In Mesopore Protein Digestion: A New Forthcoming Strategy in Proteomics


Abstract
:1. Introduction
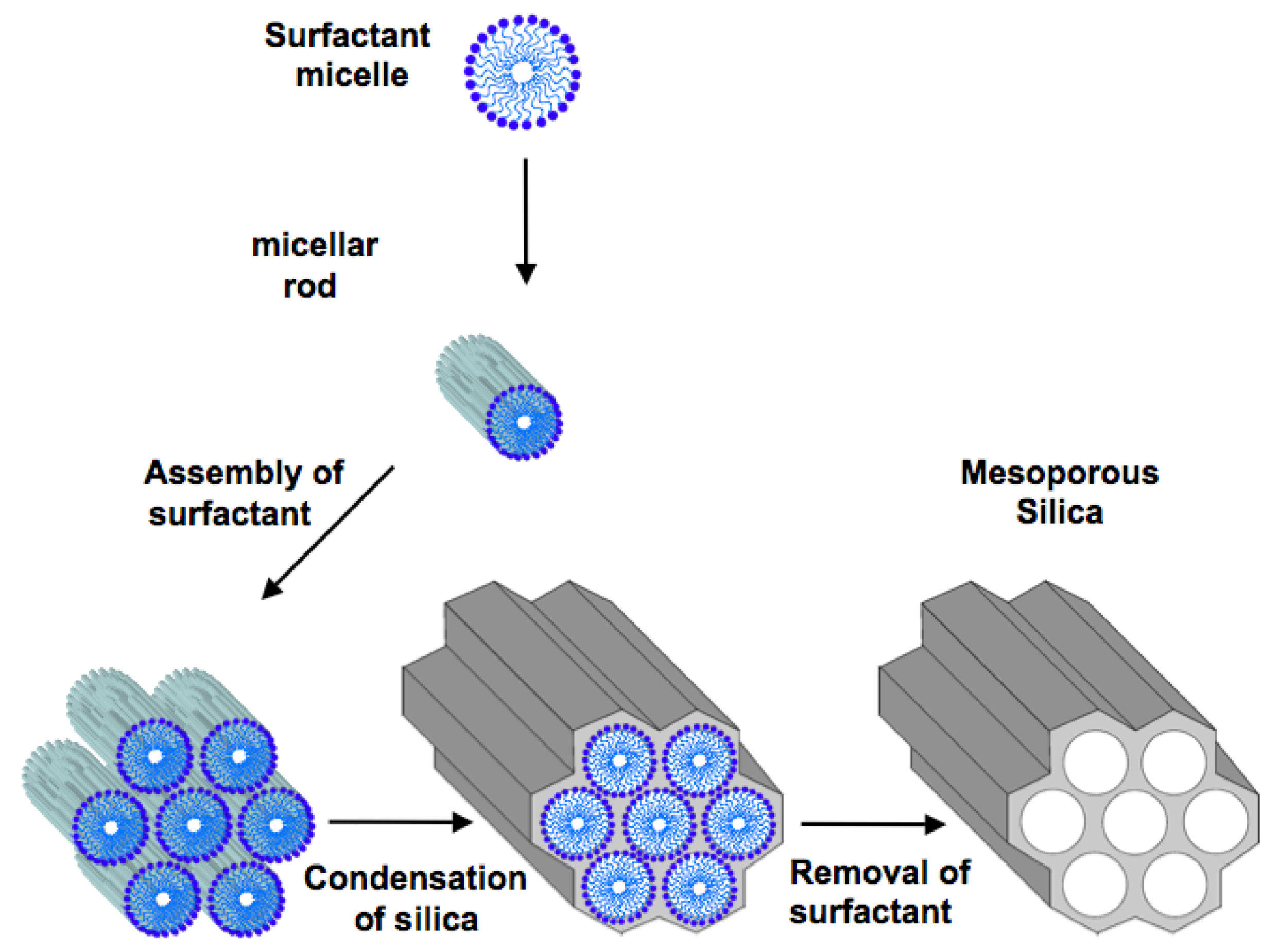
2. Ordered Mesoporous Silica
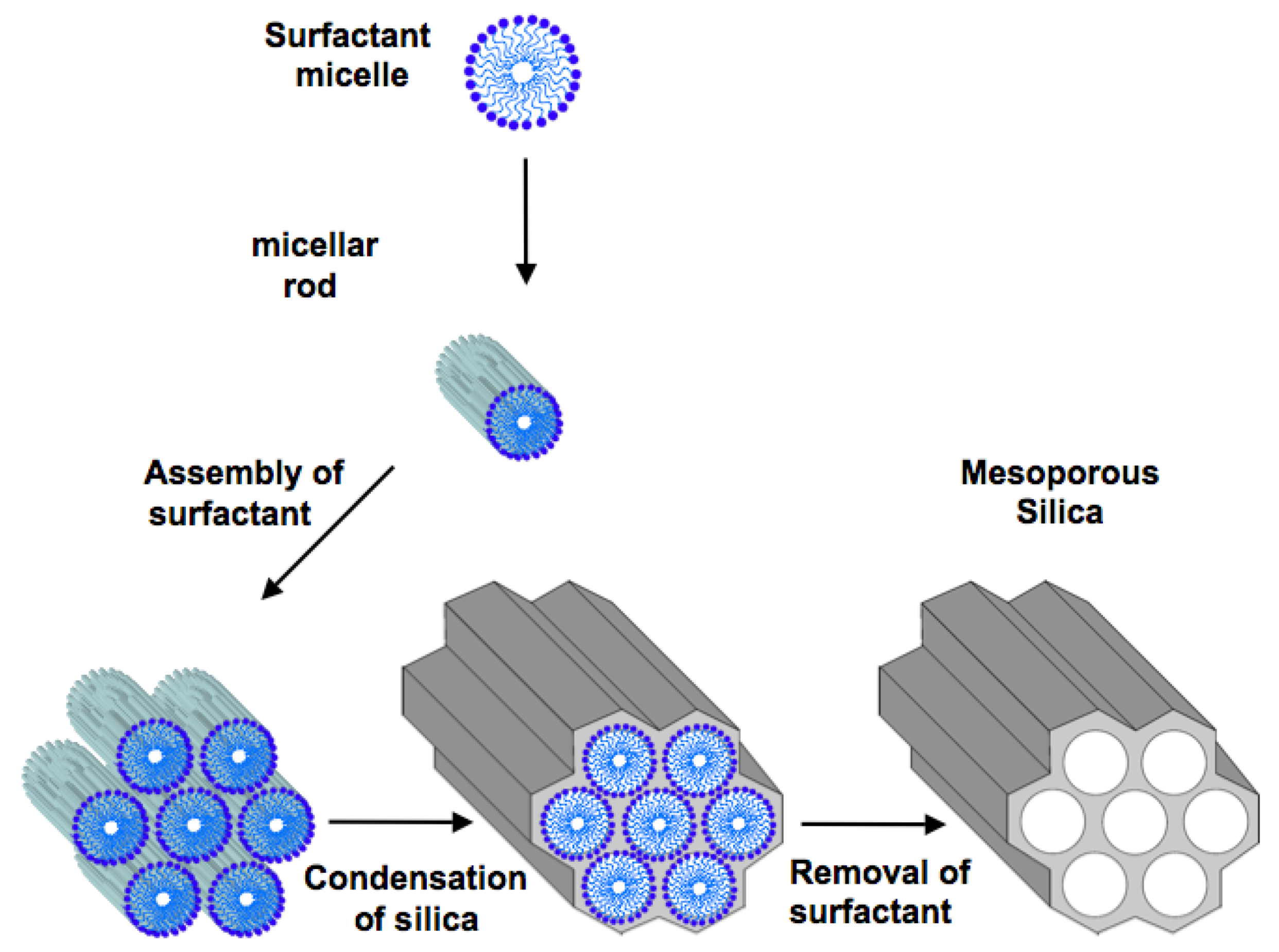
3. Mesoporous Silica for Supporting Enzyme Catalysts
4. Mesoporous Silica as Supports for Protein Digestion in Proteomics
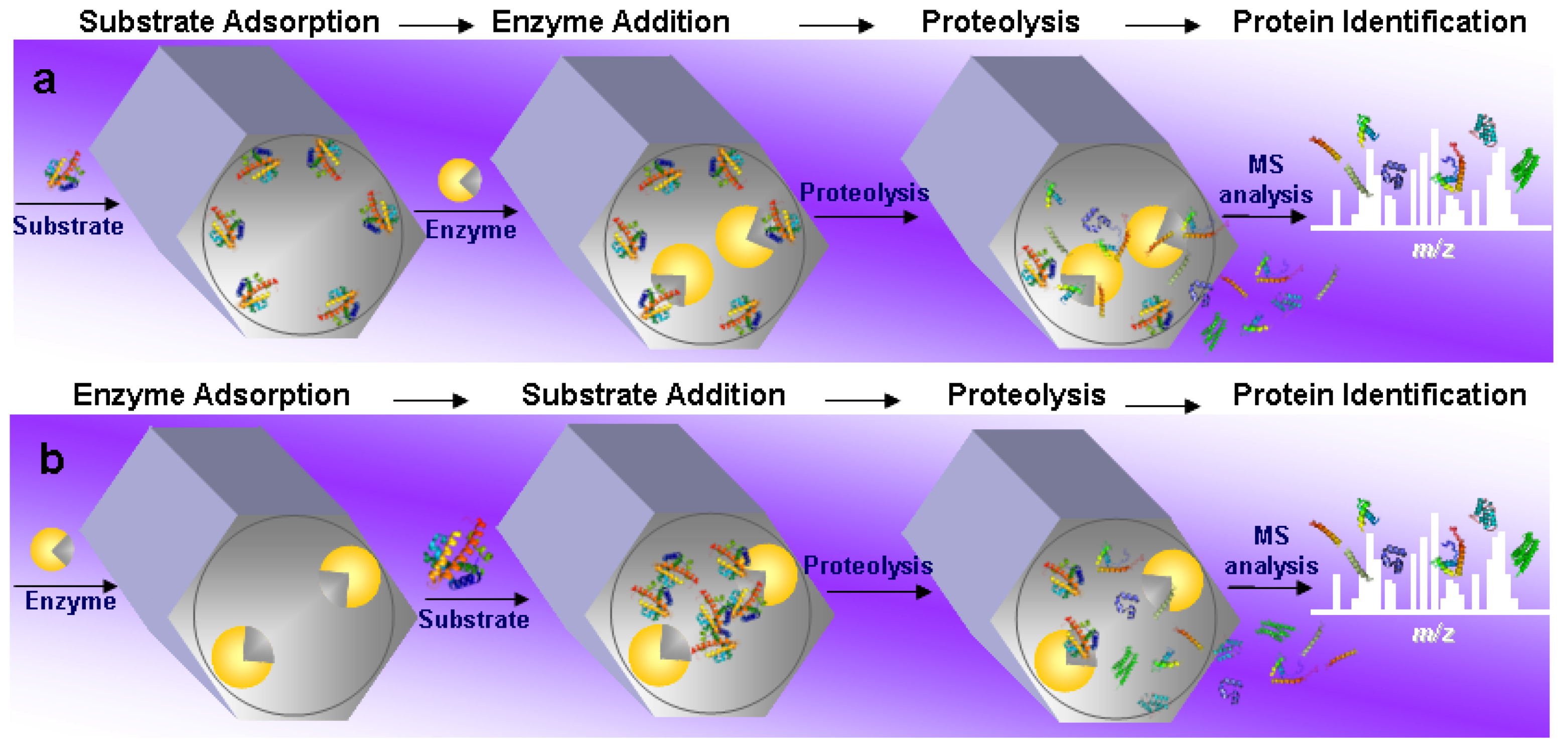
4.1. General Overview of the Field
4.2. Substrate Pre-Adsorption

{kind=link}
{kind=link}
| Protein | MW (Da) | pI | Dimension (nm) |
|---|---|---|---|
| Myoglobin | 17000 | 7.0 | 2.1 × 3.5 × 4.4 a |
| Cytochrome c | 12384 | 10.0 | 2.6 × 3.2 × 3.3 a |
| Bovine Serum Albumin | 66400 | 4.7 | 5.0 × 7.0 × 7.0 a |
| Ovalbumin | 42700 | 4.9 | 7.0 × 4.0 × 5.0 b |
| Conalbumin | 76000 | 6.0 | 5.0 × 5.6 × 9.5 a |
| Lysozyme | 14388 | 10.8 | 1.9 × 2.5 × 4.3 a |
| Trypsin | 23400 | 10.5 | ca. 3.8 b |
| MPS Material (mg) | Pore size (nm) | Substrate (amount digested) | E/S (w/w) | Digestion time | N of prot.frag (S/N) | Seq. Cov. | Ref. |
|---|---|---|---|---|---|---|---|
| SBA-15-SH (2.5) | 8 | Myoglobin (50 μg) | 1:2 | 10 min | 8 (70) | 58% | [25] |
| SBA-15 | 7 | Myoglobin (179 ng) | 1:2(2:1) | 15 min | 19 (20) | 98% | [26] |
| (2.5) | Cyt c (124 ng) | 1:2(2:1) | 15 min | 14 (20) | 84% | ||
| FDU-12 | 17 a | Myoglobin (50 μg) | 1:2 | 15 min | 12 (>80) | 84% | [27] |
| SBA-15 | 8 | Myoglobin (50 μg) | 1:2 | 15 min | NR(>80) | 31% | |
| MCF | 15 b | Myoglobin (50 μg) | 1:2 | 15 min | NR(>80) | 22% | |
| (2.5) | Ovalbumin (45 μg) /Conalbumin (15 μg) /Cyt c (1 μg) | 1:2 | 15 min | 42% | |||
| Nuclear proteins mouse liver cell (10 μg) | 26% | ||||||
| 58% | |||||||
| 1:8 | 30 min | ||||||
| HMS(CNS) | 18 | Cyt c (600 μg) | 1:3 | 20 min | 11 (NR) | 63% | [29] |
| (1) | Myoglobin (600 μg) | 1:3 | 20 min | 12 (NR) | NR | ||
| Cytoplasm of human liver tissue (5 μg) | 1:3 | 20 min | |||||
| SBA-15 SH | 5.7 | Cyt c/BSA c | 1:15 | 10 min c | 8 (NR) | 46% | [30] |
| (3) | Lysozyme/BSA d | 1:15 | |||||
| Myoglobin/BSA d | 1:15 | ||||||
| Human serum (150 μg) | 1:15 | 60 min | |||||
| SBA-15 (1) | 4.3 | Myoglobin (20) | 1:3 | 1 min | 7 (>50) | 43% | [31] |
| SBA-15 (1) | 6.3 | Myoglobin (18) | 1:3 | 1 min | 2 (>50) | 17% | [31] |
| SBA-15 APTES (1) | 4.3 | Myoglobin (16.9) | 1:3 | 1 min | 7 (>50) | 60% | [31] |
| SBA-15 AAPTES (1) | 4.4 | Myoglobin (18) | 1:3 | 1 min | 21 (>50) | 100% | [31] |
4.3. Enzyme Pre-Adsorption
4.4. Factors Affecting in Mesopore Proteolytic Efficiency
5. Conclusions
Acknowledgements
References and Notes
- Walther, T.C.; Mann, M. Mass spectrometry-based proteomics in cell biology. J. Cell Biol. 2010, 190, 491–500. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd.; Gilchrist, A.; Howell, K.E.; Bergeron, J.J. Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 2005, 6, 702–714. [Google Scholar]
- Au, C.E.; Bell, A.W.; Gilchrist, A.; Hinding, J.; Nilsson, T.; Bergeron, J.J. Organellar proteomics to create the cell map. Curr. Opin. Cell Biol. 2007, 19, 376–385. [Google Scholar] [CrossRef]
- Collins, S.R.; Kemmeren, P.; Zhao, X.C.; Greenblatt, J.F.; Spencer, F.; Holstege, F.C.; Weissman, J.S.; Krogan, N.J. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteomics 2007, 6, 439–450. [Google Scholar]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R., 3rd. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef]
- Washburn, M.P.; Wolters, D.; Yates, J.R., 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar]
- Wilm, M.; Shevchenko, A.; Houthaeve, T.; Breit, S.; Schweigerer, L.; Fotsis, T.; Mann, M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 1996, 379, 466–469. [Google Scholar]
- Flannery, A.V.; Beynon, R.J.; Bond, J.S. Proteolysis of protein for sequencing analysis and peptide mapping. In Proteolytic Enzymes: A Practical Approach; Beynon, R.J., Bond, J.S., Eds.; Oxford University Press: Oxford, UK, 1989; pp. 145–162. [Google Scholar]
- Smith, B.J. Protein sequencing protocols. In Methods in Molecular Biology, 2nd; Smith, B.J., Ed.; Humana Press Inc: Totowa, NJ, USA, 2002; Volume 211. [Google Scholar]
- Pramanik, B.N.; Mirza, U.A.; Ing, Y.H.; Liu, Y.H.; Bartner, P.L.; Webwe, P.C.; Bose, A.K. Microwave-enhanced enzyme reaction for protein mapping by mass spectrometry: A new approach to protein digestion in minutes. Protein Sci. 2002, 11, 2676–2687. [Google Scholar]
- Juan, H.F.; Chang, S.C.; Huang, H.C.; Chen, S.T. A new application of microwave technology to proteomics. Proteomics 2005, 5, 840–842. [Google Scholar] [CrossRef]
- Lopez-Ferrer, D.; Capelo, J.L.; Vazquez, J. Ultra fast trypsin digestion of proteins by high intensity focused ultrasound. J. Proteome Res. 2005, 4, 1569–1574. [Google Scholar] [CrossRef]
- Lopez-Ferrer, D.; Petritis, K.; Hixson, K.K.; Heibeck, T.H.; Moore, R.J.; Belov, M.E.; Camp, G.C.; Smith, R.D. Application of pressurized solvents for ultrafast trypsin hydrolysis in proteomics: Proteomics on the fly. J. Proteome Res. 2008, 7, 3276–3281. [Google Scholar] [CrossRef]
- Monzo, A.; Sperling, E.; Guttman, A. Proteolytic enzyme-immobilization techniques for MS-based protein analysis. TrAC Trends Anal. Chem. 2009, 28, 854–864. [Google Scholar] [CrossRef]
- Kim, J.; Kim, B.C.; Lopez-Ferrer, D.; Petritis, K.; Smith, R.D. Nanobiocatalysis for protein digestion in proteomic analysis. Proteomics 2010, 10, 687–699. [Google Scholar] [CrossRef]
- Cañas, B.; Piñeiro, C.; Calvo, E.; López-Ferrer, D.; Gallardo, J.M. Trends in sample preparation for classical and second generation proteomics. J. Chromatogr. A 2007, 1153, 235–258. [Google Scholar] [CrossRef]
- Wang, C.; Oleschuk, R.; Ouchen, F.; Li, J.; Thibault, P.; Harrison, D.J. Integration of immobilized trypsin bead beds for protein digestion within a microfluidic chip incorporating capillary electrophoresis separations and an electrospray mass spectrometry interface. Rapid Commun. Mass Spectrom. 2000, 14, 1377–1383. [Google Scholar]
- Koh, W.G.; Pishkoa, M. Immobilization of multienzyme microreactors inside microfluidic devices. Sens. Actuato. B 2005, 106, 335–342. [Google Scholar] [CrossRef]
- Massolini, G.; Calleri, E. Immobilized trypsin systems coupled on-line to separation methods: Recent developments and analytical applications. J. Sep. Sci. 2007, 28, 7–21. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.; Liang, Z.; Zhang, W.; Zhang, Y. Recent advances in immobilized enzymatic reactors and their applications in proteome analysis. Anal. Chim. Acta 2009, 632, 1–8. [Google Scholar] [CrossRef]
- Temporini, C.; Perani, E.; Mancini, F.; Bartolini, M.; Calleri, E.; Lubda, D.; Felix, G.; Andrisano, V.; Massolini, G. Optimization of a trypsin-bioreactor coupled with high-performance liquid chromatography-electrospray ionization tandem mass spectrometry for quality control of biotechnological drugs. J. Chromatogr. A 2006, 1120, 121–131. [Google Scholar] [CrossRef]
- Yiu, H.H.P.; Wright, P.A.; Botting, N.P. Enzyme immobilisation using siliceous mesoporous molecular sieves. Microporous Mesoporous Mat. 2001, 44-45, 763–768. [Google Scholar] [CrossRef]
- Fan, J.; Shui, W.; Yang, P.; Wang, X.; Xu, Y.; Wang, H.; Chen, X.; Zhao, D. Mesoporous silica nanoreactors for highly efficient proteolysis. Chem.-Eur. J. 2005, 11, 5391–5396. [Google Scholar] [CrossRef]
- Zuo, C.; Yu, W.; Zhou, X.; Zhao, D.; Yang, P. Highly efficient enrichment and subsequent digestion of proteins in the mesoporous molecular sieve silicate SBA-15 for matrix-assisted laser desorption/ionization mass spectrometry with time-of-flight/time-of-flight analyzer peptide mapping. Rapid Commun. Mass Spectrom. 2006, 20, 3139–3144. [Google Scholar] [CrossRef]
- Shui, W.; Fan, J.; Yang, P.; Liu, C.; Zhai, J.; Lei, J.; Yan, Y.; Zhao, D.; Chen, X. Nanopore-based proteolytic reactor for sensitive and comprehensive proteomic analyses. Anal. Chem. 2006, 78, 4811–4819. [Google Scholar] [CrossRef]
- Jang, S.; Kim, D.; Choi, J.; Row, K.; Ahn, W. Trypsin immobilization on mesoporous silica with or without thiol functionalization. J. Porous Mater. 2006, 13, 385–391. [Google Scholar] [CrossRef]
- Qiao, L.; Liu, Y.; Hudson, S.P.; Yang, P.; Magner, E.; Liu, B. Nanoporous reactor for efficient proteolysis. Chem.-Eur. J. 2008, 14, 151–157. [Google Scholar] [CrossRef]
- Min, Q.; Wu, R.; Zhao, L.; Qin, H.; Ye, M.; Zhu, J.J.; Zou, H. Size-selective proteolysis on mesoporous silica-based trypsin nanoreactor for low-MW proteome analysis. Chem. Commun. 2010, 46, 6144–6146. [Google Scholar]
- Casadonte, F.; Pasqua, L.; Savino, R.; Terracciano, R. Smart trypsin adsorption into N-(2-aminoethyl)-3-aminopropyl-modified mesoporous silica for ultra fast protein digestion. Chem.-Eur. J. 2010, 16, 8998–9001. [Google Scholar]
- Gallis, K.W.; Araujo, J.T.; Duff, K.J.; Moore, J.G.; Landry, C.C. The use of mesoporous silica in liquid chromatography. Adv. Mater. 1999, 17, 1452–1455. [Google Scholar]
- Zhao, X.S.; Bao, X.Y.; Guo, W.; Lee, F.Y. Immobilizing catalysts on porous materials. Materials Today 2006, 3, 32–39. [Google Scholar]
- Hartmann, M. Ordered mesoporous materials for bioadsorption and biocatalysis. Chem. Mater. 2005, 17, 4577–4593. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar]
- Ng, L.V.; Thompson, P.; Sanchez, J.; Macosko, C.W.; McCormick, A.V. Formation of cagelike intermediates from nonrandom cyclization during acid-catalyzed sol-gel polymerization of tetraethyl orthosilicate. Macromolecules 1995, 28, 6471–6476. [Google Scholar] [CrossRef]
- Lee, Y.S.; Surjadi, D.; Rathman, J.F. Effects of aluminate and silicate on the structure of quaternary ammonium surfactant aggregates. Langmuir 1996, 12, 6202–6210. [Google Scholar] [CrossRef]
- Lee, Y.S.; Surjadi, D.; Rathman, J.F. Compositional effects and hydrothermal reorganization of mesoporous silicates synthesized in surfactant solutions. Langmuir 2000, 16, 195–202. [Google Scholar]
- Vartuli, J.C.; Malek, A.; Roth, W.J.; Kresge, C.T.; McCullen, S.B. The sorption properties of as-synthesized and calcined MCM-41 and MCM-48. Microporous Mesoporous Mat. 2001, 44-45, 691–695. [Google Scholar]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Matos, J.R.; Kruk, M.; Mercuri, L.P.; Jaroniec, M.; Zhao, L.; Kamiyama, T.; Terasaki, O.; Pinnavaia, T.J.; Liu, Y. Ordered mesoporous silica with large cage-like pores: Structural identification and pore connectivity design by controlling the synthesis temperature and time. J. Am. Chem. Soc. 2003, 125, 821–829. [Google Scholar]
- Wan, Y.; Zhao, D. On the controllable soft-templating approach to mesoporous silicates. Chem. Rev. 2007, 107, 2821–2860. [Google Scholar] [CrossRef]
- Ulagappan, N.; Rao, C.N.R. Evidence for supramolecular organization of alkane and surfactant molecules in the process of forming mesoporous silica. Chem. Commun. 1996, 24, 2759–2760. [Google Scholar]
- Naik, B.; Ghosh, N.N. A Review on chemical methodologies for preparation of mesoporous silica and alumina based materials. Recent Pat. Nanotechnol. 2009, 3, 213–224. [Google Scholar] [CrossRef]
- Wight, A.P.; Davis, M.E. Design and preparation of organic-inorganic hybrid catalysts. Chem. Rev. 2002, 102, 3589–3614. [Google Scholar] [CrossRef]
- Vinu, A.; Hossain, K.Z.; Ariga, K. Recent advances in functionalization of mesoporous silica. J. Nanosci. Nanotechnol. 2005, 5, 347–371. [Google Scholar] [CrossRef]
- Melero, J.A.; van Grieken, R.; Morales, G. Advances in the synthesis and catalytic applications of organosulfonic-functionalized mesostructured materials. Chem. Rev. 2006, 106, 3790–3812. [Google Scholar]
- Vallet-Regi, M.; Ruiz-Gonzales, L.; Izquierdo-Barba, I.; Gonzales-Cablet, J.M. Revisiting silica based ordered mesoporous materials: medical applications. J. Mater. Chem. 2006, 16, 26–31. [Google Scholar] [CrossRef]
- Park, S.E.; Ryoo, R.; Ahn, W.S.; Lee, C.W.; Chang, R. Nanotechnology in Mesostructured Materials; Park, S.E., Ryoo, R., Ahn, W.S., Lee, C.W., Chang, R., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Popat, A.; Hartono, S.B.; Stahr, F.; Liu, J.; Qiao, S.Z.; Lu, G.Q. Mesoporous silica nanoparticles for bioadsorption, enzyme immobilization, and delivery carriers. Nanoscale 2011. [Google Scholar] [CrossRef]
- Terracciano, R.; Gaspari, M.; Testa, F.; Pasqua, L.; Cuda, G.; Tagliaferri, P.; Cheng, M.M.C; Nijdam, A.J.; Petricoin, E.F.; Liotta, L.A.; et al. Selective binding and enrichment for low-molecular weight biomarker molecules in human plasma after exposure to nanoporous silica particles. Proteomics 2006, 6, 3243–3250. [Google Scholar] [CrossRef]
- Terracciano, R.; Pasqua, L.; Casadonte, F.; Frascà, S.; Preianò, M.; Falcone, D.; Savino, R. Derivatized mesoporous silica beads for MALDI-TOF MS profiling of human plasma and urine. Bioconjugate Chem. 2009, 20, 913–923. [Google Scholar] [CrossRef]
- Terracciano, R.; Casadonte, F.; Pasqua, L.; Candeloro, P.; Di Fabrizio, E.; Urbani, A.; Savino, R. Enhancing plasma peptide MALDI-TOF-MS profiling by mesoporous silica assisted crystallization. Talanta 2010, 80, 1532–1538. [Google Scholar] [CrossRef]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef]
- Joo, S.H.; Choi, S.J.; Oh, I.; Kwak, J.; Liu, Z.; Terasaki, O.; Ryoo, R. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles. Nature 2001, 412, 169–172. [Google Scholar] [CrossRef]
- Schmid, A.; Dordick, J.S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [Google Scholar] [CrossRef]
- Yiu, H.H.P.; Wright, P.A. Enzymes supported on ordered mesoporous solids: A special case of an inorganic-organic hybrid. J. Mater. Chem. 2005, 15, 3690–3700. [Google Scholar]
- Diaz, J.F.; Balkus, K.J., Jr. Enzyme immobilization in MCM-41 molecular sieve. J. Mol. Catal. B: Enzym. 1996, 2, 115–126. [Google Scholar] [CrossRef]
- Hudson, S.; Cooney, J.; Magner, E. Proteins in mesoporous silicates. Angew. Chem. Int. Ed. 2008, 47, 8582–8594. [Google Scholar] [CrossRef]
- Terrés, E.; Montiel, M.; Le Borgne, S.; Torres, E. Immobilization of chloroperoxidase on meso-porous materials for the oxidation of 4,6-dimethyldibenzothiophene, a recalcitrant organic sulfur compound present in petroleum fractions. Biotechnol. Lett. 2008, 30, 173–179. [Google Scholar]
- Baker, S.E.; Sawvel, A.M.; Fan, J.; Shi, Q.; Strandwitz, N.; Stucky, G.D. Blood clot initiation by mesocellular foams: dependence on nanopore size and enzyme immobilization. Langmuir 2008, 24, 14254–14260. [Google Scholar] [CrossRef]
- Yu, A.; Gentle, I.R.; Lu, G.Q. Biocompatible polypeptide microcapsules via templating mesoporous silica spheres. J. Colloid Interface Sci. 2009, 333, 341–345. [Google Scholar] [CrossRef]
- Budi Hartono, S.; Qiao, S.Z.; Jack, K.; Ladewig, B.P.; Hao, Z.; Lu, G.Q. Improving adsorbent properties of cage-like ordered amine functionalized mesoporous silica with very large pores for bioadsorption. Langmuir 2009, 25, 413–424. [Google Scholar]
- Lin, J.; He, C.; Zhang, S. Immunoassay channels for alpha-fetoprotein based on encapsulation of biorecognition molecules into SBA-15 mesopores. Anal. Chim. Acta 2009, 643, 90–94. [Google Scholar] [CrossRef]
- Kato, K.; Seelan, S.; Saito, T. Catalytic activity of aryl alcohol oxidase immobilized in 3D-mesoporous silicates. J. Biosci. Bioong. 2009, 108, 310–313. [Google Scholar] [CrossRef]
- Itoh, T.; Ishii, R.; Matsuura, S.; Mizuguchi, J.; Hamakawa, S.; Hanaoka, T.A.; Tsunoda, T.; Mizukami, F. Enhancement in thermal stability and resistance to denaturants of lipase encapsulated in mesoporous silica with alkyltrimethylammonium (CTAB). Colloids Surf. B 2010, 75, 478–482. [Google Scholar] [CrossRef]
- Sörensen, M.H.; Ng, J.B.; Bergström, L.; Alberius, P.C. Improved enzymatic activity of Thermomyces lanuginosus lipase immobilized in a hydrophobic particulate mesoporous carrier. J. Colloid Interface Sci. 2010, 343, 359–365. [Google Scholar] [CrossRef]
- Gao, S.; Wang, Y.; Diao, X.; Luo, G.; Dai, Y. Effect of pore diameter and cross-linking method on the immobilization efficiency of Candida rugosa lipase in SBA-15. Bioresour. Technol. 2010, 101, 3830–3837. [Google Scholar] [CrossRef]
- Matsuura, S.; Tsunoda, T.; Shiomi, T.; Sakaguchi, K.; Hanaoka, T.; Mizukami, F. Detection of hetero-proteins-mesoporous silica assembly by BRET. Chem. Commun. 2010, 46, 2941–2943. [Google Scholar] [CrossRef]
- Kato, K.; Seelan, S. Enhancing activity and stability of Burkholderia cepacia lipase by immobilization on surface-functionalized mesoporous silicates. J. Biosci. Bioeng. 2010, 109, 615–617. [Google Scholar] [CrossRef]
- Wang, F.; Guo, C.; Yang, L.R.; Liu, C.Z. Magnetic mesoporous silica nanoparticles: Fabrication and their laccase immobilization performance. Bioresour. Technol. 2010, 101, 8931–8935. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Z.; Shi, Z.; Xue, P.; Chang, P.; Yan, R. Application of a novel co-enzyme reactor in chemiluminescence flow-through biosensor for determination of lactose. Talanta 2010, 82, 319–324. [Google Scholar] [CrossRef]
- Vittorini, M.; Dumitriu, E.; Barletta, G.; Secundo, F. Immobilization of Thermoanaerobium brockii alcohol dehydrogenase on SBA-15. Bioprocess Biosyst. Eng. 2011, 34, 247–251. [Google Scholar] [CrossRef]
- Noji, T.; Kamidaki, C.; Kawakami, K.; Shen, J.R.; Kajino, T.; Fukushima, Y.; Sekitoh, T.; Itoh, S. Photosynthetic oxygen evolution in mesoporous silica material: Adsorption of photosystem II reaction center complex into 23 nm nanopores in SBA. Langmuir 2011, 27, 705–713. [Google Scholar] [CrossRef]
- Xu, L.Q.; Wen, X.H.; Ding, H.J. Immobilization of lignin peroxidase on spherical mesoporous material. Huan Jing Ke Xue 2010, 31, 2493–2499. [Google Scholar]
- Wu, S.; Wang, H.; Tao, S.; Wang, C.; Zhang, L.; Liu, Z.; Meng, C. Magnetic loading of tyrosinase-Fe3O4/mesoporous silica core/shell microspheres for high sensitive electrochemical biosensing. Anal. Chim. Acta 2011, 686, 81–86. [Google Scholar] [CrossRef]
- Prasad, J.; Joshi, A.; Jayant, R.D.; Srivastava, R. Cholesterol biosensors based on oxygen sensing alginate-silica microspheres. Biotechnol. Bioeng. 2011. [Google Scholar] [CrossRef]
- Falahati, M.; Ma'mani, L.; Saboury, A.A.; Shafiee, A.; Badiei, A.R. Aminopropyl-functionalized cubic Ia3d mesoporous silica as an efficient support for immobilization of superoxide dismutase. Biochim. Biophys. Acta Proteins Proteomics 2011. [Google Scholar] [CrossRef]
- Sigala, P.A.; Kraut, D.A.; Caaveiro, J.M.M.; Pybus, B.; Ruben, E.A.; Ringe, D.; Petsko, G.A.; Herschlag, D. Testing geometrical discrimination within an enzyme active site: constrained hydrogen bonding in the ketosteroid isomerase oxyanion hole. J. Am. Chem. Soc. 2008, 130, 13696–13708. [Google Scholar]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Di Cera, E. Serine proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Cheng, M.M.C.; Cuda, G.; Bunimovich, Y.L.; Gaspari, M.; Heath, J.R.; Hill, H.D.; Mirkin, C.A; Nijdam, A.J.; Terracciano, R.; Thundat, T.; et al. Nanotechnologies for biomolecular detection and medical diagnostics. Curr. Opin. Chem. Biol. 2006, 10, 11–19. [Google Scholar] [CrossRef]
- Fan, J.; Yu, C.Z.; Gao, T.; Lei, J.; Tian, B.Z.; Wang, L.M.; Luo, Q.; Tu, B.; Zhou, W.Z.; Zhao, D.Y. Cubic mesoporous silica with large controllable entrance sizes and advanced adsorption properties. Angew. Chem. Int. Ed. 2003, 42, 3146–3150. [Google Scholar] [CrossRef]
- Fan, J.; Yu, C.Z.; Lei, J.; Zhang, Q.; Li, T.C.; Tu, B.; Zhou, W.Z.; Zhao, D.Y. Low-temperature strategy to synthesize highly ordered mesoporous silicas with very large pores. J. Am. Chem. Soc. 2005, 127, 10794–10795. [Google Scholar]
- Zhao, D.Y.; Feng, J.L.; Huo, Q.S.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar]
- Schmidt-Winkel, P.; Lukens, W.W.; Zhao, D.Y.; Yang, P.D.; Chmelka, B.F.; Stucky, G.D. Mesocellular siliceous foams with uniformly sized cells and windows. J. Am. Chem. Soc. 1999, 121, 254–255. [Google Scholar] [CrossRef]
- Goradia, D.; Cooney, J.; Hodnett, B.K.; Magner, E. The adsorption characteristics, activity and stability of trypsin onto mesoporous silicates. J. Mol. Catal. B: Enzym. 2005, 32, 231–239. [Google Scholar] [CrossRef]
- Yiu, H.H.P.; Botting, C.H.; Botting, N.P.; Wright, P.A. Size selective protein adsorption on thiol-functionalised SBA-15 mesoporous molecular sieve. Phys. Chem. Chem. Phys. 2001, 3, 2983–2985. [Google Scholar]
- Gaspari, M.; Cheng, M.M.C.; Terracciano, R.; Liu, X.; Nijdam, A.J.; Vaccari, L.; di Fabrizio, E.; Petricoin, E.F.; Liotta, L.A.; Cuda, G.; et al. Nanoporous surface as harvesting agents for mass spectrometric analysis of peptides in human plasma. J. Proteome Res. 2006, 5, 1261–1266. [Google Scholar]
- Urabe, Y.; Shiomi, T.; Itoh, T.; Kawai, A.; Tsunoda, T.; Mizukami, F.; Sakaguchi, K. Encapsulation of hemoglobin in mesoporous silica (FSM)-enhanced thermal stabilita nd resistance to denaturants. ChemBioChem 2007, 8, 668–674. [Google Scholar] [CrossRef]
- Buijs, J.; Ramstrom, M.; Danfelter, M.; Larsericsdotter, H.; Hakansson, P.; Oscarsson, S. Localized changes in the structural stability of myoglobin upon adsorption onto silica particles, as studied with hydrogen/deuterium exchange mass spectrometry. J. Colloid Interface Sci. 2003, 263, 441–448. [Google Scholar] [CrossRef]
- Transue, T.R.; Krahn, J.M.; Gabel, S.A.; DeRose, E.F.; London, R.E. X-ray and NMR characterization of covalent complexes of trypsin, borate, and alcohols. Biochemistry 2004, 43, 2829–2839. [Google Scholar] [CrossRef]
- Shylesh, S.; Wagner, A.; Seifert, A.; Ernst, S.; Thiel, W.R. Cooperative acid-base effects with functionalized mesoporous silica nanoparticles: Applications in carbon-carbon bond-formation reactions. Chem.-Eur. J. 2009, 15, 7052–7062. [Google Scholar] [CrossRef]
- Qian, K.; Wan, J.; Qiao, L.; Huang, X.; Tang, J.; Wang, Y.; Kong, J.; Yang, P.; Yu, C.; Liu, B. Macroporous material as novel catalysts for efficient and controllable proteolysis. Anal. Chem. 2009, 81, 5749–5756. [Google Scholar] [CrossRef]
- Qian, K.; Wan, J.; Huang, X.; Yang, P.; Liu, B.; Yu, C. A smart glycol-directed nanodevice from rationally designed macroporous materials. Chem.-Eur. J. 2010, 16, 822–828. [Google Scholar]
- Bi, H.; Qiao, L.; Busnel, J.M.; Liu, B.; Girault, H.H. Kinetics of proteolytic reactions in nanoporous materials. J. Proteome Res. 2009, 8, 4685–4692. [Google Scholar] [CrossRef]
- Shuler, M.L.; Kargi, F. Bioprocess Engineering: Basic Concepts, 2nd ed; Prentice Hall: Upper Saddle River, NJ, USA, 2002. [Google Scholar]
- Manyar, G.H.; Gianotti, E.; Sakamoto, Y.; Terasaki, O.; Coluccia, S.; Tumbiolo, S. Active biocatalysts based on pepsin immobilized in mesoporous SBA-15. J. Phys. Chem. C 2008, 112, 18110–18116. [Google Scholar]
- Kim, M.I.; Kim, J.; Lee, J.; Jia, H.; Na, H.B.; Youn, J.K.; Dohnalkova, A.; Grate, J.W.; Wang, P.; Hyeon, T.; et al. Crosslinked enzyme aggregates in hierarchically-ordered mesoporous silica: A simple and effective method for enzyme stabilization. Biotechnol. Bioeng. 2007, 96, 210–218. [Google Scholar]
- Zhao, B.; Shi, B.; Ma, R. Immobilization of papain on the mesoporous molecular sieve MCM-48. Eng. Life Sci. 2005, 5, 436–441. [Google Scholar] [CrossRef]
- Ritter, H.; Ramm, J.H.; Brühwiler, D. Influence of the structural properties of mesoporous silica on the adsorption of guest molecules. Materials 2010, 3, 4500–4509. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Zhang, H.; Ye, M.; Jiang, X.; Hu, L.; Li, X.; Bao, X.; Zou, H. Selective extraction of peptides from human plasma by highly ordered mesoporous silica particles for peptidome analysis. Angew. Chem. Int. Ed. Engl. 2007, 46, 962–965. [Google Scholar]
- Tian, R.; Ye, M.; Hu, L.; Li, X.; Zou, H. Selective extraction of peptides in acidic human plasma by porous silica nanoparticles for peptidome analysis with 2-D LC-MS/MS. J. Sep. Sci. 2007, 30, 2204–2209. [Google Scholar]
- Qi, Y.; Wei, J.; Wang, H.; Zhang, Y.; Xu, J.; Qian, X.; Guan, Y. Improved selection of LMW over HMW proteins from human plasma by mesoporous silica particles with external modification. Talanta 2009, 80, 703–709. [Google Scholar] [CrossRef]
- Qi, Y.; Wu, D.; Wei, J.; Ding, K.; Wang, H.; Zhang, Y.; Qian, X.; Guan, Y. Selective extraction of low molecular weight proteins by mesoporous silica particles with modified internal and external surfaces. Anal. Bioanal. Chem. 2010, 398, 1715–1722. [Google Scholar] [CrossRef]
- Bouamrani, A.; Hu, Y.; Tasciotti, E.; Li, L.; Chiappini, C.; Liu, X.; Ferrari, M. Mesoporous silica chips for selective enrichment and stabilization of low molecular weight proteome. Proteomics 2010, 10, 496–505. [Google Scholar] [CrossRef]
- Terracciano, R.; Preianó, M.; Palladino, G.P.; Carpagnano, G.E.; Foschino Barbaro, M.P; Pelaia, G.; Savino, R.; Maselli, R. Peptidome profiling of induced sputum by mesoporous silica beads and MALDI-TOF MS for non-invasive biomarker discovery of chronic inflammatory lung diseases. Proteomics 2011. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Savino, R.; Casadonte, F.; Terracciano, R. In Mesopore Protein Digestion: A New Forthcoming Strategy in Proteomics. Molecules 2011, 16, 5938-5962. https://doi.org/10.3390/molecules16075938
Savino R, Casadonte F, Terracciano R. In Mesopore Protein Digestion: A New Forthcoming Strategy in Proteomics. Molecules. 2011; 16(7):5938-5962. https://doi.org/10.3390/molecules16075938
Chicago/Turabian StyleSavino, Rocco, Francesca Casadonte, and Rosa Terracciano. 2011. "In Mesopore Protein Digestion: A New Forthcoming Strategy in Proteomics" Molecules 16, no. 7: 5938-5962. https://doi.org/10.3390/molecules16075938
APA StyleSavino, R., Casadonte, F., & Terracciano, R. (2011). In Mesopore Protein Digestion: A New Forthcoming Strategy in Proteomics. Molecules, 16(7), 5938-5962. https://doi.org/10.3390/molecules16075938