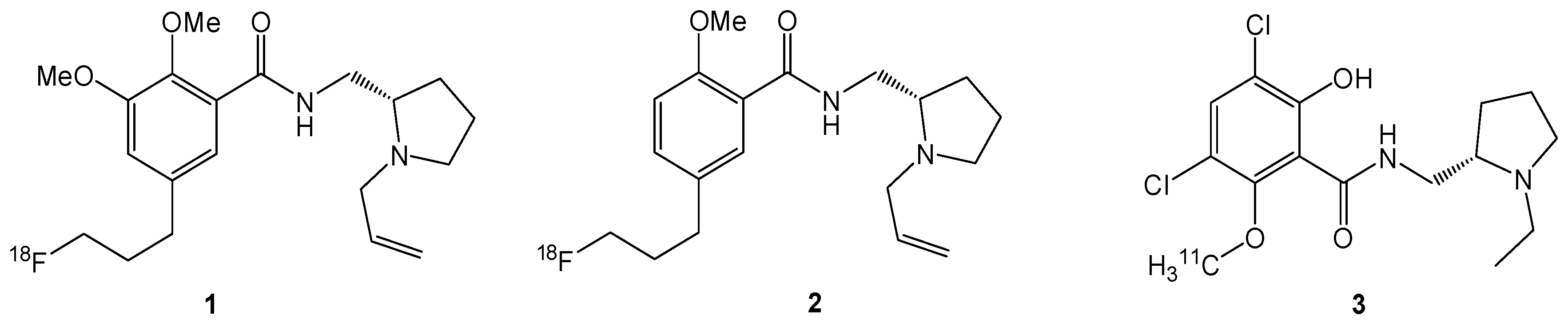
3.3. Chemistry
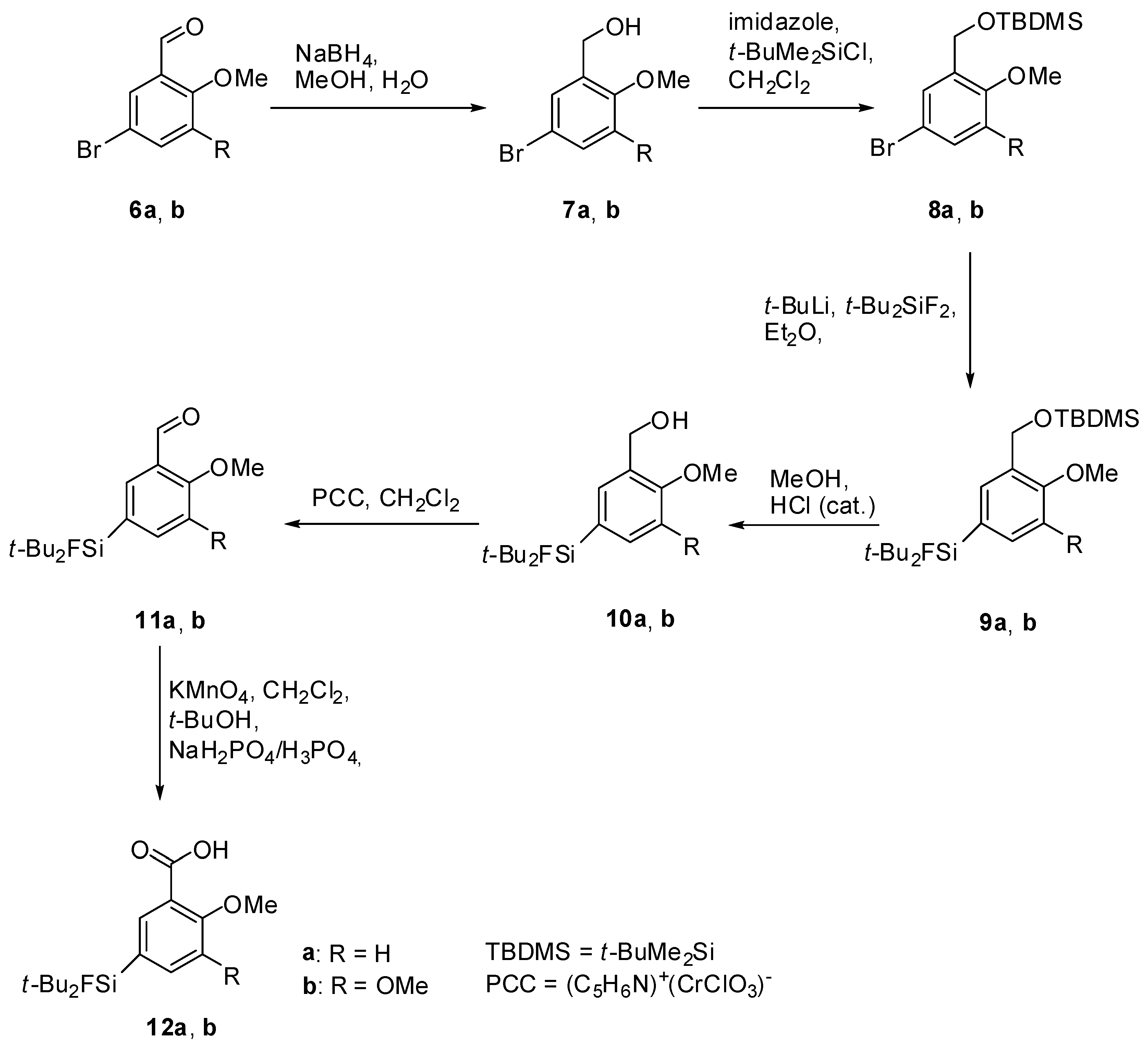
(5-Bromo-2-methoxyphenyl)methanol (7a). A solution of sodium borohydride (0.94 g, 24.8 mmol, 1.0 equiv.) in H2O (30 mL) was added to a solution of the benzaldehyde derivative 6a (5.33 g, 24.8 mmol) in methanol (200 mL). After stirring at room temperature for 24 h, methanol was evaporated and the aqueous residue was diluted with diethyl ether (200 mL). The aqueous phase was extracted with diethyl ether (3 × 100 mL), the combined organic layers were dried with MgSO4, filtered and the filtrate was evaporated to afford 7a (5.06 g, 23.31 mmol, 94%) as colourless oil. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 7.39 (d, 4J(1H-1H) = 2.4 Hz, 1H, C(6)H), 7.33 (dd, 3J(1H-1H) = 8.8 Hz, 4J(1H-1H) = 2.4 Hz, 1H, C(4)H), 6.72 (d, 3J(1H-1H) = 8.8 Hz, 1H, C(3)H), 4.61 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 2.35 (s, 1H, OH).
(5-Bromo-2,3-dimethoxyphenyl)methanol (7b). The procedure was analogous to the synthesis of 7a. The benzaldehyde derivative 6b (5.35 g, 21.83 mmol) gave alcohol 7b (5.04 g, 20.40 mmol, 93%) as a colourless oil. 1H-NMR (200.13 MHz, CDCl3): δ (ppm) = 7.05 (d, 4J(1H-1H) = 2.3 Hz, 1H, C(4)H), 6.93 (d, 4J(1H-1H) = 2.3 Hz, 1H, C(6)H), 4.59 (d, 3J(1H-1H) = 5.6 Hz, 2H, CH2), 3.80 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 2.93 (t, 3J(1H-1H) = 5.6 Hz, 1H, OH).
(5-Bromo-2-methoxybenzyloxy)(tert-butyl)dimethylsilane (8a). To a solution of 7a (5.00 g, 23.00 mmol) in dichloromethane (150 mL) imidazole (2.03 g, 29.89 mmol, 1.3 equiv.) was added and the suspension was stirred at ambient temperature for 10 min. t-Butyldimethylchlorosilane (t-BuMe2SiCl, 4.16 g, 27.59 mmol, 1.2 equiv.) was added and stirring was continued for 20 h. For work-up the mixture was diluted with H2O (40 mL) and the aqueous phase was extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried with MgSO4, filtered and the solvent was evaporated to afford 8a (7.10 g, 21.43 mmol, 93%) as a red oil. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 7.57 (d, 4J(1H-1H) = 2.5 Hz, 1H, C(6)H), 7.29 (dd, 4J(1H-1H) = 2.5 Hz, 3J(1H-1H) = 8.6 Hz, 1H, C(4)H), 6.66 (d, 3J(1H-1H) = 8.6 Hz, 1H, C(3)H), 4.71 (s, 2H, CH2), 3.77 (s, 3H, OCH3), 0.97 (s, 9H, CCH3), 0.13 (s, 6H, SiCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 154.9 (s, C(2)), 132.2 (s, C(6)), 130.0 (s, C(4)), 129.5 (s, C(1)), 113.0 (s, C(3)), 111.1 (s, C(5)), 59.7 (s, CH2), 55.3 (s, OCH3), 26.0 (s, CCH3), 18.5 (s, C(CH3)3, −5.3 (s, SiCH3).
(5-Bromo-2,3-dimethoxybenzyloxy)(tert-butyl)dimethylsilane (8b). The procedure was analogous to the synthesis of 8a. The benzyl alcohol derivative 7b (4.93, 19.95 mmol) gave the silylated alcohol 8b (5.88g, 16.27 mmol, 82%) as a slightly yellowish oil. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 7.08 (d, 4J(1H-1H) = 2.3 Hz, 1H, C(4)H), 6.82 (d, 4J(1H-1H) = 2.2 Hz, 1H, C(6)H), 4.62 (s, 2 H, CH2), 3.72 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 0.83 (s, 9H, CCH3), 0.00 (s, 6H, SiCH3).
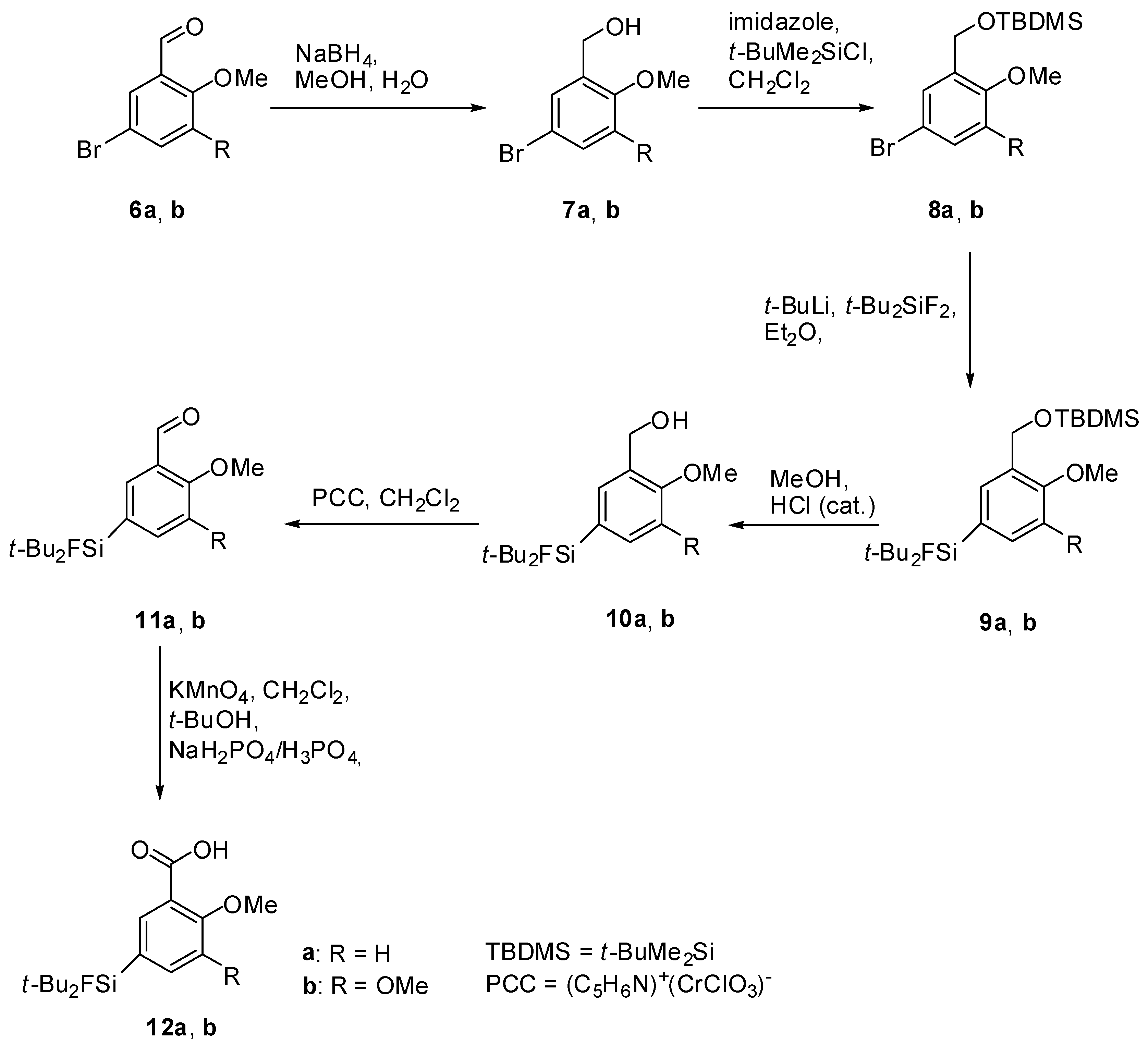
Di-tert-Butyl(3-((tert-butyldimethylsilyloxy)methyl)-4-methoxyphenyl)fluorosilane (9a). To a stirred solution of 8a (5.00 g, 15.09 mmol) in dry diethyl ether (150 mL), t-BuLi (17.8 mL, 1.7 mol/L, 30.18 mmol, 2.0 equiv.) was added dropwise at −78 °C. After stirring for 10 min t-Bu2SiF2 (3.00 g, 16.60 mmol, 1.1 equiv.) was added and stirring was continued for 19 h while the reaction mixture was allowed to warm to ambient temperature. The mixture was washed with H2O (50 mL) and the aqueous phase was extracted with diethylether (3 × 50 mL). The combined organic layers were dried with MgSO4, filtered and the solvent was evaporated to afford 9a (5.77 g, 13.98 mmol, 93%) as a colourless oil. 1H-NMR (300.13 MHz, CDCl3): δ (ppm) = 7.64 (s, 1H, C(6)H), 7.38 (d, 3J(1H-1H) = 8.1 Hz, 1H, C(4)H), 6.73 (d, 3J(1H-1H) = 8.6 Hz, 1H, C(3)H), 4.69 (s, 2H, CH2), 3.70 (s, 3H, OCH3), 0.96 (s, 18H, CCH3), 0.85 (s, 9H, CCH3), 0.00 (s, 6H, SiCH3). 13C[1H]-NMR (75.48 MHz, CDCl3): δ (ppm) = 157.7 (s, C(2)), 134.4 (d, 3J(13C-19F) = 3.9 Hz, C(6)), 132.4 (d, 3J(13C-19F) = 4.5 Hz, C(4)), 129.3 (s, C(1)), 124.3 (d, 2J(13C-19F) = 13.9 Hz, C(5)), 109.2 (s, C(3)), 60.5 (s, CH2), 55.3 (s, OCH3), 27.8 (s, CCH3), 26.4 (s, CCH3), 20.8 (d, 2J(13C-19F) = 12.5 Hz, C(CH3)3), 18.8 (s, C(CH3)3, −4.9 (s, SiCH3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −189.1 (s, 1J(19F-29Si) = 297 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 20.6 (s, Si(CH3)2tBu), 14.7 (d, 1J(29Si-19F) = 297 Hz, SiFtBu2). Elemental analysis calculated (%) for C22H41FO2Si2 (412.73 g/mol): C 64.0, H 10.0; found (%): C 63.8, H 9.6. HR-MS (GC-EI): calculated for C22H41O2F28SNa+ 435.2521, found 435.2530 [M+Na+].
Di-tert-Butyl(3-((tert-butyldimethylsilyloxy)methyl)-4,5-dimethoxyphenyl)-fluorosilane (9b). To a stirred solution of 8b (5.70 g, 15.77 mmol) in dry diethylether (150 mL) and dry THF (50 mL) t-BuLi (18.6 mL, 1.7 mol/L, 31.55 mmol, 2.0 equiv.) was added dropwise at −78 °C. After stirring for 5 min t-Bu2SiF2 (3.13 g, 17.35 mmol, 1.1 equiv.) was added and stirring was continued for 19 h while the reaction mixture was allowed to warm to ambient temperature. The mixture was concentrated in vacuo and the aqueous residue dissolved in chloroform. The aqueous phase was extracted with chloroform (4 × 100 mL) and the combined organic layers were concentrated in vacuo, dried with MgSO4 and filtered. The solvent was evaporated and the crude product was purified by column chromatography (hexane/diethylether = 20/1) to afford 9b (4.74 g, 10.71 mmol, 68%) as a colorless oil. 1H-NMR (200.13 MHz, CDCl3): δ (ppm) = 7.33 (s, 1H, C(6)H), 7.07 (s, 1H, C(4)H), 4.82 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 1.08 (s, 18H, CCH3), 0.95 (s, 9H, CCH3), 0.11 (s, 6H, SiCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 151.5 (s, C(2)), 147.0 (s, C(3)), 134.4 (s, C(1)), 128.4 (d, 2J(13C-19F) = 13.7 Hz, C(5)), 125.1 (d, 3J(13C-19F) = 4.5 Hz, C(6)), 116.5 (d, 3J(13C-19F) = 3.9 Hz, C(4)), 60.5 (s, CH2 or OCH3), 60.1 (s, CH2 or OCH3), 55.8 (s, OCH3), 27.4 (s, CCH3), 25.9 (s, CCH3), 20.3 (d, 2J(13C-19F) = 12.4 Hz, C(CH3)3), 18.3 (s, C(CH3)3, −5.3 (s, SiCH3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −189.1 (s, 1J(19F-29Si) = 298 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 20.7 (s, Si(CH3)2tBu), 14.3 (d, 1J(29Si-19F) = 298 Hz, SiFtBu2). Elemental analysis calculated (%) for C23H43FO3Si2 (412.73 g/mol): C 62.4, H 9.8; found (%): C 62.2, H 9.6. HR-MS (GC-EI): calculated for C23H43O3F28SiNa+ 465.2627, found 465.2638 [M+Na+].
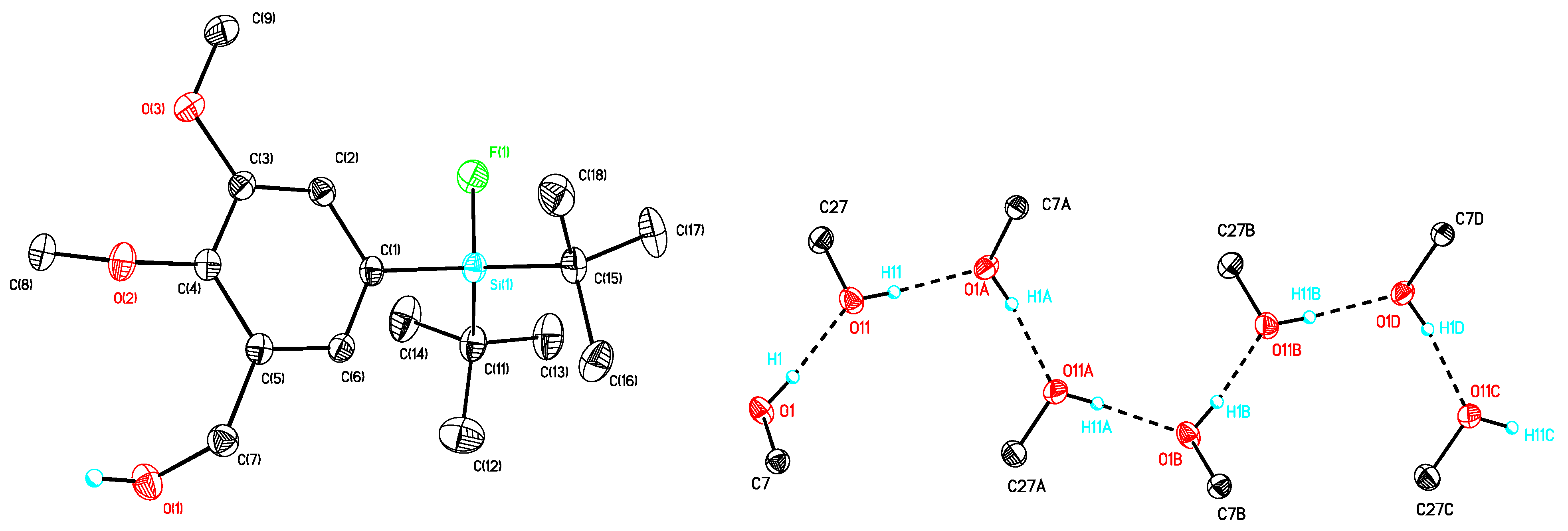
(5-(di-tert-Butylfluorosilyl)-2-methoxyphenyl)methanol (10a). To a stirred solution of 9a (4.90 g, 11.88 mmol) in methanol (250 mL) catalytic amounts of concentrated HCl was added. After stirring at room temperature for 19 h, methanol was evaporated and the residue dissolved in H2O and diethyl ether. The aqueous phase was extracted with diethyl ether (3 × 50 mL) and the combined organic layers were washed with saturated NaHCO3-solution (50 mL), dried with MgSO4, and filtrated. The solvent was evaporated to afford 10a (3.05 g, 10.22 mmol, 86%) as a colorless oil. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 7.50 (d, 3J(1H-1H) = 8.1 Hz, 1H, C(4)H), 7.47 (s, 1H, C(6)H), 6.89 (d, 3J(1H-1H) = 8.1 Hz, 1H, C(3)H), 4.67 (d, 3J(1H-1H) = 6.2 Hz, 2H, CH2), 3.85 (s, 3H, OCH3), 2.51 (t, 3J(1H-1H) = 6.2 Hz, 1H, OH), 1.03 (s, 18H, CCH3). 13C[1H]-NMR (400.13 MHz, CDCl3): δ (ppm) = 158.7 (s, C(2)), 135.2 (d, 3J(13C-19F) = 4.3 Hz, C(6)), 134.3 (d, 3J(13C-19F) = 4.1 Hz, C(4)), 128.3 (s, C(1)), 124.5 (d, 2J(13C-19F) = 13.9 Hz, C(5)), 109.6 (s, C(3)), 62.2 (s, CH2), 55.0 (s, OCH3), 27.3 (s, CCH3), 20.2 (d, 2J(13C-19F) = 12.4 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −189.0 (s, 1J(19F-29Si) = 297 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.6 (d, 1J(29Si-19F) = 297 Hz). Elemental analysis calculated (%) for C16H27FO2Si (298.47 g/mol): C 64.4, H 9.1; found (%): C 64.1, H 9.2. IR (KBr): ν (cm−1) = 3326 (ν(OH)). HR-MS (GC-EI): calculated for C16H27O2F28Si+ 298.1759, found 298.1761 [M+].
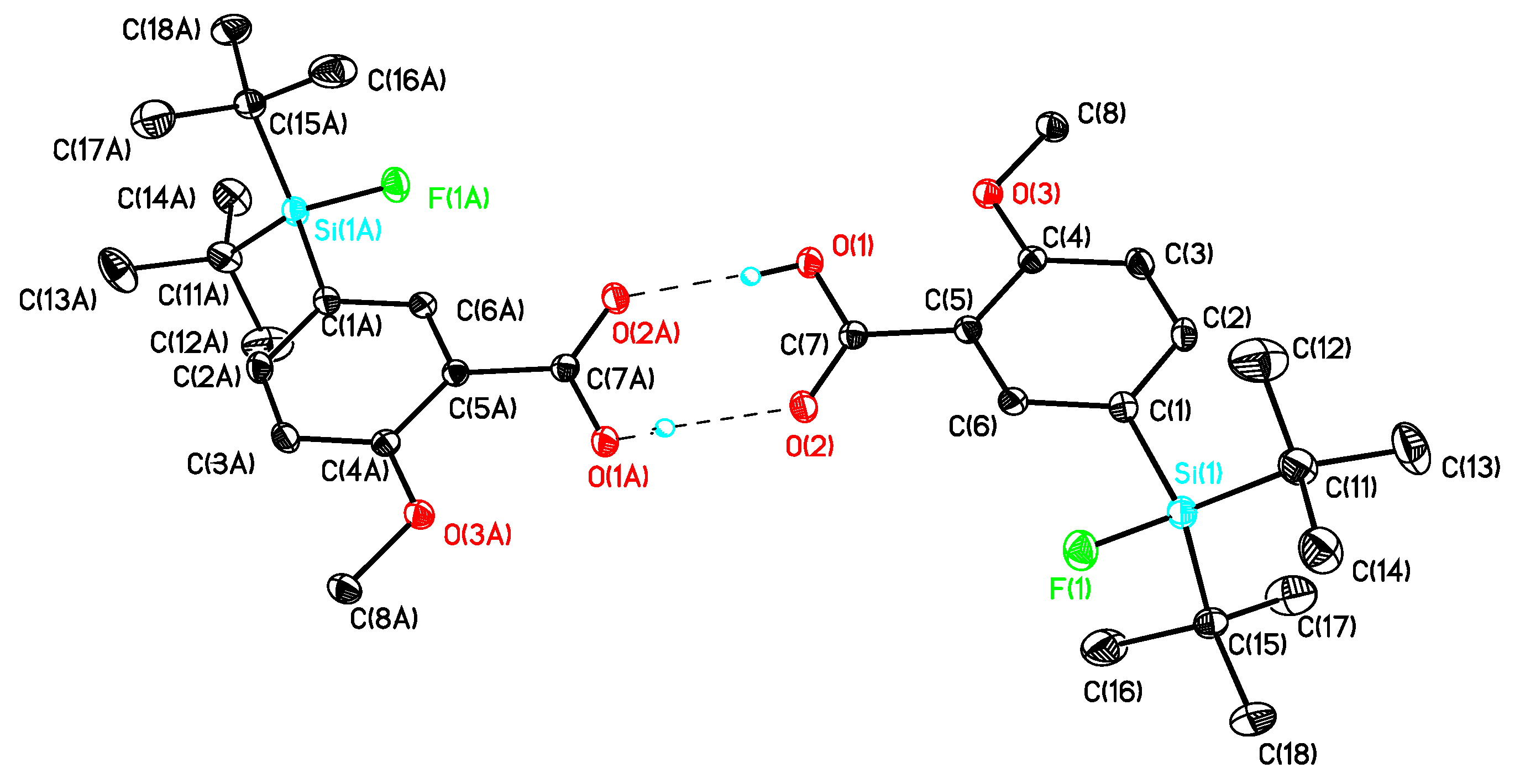
(5-(di-tert-Butylfluorosilyl)-2,3-dimethoxyphenyl)methanol (10b). The procedure was analogous to the synthesis of 10a starting from the protected alcohol 9b (4.69, 10.59 mmol). The crude product was purified by column chromatography (hexane/diethylether = 3/1 → hexane/diethylether = 2/1 → hexane/diethylether = 1/1 → hexane/diethylether) to afford 10b (3.13, 9.53 mmol, 90%) as a white crystalline solid of m.p. 80 °C. 1H-NMR (200.13 MHz, CDCl3): δ (ppm) = 7.14 (d, 4J(1H-1H) = 0.9 Hz, 1H, (C4)H), 7.03 (d, 4J(1H-1H) = 0.9 Hz, 1H, (C6)H), 4.61 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.17 (s, 1H, OH), 1.00 (s, 18H, CCH3). 13C[1H]-NMR (75.48 MHz, CDCl3): δ (ppm) = 152.1 (s, (C2)), 148.6 (s, (C3)), 134.5 (s, (C1)), 129.3 (d, 2J(13C-19F) = 13.6 Hz, (C5)), 126.9 (d, 3J(13C-19F) = 4.1 Hz, (C4)), 117.7 (d, 3J(13C-19F) = 3.9 Hz, (C6)), 61.3 (s, CH2), 61.1 (s, OCH3), 56.1 (s, OCH3), 27.8 (s, CCH3), 20.6 (d, 2J(13C-19F) = 12.4 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −188.5 (s, 1J(19F-29Si) = 298 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.1 (d, 1J(29Si-19F) = 298 Hz). Elemental analysis calculated (%) for C17H29FO3Si (328.19 g/mol): C 62.2, H 8.9; found (%): C 62.0, H 8.9. IR (KBr): ν (cm−1) = 3294 (ν(OH)). HR-MS (GC-EI): calculated for C17H29O3F28Si+ 328.1865, found 328.1869 [M+].
(3-(Bromomethyl)-4-methoxyphenyl)di-tert-butylfluorosilane (10c). The procedure was analogous to the synthesis of 10a starting from 9a (2.80 g, 6.78 mmol) and concentrated HBr (50 mL). After crystallisation from diethyl ether/hexane, 10c (1.80 g, 4.98 mmol, 73%) was obtained as white crystalline solid of m.p. 61 °C. 1H-NMR (300.13 MHz, CDCl3): δ (ppm) = 7.54–7.57 (m, 2 H, C(4)H und C(6)H), 6.93 (d, 3J(1H-1H) = 8.7 Hz, 1H, C(3)H), 4.61 (s, 2H, CH2), 3.94 (s, 3H, OCH3), 1.08 (s, 18H, CCH3). 13C[1H]-NMR (75.48 MHz, CDCl3): δ (ppm) = 159.1 (s, C(2)), 137.1 (d, 3J(13C-19F) = 4.1 Hz, C(4)), 136.6 (d, 3J(13C-19F) = 4.2 Hz, C(6)), 126.1 (s, C(1)), 125.2 (d, 2J(13C-19F) = 14.1 Hz, C(5)), 110.8 (s, C(3)), 55.9 (s, OCH3), 29.5 (s, CH2), 27.8 (s, CCH3), 20.7 (d, 2J(13C-19F) = 12.4 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −189.0 (s, 1J(19F-29Si) = 297 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.4 (d, 1J(29Si-19F) = 297 Hz). Elemental analysis calculated (%) for C16H26BrFOSi (360.09 g/mol): C 53.2, H 7.3; found (%): C 53.3, H 6.9. HR-MS (LC-ESI): calculated for C16H26O79BrF28Si [M]+ (m/z) 360.0915, found 360.0922; for C16H26O81BrF28Si [M]+ (m/z) 362.0894, found 362.0907.
3-(Chloromethyl)-4-methoxyphenyl)-di-tert-butylfluorosilane (10d). The procedure was analogous to the synthesis of 10a starting from 9a (1.99 g, 4.82 mmol, 1.0 equiv.) and concentrated HCl (50 mL). Crystallisation from diethylether/hexane gave the product 10d (1.16 g, 3.66 mmol, 76%) as white crystalline solid of m.p. 64 °C. 1H-NMR (300.13 MHz, CDCl3): δ (ppm) = 7.54–7.49 (m, 2H, C(4)H und C(6)H), 6.89 (d, 3J(1H-1H) = 7.9 Hz, 1H, C(3)H), 4.64 (s, 2H, CH2), 3.85 (s, 3H, OCH3), 1.03 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 158.5 (s, C(2)), 136.2 (d, 3J(13C-19F) = 4.1 Hz, C(4)), 136.1 (d, 3J(13C-19F) = 4.2 Hz, C(6)), 125.2 (s, C(1)), 124.6 (d, 2J(13C-19F) = 14.1 Hz, C(5)), 110.1 (s, C(3)), 55.3 (s, OCH3), 41.6 (s, CH2), 27.3 (s, CCH3), 20.2 (d, 2J(13C-19F) = 12.5 Hz, C(CH3)3). Elemental analysis calculated (%) for C16H26ClFOSi (316.91 g/mol): C 60.6, H 8.3; found (%): C 60.8. HR-MS (LC-ESI): calculated for calculated for C16H26OF28Si [M−Cl−]+ (m/z) 281.1732, found 281.1733.
5-(di-tert-Butylfluorosilyl)-2-methoxybenzaldehye (11a). To an ice-cooled and stirred suspension of pyridinium chlorochromate (2.86 g, 13.27 mmol, 3.0 equiv.) in dry CH2Cl2 (150 mL) was added drop-wise within 10 min a solution of 10a (1.32 g, 4.42 mmol) in dry CH2Cl2 (30 mL). The reaction mixture was stirred at room temperature for 2 h. After the reaction mixture had been diluted with diethyl ether (300 mL), the supernatant solution was decanted and the residue was washed with diethyl ether (2 × 100 mL). Filtration of the combined organic layers over a pad of silica and evaporation of the solvent afforded 11a (1.31 g, 4.42 mmol, quantitative) as a slightly yellowish amorphous solid, m.p. 38 °C. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 10.43 (s, 1H, CHO), 8.00 (s, 1H, C(6)H), 7.73 (d, 3J(1H-1H) = 8.3 Hz, 1H, C(4)H), 6.98 (d, 3J(1H-1H) = 8.3 Hz, 1H, C(3)H), 3.88 (s, 3H, OCH3), 0.98 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 189.6 (s, CHO), 162.7 (s, C(2)), 141.6 (d, 3J(13C-19F) = 4.1 Hz, C(4)), 134.1 (d, 3J(13C-19F) = 4.3 Hz, C(6)), 129.9 (s, C(1)), 124.8 (d, 2J(13C-19F) = 14.1 Hz, C(5)), 111.1 (s, C(3)), 55.4 (s, OCH3), 27.2 (s, CCH3), 20.1 (d, 2J(13C-19F) = 12.4 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −188.6 (s, 1J(19F-29Si) = 298 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.3 (d, 1J(29Si-19F) = 298 Hz). Elemental analysis calculated (%) for C16H27FO2Si (296.45 g/mol): C 64.8, H 8.5; found (%): C 64.0, H 8.7. HR-MS (GC-EI): calculated for C16H25O2F28Si [M]+ (m/z): 296.1602; found 296.1601.
5-(di-tert-Butylfluorosilyl)-2,3-dimethoxybenzaldehyde (11b). The procedure was analogous to the synthesis of 11a. The alcohol 10b (3.04 g, 9.25 mmol) gave the aldehyde 11b (3.12 g, 9.25 mmol, quantitative) as a slightly yellowish amorphous solid of m.p. 73 °C. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 10.36 (s, 1H, CHO), 7.55 (s, 1H, C(6)H), 7.28 (s, 1H, C(4)H), 3.93 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 0.97 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 189.9 (s, CHO), 153.6.7 (s, C(2)), 152.3 (s, C(3)), 129.2 (d, 2J(13C-19F) = 14.2 Hz, C(5)), 128.9 (s, C(1)), 124.7 (d, 3J(13C-19F) = 4.4 Hz, C(4)), 122.9 (d, 3J(13C-19F) = 4.2 Hz, C(6)), 62.0 (s, OCH3), 55.4 (s, OCH3), 27.1 (s, CCH3), 20.1 (d, 2J(13C-19F) = 12.2 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −188.2 (s, 1J(19F-29Si) = 298.9 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 13.9 (d, 1J(29Si-19F) = 299.0 Hz). Elemental analysis calculated (%) for C17H27FO3Si (326.48 g/mol): C 62.5, H 8.3; found (%): C 62.4, H 8.4. IR (KBr): ν (cm-1) = 1692 (ν (C=O)). HR-MS (GC-EI): calculated for C17H27O3FSi [M]+ (m/z): 326.1708; found 326.1697.
5-(di-tert-Butylfluorosilyl)-2-methoxybenzoic acid (12a). To a stirred solution of 11a (1.31 g, 4.42 mmol) in CH2Cl2 (5 mL) and t-BuOH (30 mL) one after another a buffered solution (30 mL, pH = 3) of NaH2PO4 (1.25 M), concentrated H3PO4 and an aqueous solution of KMnO4 (50 mL, 1 M) were added. Stirring was continued at room temperature for 3 h. The reaction mixture was quenched with saturated Na2SO3-solution (150 mL) and HCl (2 M). The latter reagents were added until the mixture turned colorless. After extraction with diethylether (3 × 200 mL) the combined organic layers were dried with MgSO4, filtered and the solvent was evaporated to afford 12a (1.01 g, 3.23 mmol, 73%) as a white amorphous solid that was re-crystallised from diethyl ether/hexane to give colorless crystals of m.p. 123 °C. 1H-NMR (200 MHz, CDCl3): δ (ppm) = 8.40 (d, 4J(1H-1H) = 1.8 Hz, 1H, C(6)H), 7.81 (dd, 3J(1H-1H) = 8.3 Hz, 4J(1H-1H) = 1.8 Hz, 1H, C(4)H), 7.09 (d, 3J(1H-1H) = 8.3 Hz, 1H, C(3)H), 4.09 (s, 3H, OCH3), 1.04 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 165.5 (s, COOH), 159.8 (s, C(2)), 140.8 (d, 3J(13C-19F) = 4.0 Hz, C(4)), 139.2 (d, 3J(13C-19F) = 4.4 Hz, C(6)), 127.1 (d, 2J(13C-19F) = 14.3 Hz, C(5)), 116.9 (s, C(1)), 111.1 (s, C(3)), 56.5 (s, OCH3), 27.2 (s, CCH3), 20.1 (d, 2J(13C-19F) = 12.2 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −188.5 (s, 1J(19F-29Si) = 298 Hz). Elemental analysis calculated (%) for C16H25FO3Si (312.45 g/mol): C 61.5, H 8.1; found (%): C 61.1, H 7.9. HR-MS (GC-EI): calculated for C16H25O3FSi [M]+ (m/z): 312.1552; found 312.1563. IR (KBr): ν (cm−1) = 2989 (ν(OH)), 1701 (ν (C=O)).
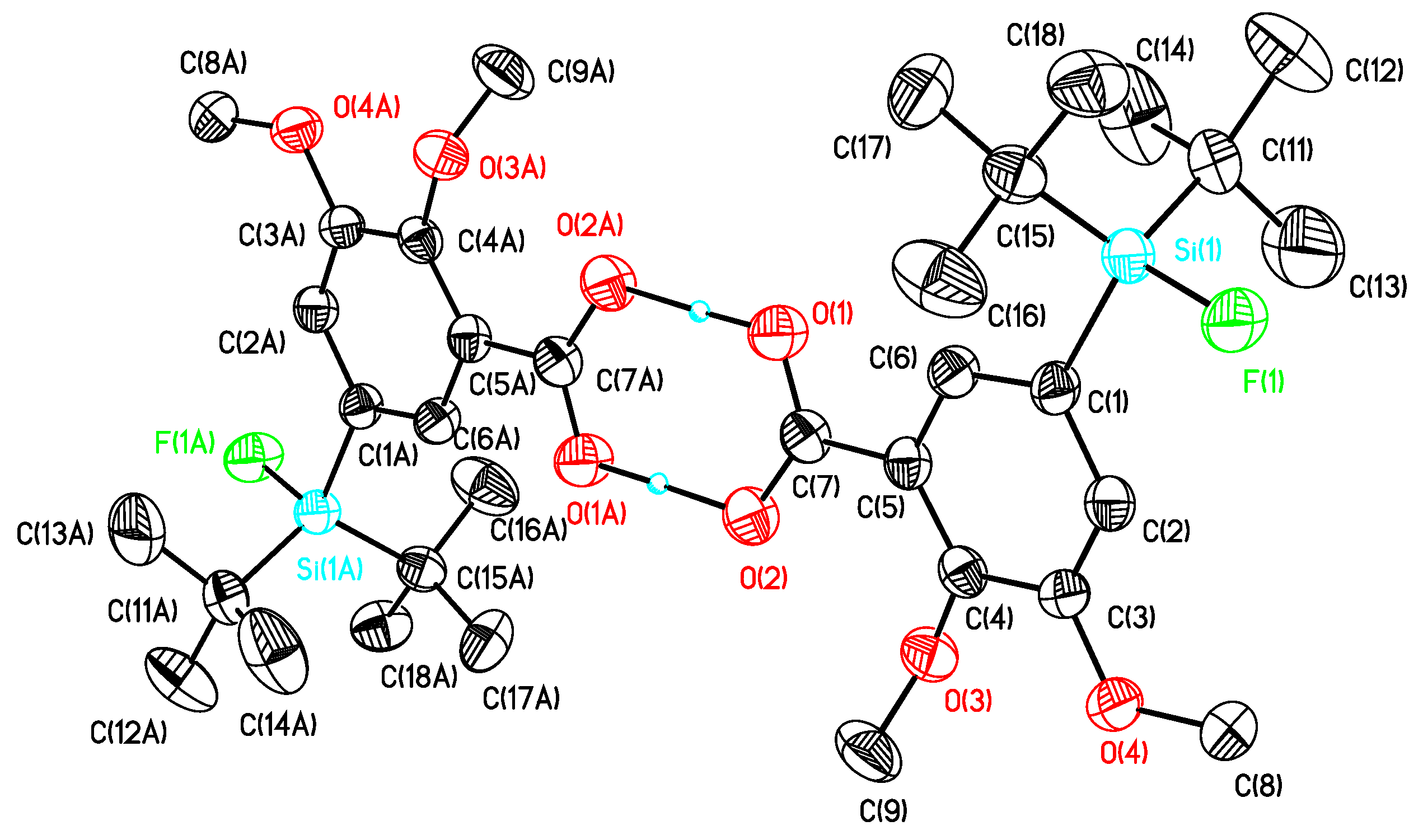
5-(di-tert-Butylfluorosilyl)-2,3-dimethoxybenzoic acid (12b). The procedure was analogous to the synthesis of 12a. The aldehyde 11b (2.00 g, 6.13 mmol) gave after re-crystallization from diethylether/hexane the carboxylic acid 12b (2.02 g, 5.90 mmol, 96%) as colorless crystals of m.p. 118 °C. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 7.91 (s, 1H, C(6)H), 7.34 (s, 1H, C(4)H), 4.10 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 1.04 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, C6D6): δ (ppm) = 166.8 (s, COOH), 152.4 (s, C(2)), 150.2 (s, C(3)), 130.1 (d, 2J(13C-19F) = 14.2 Hz, C(5)), 129.3 (d, 3J(13C-19F) = 4.7 Hz, C(4)), 123.3 (s, C(1)), 122.0 (d, 3J(13C-19F) = 3.9 Hz, C(6)), 61.1 (s, OCH3), 55.2 (s, OCH3), 27.1 (s, CCH3), 20.1 (d, 2J(13C-19F) = 12.1 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −187.7 (s, 1J(19F-29Si) = 299 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.3 (d, 1J(29Si-19F) = 299 Hz). Elemental analysis calculated (%) for C17H27O4FSi (342.17 g/mol): C 59.6, H 8.0; found (%): C 59.4, H 7.6. HR-MS (GC-EI): calculated for C17H27O4FSi [M]+ (m/z): 342.1657; found 342.1641. IR (KBr): ν (cm−1) = 2974 (ν(OH)), 1685 (ν (C=O)).
(S)-1-Allylpyrrolidine-2-carboxamide (
14). The synthesis followed the same procedure as described in the literature [
12]. The reaction of (S)-pyrrolinamide
13 (1.03 g, 9.02 mmol) with allyl iodide (0.83 mL, 9.02 mmol, 1.0 equiv.) gave compound
14 (1.29 g, 8.37 mmol, 93%) as white amorphous solid.
1H-NMR (400.13 MHz, CDCl
3): δ (ppm) = 7.19 (s, 1H, NH–H), 6.00 (s, 1H, NH–H), 5.87–5.76 (m, 1H, CH=CH
2), 5.20–5.04 (m, 2H, CH=CH
2), 3.28 (dd,
3J1(
1H-
1H) = 13.6 Hz,
3J2(
1H-
1H) = 5.9 Hz, 1H, CH), 3.14–3.07 (m, 1H, CH–H), 3.06–2.99 (m, 2H, N–CH
2–CH), 2.37–2.27 (m, 1H, CH–H), 2.21–2.09 (m, 1H, CH–H), 1.91–1.81 (m, 1H, CH–H), 1.78–1.69 (m, 2H, CH
2).
13C[
1H]-NMR (100.63 MHz, CDCl
3): δ (ppm) = 178.3 (s, CONH
2), 135.1 (s, CH=CH
2), 117.3 (s, CH=CH
2), 66.7 (s, CH), 58.2 (s, CH
2), 53.9 (s, CH
2), 30.6 (s, CH
2), 24.2 (s, CH
2). HR-MS (LC-ESI): calculated for C
8H
15ON
2 [M+H]
+ (
m/z): 155.1; found 155.2.
(S)-(1-Allylpyrrolidine-2-yl)methanamine (
15). The synthesis followed the same procedure as described in the literature [
12]. The reaction of (
S)-1-Allylpyrrolidine-2-carboxamide
14 (2.57 g, 16.67 mmol) with DIBAL-solution (100 mL, 1 M in THF, 6.0 eq.) gave compound
15 (1.68 g, 11.98 mmol, 72%) as slightly yellowish oil.
1H-NMR (400.13 MHz, CDCl
3): δ (ppm) = 5.71–5.57 (m, 1H, CH=CH
2), 4.98–4.80 (m, 2H, CH=CH
2), 3.16 (dd,
2J1(
1H-
1H) = 13.4 Hz,
3J(
1H-
1H) = 5.5 Hz, 1H, NH
2CH–H), 2.86–2.79 (m, 1H, CH), 2.60 (dd,
2J(
1H-
1H) = 13.5 Hz,
3J(
1H-
1H) = 7.5 Hz, 1H, NH
2CH–H), 2.48–2.42 (m, 2H, CH
2), 2.22–1.92 (m, 2H, NH
2), 1.69–1.41 (m, 6H, CH
2CH
2CH
2).
13C[
1H]-NMR (100.63 MHz, CDCl
3): δ (ppm) = 136.1 (s, CH=CH
2), 116.2 (s, CH=CH
2), 65.1 (s, CH), 57.5 (s, CH
2), 54.1 (s, CH
2), 44.2 (s, CH
2NH
2), 27.9 (s, CH
2), 22.6 (s, CH
2).
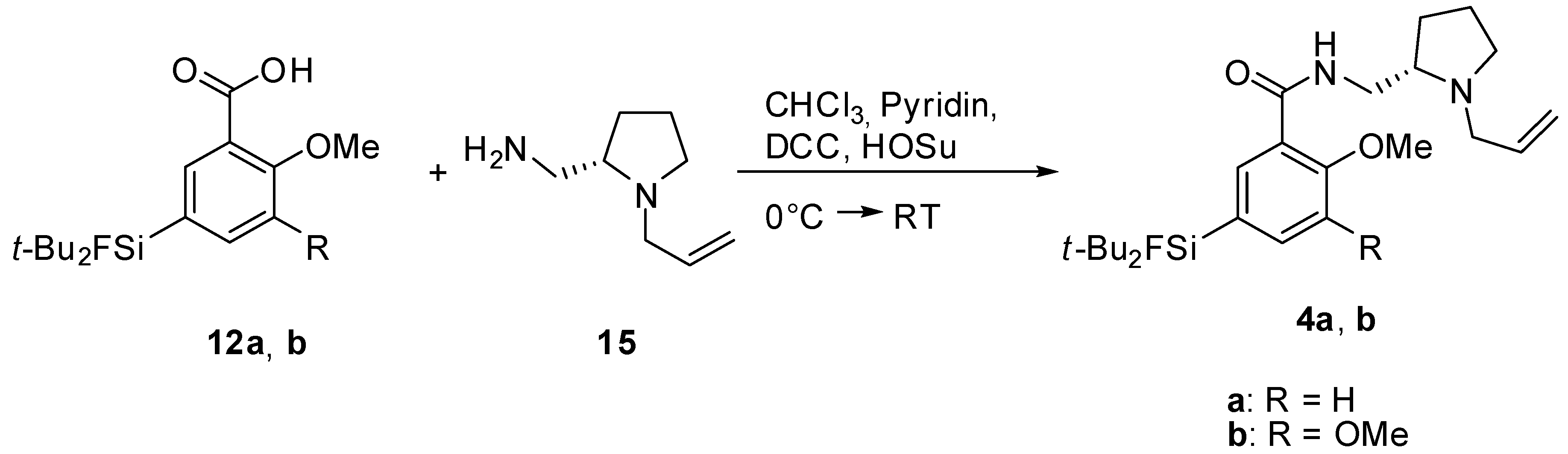
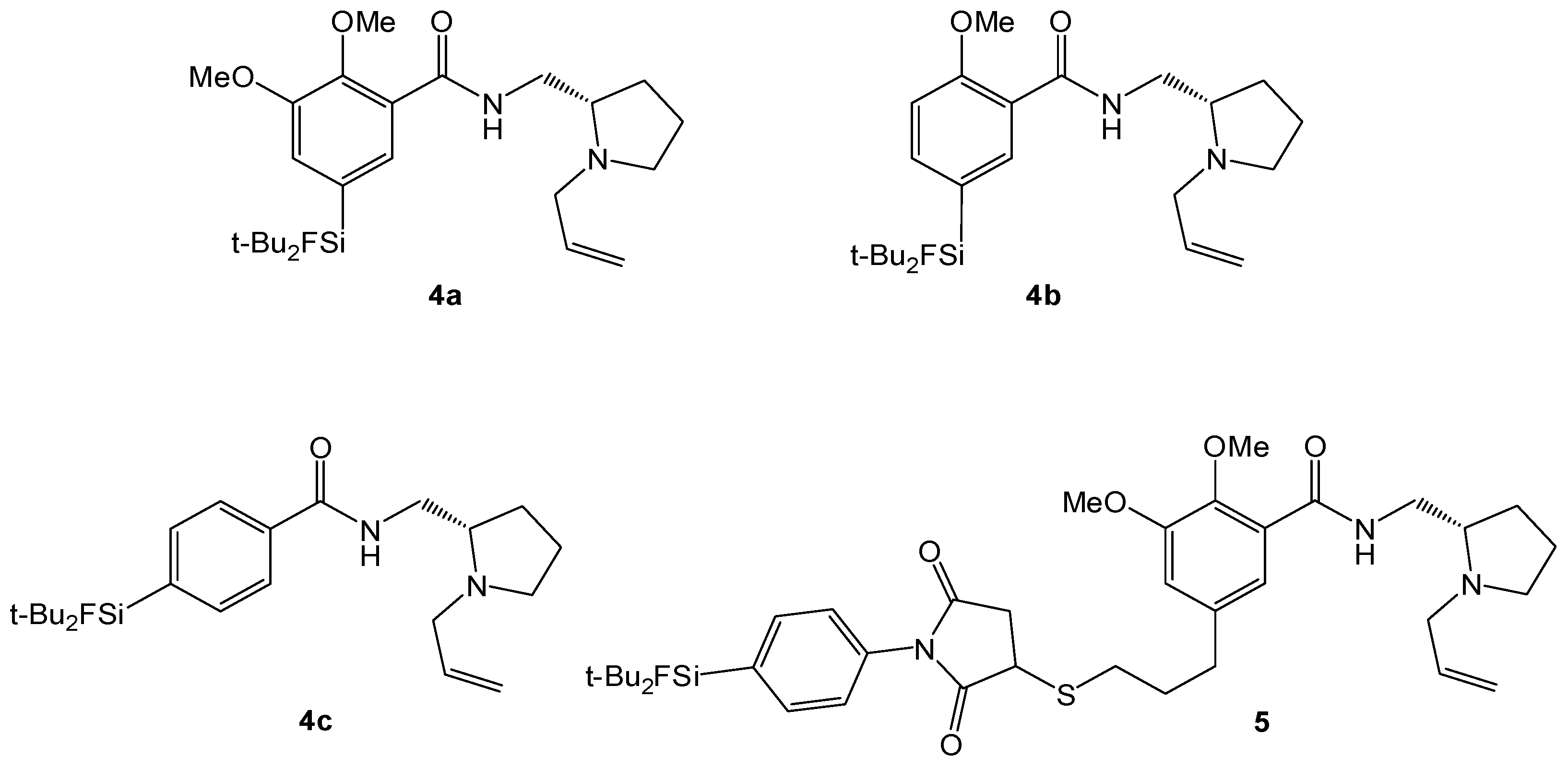
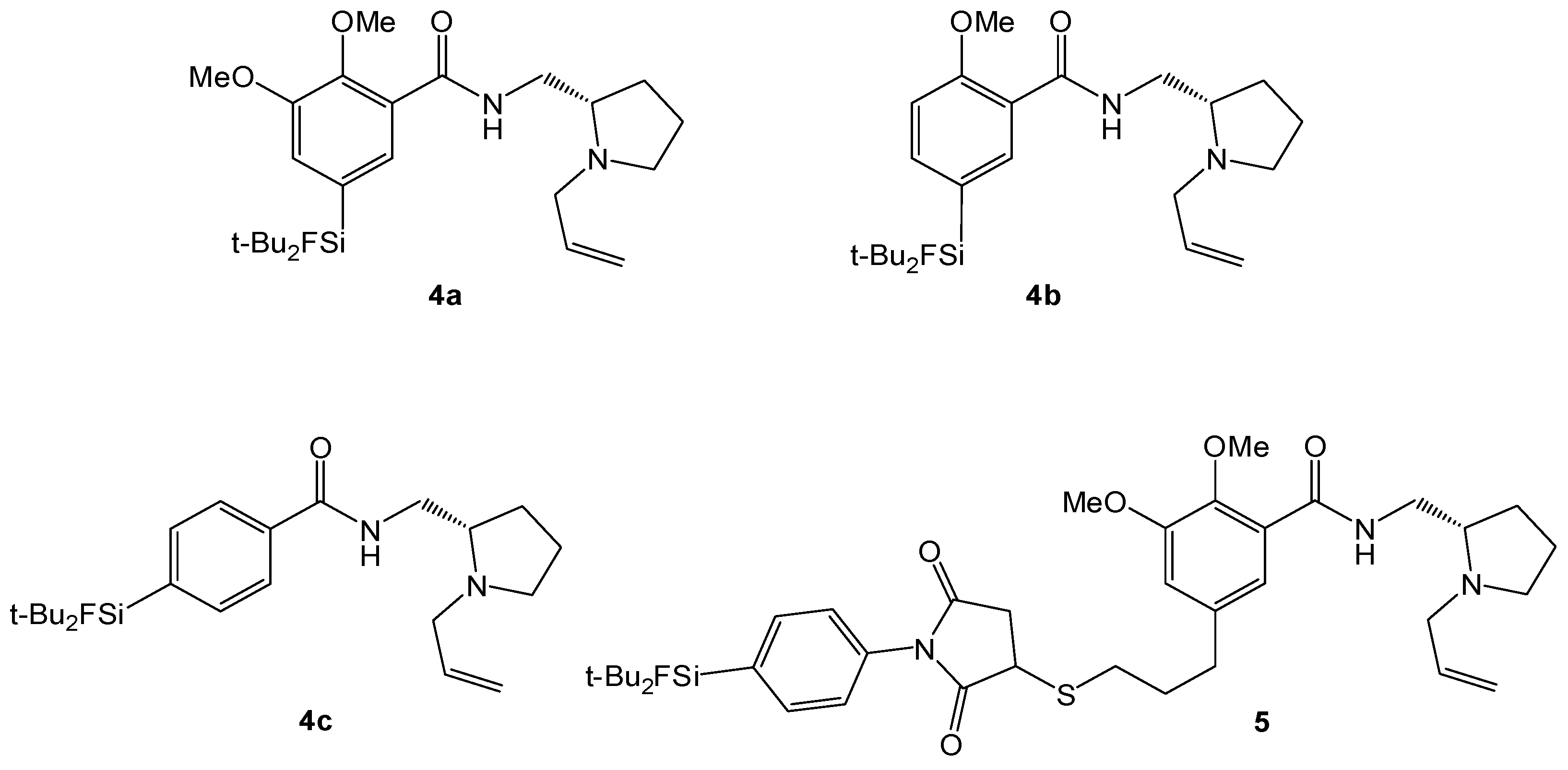
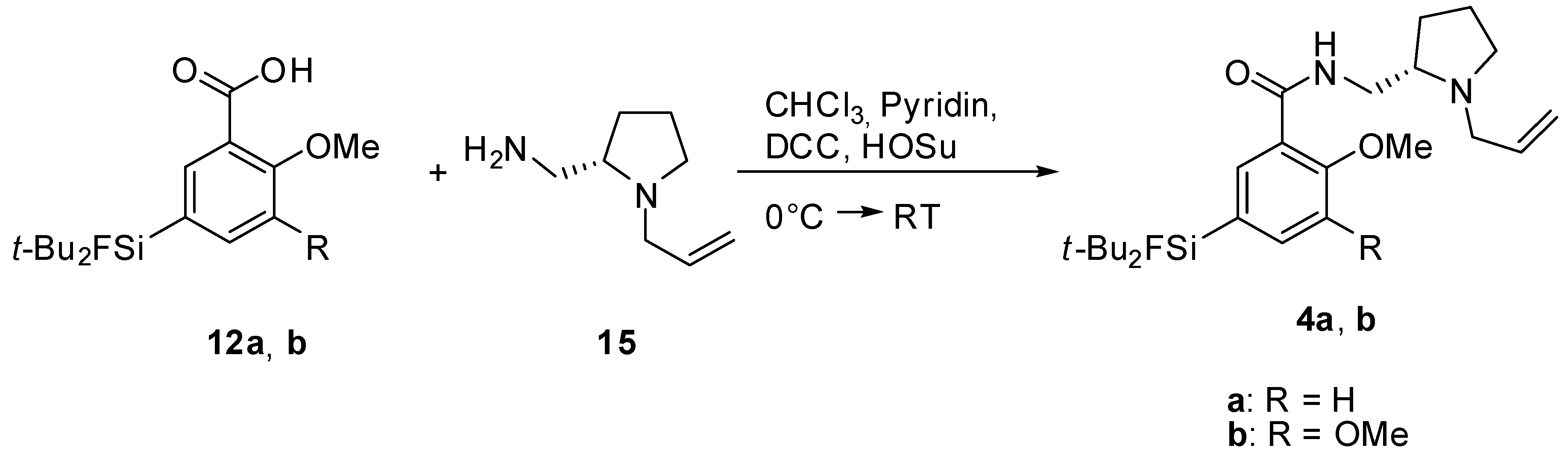
(S)-N-((1-Allylpyrrolidine-2-yl)methyl)-5-(di-tert-butylfluorosilyl)-2-methoxybenzamide (SiFA-DMFP 4a). To an ice-cooled solution in dry CHCl3 containing the substituted benzoic acid 12a (0.97 g, 3.10 mmol), (S)-(1-Allylpyrrolidine-2-yl)methanamine 15 (0.43 g, 3.10 mmol, 1.0 equiv.) and pyridine (0.25 mL, 3.10 mmol, 1.0 equiv.) dicyclohexylcarbodiimide (0.64 g, 3.10 mmol, 1.0 equiv.) and N-hydroxysuccinimide (0.36 g, 3.10 mmol, 1.0 equiv.) were added under stirring. The mixture was stirred at 0 °C for 5 h and 17 h at ambient temperature. After the white precipitate had been filtered, the filtrate was washed with saturated NaHCO3-solution (20 mL) and subsequently with H2O (20 mL). After extracting the aqueous phase with diethyl ether (20 mL) the combined organic layers were dried with MgSO4, filtered and the solvent was evaporated to give an oily residue. The latter was purified by column chromatography (CHCl3/Ethanol = 20/1 → CHCl3/Ethanol = 10/1) to afford benzamide 4a (0.55 g, 1.27 mmol, 41%) as a yellowish oil. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 8.44 (d, 4J(1H-1H) = 1.4 Hz, 1H, C(6)H), 8.37 (d, 3J(1H-1H) = 3.8 Hz, 1H, NH), 7.68 (dd, 3J(1H-1H) = 8.2 Hz, 4J(1H-1H) = 1.3 Hz, 1H, C(4)H), 7.00 (d, 3J(1H-1H) = 8.2 Hz, 1H, C(3)H), 5.96–5.84 (m, 1H, CH=CH2), 5.24-5.06 (m, 2H, CH=CH2), 3.95 (s, 3H, OCH3), 3.75 (ddd, 2J(1H-1H) = 13.8 Hz, 3J(1H-1H) = 7.1 Hz, 3J(1H-1H) = 3.1 Hz, 1H, NHCH–H), 3.48 (dd, 2J(1H-1H) = 13.5 Hz, 3J(1H-1H) = 5.3 Hz, 1H, CH2=CHCH-H), 3.35 (ddd, 2J(1H-1H) = 13.9 Hz, 3J(1H-1H) = 3.7 Hz, 3J(1H-1H) = 3.7 Hz, 1H, NHCH–H), 3.19–3.13 (m, 1H, NCH–H), 2.91 (dd, 2J(1H-1H) = 13.5 Hz, 3J(1H-1H) = 7.5 Hz, 1H, CH2=CHCH–H), 2.75 (s, 1H, CH), 2.33–2.22 (m, 1H, NCH–H), 1.99–1.85 (m, 1H, CH2CH2CH2), 1.79–1.61 (m, 3H, CHCH2CH–H), 1.02 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 165.5 (s, CONH), 158.7 (s, C(2)), 138.6 (d, 3J(13C-19F) = 3.8 Hz, C(4)), 137.7 (d, 3J(13C-19F) = 4.7 Hz, C(6)), 135.8 (s, CH=CH2), 125.3 (d, 2J(13C-19F) = 14.5 Hz, C(5)), 120.9 (s, C(1)), 116.2 (s, CH=CH2), 110.7 (s, C(3)), 61.9 (s, CH), 57.0 (s, CH2CH=CH2), 55.5 (s, OCH3), 54.2 (s, NCH2), 41.3 (s, CH2NH), 28.5 (s, CH2), 27.3 (s, CCH3), 22.9 (s, CH2), 20.2 (d, 2J(13C-19F) = 12.0 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −188.8 (s, 1J(19F-29Si) = 298 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.4 (d, 1J(29Si-19F) = 298 Hz). Elemental analysis calculated (%) for C24H39FN2O2Si∙H2O (452.68 g/mol): C 63.7, H 9.1, N 6.2; found: C 64.1, H 9.1, N 6.3. HR-MS (LC-ESI): calculated for C24H40O2N2FSi [M+H]+ (m/z): 435.2838; found: 435.2832.
(S)-N-((1-Allylpyrrolidine-2-yl)methyl)-5-(di-tert-butylfluorosilyl)-2,3-dimethoxybenzamide (SiFA-FP, 4b). The procedure was analogous to the synthesis of 4a. The reaction of the benzoic acid derivative 12b (1.47 g, 4.28 mmol) with (S)-(1-Allylpyrrolidine-2-yl)methanamine 15 (0.60 g, 3.10 mmol, 1.0 equiv.) gave SiFA-FP 4b (0.81 g, 1.74 mmol, 41%) as a yellowish oil. 1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 8.51 (s, br, 1H, NH), 7.93 (s, 1H, C(4)H), 7.24 (s, 1H, C(6)H), 5.99–5.87 (m, 1H, CH=CH2), 5.27-5.09 (m, 2H, CH=CH2), 3.93 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.80 (ddd, 2J(1H-1H) = 13.9 Hz, 3J(1H-1H) = 6.9 Hz, 3J(1H-1H) = 3.6 Hz, 1H, NHCH–H), 3.54 (dd, 2J(1H-1H) = 13.5 Hz, 3J(1H-1H) = 5.3 Hz, 1H, CH2=CHCH–H), 3.47–3.37 (m, 1H, NHCH–H), 3.19–3.13 (m, 1H, NCH–H), 2.98 (dd, 2J(1H-1H) = 13.2 Hz, 3J(1H-1H) = 7.6 Hz, 1H, CH2=CHCH–H), 2.86 (s, 1H, CH), 2.39–2.29 (m, 1H, NCH–H), 2.03–1.91 (m, 1H, CH2CH2CH2), 1.87–1.67 (m, 3H, CHCH2CH–H), 1.05 (s, 18H, CCH3). 13C[1H]-NMR (100.63 MHz, CDCl3): δ (ppm) = 165.5 (s, CONH), 151.9 (s, C(2)), 148.8 (s, C(3)), 134.6 (s, CH=CH2), 129.4 (d, 2J(13C-19F) = 14.0 Hz, C(5)), 128.4 (d, 3J(13C-19F) = 4.7 Hz, C(4)), 125.8 (s, C(1)), 120.3 (d, 3J(13C-19F) = 3.9 Hz, C(6)), 117.9 (s, CH=CH2), 62.2 (s, CH), 61.2 (s, OCH3), 57.0 (s, CH2CH=CH2), 56.0 (s, OCH3), 53.9 (s, NCH2), 41.0 (s, CH2NH), 28.4 (s, CH2), 27.3 (s, CCH3), 22.6 (s, CH2), 20.2 (d, 2J(13C-19F) = 12.2 Hz, C(CH3)3). 19F-NMR (282.38 MHz, CDCl3): δ (ppm) = −188.5 (s, 1J(19F-29Si) = 298 Hz). 29Si-NMR (59.63 MHz, CDCl3): δ (ppm) = 14.2 (d, 1J(29Si-19F) = 299 Hz). Elemental analysis calculated (%) for C25H41FN2O3Si∙H2O (482.70 g/mol): C 62.2, H 9.0, N 5.8; found (%): C 61.9, H 8.6, N 4.9. HR-MS (LC-ESI): calculated for C25H42O3N2FSi [M+H]+ (m/z): 465.2943; found: 465.2924.
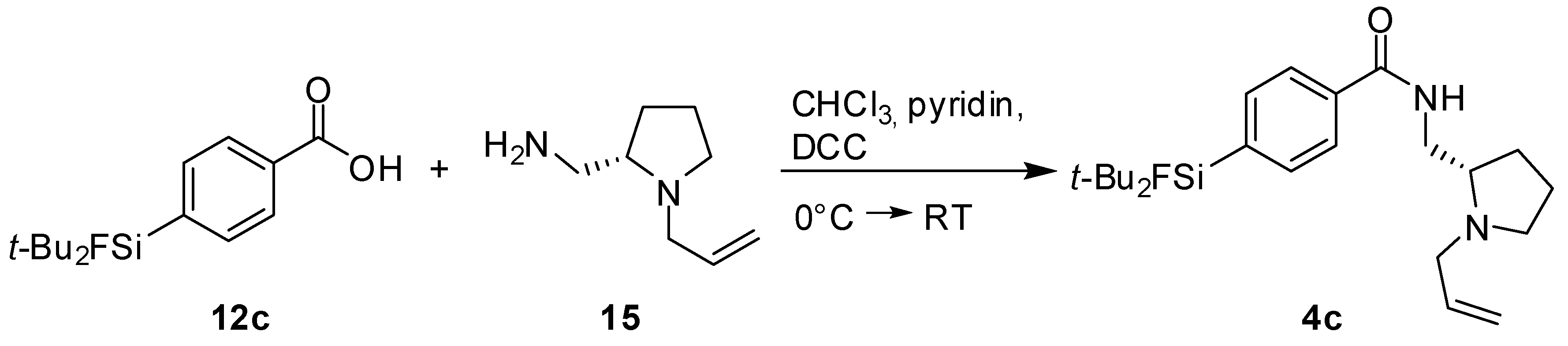
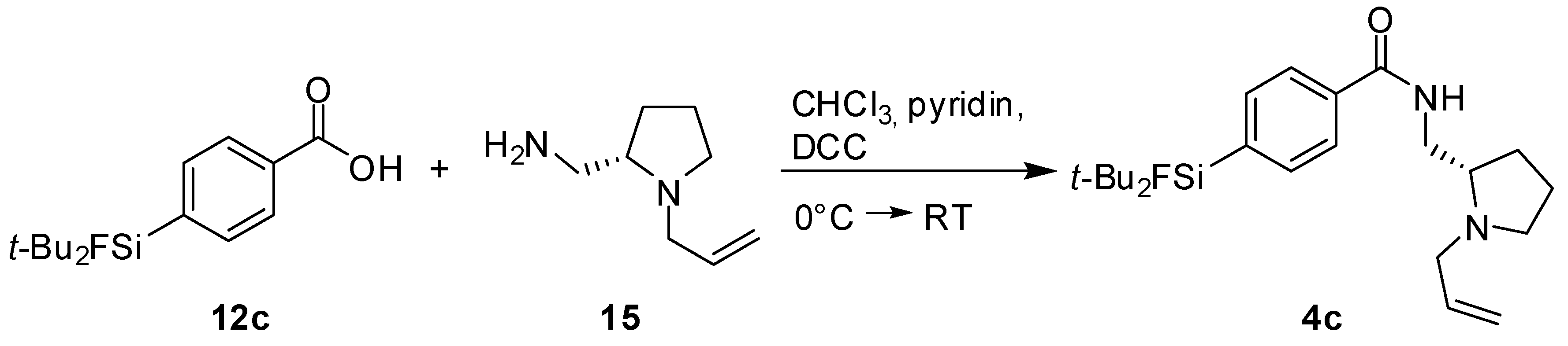
(S)-N-((1-Allylpyrrolidine-2-yl)methyl)-4-(di-tert-butylfluorosilyl) benzamide (SiFA-DDMFP,
4c). The procedure was analogous to the synthesis of
4a. The reaction of the benzoic acid derivative
12c [
17] (0.50 g, 1.77 mmol) with (
S)-(1-Allylpyrrolidine-2-yl)methanamine
15 (0.25 g, 1.77 mmol, 1.0 equiv.) gave SiFA-DDMFP
11c (0.08 g, 0.20 mmol, 11%) as a yellowish oil.
1H-NMR (400.13 MHz, CDCl
3): δ (ppm) = 7.80 (d,
3J(
1H-
1H) = 7.7 Hz, 2H, Ho), 7.69 (d,
3J(
1H-
1H) = 8.0 Hz, 2H, Hm), 7.00 (s, 1H, NH) 5.99–5.86 (m, 1H, CH=CH
2), 5.29–5.12 (m, 2H, CH=CH
2), 3.73 (ddd,
2J(
1H-
1H) = 13.7 Hz,
3J(
1H-
1H) = 7.4 Hz,
3J(
1H-
1H) = 3.2 Hz, 1H, NHCH–H), 3.50–3.43 (m, 2H), 3.2 (s, 1H), 3.04–2.76 (m, 2H), 2.42–2.28 (m, 1H), 1.92–1.90 (m, 1H), 1.64–1.54 (m, 3H), 1.06 (s, 18H, CCH
3).
13C[
1H]-NMR (100.63 MHz, CDCl
3): δ (ppm) = 167.8 (s, CONH), 137.9 (d,
2J(
13C-
19F) = 13.7 Hz, Cp), 135.2 (s, Ci), 134.1 (d,
3J(
13C-
19F) = 4.7 Hz, Cm), 134.1 (s, CH=CH
2), 126.0 (s, Co), 117.4 (s, CH=CH
2), 62.8 (s, CH), 57.4 (s, CH
2CH=CH
2), 54.1 (s, NCH
2), 40.7 (s, CH
2NH), 28.2 (s, CH
2), 27.2 (s, CCH
3), 23.1 (s, CH
2), 20.2 (d,
2J(
13C-
19F) = 12.3 Hz, C(CH
3)
3).
19F-NMR (282.38 MHz, CDCl
3): δ (ppm) = −189.2 (s,
1J(
19F-
29Si) = 299 Hz).
29Si-NMR (59.63 MHz, CDCl
3): δ (ppm) = 14.0 (d,
1J(
29Si-
19F) = 299 Hz). Elemental analysis calculated (%) for C
23H
37FN
2OSi∙H
2O + C
8H
16N
2 (562.88 g/mol): C 66.2, H 9.5, N 8.4; found (%): C 66.0, H 9.2, N 8.2. HR-MS (LC-ESI): calculated for C
23H
37ON
2FSi [M+H]
+ (
m/z): 405.2732; found: 405.2726.
(S)-N-((1-Allylpyrrolidin-2-yl)methyl)-5-(3-(1-(4-(di-tert-butylfluorosilyl)phenyl)-2,5-dioxopyrrolidin-3-ylthio)propyl)-2,3-dimethoxybenzamide (SiFA-M-FP,
5). To a freshly prepared solution of 1-(4-(di-
tert-butylfluorosilyl)phenyl)-1
H-pyrrole-2,5-dione (SiFA-maleimide, [
17]) (3 mg, 9 µmol) in phosphate buffer (PB, 0.1 M, pH 6.0) and acetonitrile (1:1) was added a solution of (
S)-
N-((1-allylpyrrolidin-2-yl)methyl)-5-(3-mercaptopropyl)-2,3-dimethoxybenzamide (FP-thiol) (3.4 mg, 9 µmol) in acetonitrile (100 µL) and the pH of the solution was adjusted to 7.2 using PB (0.1M, pH = 7.2, 200 µL). After 10 min, the product was isolated by semi-preparative HPLC applying a gradient of 40%–80% acetonitrile in 10 minutes. The product was obtained as white powder after lyophilization (3.1 mg, 4.4 µmol, 49% yield). ESI-MS calculated for [M+H]
+ (
m/z): 712.35, found: 712.36.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}