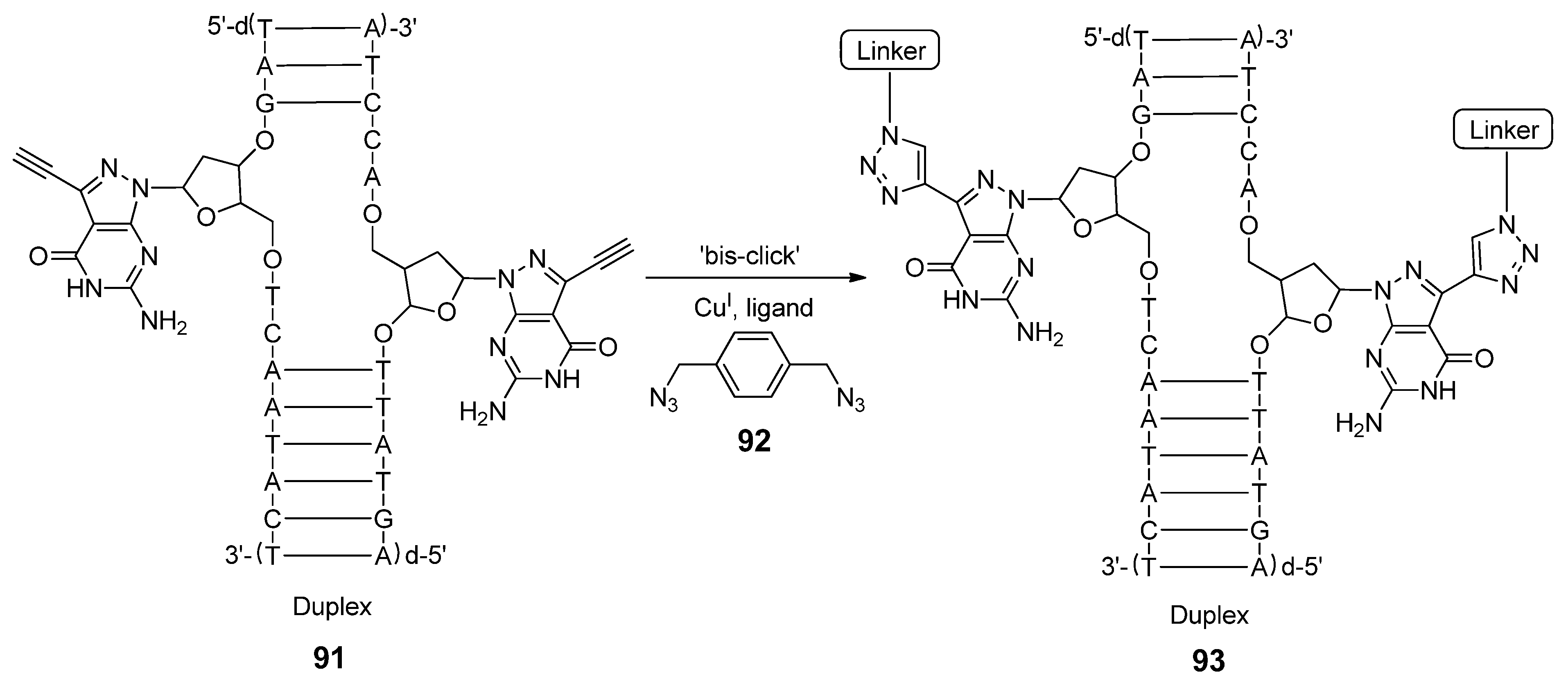
3. Conclusions
Acknowledgments
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu(I)-catalyzed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 2009, 109, 4207–4220. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Meldal, M.; Tornoe, C.W. Cu-Catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- de Miguel, G.; Wielopolski, M.; Schuster, D.I.; Fazio, M.A.; Lee, O.P.; Haley, C.K.; Ortiz, A.L.; Echegoyen, L.; Clark, T.; Guldi, D.M. Triazole bridges as versatile linkers in electron donor-acceptor conjugates. J. Am. Chem. Soc. 2011, 133, 13036–13054. [Google Scholar]
- van Dijk, M.; Nollet, M.L.; Weijers, P.; Dechesne, A.C.; van Nostrum, C.F.; Hennink, W.E.; Rijkers, D.T.S.; Liskamp, R.M.J. Synthesis and characterization of biodegradable peptide-based polymers prepared by microwave-assisted click chemistry. Biomacromolecules 2008, 9, 2834–2843. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Brown, T. Click chemistry with DNA. Chem. Soc. Rev. 2010, 39, 1388–1405. [Google Scholar] [CrossRef]
- Cho, I.S.; Kim, J.; Lim, D.H.; Ahn, H.C.; Kim, H.; Lee, K.B.; Lee, Y.S. Improved serum stability and biophysical properties of siRNAs following chemical modifications. Biotechnol. Lett. 2008, 30, 1901–1908. [Google Scholar] [CrossRef]
- Miller, M.B.; Tang, Y.W. Basic concepts of microarrays and potential applications in clinical microbiology. Clin. Microbiol. Rev. 2009, 22, 611–633. [Google Scholar] [CrossRef]
- Seo, T.S.; Bai, X.P.; Ruparel, H.; Li, Z.M.; Turro, N.J.; Ju, J.Y. Photocleavable fluorescent nucleotides for DNA sequencing on a chip constructed by site-specific coupling chemistry. Proc. Natl. Acad. Sci. USA 2004, 101, 5488–5493. [Google Scholar] [CrossRef]
- Chevolot, Y.; Bouillon, C.; Vidal, S.; Morvan, F.; Meyer, A.; Cloarec, J.-P.; Jochum, A.; Praly, J.-P.; Vasseur, J.-J.; Souteyrand, E. DNA-Based carbohydrate biochips: A platform for surface glyco-engineering. Angew. Chem. Int. Ed. Engl. 2007, 46, 2398–2402. [Google Scholar]
- Liu, X.; Farmerie, W.; Schuster, S.; Tan, W. Molecular beacons for DNA biosensors with micrometer to submicrometer dimensions. Anal. Biochem. 2000, 283, 56–63. [Google Scholar]
- Seio, K.; Takaku, Y.; Miyazaki, K.; Kurohagi, S.; Masaki, Y.; Ohkubo, A.; Sekine, M. Synthesis of terminally modified oligonucleotides and their hybridization dependence on the size of the target RNAs. Org. Biomol. Chem. 2009, 7, 2440–2451. [Google Scholar] [CrossRef]
- Izant, J.G.; Weintraub, H. Inhibition of thymidine kinase gene expression by anti-sense RNA: A molecular approach to genetic analysis. Cell 1984, 36, 1007–1016. [Google Scholar] [CrossRef]
- Uhlmann, E.; Peyman, A. Antisense oligonucleotides: A new therapeutic principle. Chem. Rev. 1990, 90, 543–584. [Google Scholar] [CrossRef]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.Q.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Watts, J.K.; Deleavey, G.F.; Damha, M.J. Chemically modified siRNA: Tools and applications. Drug Discov. Today 2008, 13, 842–855. [Google Scholar] [CrossRef]
- Kim, S.I.; Shin, D.; Lee, H.; Ahn, B.Y.; Yoon, Y.; Kim, M. Targeted delivery of siRNA against hepatitis C virus by apolipoprotein A-I-bound cationic liposomes. J. Hepatol. 2009, 50, 479–488. [Google Scholar] [CrossRef]
- Khaliq, S.; Khaliq, S.A.; Zahur, M.; Ijaz, B.; Jahan, S.; Ansar, M.; Riazuddin, S.; Hassan, S. RNAi as a new therapeutic strategy against HCV. Biotechnol. Adv. 2010, 28, 27–34. [Google Scholar] [CrossRef]
- Elayadi, H.; Smietana, M.; Pannecouque, C.; Leyssen, P.; Neyts, J.; Vasseur, J.-J.; Lazrek, H.B. Straightforward synthesis of triazoloacyclonucleotide phosphonates as potential HCV inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 7365–7368. [Google Scholar] [CrossRef]
- Corey, D.R. Chemical modification: The key to clinical application of RNA interference? J. Clin. Invest. 2007, 117, 3615–3622. [Google Scholar] [CrossRef]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen—Clinical pharmacology and potential drug interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [Google Scholar] [CrossRef]
- Huffman, J.H.; Sidwell, R.W.; Khare, G.P.; Witkowski, J.T.; Allen, L.B.; Robins, R.K. In-vitro effect of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole, ICN-1229) on deoxyribonucleic acid and ribonucleic acid viruses. Antimicrob. Agents Ch. 1973, 3, 235–241. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Brown, T. New strategy for the synthesis of chemically modified RNA constructs exemplified by hairpin and hammerhead ribozymes. Proc. Natl. Acad. Sci. USA 2010, 107, 15329–15334. [Google Scholar] [CrossRef]
- Mascini, M.; Palchetti, I.; Tombelli, S. Nucleic acid and peptide aptamers: Fundamentals and bioanalytical aspects. Angew. Chem. Int. Ed. Engl. 2012, 51, 1316–1332. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Kaihatsu, K.; Janowski, B.A.; Corey, D.R. Recognition of chromosomal DNA by PNAs. Chem. Biol. 2004, 11, 749–758. [Google Scholar] [CrossRef]
- Saito, Y.; Escuret, V.; Durantel, D.; Zoulim, F.; Schinazi, R.F.; Agrofoglio, L.A. Synthesis of 1,2,3-triazolo-carbanucleoside analogues of ribavirin targeting an HCV in replicon. Bioorg. Med. Chem. 2003, 11, 3633–3639. [Google Scholar] [CrossRef]
- El Akri, K.; Bougrin, K.; Balzarini, J.; Faraj, A.; Benhida, R. Efficient synthesis and in vitro cytostatic activity of 4-substituted triazolyl-nucleosides. Bioorg. Med. Chem. Lett. 2007, 17, 6656–6659. [Google Scholar] [CrossRef]
- Ming, X.; Seela, F. A nucleobase-discriminating pyrrolo-dC click adduct designed for DNA fluorescence mismatch sensing. Chem. Eur. J. 2012, 18, 9590–9600. [Google Scholar] [CrossRef]
- Xiong, Z.; Qiu, X.-L.; Huang, Y.; Qing, F.-L. Regioselective synthesis of 5-trifluoromethyl-1,2,3-triazole nucleoside analogues via TBS-directed 1,3-dipolar cycloaddition reaction. J. Fluorine Chem. 2011, 132, 166–174. [Google Scholar] [CrossRef]
- Gramlich, P.M.E.; Warncke, S.; Gierlich, J.; Carell, T. Click-click-click: Single to triple modification of DNA. Angew. Chem. Int. Ed. Engl. 2008, 47, 3442–3444. [Google Scholar] [CrossRef]
- Ostergaard, M.E.; Guenther, D.C.; Kumar, P.; Baral, B.; Deobald, L.; Paszczynski, A.J.; Sharma, P.K.; Hrdlicka, P.J. Pyrene-functionalized triazole-linked 2′-deoxyuridines—probes for discrimination of single nucleotide polymorphisms (SNPs). Chem. Commun. 2010, 46, 4929–4931. [Google Scholar]
- Dodd, D.W.; Swanick, K.N.; Price, J.T.; Brazeau, A.L.; Ferguson, M.J.; Jones, N.D.; Hudson, R.H.E. Blue fluorescent deoxycytidine analogues: Convergent synthesis, solid-state and electronic structure, and solvatochromism. Org. Biomol. Chem. 2010, 8, 663–666. [Google Scholar] [CrossRef]
- Seela, F.; Ingale, S.A. Double click” reaction on 7-deazaguanine DNA: Synthesis and excimer fluorescence of nucleosides and oligonucleotides with branched side chains decorated with proximal pyrenes. J. Org. Chem. 2010, 75, 284–295. [Google Scholar] [CrossRef]
- Xiong, H.; Seela, F. Cross-linked DNA: Site-selective “click” ligation in duplexes with bis-azides and stability changes caused by internal cross-links. Bioconjug. Chem. 2012, 23, 1230–1243. [Google Scholar] [CrossRef]
- El-Sagheer, A.H. Very stable end-sealed double stranded DNA by click chemistry. Nucleos. Nucleot. Nucl. 2009, 28, 315–323. [Google Scholar] [CrossRef]
- Jacobsen, M.F.; Ravnsbaek, J.B.; Gothelf, K.V. Small molecule induced control in duplex and triplex DNA-directed chemical reactions. Org. Biomol. Chem. 2010, 8, 50–52. [Google Scholar] [CrossRef]
- Seela, F.; Xiong, H.; Budow, S. Synthesis and ‘double click’ density functionalization of 8-aza-7-deazaguanine DNA bearing branched side chains with terminal triple bonds. Tetrahedron 2010, 66, 3930–3943. [Google Scholar] [CrossRef]
- Krishna, H.; Caruthers, M.H. Alkynyl Phosphonate DNA: A versatile “click” able backbone for DNA-based biological applications. J. Am. Chem. Soc. 2012, 134, 11618–11631. [Google Scholar] [CrossRef]
- Peacock, H.; Maydanovych, O.; Beal, P.A. N2-Modified 2-aminopurine ribonucleosides as minor-groove-modulating adenosine replacements in duplex RNA. Org. Lett. 2010, 12, 1044–1047. [Google Scholar] [CrossRef]
- Ohtsuka, E.; Ikehara, M.; Soll, D. Recent developments in the chemical synthesis of polynucleotides. Nucleic Acids Res. 1982, 10, 6553–6570. [Google Scholar] [CrossRef]
- Caruthers, M.H.; Barone, A.D.; Beaucage, S.L.; Dodds, D.R.; Fisher, E.F.; McBride, L.J.; Matteucci, M.; Stabinsky, Z.; Tang, J.Y. Chemical synthesis of deoxyoligonucleotides by the phosphoramidite method. Method. Enzymol. 1987, 154, 287–313. [Google Scholar]
- Wilson, J.N.; Kool, E.T. Fluorescent DNA base replacements: reporters and sensors for biological systems. Org. Biomol. Chem. 2006, 4, 4265–4274. [Google Scholar] [CrossRef]
- Peacock, H.; Fostvedt, E.; Beal, P.A. Minor-groove-modulating adenosine replacements control protein binding and RNAi activity in siRNAs. ACS Chem. Biol. 2010, 5, 1115–1124. [Google Scholar] [CrossRef]
- Jorgensen, A.S.; Shaikh, K.I.; Enderlin, G.; Ivarsen, E.; Kumar, S.; Nielsen, P. The synthesis of double-headed nucleosides by the CuAAC reaction and their effect in secondary nucleic acid structures. Org. Biomol. Chem. 2011, 9, 1381–1388. [Google Scholar] [CrossRef]
- Okamoto, A.; Saito, Y.; Saito, I. Design of base-discriminating fluorescent nucleosides. J. Photoch. Photobio. C 2005, 6, 108–122. [Google Scholar] [CrossRef]
- Addepalli, H.; Meena; Peng, C.G.; Wang, G.; Fan, Y.; Charisse, K.; Jayaprakash, K.N.; Rajeev, K.G.; Pandey, R.K.; Lavine, G.; Zhang, L.; et al. Modulation of thermal stability can enhance the potency of siRNA. Nucleic Acids Res. 2010, 38, 7320–7331. [Google Scholar]
- Chittepu, P.; Sirivolu, V.R.; Seela, F. Nucleosides and oligonucleotides containing 1,2,3-triazole residues with nucleobase tethers: Synthesis via the azide-alkyne ‘click’ reaction. Bioorg. Med. Chem. 2008, 16, 8427–8439. [Google Scholar]
- Ding, P.; Wunnicke, D.; Steinhoff, H.-J.; Seela, F. Site-directed spin-labeling of DNA by the azide-alkyne ‘click’ reaction: Nanometer distance measurements on 7-deaza-2′-deoxyadenosine and 2′-deoxyuridine nitroxide conjugates spatially separated or linked to a ‘dA-dT’ base pair. Chem. Eur. J. 2010, 16, 14385–14396. [Google Scholar] [CrossRef]
- Beyer, C.; Wagenknecht, H.-A. In situ azide formation and “click” reaction of nile red with DNA as an alternative postsynthetic route. Chem. Commun. 2010, 46, 2230–2231. [Google Scholar] [CrossRef]
- Park, S.M.; Yang, H.; Park, S.-K.; Kim, H.M.; Kim, B.H. Design, synthesis, and anticancer activities of novel perfluoroalkyltriazole-appended 2′-deoxyuridines. Bioorg. Med. Chem. Lett. 2010, 20, 5831–5834. [Google Scholar]
- Montagu, A.; Roy, V.; Balzarini, J.; Snoeck, R.; Andrei, G.; Agrofoglio, L.A. Synthesis of new C5-(1-substituted-1,2,3-triazol-4 or 5-yl)-2′-deoxyuridines and their antiviral evaluation. Eur. J. Med. Chem. 2011, 46, 778–786. [Google Scholar] [CrossRef]
- Streeter, D.G.; Witkowski, J.T.; Khare, G.P.; Sidwell, R.W.; Bauer, R.J.; Robins, R.K.; Simon, L.N. Mechanism of action of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc. Natl. Acad. Sci. USA 1973, 70, 1174–1178. [Google Scholar] [CrossRef]
- Lin, J.; Roy, V.; Wang, L.; You, L.; Agrofoglio, L.A.; Deville-Bonne, D.; McBrayer, T.R.; Coats, S.J.; Schinazi, R.F.; Eriksson, S. 3′-(1,2,3-Triazol-1-yl)-3′-deoxythymidine analogs as substrates for human and Ureaplasma parvum thymidine kinase for structure-activity investigations. Bioorg. Med. Chem. 2010, 18, 3261–3269. [Google Scholar] [CrossRef]
- Yu, B.; Zhao, X.; Lee, L.J.; Lee, R.J. Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009, 11, 195–203. [Google Scholar] [CrossRef]
- Crooke, S.T. Basic Principles of Antisense Therapeutics; Springer: Berlin/Heidelberg, Germany, 1998; pp. 1–50. [Google Scholar]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef]
- Andersen, N.K.; Dossing, H.; Jensen, F.; Vester, B.; Nielsen, P. Duplex and triplex formation of mixed pyrimidine oligonucleotides with stacking of phenyl-triazole moieties in the major groove. J. Org. Chem. 2011, 76, 6177–6187. [Google Scholar] [CrossRef]
- Wagner, R.W.; Matteucci, M.D.; Lewis, J.G.; Gutierrez, A.J.; Moulds, C.; Froehler, B.C. Antisense gene inhibition by oligonucleotides containing C-5 propyne pyrimidines. Science 1993, 260, 1510–1513. [Google Scholar]
- Gutierrez, A.J.; Matteucci, M.D.; Grant, D.; Matsumura, S.; Wagner, R.W.; Froehler, B.C. Antisense gene inhibition by C-5-substituted deoxyuridine-containing oligodeoxynucleotides. Biochemistry 1997, 36, 743–748. [Google Scholar]
- Kumar, P.; Chandak, N.; Nielsen, P.; Sharma, P.K. Sulfonamide bearing oligonucleotides: Simple synthesis and efficient RNA recognition. Bioorg. Med. Chem. 2012, 20, 3843–3849. [Google Scholar] [CrossRef]
- Kocalka, P.; Andersen, N.K.; Jensen, F.; Nielsen, P. Synthesis of 5-(1,2,3-triazol-4-yl)-2′-deoxyuridines by a click chemistry approach: Stacking of triazoles in the major groove gives increased nucleic acid duplex stability. ChemBioChem 2007, 8, 2106–2116. [Google Scholar] [CrossRef]
- Whiting, M.; Muldoon, J.; Lin, Y.C.; Silverman, S.M.; Lindstrom, W.; Olson, A.J.; Kolb, H.C.; Finn, M.G.; Sharpless, K.B.; Elder, J.H.; et al. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew. Chem. Int. Ed. Engl. 2006, 45, 1435–1439. [Google Scholar]
- Robins, M.J.; Barr, P.J. Nucleic acid related compounds. 39. Efficient conversion of 5-iodo to 5-alkynyl and derived 5-substituted uracil bases and nucleosides. J. Org. Chem. 1983, 48, 1854–1862. [Google Scholar]
- Bramsen, J.B.; Laursen, M.B.; Nielsen, A.F.; Hansen, T.B.; Bus, C.; Langkjaer, N.; Babu, B.R.; Hojland, T.; Abramov, M.; Van Aerschot, A.; et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009, 37, 2867–2881. [Google Scholar]
- Robbins, M.; Judge, A.; MacLachlan, I. siRNA and innate immunity. Oligonucleotides 2009, 19, 89–101. [Google Scholar] [CrossRef]
- Chang, K.Y.; Ramos, A. The double-stranded RNA-binding motif, a versatile macromolecular docking platform. FEBS J. 2005, 272, 2109–2117. [Google Scholar] [CrossRef]
- Tian, B.; Bevilacqua, P.C.; Diegelman-Parente, A.; Mathews, M.B. The double-stranded-RNA-binding motif: Interference and much more. Nat. Rev. Mol. Cell Biol. 2004, 5, 1013–1023. [Google Scholar] [CrossRef]
- Qing, G.; Xiong, H.; Seela, F.; Sun, T. Spatially controlled DNA nanopatterns by “click” chemistry using oligonucleotides with different anchoring sites. J. Am. Chem. Soc. 2010, 132, 15228–15232. [Google Scholar]
- Seela, F.; Xiong, H.; Leonard, P.; Budow, S. 8-Aza-7-deazaguanine nucleosides and oligonucleotides with octadiynyl side chains: Synthesis, functionalization by the azide-alkyne ‘click’ reaction and nucleobase specific fluorescence quenching of coumarin dye conjugates. Org. Biomol. Chem. 2009, 7, 1374–1387. [Google Scholar] [CrossRef]
- Manalo, M.N.; Perez, L.M.; LiWang, A. Hydrogen-bonding and π-π base-stacking interactions are coupled in DNA, as suggested by calculated and experimental trans H-bond deuterium isotope shifts. J. Am. Chem. Soc. 2007, 129, 11298–11299. [Google Scholar] [CrossRef]
- Kellner, S.; Seidu-Larry, S.; Burhenne, J.; Motorin, Y.; Helm, M. A multifunctional bioconjugate module for versatile photoaffinity labeling and click chemistry of RNA. Nucleic Acids Res. 2011, 39, 7348–7360. [Google Scholar] [CrossRef]
- Onizuka, K.; Shibata, A.; Taniguchi, Y.; Sasaki, S. Pin-point chemical modification of RNA with diverse molecules through the functionality transfer reaction and the copper-catalyzed azide-alkyne cycloaddition reaction. Chem. Commun. 2011, 47, 5004–5006. [Google Scholar] [CrossRef]
- Schiemann, O.; Prisner, T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q Rev. Biophys. 2007, 40, 1–53. [Google Scholar] [CrossRef]
- Bushmakina, N.G.; Misharin, A.Y. A simple synthesis of 4-amino-2,2,6,6-tetramethyl-1-piperidinyloxy radical. Synthesis-Stuttgart 1986, 11, 966. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of 11C, 18F, 15O, and 13N Radiolabels for Positron Emission Tomograpy. Angew. Chem. Int. Ed. Engl. 2008, 47, 8998–9033. [Google Scholar] [CrossRef]
- Kuboyama, T.; Nakahara, M.; Yoshino, M.; Cui, Y.; Sako, T.; Wada, Y.; Imanishi, T.; Obika, S.; Watanabe, Y.; Suzuki, M.; et al. Stoichiometry-focused 18F-labeling of alkyne-substituted oligodeoxynucleotides using azido([18F]fluoromethyl)benzenes by Cu-catalyzed Huisgen reaction. Bioorg. Med. Chem. 2011, 19, 249–255. [Google Scholar]
- Inoue, H.; Imura, A.; Ohtsuka, E. Synthesis of dodecadeoxyribonucleotides containing a pyrrolo[2,3-d]pyrimidine nucleoside and their base-pairing ability. Nippon Kagaku Kaishi 1987, 1214–1220. [Google Scholar]
- Dierckx, A.; Diner, P.; El-Sagheer, A.H.; Kumar, J.D.; Brown, T.; Grotli, M.; Wilhelmsson, L.M. Characterization of photophysical and base-mimicking properties of a novel fluorescent adenine analogue in DNA. Nucleic Acids Res. 2011, 39, 4513–4524. [Google Scholar]
- Dyrager, C.; Borjesson, K.; Diner, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L.M.; Grotli, M. Synthesis and photophysical characterisation of fluorescent 8-(1H-1,2,3-triazol-4-yl)adenosine derivatives. Eur. J. Org. Chem. 2009, 1515–1521. [Google Scholar]
- Seela, F.; Peng, X.; Budow, S. Advances in the synthesis of 7-deazapurine-pyrrolo[2,3-d]pyrimidine-2 ′-deoxyribonucleosides including D- and L-enantiomers, fluoro derivatives and 2′,3′-dideoxyribonucleosides. Curr. Org. Chem. 2007, 11, 427–462. [Google Scholar] [CrossRef]
- Winnik, F.M. Photophysics of preassociated pyrenes in aqueous polymer-solutions and in other organized media. Chem. Rev. 1993, 93, 587–614. [Google Scholar] [CrossRef]
- Ming, X.; Ding, P.; Leonard, P.; Budow, S.; Seela, F. Parallel-stranded DNA: Enhancing duplex stability by the ‘G-clamp ’ and a pyrrolo-dC derivative. Org. Biomol. Chem. 2012, 10, 1861–1869. [Google Scholar] [CrossRef]
- Lin, K.Y.; Matteucci, M.D. A cytosine analogue capable of clamp-like binding to a guanine in helical nucleic acids. J. Am. Chem. Soc. 1998, 120, 8531–8532. [Google Scholar] [CrossRef]
- Koszytkowska-Stawinska, M.; Mironiuk-Puchalska, E.; Rowicki, T. Synthesis of 1,2,3-triazolo-nucleosides via the post-triazole N-alkylation. Tetrahedron 2012, 68, 214–225. [Google Scholar] [CrossRef]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Ganesan, M.; Muraleedharan, K.M. Synthesis of β-hydroxyphosphonate and 1,2-dihydroxy acyclic nucleoside analogs via 1,3-dipolar cycloaddition strategy. Nucleos. Nucleot. Nucl. 2010, 29, 91–96. [Google Scholar] [CrossRef]
- Trakossas, S.; Coutouli-Argyropoulou, E.; Hadjipavlou-Litina, D.J. Synthesis of modified triazole nucleosides possessing one or two base moieties via a click chemistry approach. Tetrahedron Lett. 2011, 52, 1673–1676. [Google Scholar] [CrossRef]
- Duschinsky, R.; Pleven, E.; Heidelberger, C. The synthesis of 5-fluoropyrimidines. J. Am. Chem. Soc. 1957, 79, 4559–4560. [Google Scholar]
- Lakshman, M.K.; Singh, M.K.; Parrish, D.; Balachandran, R.; Day, B.W. Azide-tetrazole equilibrium of C-6 azidopurine nucleosides and their ligation reactions with alkynes. J. Org. Chem. 2010, 75, 2461–2473. [Google Scholar] [CrossRef]
- Mathew, S.C.; By, Y.; Berthault, A.; Virolleaud, M.-A.; Carrega, L.; Chouraqui, G.; Commeiras, L.; Condo, J.; Attolini, M.; Gaudel-Siri, A.; et al. Expeditious synthesis and biological evaluation of new C-6 1,2,3-triazole adenosine derivatives A1 receptor antagonists or agonists. Org. Biomol. Chem. 2010, 8, 3874–3881. [Google Scholar] [CrossRef]
- Driowya, M.; Puissant, A.; Robert, G.; Auberger, P.; Benhida, R.; Bougrin, K. Ultrasound-assisted one-pot synthesis of anti-CML nucleosides featuring 1,2,3-triazole nucleobase under iron-copper catalysis. Ultrason. Sonochem. 2012, 19, 1132–1138. [Google Scholar] [CrossRef]
- Kolganova, N.A.; Florentiev, V.L.; Chudinov, A.V.; Zasedatelev, A.S.; Timofeev, E.N. Simple and stereoselective preparation of an 4-(aminomethyl)-1,2,3-triazolyl nucleoside phosphoramidite. Chem. Biodivers. 2011, 8, 568–576. [Google Scholar] [CrossRef]
- Hou, S.; Liu, W.; Ji, D.; Zhao, Z. Efficient synthesis of triazole moiety-containing nucleotide analogs and their inhibitory effects on a malic enzyme. Bioorg. Med. Chem. Lett. 2011, 21, 1667–1669. [Google Scholar] [CrossRef]
- Bhatt, B.; Thomson, R.J.; von Itzstein, M. An efficient approach to 2,5-anhydro-glucitol-based 1′-homo-N-nucleoside mimetics. Tetrahedron Lett. 2011, 52, 2741–2743. [Google Scholar] [CrossRef]
- Alvarez, R.; Velazquez, S.; Sanfelix, A.; Aquaro, S.; Declercq, E.; Perno, C.F.; Karlsson, A.; Balzarini, J.; Camarasa, M.J. 1,2,3-Triazole-[2′,5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]-3′-spiro-5′′-(4′′-amino-1′′,2′′-oxathiol 2′′,2′′-dioxide) (TSAO) analogs: Synthesis and anti-HIV-1 activity. J. Med. Chem. 1994, 37, 4185–4194. [Google Scholar] [CrossRef]
- Singh, S.K.; Nielsen, P.; Koshkin, A.A.; Wengel, J. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 455–456. [Google Scholar]
- Van Poecke, S.; Negri, A.; Gago, F.; Van Daele, I.; Solaroli, N.; Karlsson, A.; Balzarini, J.; Van Calenbergh, S. 3′-[4-Aryl-(1,2,3-triazol-1-yl)]-3′-deoxythymidine Analogues as Potent and Selective Inhibitors of Human Mitochondrial Thymidine Kinase. J. Med. Chem. 2010, 53, 2902–2912. [Google Scholar] [CrossRef]
- Yu, J.-L.; Wu, Q.-P.; Zhang, Q.-S.; Xi, X.-D.; Liu, N.-N.; Li, Y.-Z.; Liu, Y.-H.; Yin, H.-Q. Synthesis and antitumor activity of novel 2′,3′-diethanethio-2′,3′,5′-trideoxy-5′-triazolonucleoside analogues. Eur. J. Med. Chem. 2010, 45, 3219–3222. [Google Scholar]
- Baraniak, D.; Kacprzak, K.; Celewicz, L. Synthesis of 3′-azido-3′-deoxythymidine (AZT)–Cinchona alkaloid conjugates via click chemistry: Toward novel fluorescent markers and cytostatic agents. Bioorg. Med. Chem. Lett. 2011, 21, 723–726. [Google Scholar] [CrossRef]
- Yamada, T.; Peng, C.G.; Matsuda, S.; Addepalli, H.; Jayaprakash, K.N.; Alam, M.R.; Mills, K.; Maier, M.A.; Charisse, K.; Sekine, M.; et al. Versatile site-specific conjugation of small molecules to siRNA using click chemistry. J. Org. Chem. 2011, 76, 1198–1211. [Google Scholar]
- Roy, V.; Obikhod, A.; Zhang, H.-W.; Coats, S.J.; Herman, B.D.; Sluis-Cremer, N.; Agrofoglio, L.A.; Schinazi, R.F. Synthesis and anti-HIV evaluation of 3′-triazolo nucleosides. Nucleos. Nucleot. Nucl. 2011, 30, 264–270. [Google Scholar] [CrossRef]
- Fauster, K.; Hartl, M.; Santner, T.; Aigner, M.; Kreutz, C.; Bister, K.; Ennifar, E.; Micura, R. 2′-Azido RNA, a versatile tool for chemical biology: Synthesis, X-ray structure, siRNA applications, click labeling. ACS Chem. Biol. 2012, 7, 581–589. [Google Scholar] [CrossRef]
- Kaczmarek, O.; Scheidt, H.A.; Bunge, A.; Foese, D.; Karsten, S.; Arbuzova, A.; Huster, D.; Liebscher, J. 2′-Linking of lipids and other functions to uridine through 1,2,3-triazoles and membrane anchoring of the amphiphilic products. Eur. J. Org. Chem. 2010, 1579–1586. [Google Scholar]
- Sau, S.P.; Hrdlicka, P.J. C2′-Pyrene-functionalized triazole-linked DNA: Universal DNA/RNA hybridization probes. J. Org. Chem. 2012, 77, 5–16. [Google Scholar] [CrossRef]
- Johansson, K.; Ramaswamy, S.; Ljungcrantz, C.; Knecht, W.; Piskur, J.; Munch-Petersen, B.; Eriksson, S.; Eklund, H. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nat. Struct. Biol. 2001, 8, 616–620. [Google Scholar] [CrossRef]
- Hallek, M.; Wanders, L.; Strohmeyer, S.; Emmerich, B. Thymidine kinase: A tumor-marker with prognostic value for non-Hodgkin’s lymphoma and a broad range of potential clinical applications. Ann. Hematol. 1992, 65, 1–5. [Google Scholar] [CrossRef]
- Eriksson, S.; Munch-Petersen, B.; Johansson, K.; Eklund, H. Structure and function of cellular deoxyribonucleoside kinases. Cell. Mol. Life Sci. 2002, 59, 1327–1346. [Google Scholar] [CrossRef]
- Christensen, M.S.; Madsen, C.M.; Nielsen, P. Synthesis and modelling of DNA junction and minor groove zipper motifs incorporating the double-headed nucleoside 5′(S)-C-(thymine-1-ylmethyl)thymidine. Org. Biomol. Chem. 2007, 5, 1586–1594. [Google Scholar] [CrossRef]
- Aigner, M.; Hartl, M.; Fauster, K.; Steger, J.; Bister, K.; Micura, R. Chemical synthesis of site-specifically 2′-azido-modified RNA and potential applications for bioconjugation and RNA interference. ChemBioChem 2011, 12, 47–51. [Google Scholar] [CrossRef]
- Staudinger, H.; Meyer, J. On new organic phosphorus bonding III phosphine methylene derivatives and phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar] [CrossRef]
- Micura, R. Cyclic Oligoribonucleotides (RNA) by solid-phase synthesis. Chem. Eur. J. 1999, 5, 2077–2082. [Google Scholar] [CrossRef]
- Birks, J.B. Excimers. Rep. Prog. Phys. 1975, 38, 903–974. [Google Scholar] [CrossRef]
- Caruthers, M.H. Chemical synthesis of DNA and DNA analogs. Accounts Chem. Res. 1991, 24, 278–284. [Google Scholar] [CrossRef]
- Paredes, E.; Das, S.R. Click chemistry for rapid labeling and ligation of RNA. ChemBioChem 2011, 12, 125–131. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Srihari, P.; Nagesh, C.; Kiranmai, N.; Nagesh, N.; Idris, M.M. Synthesis of readily accessible triazole-linked dimer deoxynucleoside phosphoramidite for solid-phase oligonucleotide synthesis. Synthesis-Stuttgart 2010, 3710–3714. [Google Scholar]
- Chandrasekhar, S.; Rambabu, C.; Reddy, A.S. Spirastrellolide B: the synthesis of southern (C9–C25) region. Org. Lett. 2008, 10, 4355–4357. [Google Scholar] [CrossRef]
- Corey, E.J.; Fuchs, P.L. Synthetic method for formyl->ethynyl conversion (RCHO->RC=CH or RC=CR′). Tetrahedron Lett. 1972, 3769–3772. [Google Scholar]
- Varizhuk, A.; Chizhov, A.; Smirnov, I.; Kaluzhny, D.; Florentiev, V. Triazole-Linked Oligonucleotides with Mixed-Base Sequences: Synthesis and Hybridization Properties. Eur. J. Org. Chem. 2012, 11, 2173–2179. [Google Scholar]
- El-Sagheer, A.H.; Brown, T. Synthesis and Polymerase Chain Reaction Amplification of DNA Strands Containing an Unnatural Triazole Linkage. J. Am. Chem. Soc. 2009, 131, 3958–3964. [Google Scholar] [CrossRef]
- Montevecchi, P.C.; Manetto, A.; Navacchia, M.L.; Chatgilialoglu, C. Thermal decomposition of the tert-butyl perester of thymidine-5′-carboxylic acid. Formation and fate of the pseudo-C4′ radical. Tetrahedron 2004, 60, 4303–4308. [Google Scholar]
- Zhang, J.C.; Matteucci, M.D. Synthesis of a N-acylsulfamide linked dinucleoside and its incorporation into an oligonucleotide. Bioorg. Med. Chem. Lett. 1999, 9, 2213–2216. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Sanzone, A.P.; Gao, R.; Tavassoli, A.; Brown, T. Biocompatible artificial DNA linker that is read through by DNA polymerases and is functional in Escherichia coli. Proc. Natl. Acad. Sci. USA 2011, 108, 11338–11343. [Google Scholar]
- Dallmann, A.; El-Sagheer, A.H.; Dehmel, L.; Mugge, C.; Griesinger, C.; Ernsting, N.P.; Brown, T. Structure and Dynamics of Triazole-Linked DNA: Biocompatibility Explained. Chem. Eur. J. 2011, 17, 14714–14717. [Google Scholar] [CrossRef]
- Mutisya, D.; Selvam, C.; Kennedy, S.D.; Rozners, E. Synthesis and properties of triazole-linked RNA. Bioorg. Med. Chem. Lett. 2011, 21, 3420–3422. [Google Scholar] [CrossRef]
- Isobe, H.; Fujino, T.; Yamazaki, N.; Guillot-Nieckowski, M.; Nakamura, E. Triazole-linked analogue of deoxyribonucleic acid (TLDNA): Design, synthesis, and double-strand formation with natural DNA. Org. Lett. 2008, 10, 3729–3732. [Google Scholar] [CrossRef]
- Fujino, T.; Endo, K.; Yamazaki, N.; Isobe, H. Synthesis of triazole-linked analogues of RNA (TLRNA). Chem. Lett. 2012, 41, 403–405. [Google Scholar] [CrossRef]
- Efthymiou, T.C.; Desaulniers, J.-P. Synthesis and properties of oligonucleotides that contain a triazole-linked nucleic acid dimer. J. Heterocycl. Chem. 2011, 48, 533–539. [Google Scholar] [CrossRef]
- Efthymiou, T.C.; Vanthi, H.; Oentoro, J.; Peel, B.; Desaulniers, J.-P. Efficient synthesis and cell-based silencing activity of siRNAs that contain triazole backbone linkages. Bioorg. Med. Chem. Lett. 2012, 22, 1722–1726. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar]
- Chouikhi, D.; Barluenga, S.; Winssinger, N. Clickable peptide nucleic acids (cPNA) with tunable affinity. Chem. Commun. 2010, 46, 5476–5478. [Google Scholar] [CrossRef]
- Thomson, S.A.; Josey, J.A.; Cadilla, R.; Gaul, M.D.; Hassman, C.F.; Luzzio, M.J.; Pipe, A.J.; Reed, K.L.; Ricca, D.J.; Wiethe, R.W.; et al. Fmoc mediated synthesis of peptide nucleic acids. Tetrahedron 1995, 51, 6179–6194. [Google Scholar] [CrossRef]
- Kumar, R.; El-Sagheer, A.; Tumpane, J.; Lincoln, P.; Wilhelmsson, L.M.; Brown, T. Template-directed oligonucleotide strand ligation, covalent intramolecular DNA circularization and catenation using click chemistry. J. Am. Chem. Soc. 2007, 129, 6859–6864. [Google Scholar]
- Eckstein, F. Nucleoside phosphorothioates. J. Am. Chem. Soc. 1966, 88, 4292–4294. [Google Scholar] [CrossRef]
- Singh, Y.; Spinelli, N.; Defrancq, E. Chemical strategies for oligonucleotide-conjugates synthesis. Curr. Org. Chem. 2008, 12, 263–290. [Google Scholar] [CrossRef]
- Sameiro, M.; Goncalves, T. Fluorescent labeling of biomolecules with organic probes. Chem. Rev. 2009, 109, 190–212. [Google Scholar] [CrossRef]
- Best, M.D. Click chemistry and bioorthogonal reactions: Unprecedented selectivity in the labeling of biological molecules. Biochemistry 2009, 48, 6571–6584. [Google Scholar] [CrossRef]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef]
- Englisch, U.; Gauss, D.H. Chemically modified oligonucleotides as probes and inhibitors. Angew. Chem. Int. Ed. Engl. 1991, 30, 613–629. [Google Scholar] [CrossRef]
- Nitin, N.; Santangelo, P.J.; Kim, G.; Nie, S.M.; Bao, G. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004, 32, 1–8. [Google Scholar]
- Eguchi, A.; Akuta, T.; Okuyama, H.; Senda, T.; Yokoi, H.; Inokuchi, H.; Fujita, S.; Hayakawa, T.; Takeda, K.; Hasegawa, M.; et al. Protein transduction domain of HIV-1 tat protein promotes efficient delivery of DNA into mammalian cells. J. Biol. Chem. 2001, 276, 26204–26210. [Google Scholar]
- Brown, S.D.; Graham, D. Conjugation of an oligonucleotide to Tat, a cell-penetrating peptide, via click chemistry. Tetrahedron Lett. 2010, 51, 5032–5034. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P.; Lonnberg, H. Solid-supported synthesis and click conjugation of 4′-C-alkyne functionalized oligodeoxyribonucleotides. Bioconjug. Chem. 2010, 21, 1890–1901. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom transfer radical polymerization: From mechanisms to applications. Isr. J. Chem. 2012, 52, 206–220. [Google Scholar] [CrossRef]
- Pan, P.J.; Fujita, M.; Ooi, W.Y.; Sudesh, K.; Takarada, T.; Goto, A.; Maeda, M. DNA-functionalized thermoresponsive bioconjugates synthesized via ATRP and click chemistry. Polymer 2011, 52, 895–900. [Google Scholar]
- Averick, S.; Paredes, E.; Li, W.W.; Matyjaszewski, K.; Das, S.R. Direct DNA conjugation to star polymers for controlled reversible assemblies. Bioconjug. Chem. 2011, 22, 2030–2037. [Google Scholar] [CrossRef]
- Jewett, J.C.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [Google Scholar] [CrossRef]
- van Delft, P.; Meeuwenoord, N.J.; Hoogendoorn, S.; Dinkelaar, J.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V. Synthesis of oligoribonucleic acid conjugates using a cyclooctyne phosphoramidite. Org. Lett. 2010, 12, 5486–5489. [Google Scholar]
- Jayaprakash, K.N.; Peng, C.G.; Butler, D.; Varghese, J.P.; Maier, M.A.; Rajeev, K.G.; Manoharan, M. Non-nucleoside building blocks for copper-assisted and copper-free click chemistry for the efficient synthesis of RNA conjugates. Org. Lett. 2010, 12, 5410–5413. [Google Scholar]
- Boisgard, R.; Kuhnast, B.; Vonhoff, S.; Younes, C.; Hinnen, F.; Verbavatz, J.M.; Rousseau, B.; Furste, J.; Wlotzka, B.; Dolle, F.; et al. In vivo biodistribution and pharmacokinetics of 18F-labelled Spiegelmers: A new class of oligonucleotidic radiopharmaceuticals. Eur. J. Nucl. Med. Mol. I. 2005, 32, 470–477. [Google Scholar] [CrossRef]
- Viel, T.; Boisgard, R.; Kuhnast, B.; Jego, B.; Siquier-Pernet, K.; Hinnen, F.; Dolle, F.; Tavitian, B. Molecular imaging study on in vivo distribution and pharmacokinetics of modified small interfering RNAs (siRNAs). Oligonucleotides 2008, 18, 201–212. [Google Scholar] [CrossRef]
- Inkster, J.A.H.; Guerin, B.; Ruth, T.J.; Adam, M.J. Radiosynthesis and bioconjugation of [18F]FPy5yne, a prosthetic group for the 18F labeling of bioactive peptides. J. Labelled Compd. Rad. 2008, 51, 444–452. [Google Scholar] [CrossRef]
- Inkster, J.A.H.; Adam, M.J.; Storr, T.; Ruth, T.J. Labeling of an antisense oligonucleotide with [18F]FPy5yne. Nucleos. Nucleot. Nucl. 2009, 28, 1131–1143. [Google Scholar] [CrossRef]
- Mercier, F.; Paris, J.; Kaisin, G.; Thonon, D.; Flagothier, J.; Teller, N.; Lemaire, C.; Luxen, A. General method for labeling siRNA by click chemistry with fluorine-18 for the purpose of PET imaging. Bioconjug. Chem. 2011, 22, 108–114. [Google Scholar] [CrossRef]
- Lachmann, D.; Berndl, S.; Wolfbeis, O.S.; Wagenknecht, H.-A. Synthetic incorporation of Nile Blue into DNA using 2′-deoxyriboside substitutes: Representative comparison of (R)- and (S)-aminopropanediol as an acyclic linker. Beilstein J. Org. Chem. 2010, 6, 1–7. [Google Scholar]
- Takarada, T.; Tamaru, D.; Liang, X.G.; Asanuma, H.; Komiyama, M. L-Threoninol as a chiral linker of azobenzene for the effective photo-regulation of DNA triplex formation. Chem. Lett. 2001, 30, 732–733. [Google Scholar]
- Asanuma, H.; Takarada, T.; Yoshida, T.; Tamaru, D.; Liang, X.G.; Komiyama, M. Enantioselective incorporation of azobenzenes into oligodeoxyribonucleotide for effective photoregulation of duplex formation. Angew. Chem. Int. Ed. Engl. 2001, 40, 2671–2673. [Google Scholar] [CrossRef]
- Shi, Y.; Machida, K.; Kuzuya, A.; Komiyama, M. Design of phosphoramidite monomer for optimal incorporation of functional intercalator to main chain of oligonucleotide. Bioconjug. Chem. 2005, 16, 306–311. [Google Scholar] [CrossRef]
- Li, X.Y.; Liu, D.R. DNA-templated organic synthesis: Nature ’s strategy for controlling chemical reactivity applied to synthetic molecules. Angew. Chem. Int. Ed. Engl. 2004, 43, 4848–4870. [Google Scholar] [CrossRef]
- Chen, Y.; Mao, C. Reprogramming DNA-directed reactions on the basis of a DNA conformational change. J. Am. Chem. Soc. 2004, 126, 13240–13241. [Google Scholar] [CrossRef]
- Jacobsen, M.F.; Ravnsbaek, J.B.; Gothelf, K.V. Small molecule induced control in duplex and triplex DNA-directed chemical reactions. Org. Biomol. Chem. 2010, 8, 50–52. [Google Scholar] [CrossRef]
- Nikan, M.; Bare, G.A.L.; Sherman, J.C. Synthesis of a water-soluble triazole-linked cavitand-guanosine conjugate. Tetrahedron Lett. 2011, 52, 1791–1793. [Google Scholar] [CrossRef]
- Ou, T.-M.; Lu, Y.-J.; Tan, J.-H.; Huang, Z.-S.; Wong, K.-Y.; Gu, L.-Q. G-quadruplexes: Targets in anticancer drug design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef]
- Seela, F.; Sirivolu, V.R. DNA containing side chains with terminal triple bonds: Base-pair stability and functionalization of alkynylated pyrimidines and 7-deazapurines. Chem. Biodivers. 2006, 3, 509–514. [Google Scholar] [CrossRef]
- Seela, F.; Sirivolu, V.R. Nucleosides and oligonucleotides with diynyl side chains: Base pairing and functionalization of 2′-deoxyuridine derivatives by the copper(I)-catalyzed alkyne-azide ′click′ cycloaddition. Helv. Chim. Acta 2007, 90, 535–552. [Google Scholar] [CrossRef]
- Seela, F.; Sirivolu, V.R.; Chittepu, P. Modification of DNA with octadiynyl side chains: Synthesis, base pairing, and formation of fluorescent coumarin dye conjugates of four nucleobases by the alkyne–azide “click” reaction. Bioconjug. Chem. 2008, 19, 211–224. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Efthymiou, T.; Gong, W.; Desaulniers, J.-P. Chemical Architecture and Applications of Nucleic Acid Derivatives Containing 1,2,3-Triazole Functionalities Synthesized via Click Chemistry. Molecules 2012, 17, 12665-12703. https://doi.org/10.3390/molecules171112665
Efthymiou T, Gong W, Desaulniers J-P. Chemical Architecture and Applications of Nucleic Acid Derivatives Containing 1,2,3-Triazole Functionalities Synthesized via Click Chemistry. Molecules. 2012; 17(11):12665-12703. https://doi.org/10.3390/molecules171112665
Chicago/Turabian StyleEfthymiou, Tim, Wei Gong, and Jean-Paul Desaulniers. 2012. "Chemical Architecture and Applications of Nucleic Acid Derivatives Containing 1,2,3-Triazole Functionalities Synthesized via Click Chemistry" Molecules 17, no. 11: 12665-12703. https://doi.org/10.3390/molecules171112665
APA StyleEfthymiou, T., Gong, W., & Desaulniers, J.-P. (2012). Chemical Architecture and Applications of Nucleic Acid Derivatives Containing 1,2,3-Triazole Functionalities Synthesized via Click Chemistry. Molecules, 17(11), 12665-12703. https://doi.org/10.3390/molecules171112665