The Expanding Role of Electrospray Ionization Mass Spectrometry for Probing Reactive Intermediates in Solution

Abstract
:1. Introduction
2. Reactive Intermediates Occurring in Liquid-Phase Reactions
3. Challenges for Analyzing Reaction Intermediates in Complex Systems
4. Current Strategies for Detecting Reaction Intermediates by ESI-MS
4.1. Basics Principles of ESI-MS Detection of Reaction Intermediates in Solution Mixture

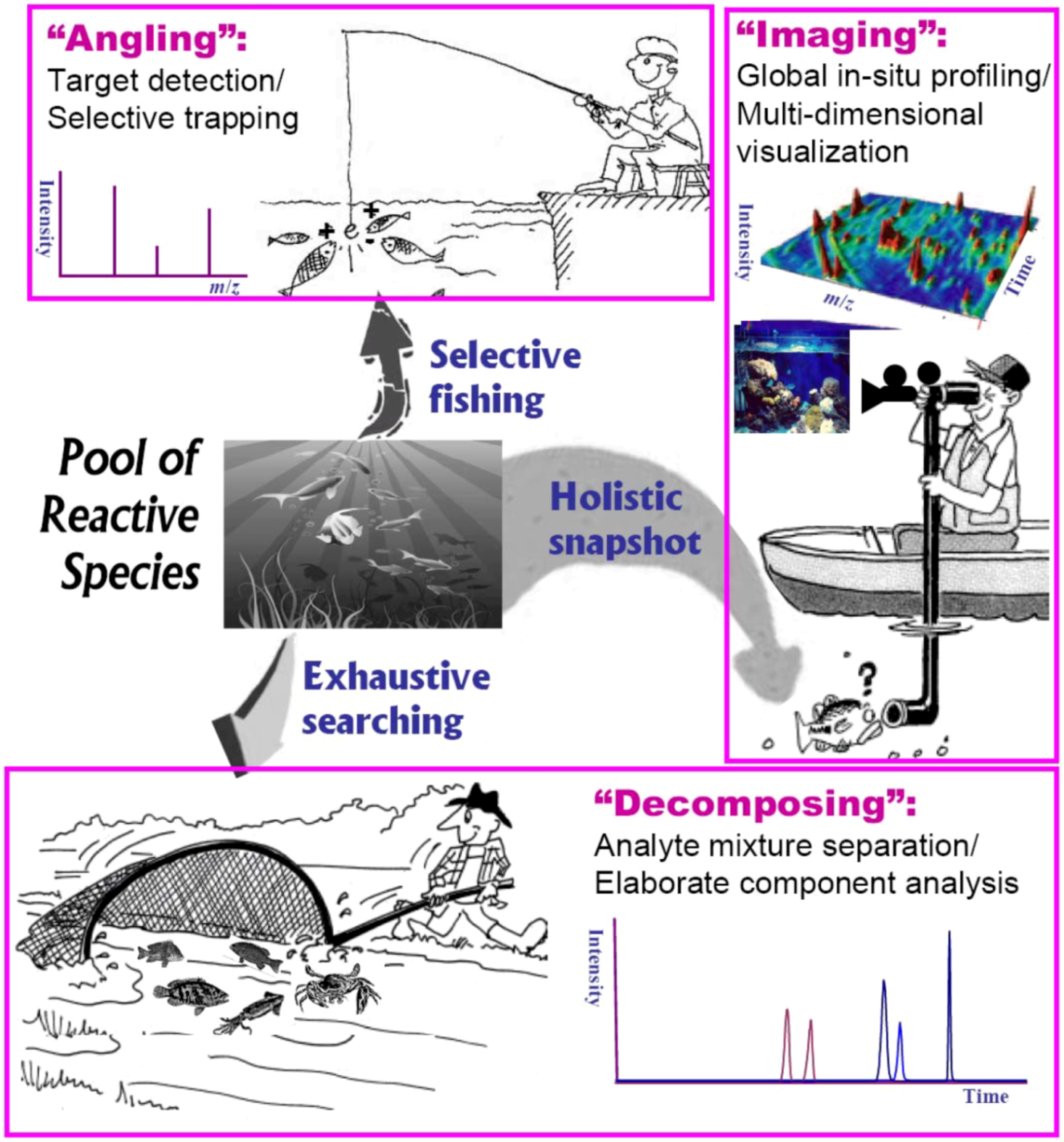
4.2. Generic Tactics for Probing Reactive Intermediates in Solution
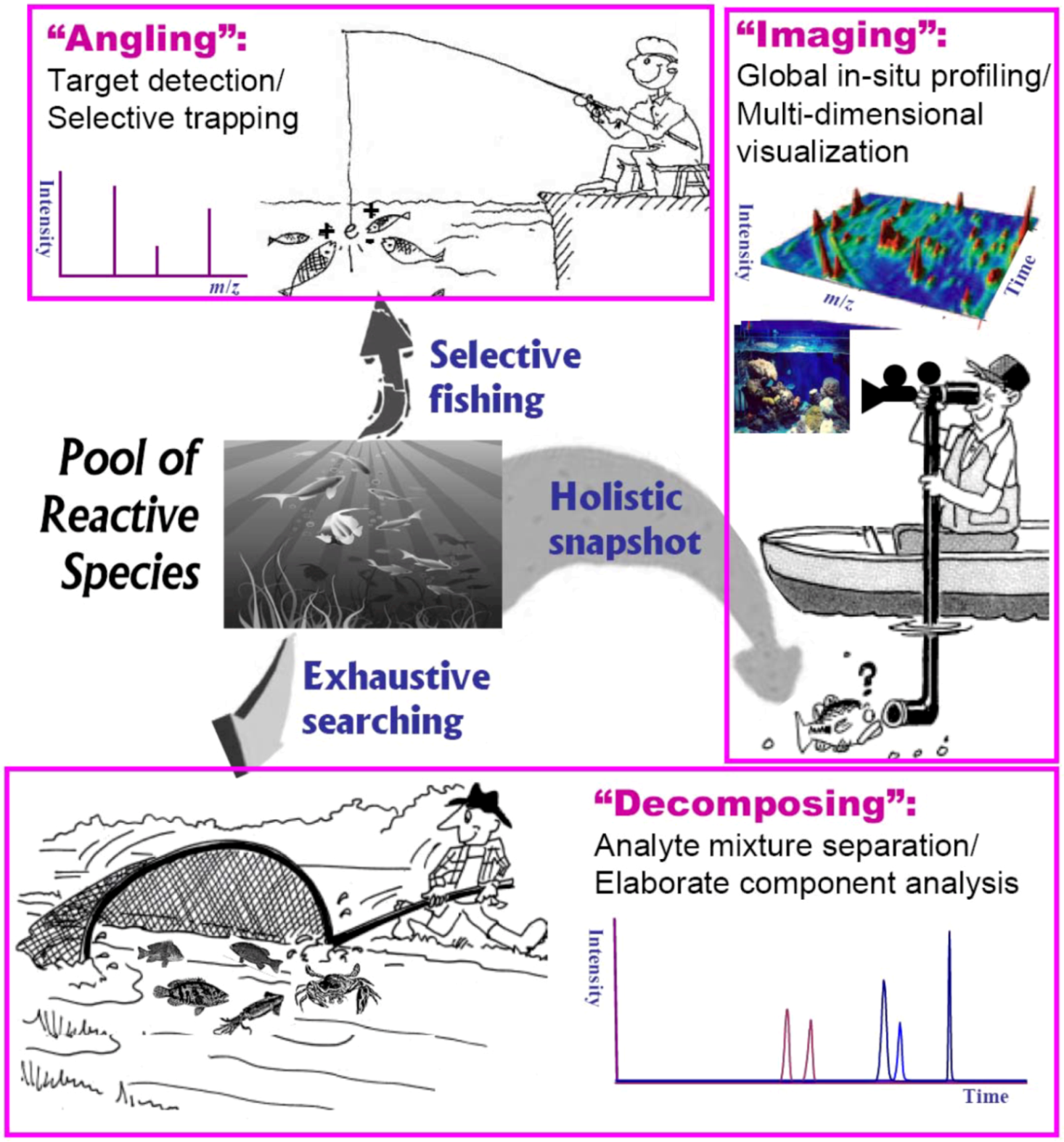
4.3. ESI-MS Monitoring of Intermediates in Reaction Process
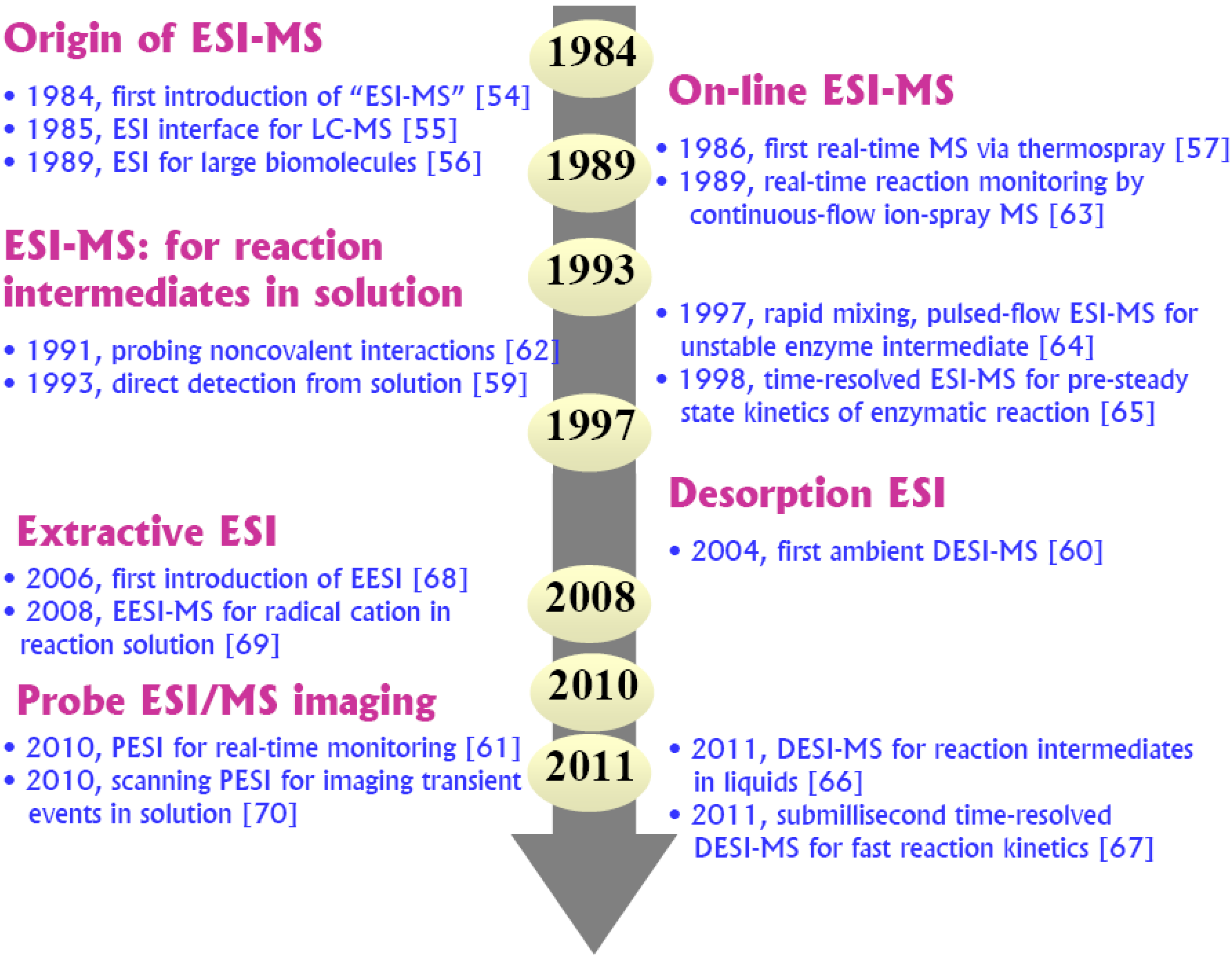
5. Recent Advances in ESI-MS Approaches to Probing Reactive Intermediates
5.1. Wet-Chemical Treatments for Extending Detection Scope

{kind=link}
{kind=link}
| Reactive species | Origin | Wet chemistry reagent | Intended use | Reference |
|---|---|---|---|---|
| Monophosphine norbornene complex | Ring-opening metathesis polymerization (ROMP) of norbornene | Norbornene-CH2P+Ph3Cl−; -CH2N+(CH3)2(CH2Ph) Cl− (as charge-tagged substrate) | Charge tag (+) | [53,72] |
| Pd-containing intermediates | Cu-free Sonogashira (Heck alkynylation) | [p-IC6H4CH2PPh3]+[PF6]− (as charge-tagged substrate) | Charge tag (+) | [100] |
| Organopalladium species | Pd-mediated (Heck) cross-coupling reactions | [p-IC6H4N(CH3)3]+[I]− (as charge-tagged substrate) | Charge tag (+) | [101] |
| Bis(phosphino) palladium species | Pd-catalysed (Sonogashira) cross-coupling reaction | [Na]+[PPh2(m-C6H4SO3)]− (as charge-tagged substrate) | Charge tag (−) | [102] |
| Palladium complex | Suzuki and Heck Phosphine-Free Reactions | Acetate anion bearing an imidazolium cation (ligands) | Charge tag (+) | [103] |
| Neutral radicals | Radical chain reactions | Lewis acids: Sc(Otf)3 | Cationization as [R∙Sc(Otf)2]+ | [12,104] |
| Ru-carbene species | Ru-carbene based olefin metathesis | Alkali-metal salts | Cationization as alkali adducts | [89] |
| Pd hydride; Neutral Pd(II) complex | Pd-catalyzed addition of allenes to organoboronic acids | CH3COOH | Facilitating ESI (–e and −H∙) to form cationic Pd complex | [97] |
| Hydroxyl-sulfonamide | Metabolic bioactivation of sulfonamide | Ascorbic acid | Stabilizer to inhibit oxidation | [99] |
| Reactive metabolites | Electrochemical simulation of oxidative metabolism | Ferrocenylpropionate (FP)-GSH | Trapping agent; Retention tag | [96] |
| Reactive drug metabolites | P450-mediated drug bioactivation | Deuterium labeled bis-methyl GSH esters (GSH(CD3)2) | Trapping agent; Ion-signal sensitizer | [80] |
| Bioactivated intermediates | Bioactivation of xenobiotics | D-Isomer of peptide: gly-tyr-pro-cys-pro-his-pro | Trapping agent | [105] |
| Radical intermediaries | (Glyco-)xidation of phosphatidylethanolamine | 5,5-dimethyl-pyrroline N-oxide (DMPO) | Spin trap | [106] |
| Electrophilic species | P450-mediated drug bioactivation | N-(2-bromocarbobenzyloxy)-GSH (GSH-Br) | Trapping agent | [95] |
| Epoxide metabolites | in vitro metabolic bioactivation | Cob(I)alamin | Trapping agent; Charge tag | [107] |
| Reactive metabolites | P450-mediated drug bioactivation | Stable isotope labeled GSH, KCN and semicarbazide | Trapping agents; Isotopic tagging | [108,109] |
| Reactive metabolites | UGT-mediated drug bioactivation | N-acetylcysteine (NAC) | Trapping agent | [110] |
| Reactive metabolites | P450-mediated drug bioactivation | quaternary ammonium GSH conjugating agent (QA-GSH) | Trapping agent; Semiquantitation tag | [93] |
5.2. Current Evolutions of Time-resolved ESI-MS for Complex Solutions
| ESI type (Acronym) | Date of origin | Ionization principle | Significant feature | Reference |
|---|---|---|---|---|
| ESI | 1984 | Electrospray ionization | API for ions in solution | [54] |
| DEP | 1999 | Direct electrospray probe | Electrospray without capillary | [117] |
| FD-ESI | 2002 | Fused-droplet electrospray ionization | Extremely high salt tolerance | [118] |
| DESI | 2004 | Desorption electrospray ionization | Direct ambient MS sampling | [60] |
| ELDI | 2005 | Electrospray laser desorption ionization | Additional selectivity and scope | [115] |
| EESI | 2006 | Extractive electrospray ionization | Liquid extraction between two sprayers | [68] |
| Reactive DESI | 2006 | Reactive desorption electrospray ionization | On-line reaction for specific identification | [114] |
| PESI | 2007 | Probe electrospray ionization | Solid needle for non-invasive ESI | [61,119] |
| MALDESI | 2007 | Matrix-assisted laser desorption electrospray ionization | Shot-to-shot reproducibility; Limitations in spatial resolution | [115] |
| LAESI | 2007 | Laser ablation electrospray ionization | 3-D imaging biomolecular distributions | [120] |
| ND-EESI | 2007 | Neutral desorption extractive electrospray ionization | Direct ionization of nonvolatile analytes inside a heterogeneous or viscous matrix | [121] |
| IR-LADESI | 2008 | Infrared laser-assisted desorption electrospray ionization | Direct analysis of water-containing samples under ambient conditions | [115] |
| DEMI | 2009 | Desorption electrospray metastable-induced ionization | Direct multimode detection of intact molecules | [116] |
5.2.1. Ambient ESI for Direct Ionization of Liquid Samples
5.2.2. Desorption ESI-MS for Monitoring Fast Reactions and Intermediates
| (Bio)chemical reaction | Intermediate species | Instrumentation (reaction-sampling-ionization-detection) | Temporal resolution | Reference |
|---|---|---|---|---|
| Synthase catalyzed reaction | Tetrahedral intermediate | Pulsed flow (rapid-mixing) device—ESI-QMS | 30 ms | [64] |
| Pre-steady state enzymatic kinetic | Transient enzyme intermediate | Two syringes in a reaction mixing tee—ESI-QMS | tens of ms | [65] |
| Chlorophyll demetalation | Specific reactive species (time profile) | Capillary mixer with adjustable reaction chamber volume—ESI-QQQ | ms | [111] |
| Pd(PPh3)4 decomposition | Pd-containing reactive species | Continuous pressurized sample infusion—ESI-QTOF | N/A | [145] |
| Sandmeyer’s cyclization | Three new cationic intermediate | Microreactor—ESI-QTOF | subs | [146] |
| Electro-oxidation | Perylene radical cation | Electrochemical cell—DESI-QTrap | N/A | [123] |
| Pyrolytic reactions | Reactive ketenes | Flow pyrolyzer-multichannel ESI-QQQ | ~0.2 s | [147] |
| Electron-transfer cata-lyzed dimerization | Distonic tetramethylene radical cation | Gas/liquid setup—EESI-QTOF; Liquid/liquid setup—EESI-QTOF | ms | [69] |
| Catalytic transfer hydrogenation | Ru-complex intermediates | Microdroplet reaction vessel for reactive DESI-IT | ms | [66] |
| Catalytic transfer hydrogenation | Transient Ru-methyl formate species | Microdroplet reaction vessel for reactive DESI-Orbitrap | subms-ms | [50] |
| Morita-Baylis-Hillman reaction | Two key MBH intermediates | Venturi easy ambient sonic-spray ionization (V-EASI)-QMS | N/A | [135] |
| KDO8P synthase reaction | Noncovalent acyclic hemiketal intermediate | Rapid mixing device—ESI-QTOF | 50−630 ms | [148] |
| Eschweiler-Clarke reaction | Reactive iminium ion; Sodiated amino alcohol | Reactive DESI-IT | ms | [137] |
| Fast oxidations of I− and S2O32− by O3 | Short-lived ISO3− and IS2O3− | Reactive DESI-QMS | ~1 ms | [149] |
| Catalytic acetylation of benzyl alcohol | Positively charged intermediates | Online ND setup—EESI-QTOF | <1 s | [126] |
| Borsche-Drechsel cyclization | Ionic intermediate (protonated hydrazone) | Electrosonic spray ionization (ESSI)-IT | N/A | [150] |
| Zemplén deprotection | Mono-deprotection intermediate | Capillary action supported sampling tool—contactless API emitter-IT | 1 min | [15] |
| Electrochemical red/oxidization | N-hydroxyl and amine labile intermediates | Electrochemical flow cell—nanoDESI-LTQ/Orbitrap | N/A | [151] |
| Schiff base formation | Hemiacetals | PESI-QTOF | 0.1−0.33 s | [61] |
| Monitoring of 3-D cell culture system | (Bio)chemical transients | Inline microdialysis—ambient nanoESI-QTOF | 100 μm (spatial) | [152] |
5.3. Emerging ESI-MS-Based Reaction Profiling and in-Situ Imaging
6. Conclusions
Acknowledgments
References
- Turecek, F. Transient intermediates of chemical reactions by neutralization-reionization mass spectrometry. Top. Curr. Chem. 2003, 225, 77–129. [Google Scholar] [CrossRef]
- Fabris, D. Mass spectrometric approaches for the investigation of dynamic processes in condensed phase. Mass Spectrom. Rev. 2005, 24, 30–54. [Google Scholar] [CrossRef]
- Santos, L.S. Online Mechanistic Investigations of Catalyzed Reactions by Electrospray Ionization Mass Spectrometry: A Tool to Intercept Transient Species in Solution. Eur. J. Org. Chem. 2008, 2, 235–253. [Google Scholar] [CrossRef]
- Park, B.K.; Boobis, A.; Clarke, S.; Goldring, C.E.; Jones, D.; Kenna, J.G.; Lambert, C.; Laverty, H.G.; Naisbitt, D.J.; Nelson, S.; et al. Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discov. 2011, 10, 292–306. [Google Scholar] [CrossRef]
- Leung, L.; Kalgutkar, A.S.; Obach, R.S. Metabolic activation in drug-induced liver injury. Drug Metabol. Rev. 2012, 44, 18–33. [Google Scholar] [CrossRef]
- Fu, P.P.; Xia, Q.; Sun, X.; Yu, H. Phototoxicity and environmental transformation of polycyclic aromatic hydrocarbons (PAHs)-light-induced reactive oxygen species, lipid peroxidation, and DNA damag. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2012, 30, 1–41. [Google Scholar]
- Zeng, X.Q.; Wang, D.X. Novel gaseous transient species: Generation and characterization. Sci. China Ser. B 2007, 50, 145–169. [Google Scholar] [CrossRef]
- Dorfman, L.M.; DePalma, V.M. Fast reaction studies of carbocations in solution. Pure Appl. Chem. 1979, 51, 123–129. [Google Scholar] [CrossRef]
- Almond, M.J.; Jenkins, S.L. Short-Lived. Intermediates. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Wang, Y.T.; Jin, K.J.; Leopold, S.H.; Wang, J.; Peng, H.L.; Platz, M.S.; Xue, J.; .Phillips, D.L.; Glover, S.A.; Novak, M. Characterization of reactive intermediates generated during photolysis of 4-acetoxy-4-aryl-2,5-cyclohexadienones: Oxenium ions and aryloxy radicals. J. Am. Chem. Soc. 2008, 130, 16021–16030. [Google Scholar]
- Sawyer, K.R.; Cahoon, J.F.; Shanoski, J.E.; Glascoe, E.A.; Kling, M.F.; Schlegel, J.P.; Zoerb, M.C.; Hapke, M.; Hartwig, J.F.; Webster, C.E.; et al. Time-resolved IR studies on the mechanism for the functionalization of primary C-H bonds by photoactivated Cp*W(CO)3(Bpin). J. Am. Chem. Soc. 2010, 132, 1848–1859. [Google Scholar]
- Griep-Raming, J.; Meyer, S.; Bruhn, T.; Metzger, J.O. Investigation of reactive intermediates of chemical reactions in solution by electrospray ionization mass spectrometry: Radical chain reactions. Angew Chem. Int. Ed. Engl. 2002, 41, 2738–2742. [Google Scholar] [CrossRef]
- Schrader, W.; Handayani, P.P.; Zhou, J.; List, B. Characterization of key intermediates in a complex organocatalytic cascade reaction using mass spectrometry. Angew Chem. Int. Ed. Engl. 2009, 48, 1463–1466. [Google Scholar] [CrossRef]
- Johansson, J.R.; Nordén, B. Sniffing out early reaction intermediates. Proc. Natl. Acad. Sci. USA 2012, 109, 2186–2187. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Chao, C.S.; Mong, K.K.; Chen, Y.C. Online monitoring of chemical reactions by contactless atmospheric pressure ionization mass spectrometry. J. Mass Spectrom. 2012, 47, 586–590. [Google Scholar] [CrossRef]
- Eric, W.; Diau, G.; Casanova, J.; Roberts, J.D.; Zewail, A.H. Chemistry: Femtosecond observation of benzyne intermediates in a molecular beam: Bergman rearrangement in the isolated molecule. Proc. Natl. Acad. Sci. USA 2000, 97, 1376–1379. [Google Scholar]
- Vivekananda, S.; Sadilek, M.; Polasek, M.; Turecek, F. Lactone enols are stable in the gas phase but highly unstable in solution. J. Am. Chem. Soc. 2002, 124, 13282–13289. [Google Scholar] [CrossRef]
- Dmitri, V.; Zagorevskii, J.; Holmes, L. Neutralization-reionization mass spectrometry applied to organometallic and coordination chemistry (update: 1994–1998). Mass Spectrom. Rev. 1999, 18, 87–118. [Google Scholar] [CrossRef]
- Coelho, F.; Eberlin, M.N. The bridge connecting gas-phase and solution chemistries. Angew Chem. Int. Ed. Engl. 2011, 50, 5261–5263. [Google Scholar] [CrossRef]
- Laiko, V.V.; Baldwin, M.A.; Burlingame, A.L. Atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2000, 72, 652–657. [Google Scholar] [CrossRef]
- Santos, L.S.; Knaack, L.; Metzger, J.O. Investigation of chemicalreactions in solution using API-MS. Int. J. Mass Spectrom. 2005, 246, 84–104. [Google Scholar] [CrossRef]
- Meurer, E.C.; da Rocha, L.L.; Pilli, R.A.; Eberlin, M.N.; Santos, L.S. Transient intermediates of the Tebbe reagent intercepted and characterized by atmospheric pressure chemical ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 2626–2629. [Google Scholar] [CrossRef]
- Meurer, E.C.; Santos, L.S.; Pilli, R.A.; Eberlin, M.N. Probing the mechanism of the Petasis olefination reaction by atmospheric pressure chemical ionization mass and tandem mass spectrometry. Org. Lett. 2003, 5, 1391–1394. [Google Scholar] [CrossRef]
- Eberlin, M.N. Electrospray ionization mass spectrometry: A major tool to investigate reaction mechanisms in both solution and the gas phase. Eur. J. Mass Spectrom. 2007, 13, 19–28. [Google Scholar] [CrossRef]
- Schröder, D. Applications of electrospray ionization mass spectrometry in mechanistic studies and catalysis research. Acc. Chem. Res. 2012, 45, 1521–1532. [Google Scholar] [CrossRef]
- Dalmazio, I.; Santos, L.S.; Lopes, R.P.; Eberlin, M.N.; Augusti, R. Advanced oxidation of caffeine in water: On-line and real-time monitoring by electrospray ionization mass spectrometry. Environ. Sci. Technol. 2005, 39, 5982–5988. [Google Scholar] [CrossRef]
- Santos, L.S. Reactive Intermediates: MS Investigations in Solution, 1st ed; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 63–198. [Google Scholar]
- Meyer, S.; Metzger, J.O. Use of electrospray ionization mass spectrometry for the investigation of radical cation chain reactions in solution: Detection of transient radical cations. Anal. Bioanal. Chem. 2003, 377, 1108–1114. [Google Scholar] [CrossRef]
- Roithova, J. Characterization of reaction intermediates by ion spectroscopy. Chem. Soc. Rev. 2012, 41, 547–559. [Google Scholar] [CrossRef]
- Bortolini, O.; Conte, V. Mass spectrometric characterization of high-valent metal-oxo, -peroxo and -peroxy intermediates of relevance in oxidation processes. Mass Spectrom. Rev. 2006, 25, 724–740. [Google Scholar]
- Guengerich, F.P. Generation of reactive intermediates. J. Biochem. Mol. Toxicol. 2005, 19, 173–174. [Google Scholar] [CrossRef]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Symposium Review: Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef]
- Ma, S.; Zhu, M. Recent advances in applications of liquid chromatography-tandem mass spectrometry to the analysis of reactive drug metabolites. Chem. Biol. Interact. 2009, 179, 25–37. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Hollenberg, P.F.; Kent, U.M.; Bumpus, N.N. Mechanism-based inactivation of human cytochromes p450s: Experimental characterization, reactive intermediates, and clinical implications. Chem. Res. Toxicol. 2008, 21, 189–205. [Google Scholar] [CrossRef]
- Ma, S.; Subramanian, R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J. Mass Spectrom. 2006, 41, 1121–1139. [Google Scholar] [CrossRef]
- Orhan, H.; Vermeulen, N.P. Conventional and novel approaches in generating and characterization of reactive intermediates from drugs/drug candidates. Curr. Drug Metab. 2011, 12, 383–394. [Google Scholar]
- Ju, C.; Uetrecht, J.P. Mechanism of idiosyncratic drug reactions: Reactive metabolite formation, protein binding and the regulation of the immune system. Curr. Drug Metab. 2002, 3, 367–377. [Google Scholar] [CrossRef]
- Liebler, D.C.; Guengerich, F.P. Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 2005, 4, 410–420. [Google Scholar] [CrossRef]
- Wen, B.; Fitch, W.L. Analytical strategies for the screening and evaluation of chemically reactive drug metabolites. Expert Opin. Drug Met. 2009, 5, 39–55. [Google Scholar] [CrossRef]
- Eichhorn, P.; Knepper, T.P. Alpha, beta-unsaturated sulfophenylcarboxylates as degradation intermediates of linear alkylbenzenesulfonates: Evidence for omega-oxygenation followed by beta-oxidations by liquid chromatography-mass spectrometry. Environ. Toxicol. Chem. 2002, 21, 1–8. [Google Scholar]
- Moura, F.C.; Araujo, M.H.; Dalmazio, I.; Alves, T.M.; Santos, L.S.; Eberlin, M.N.; Augusti, R.; Lago, R.M. Investigation of reaction mechanisms by electrospray ionization mass spectrometry: Characterization of intermediates in the degradation of phenol by a novel iron/magnetite/hydrogen peroxide heterogeneous oxidation system. Rapid Commun. Mass Spectrom. 2006, 20, 1859–1863. [Google Scholar] [CrossRef]
- Dalmazio, I.; Urzedo, A.P.; Alves, T.M.; Catharino, R.R.; Eberlin, M.N.; Nascentes, C.C.; Augusti, R. Electrospray ionization mass spectrometry monitoring of indigo carmine degradation by advanced oxidative processes. J. Mass Spectrom. 2007, 42, 1273–1278. [Google Scholar] [CrossRef]
- Matamoros, V.; Jover, E.; Bayona, J.M. Advances in the determination of degradation intermediates of personal care products in environmental matrixes: A review. Anal. Bioanal. Chem. 2009, 393, 847–860. [Google Scholar] [CrossRef]
- Rand, A.A.; Mabury, S.A. In vitro interactions of biological nucleophiles with fluorotelomer unsaturated acids and aldehydes: Fate and consequences. Environ. Sci. Technol. 2012, 46, 7398–7406. [Google Scholar]
- Colton, R.; D’Agostino, A.; Traeger, J.C. Electrospray mass spectrometry applied to inorganic and organometallic chemistry. Mass Spectrom. Rev. 1995, 14, 79–106. [Google Scholar] [CrossRef]
- Henderson, W.; Nicholson, B.K.; McCaffrey, L.J. Applications of electro-spray mass spectrometry in organometallic chemistry. Polyhedron 1998, 17, 4291–4313. [Google Scholar] [CrossRef]
- Eldin, S.; Jencks, W.P. Lifetimes of Iminium Ions in Aqueous Solution. J. Am. Chem. Soc. 1995, 117, 4851–4857. [Google Scholar] [CrossRef]
- Eldin, S.; Digits, J.A.; Huang, S.T.; Jencks, W.P. Lifetime of an Aliphatic Iminium Ion in Aqueous Solution. J. Am. Chem. Soc. 1995, 117, 6631–6632. [Google Scholar] [CrossRef]
- Perry, R.H.; Brownell, K.R.; Chingin, K.; Cahill, T.J.; Waymouth, R.M.; Zare, R.N. From the Cover: Transient Ru-methyl formate intermediates generated with bifunctional transfer hydrogenation catalysts. Proc. Natl. Acad. Sci. USA 2012, 109, 2246–2250. [Google Scholar]
- Shiraiwa, M.; Sosedova, Y.; Rouviere, A.; Yang, H.; Zhang, Y.; Abbatt, J.P.; Ammann, M.; Poschl, U. The role of long-lived reactive oxygen intermediates in the reaction of ozone with aerosol particles. Nat. Chem. 2011, 3, 291–295. [Google Scholar] [CrossRef]
- Yan, Z.; Maher, N.; Torres, R.; Caldwell, G.W.; Huebert, N. Rapid detection and characterization of minor reactive metabolites using stable-isotope trapping in combination with tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 3322–3330. [Google Scholar] [CrossRef]
- Chen, P. Electrospray ionization tandem mass spectrometry in high-throughput screening of homogeneous catalysts. Angew Chem. Int. Ed. Engl. 2003, 42, 2832–2847. [Google Scholar] [CrossRef]
- Yamashita, M.; Fenn, J.B. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984, 88, 4451–4459. [Google Scholar] [CrossRef]
- Whitehouse, C.M.; Dreyer, R.N.; Yamashita, M.; Fenn, J.B. Electrospray interface for liquid chromatographs and mass spectrometers. Anal. Chem. 1985, 57, 675–679. [Google Scholar]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar]
- Hambitzer, G.; Heitbaum, J. Electrochemical thermospray mass spectrometry. Anal. Chem. 1986, 58, 1067–1070. [Google Scholar] [CrossRef]
- Fürmeier, S.; Metzger, J.O. Detection of Transient Radical Cations in Electron Transfer-Initiated Diels-Alder Reactions by Electrospray Ionization Mass Spectrometry. J. Am. Chem. Soc. 2004, 126, 14485–14492. [Google Scholar]
- Wilson, S.R.; Perez, J.; Pasternak, A. ESI-MS detection of ionic intermediates in phosphine-mediated reactions. J. Am. Chem. Soc. 1993, 115, 1994–1997. [Google Scholar] [CrossRef]
- Takáts, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass Spectrometry Sampling Under Ambient Conditions with Desorption Electrospray Ionization. Science 2004, 306, 471–473. [Google Scholar]
- Yu, Z.; Chen, L.C.; Erra-Balsells, R.; Nonami, H.; Hiraoka, K. Real-time reaction monitoring by probe electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 1507–1513. [Google Scholar] [CrossRef]
- Ganem, B.; Li, Y.T.; Henion, J.D. Detection of noncovalent receptor-ligand complexes by mass spectrometry. J. Am. Chem. Soc. 1991, 113, 6294–6296. [Google Scholar] [CrossRef]
- Lee, E.D.; Mueck, W.; Henion, J.D.; Covey, R.T. Real-time reaction monitoring by continuous-introduction ion-spray tandem mass spectrometry. J. Am. Chem. Soc. 1989, 111, 4600–4604. [Google Scholar] [CrossRef]
- Anthony, A.; Paiva, R.F.; Tilton, J.; Crooks, G.P.; Huang, L.Q.; Anderson, K.S. Detection and Identification of Transient Enzyme Intermediates Using Rapid Mixing, Pulsed-Flow Electrospray Mass Spectrometry. Biochemistry 1997, 36, 15472–15476. [Google Scholar] [CrossRef]
- Zechel, D.L.; Konermann, L.; Withers, S.G.; Douglas, D.J. Pre-Steady State Kinetic Analysis of an Enzymatic Reaction Monitored by Time-Resolved Electrospray Ionization Mass Spectrometry. Biochemistry 1998, 37, 7664–7669. [Google Scholar] [CrossRef]
- Perry, R.H.; Splendore, M.; Chien, A.; Davis, N.K.; Zare, R.N. Detecting reaction intermediates in liquids on the millisecond time scale using desorption electrospray ionization. Angew Chem. Int. Ed. Engl. 2011, 50, 250–254. [Google Scholar]
- Miao, Z.; Chen, H.; Liu, P.; Liu, Y. Development of submillisecond time-resolved mass spectrometry using desorption electrospray ionization. Anal. Chem. 2011, 83, 3994–3997. [Google Scholar] [CrossRef]
- Chen, H.; Venter, A.; Cooks, R.G. Extractive electrospray ionization for direct analysis of undiluted urine, milk and other complex mixtures without sample preparation. Chem. Commun. (Camb.) 2006, 19, 2042–2044. [Google Scholar]
- Marquez, C.A.; Wang, H.Y.; Fabbretti, F.; Metzger, J.O. Electron-Transfer-Catalyzed Dimerization of trans-Anethole: Detection of the Distonic Tetramethylene Radical Cation Intermediate by Extractive Electrospray Ionization Mass Spectrometry. J. Am. Chem. Soc. 2008, 130, 17208–17209. [Google Scholar] [CrossRef]
- Kottke, P.A.; Degertekin, F.L.; Fedorov, A.G. Scanning mass spectrometry probe: A scanning probe electrospray ion source for imaging mass spectrometry of submerged interfaces and transient events in solution. Anal. Chem. 2010, 82, 19–22. [Google Scholar] [CrossRef]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Adlhart, C.; Chen, P. Fishing for Catalysts: Mechanism-Based Probes for Active Species in Solution. Helv. Chim. Acta 2000, 83, 2192–2196. [Google Scholar] [CrossRef]
- Li, R.; Smith, R.L.; Kenttämaa, H.I. Fluorine Substitution Enhances the Reactivity of Substituted Phenyl Radicals toward Organic Hydrogen Atom Donors. J. Am. Chem. Soc. 1996, 118, 5056–5061. [Google Scholar] [CrossRef]
- MacAleese, L.; Maitre, P. Infrared spectroscopy of organometallic ions in the gas phase: From model to real world complexes. Mass Spectrom. Rev. 2007, 26, 583–605. [Google Scholar] [CrossRef]
- Guo, B.; Chen, B.; Liu, A.; Zhu, W.; Yao, S. Liquid Chromatography-mass spectrometric multiple reaction monitoring-based strategies for expanding targeted profiling towards quantitative metabolomics. Curr. Drug Metab. 2012. [Epub ahead of print]. [Google Scholar]
- Lalli, P.M.; Rodrigues, T.S.; Arouca, A.M.; Eberlin, M.N.; Neto, B.A.D. N-heterocyclic carbenes with negative-charge tags: Direct sampling from ionic liquid solutions. RSC Adv. 2012, 2, 3201–3203. [Google Scholar] [CrossRef]
- Shneier, A.; Kleanthous, C.; Deka, R.; Coggins, J.R.; Abell, C. Observation of an imine intermediate on dehydroquinase by electrospray mass spectrometry. J. Am. Chem. Soc. 1991, 113, 9416–9418. [Google Scholar] [CrossRef]
- Mattocks, A.R.; Legg, R.F.; Jukes, R. Trapping of short-lived electrophilic metabolites of pyrrolizidine alkaloids escaping from perfused rat liver. Toxicol. Lett. 1990, 54, 93–99. [Google Scholar] [CrossRef]
- Zhu, X.; Kalyanaraman, N.; Subramanian, R. Enhanced screening of glutathione-trapped reactive metabolites by in-source collision-induced dissociation and extraction of product ion using UHPLC-high resolution mass spectrometry. Anal. Chem. 2011, 83, 9516–9523. [Google Scholar] [CrossRef]
- Defoy, D.; Dansette, P.M.; Neugebauer, W.; Wagner, J.R.; Klarskov, K. Evaluation of deuterium labeled and unlabeled bis-methyl glutathione combined with nanoliquid chromatography-mass spectrometry to screen and characterize reactive drug metabolites. Chem. Res. Toxicol. 2011, 24, 412–417. [Google Scholar] [CrossRef]
- Chughtai, K.; Heeren, R.M. Mass spectrometric imaging for biomedical tissue analysis. Chem. Rev. 2010, 110, 3237–3277. [Google Scholar] [CrossRef]
- Aliprantis, A.O.; Canary, J.W. Observation of Catalytic Intermediates in the Suzuki Reaction by Electrospray Mass Spectrometry. J. Am. Chem. Soc. 1994, 116, 6985–6986. [Google Scholar] [CrossRef]
- Koster, S.; Verpoorte, E.A. Decade of microfluidic analysis coupled with electrospray mass spectrometry: An overview. Lab Chip. 2007, 7, 1394–1412. [Google Scholar] [CrossRef]
- Rob, T.; Wilson, D.J. A versatile microfluidic chip for millisecond time-scale kinetic studies by electrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 124–130. [Google Scholar] [CrossRef]
- Mao, S.; Gao, D.; Liu, W.; Wei, H.; Lin, J.M. Imitation of drug metabolism in human liver and cytotoxicity assay using a microfluidic device coupled to mass spectrometric detection. Lab Chip. 2012, 12, 219–226. [Google Scholar] [CrossRef]
- Deng, P.; Zhan, Y.; Chen, X.; Zhong, D. Derivatization methods for quantitative bioanalysis by LC-MS/MS. Bioanalysis 2012, 4, 49–69. [Google Scholar] [CrossRef]
- Busch, K.L.; Unger, S.E.; Vincze, A.; Cooks, R.G.; Keough, T. Desorption ionization mass spectrometry: Sample preparation for secondary ion mass spectrometry, laser desorption, and field desorption. J. Am. Chem. Soc. 1982, 104, 1507–1511. [Google Scholar] [CrossRef]
- Chisholm, D.M.; McIndoe, J.S. Charged ligands for catalyst immobilisation and analy Affiliation Information. Dalton Trans. 2008, 30, 3933–3945. [Google Scholar] [CrossRef]
- Wang, H.Y.; Yim, W.L.; Kluner, T.; Metzger, J.O. ESIMS studies and calculations on alkali-metal adduct ions of ruthenium olefin metathesis catalysts and their catalytic activity in metathesis reactions. Chemistry 2009, 15, 10948–10959. [Google Scholar] [CrossRef]
- Bayer, E.; Gfrörer, P.; Rentel, C. Coordination-Ionspray-MS (CIS-MS), a Universal Detection and Characterization Method for Direct Coupling with Separation Techniques. Angew. Chem. Int. Edit. Engl. 1999, 38, 992–995. [Google Scholar] [CrossRef]
- Yan, Z.; Caldwell, G.W. Stable-isotope trapping and high-throughput screenings of reactive metabolites using the isotope MS signature. Anal. Chem. 2004, 76, 6835–6847. [Google Scholar] [CrossRef]
- Gan, J.; Harper, T.W.; Hsueh, M.M.; Qu, Q.; Humphreys, W.G. Dansyl glutathione as a trapping agent for the quantitative estimation and identification of reactive metabolites. Chem. Res. Toxicol. 2005, 18, 896–903. [Google Scholar] [CrossRef]
- Soglia, J.R.; Contillo, L.G.; Kalgutkar, A.S.; Zhao, S.; Hop, C.E.; Boyd, J.G.; Cole, M.J. A semiquantitative method for the determination of reactive metabolite conjugate levels in vitro utilizing liquid chromatography-tandem mass spectrometry and novel quaternary ammonium glutathione analogues. Chem. Res. Toxicol. 2006, 19, 480–490. [Google Scholar] [CrossRef]
- Yan, Z.; Maher, N.; Torres, R.; Huebert, N. Use of a trapping agent for simultaneous capturing and high-throughput screening of both “soft” and “hard” reactive metabolites. Anal. Chem. 2007, 79, 4206–4214. [Google Scholar] [CrossRef]
- Leblanc, A.; Shiao, T.C.; Roy, R.; Sleno, L. Improved detection of reactive metabolites with a bromine-containing glutathione analog using mass defect and isotope pattern matching. Rapid Commun. Mass Spectrom. 2010, 24, 1241–1250. [Google Scholar] [CrossRef]
- Jahn, S.; Lohmann, W.; Bomke, S.; Baumann, A.; Karst, U. A ferrocene-based reagent for the conjugation and quantification of reactive metabolites. Anal. Bioanal. Chem. 2012, 402, 461–471. [Google Scholar] [CrossRef]
- Qian, R.; Guo, H.; Liao, Y.; Guo, Y.; Ma, S. Probing the mechanism of the palladium-catalyzed addition of organoboronic acids to allenes in the presence of AcOH by ESI-FTMS. Angew. Chem. Int. Ed. Engl. 2005, 44, 4771–4774. [Google Scholar] [CrossRef]
- Guo, B.; Huang, Z.; Wang, M.; Wang, X.; Zhang, Y.; Chen, B.; Li, Y.; Yan, H.; Yao, S. Simultaneous direct analysis of benzimidazole fungicides and relevant metabolites in agricultural products based on multifunction dispersive solid-phase extraction and liquid chromatography-mass spectrometry. J. Chromatogr. A 1217, 4796–4807. [Google Scholar]
- Huang, C.; Guo, B.; Wang, X.; Li, J.; Zhu, W.; Chen, B.; Ouyang, S.; Yao, S. A generic approach for expanding homolog-targeted residue screening of sulfonamides using a fast matrix separation and class-specific fragmentation-dependent acquisition with a hybrid quadrupole-linear ion trap mass spectrometer. Anal. Chim. Acta 2012, 737, 83–98. [Google Scholar] [CrossRef]
- Vikse, K.L.; Ahmadi, Z.; Manning, C.C.; Harrington, D.A.; McIndoe, J. Powerful insight into catalytic mechanisms through simultaneous monitoring of reactants, products, and intermediates. Angew. Chem. Int. Ed. Engl. 2011, 50, 8304–8306. [Google Scholar]
- Schade, M.A.; Fleckenstein, J.E.; Knochel, P.; Koszinowski, K. Charged tags as probes for analyzing organometallic intermediates and monitoring cross-coupling reactions by electrospray-ionization mass spectrometry. J. Org. Chem. 2010, 75, 6848–6857. [Google Scholar] [CrossRef]
- Vikse, K.L.; Henderson, M.A.; Oliver, A.G.; McIndoe, J.S. Direct observation of key intermediates by negative-ion electrospray ionisation mass spectrometry in palladium-catalysed cross-coupling. Chem. Commun. (Camb.) 2010, 46, 7412–7414. [Google Scholar] [CrossRef]
- Oliveira, F.F.; Santos, M.R.; Lalli, P.M.; Schmidt, E.M.; Bakuzis, P.; Lapis, A.A.; Monteiro, A.L.; Eberlin, M.N.; Neto, B.A. Charge-tagged acetate ligands as mass spectrometry probes for metal complexes investigations: Applications in Suzuki and Heck phosphine-free reactions. J. Org. Chem. 2011, 76, 10140–10147. [Google Scholar] [CrossRef]
- Furmeier, S.; Griep-Raming, J.; Hayen, A.; Metzger, J.O. Chelation-controlled radical chain reactions studied by electrospray ionization mass spectrometry. Chemistry 2005, 11, 5545–5554. [Google Scholar] [CrossRef]
- Laine, J.E.; Auriola, S.; Pasanen, M.; Juvonen, R.O. D-Isomer of gly-tyr-pro-cys-pro-his-pro peptide: A novel and sensitive in vitro trapping agent to detect reactive metabolites by electrospray mass spectrometry. Toxicol. In Vitro 2011, 25, 411–425. [Google Scholar] [CrossRef]
- Simoes, C.; Domingues, P.; Domingues, M.R. Identification of free radicals in oxidized and glycoxidized phosphatidylethanolamines by spin trapping combined with tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 931–939. [Google Scholar]
- Motwani, H.V.; Fred, C.; Haglund, J.; Golding, B.T.; Tornqvist, M. Cob(I)alamin for trapping butadiene epoxides in metabolism with rat S9 and for determining associated kinetic parameters. Chem. Res. Toxicol. 2009, 22, 1509–1516. [Google Scholar] [CrossRef]
- Rousu, T.; Pelkonen, O.; Tolonen, A. Rapid detection and characterization of reactive drug metabolites in vitro using several isotope-labeled trapping agents and ultra-performance liquid chromatography/time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 843–855. [Google Scholar]
- Rousu, T.; Tolonen, A. Characterization of cyanide-trapped methylated metabonates formed during reactive drug metabolite screening in vitro. Rapid Commun. Mass Spectrom. 2011, 25, 1382–1390. [Google Scholar]
- Harada, H.; Endo, T.; Momose, Y.; Kusama, H. A liquid chromatography/tandem mass spectrometry method for detecting UGT-mediated bioactivation of drugs as their N-acetylcysteine adducts in human liver microsomes. Rapid Commun. Mass Spectrom. 2009, 23, 564–570. [Google Scholar]
- Wilson, D.J.; Konermann, L. A capillary mixer with adjustable reaction chamber volume for millisecond time-resolved studies by electrospray mass spectrometry. Anal. Chem. 2003, 75, 6408–6414. [Google Scholar] [CrossRef]
- Agrawal, D.; Schöder, D. Insight into Solution Chemistry from Gas-Phase Experiments. Organometallics 2011, 30, 32–35. [Google Scholar]
- Ifa, D.R.; Wu, C.; Ouyang, Z.; Cooks, R.G. Desorption electrospray ionization and other ambient ionization methods: Current progress and preview. Analyst 2010, 135, 669–681. [Google Scholar] [CrossRef]
- Cooks, R.G.; Ouyang, Z.; Takats, Z.; Wiseman, J.M. Ambient Mass Spectrometry. Science 2006, 311, 1566–1570. [Google Scholar]
- Weston, D.J. Ambient Ionization Mass Spectrometry: Current Understanding of Mechanistic Theory; Analytical Performance and Application Areas. Analyst 2010, 135, 661–668. [Google Scholar] [CrossRef]
- Harris, G.A.; Galhena, A.S.; Fernandez, F.M. Ambient sampling/ionization mass spectrometry: Applications and current trends. Anal. Chem. 2011, 83, 4508–4538. [Google Scholar]
- Kuo, C.P.; Shiea, J. Application of direct electrospray probe to analyze biological compounds and to couple to solid-phase microextraction to detect trace surfactants in aqueous solution. Anal. Chem. 1999, 71, 4413–4417. [Google Scholar] [CrossRef]
- Chang, D.Y.; Lee, C.C.; Shiea, J. Detecting large biomolecules from high-salt solutions by fused-droplet electrospray ionization mass spectrometry. Anal. Chem. 2002, 74, 2465–2469. [Google Scholar] [CrossRef]
- Hiraoka, K.; Nishidate, K.; Mori, K.; Asakawa, D.; Suzuki, S. Development of probe electrospray using a solid needle. Rapid Commun. Mass Spectrom. 2007, 21, 3139–3144. [Google Scholar] [CrossRef]
- Nemes, P.; Vertes, A. Laser ablation electrospray ionization for atmospheric pressure, in vivo, and imaging mass spectrometry. Anal. Chem. 2007, 79, 8098–8106. [Google Scholar] [CrossRef]
- Chen, H.; Yang, S.; Wortmann, A.; Zenobi, R. Neutral desorption sampling of living objects for rapid analysis by extractive electrospray ionization mass spectrometry. Angew. Chem. Int. Ed. Engl. 2007, 46, 7591–7594. [Google Scholar] [CrossRef]
- Mulligan, C.C.; MacMillan, D.K.; Noll, R.J.; Cooks, R.G. Fast analysis of high-energy compounds and agricultural chemicals in water with desorption electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 3729–3736. [Google Scholar] [CrossRef]
- Miao, Z.; Chen, H. Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS). J. Am. Soc. Mass Spectrom. 2009, 20, 10–19. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, M.; Lin, Z.; Zhang, S.; Yang, C.; Zhang, X. Versatile platform employing desorption electrospray ionization mass spectrometry for high-throughput analysis. Anal. Chem. 2008, 80, 6131–6136. [Google Scholar] [CrossRef]
- Luo, M.; Hu, B.; Zhang, X.; Peng, D.; Chen, H.; Zhang, L.; Huan, Y. Extractive electrospray ionization mass spectrometry for sensitive detection of uranyl species in natural water samples. Anal. Chem. 2010, 82, 282–289. [Google Scholar] [CrossRef]
- Zhu, L.; Gamez, G.; Chen, H.W.; Huang, H.X.; Chingin, K.; Zenobi, R. Real-time, on-line monitoring of organic chemical reactions using extractive electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 2993–2998. [Google Scholar] [CrossRef]
- McCullough, B.J.; Bristow, T.; O’Connor, G.; Hopley, C. On-line reaction monitoring by extractive electrospray ionisation. Rapid Commun. Mass Spectrom. 2011, 25, 1445–1451. [Google Scholar]
- Chen, H.; Zenobi, R. Neutral desorption sampling of biological surfaces for rapid chemical characterization by extractive electrospray ionization mass spectrometry. Nat. Protoc. 2008, 3, 1467–1475. [Google Scholar]
- Li, X.; Hu, B.; Ding, J.; Chen, H. Rapid characterization of complex viscous samples at molecular levels by neutral desorption extractive electrospray ionization mass spectrometry. Nat. Protoc. 2011, 6, 1010–1025. [Google Scholar] [CrossRef]
- Chen, H.; Gamez, G.; Zenobi, R. What can we learn from ambient ionization techniques? J. Am. Soc. Mass Spectrom. 2009, 20, 1947–1963. [Google Scholar] [CrossRef]
- Kass, S.R.; Broadus, K.M. Reactive intermediates via Fourier transform mass spectrometry. J. Phys. Org. Chem. 2002, 15, 461–468. [Google Scholar]
- Schäfer, M.; Drayß, M.; Springer, A.; Zacharias, P.; Meerholz, K. Radical cations in electrospray mass spectrometry: Formation of open-shell species, examination of the fragmentation behaviour in ESI-MSn and reaction mechanism studies by detection of transient radical cations. Eur. J. Org. Chem. 2007, 31, 5162–5174. [Google Scholar]
- Nour, H.F.; Lopez-Periago, A.M.; Kuhnert, N. Probing the mechanism and dynamic reversibility of trianglimine formation using real-time electrospray ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 1070–1080. [Google Scholar] [CrossRef]
- Gamez, G.; Zhu, L.; Disko, A.; Chen, H.; Azov, V.; Chingin, K.; Kramer, G.; Zenobi, R. Real-time, in vivo monitoring and pharmacokinetics of valproic acid via a novel biomarker in exhaled breath. Chem. Commun. (Camb.) 2011, 47, 4884–4886. [Google Scholar]
- Santos, V.G.; Regiani, T.; Dias, F.F.; Romao, W.; Jara, J.L.; Klitzke, C.F.; Coelho, F.; Eberlin, M. N. Venturi easy ambient sonic-spray ionization. Anal. Chem. 2011, 83, 1375–1380. [Google Scholar]
- Cotte-Rodriguez, I.; Takats, Z.; Talaty, N.; Chen, H.; Cooks, R.G. Desorption electrospray ionization of explosives on surfaces: Sensitivity and selectivity enhancement by reactive desorption electrospray ionization. Anal. Chem. 2005, 77, 6755–6764. [Google Scholar] [CrossRef]
- Xu, G.; Chen, B.; Guo, B.; He, D.; Yao, S. Detection of intermediates for the Eschweiler-Clarke reaction by liquid-phase reactive desorption electrospray ionization mass spectrometry. Analyst 2011, 136, 2385–2390. [Google Scholar] [CrossRef]
- Huang, G.; Chen, H.; Zhang, X.; Cooks, R.G.; Ouyang, Z. Rapid screening of anabolic steroids in urine by reactive desorption electrospray ionization. Anal. Chem. 2007, 79, 8327–32. [Google Scholar] [CrossRef]
- Ding, J.; Gu, H.; Yang, S.; Li, M.; Li, J.; Chen, H. Selective detection of diethylene glycol in toothpaste products using neutral desorption reactive extractive electrospray ionization tandem mass spectrometry. Anal. Chem. 2009, 81, 8632–8638. [Google Scholar] [CrossRef]
- Venter, A.; Sojka, P.E.; Cooks, R.G. Droplet dynamics and ionization mechanisms in desorption electrospray ionization mass spectrometry. Anal. Chem. 2006, 78, 8549–8555. [Google Scholar] [CrossRef]
- Wortmann, A.; Kistler-Momotova, A.; Zenobi, R.; Heine, M.C.; Wilhelm, O.; Pratsinis, S.E. Shrinking droplets in electrospray ionization and their influence on chemical equilibria. J. Am. Soc. Mass Spectrom. 2007, 18, 385–393. [Google Scholar] [CrossRef]
- Girod, M.; Moyano, E.; Campbell, D.I.; Cooks, R.G. Accelerated bimolecular reactions in microdroplets studied by desorption electrospray ionization mass spectrometry. Chem. Sci. 2011, 2, 501–510. [Google Scholar] [CrossRef]
- Pasilis, S.P.; Kertesz, V.; van Berkel, G.J. Unexpected analyte oxidation during desorption electrospray ionization-mass spectrometry. Anal. Chem. 2008, 80, 1208–1214. [Google Scholar] [CrossRef]
- Nefliu, M.; Smith, J.N.; Venter, A.; Cooks, R.G. Internal energy distributions in desorption electrospray ionization (DESI). J. Am. Soc. Mass Spectrom. 2008, 19, 420–427. [Google Scholar] [CrossRef]
- Vikse, K.L.; Woods, M.P.; McIndoe, J.S. Pressurized sample infusion for the continuous analysis of air- and moisture-sensitive reactions using electrospray ionization mass spectrometry. Organometallics 2010, 29, 6615–6618. [Google Scholar] [CrossRef]
- Silva, B.V.; Violante, F.A.; Pinto, A.C.; Santos, L.S. The mechanism of Sandmeyer's cyclization reaction by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 423–428. [Google Scholar] [CrossRef]
- Hong, C.M.; Tsai, F.C.; Shiea, J. A multiple channel electrospray source used to detect highly reactive ketenes from a flow pyrolyzer. Anal. Chem. 2000, 72, 1175–1178. [Google Scholar] [CrossRef]
- Roberts, A.; Furdui, C.; Anderson, K.S. Observation of a chemically labile, noncovalent enzyme intermediate in the reaction of metal-dependent Aquifex pyrophilus KDO8PS by time-resolved mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 1919–1924. [Google Scholar] [CrossRef]
- Enami, S.; Vecitis, C.D.; Cheng, J.; Hoffmann, M.R.; Colussi, A.J. Electrospray mass spectrometric detection of products and short-lived intermediates in aqueous aerosol microdroplets exposed to a reactive gas. J. Phys. Chem. A 2007, 111, 13032–13037. [Google Scholar]
- Chen, H.; Eberlin, L.S.; Nefliu, M.; Augusti, R.; Cooks, R.G. Organic reactions of ionic intermediates promoted by atmospheric-pressure thermal activation. Angew. Chem. Int. Ed. Engl. 2008, 47, 3422–3425. [Google Scholar] [CrossRef]
- Liu, P.; Lanekoff, I.T.; Laskin, J.; Dewald, H.D.; Chen, H. Study of electrochemical reactions using nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 2012, 84, 5737–5743. [Google Scholar] [CrossRef]
- Olivero, D.; LaPlaca, M.; Kottke, P.A. Ambient nanoelectrospray ionization with in-line microdialysis for spatially resolved transient biochemical monitoring within cell culture environments. Anal. Chem. 2012, 84, 2072–2075. [Google Scholar] [CrossRef]
- Li, F.; Lu, J.; Ma, X. Profiling the reactive metabolites of xenobiotics using metabolomic technologies. Chem. Res. Toxicol. 2011, 24, 744–751. [Google Scholar] [CrossRef]
- Chen, H.; Wortmann, A.; Zhang, W.; Zenobi, R. Rapid in vivo fingerprinting of nonvolatile compounds in breath by extractive electrospray ionization quadrupole time-of-flight mass spectrometry. Angew. Chem. Int. Ed. Engl. 2007, 46, 580–583. [Google Scholar] [CrossRef]
- Laskin, J.; Heath, B.S.; Roach, P.J.; Cazares, L.; Semmes, O.J. Tissue imaging using nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 2012, 84, 141–148. [Google Scholar] [CrossRef]
- Huang, G.; Li, G.; Cooks, R.G. Induced nanoelectrospray ionization for matrix-tolerant and high-throughput mass spectrometry. Angew. Chem. Int. Ed. Engl. 2011, 50, 9907–9910. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, S.; Wen, F.; Ma, X.; Yang, C.; Zhang, X. In vivo nanoelectrospray for the localization of bioactive molecules in plants by mass spectrometry. Anal. Chem. 2012, 84, 3058–3062. [Google Scholar] [CrossRef]
- Lambert, J.B. A tamed reactive intermediate. Science 2008, 322, 1333–1334. [Google Scholar] [CrossRef]
- Robbins, D.W.; Hartwig, J.F. A simple, multidimensional approach to high-throughput discovery of catalytic reactions. Science 2011, 333, 1423–1427. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, W.; Yuan, Y.; Zhou, P.; Zeng, L.; Wang, H.; Tang, L.; Guo, B.; Chen, B. The Expanding Role of Electrospray Ionization Mass Spectrometry for Probing Reactive Intermediates in Solution. Molecules 2012, 17, 11507-11537. https://doi.org/10.3390/molecules171011507
Zhu W, Yuan Y, Zhou P, Zeng L, Wang H, Tang L, Guo B, Chen B. The Expanding Role of Electrospray Ionization Mass Spectrometry for Probing Reactive Intermediates in Solution. Molecules. 2012; 17(10):11507-11537. https://doi.org/10.3390/molecules171011507
Chicago/Turabian StyleZhu, Weitao, Yu Yuan, Peng Zhou, Le Zeng, Hua Wang, Ling Tang, Bin Guo, and Bo Chen. 2012. "The Expanding Role of Electrospray Ionization Mass Spectrometry for Probing Reactive Intermediates in Solution" Molecules 17, no. 10: 11507-11537. https://doi.org/10.3390/molecules171011507
APA StyleZhu, W., Yuan, Y., Zhou, P., Zeng, L., Wang, H., Tang, L., Guo, B., & Chen, B. (2012). The Expanding Role of Electrospray Ionization Mass Spectrometry for Probing Reactive Intermediates in Solution. Molecules, 17(10), 11507-11537. https://doi.org/10.3390/molecules171011507