General
All reactions were carried out under an argon atmosphere unless otherwise noted. When necessary, solvents were dried prior to use. Dry THF, dry Et2O and dry CH2Cl2 were purchased from Kanto Chemical Co., Inc. Optical rotations were measured on a JASCO P-2200 digital polarimeter with a sodium (D line) lamp. IR spectra were recorded on a Jasco Model A-202 spectrophotometer. 1H-NMR spectra and 13C-NMR spectra were obtained on JEOL JNM-EX270, JNM-GX400, JNM-α400, JNM-AL400 and JNM-ECX400 spectrometers in deuterated solvent using tetramethylsilane as an internal standard. Deuteriochloroform was used as a solvent, unless otherwise stated. Optical purity was determined by HPLC using an OJ-H column. High-resolution mass spectra were obtained on a Waters LCT Piemier XE (ESI) or JEOL JMS-700 (FAB). Preparative and analytical TLC were carried out on silica gel plate (Kieselgel 60 F254, E. Merck AG., Germany) using UV light and/or 5% molybdophosphoric acid in ethanol for detection. Kanto silica 60N (spherical, neutral, 105–210 μm) was used for column chromatography.
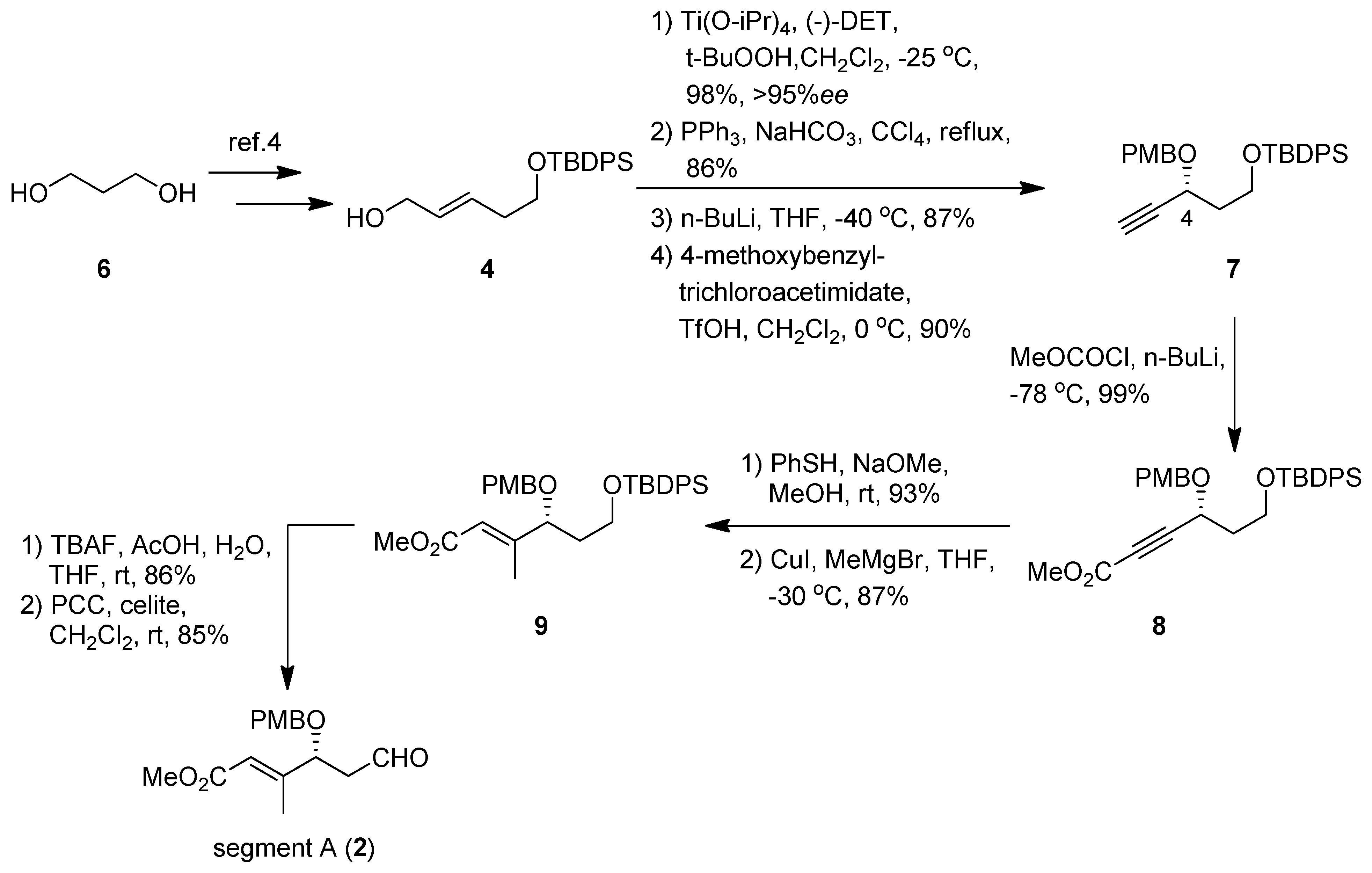
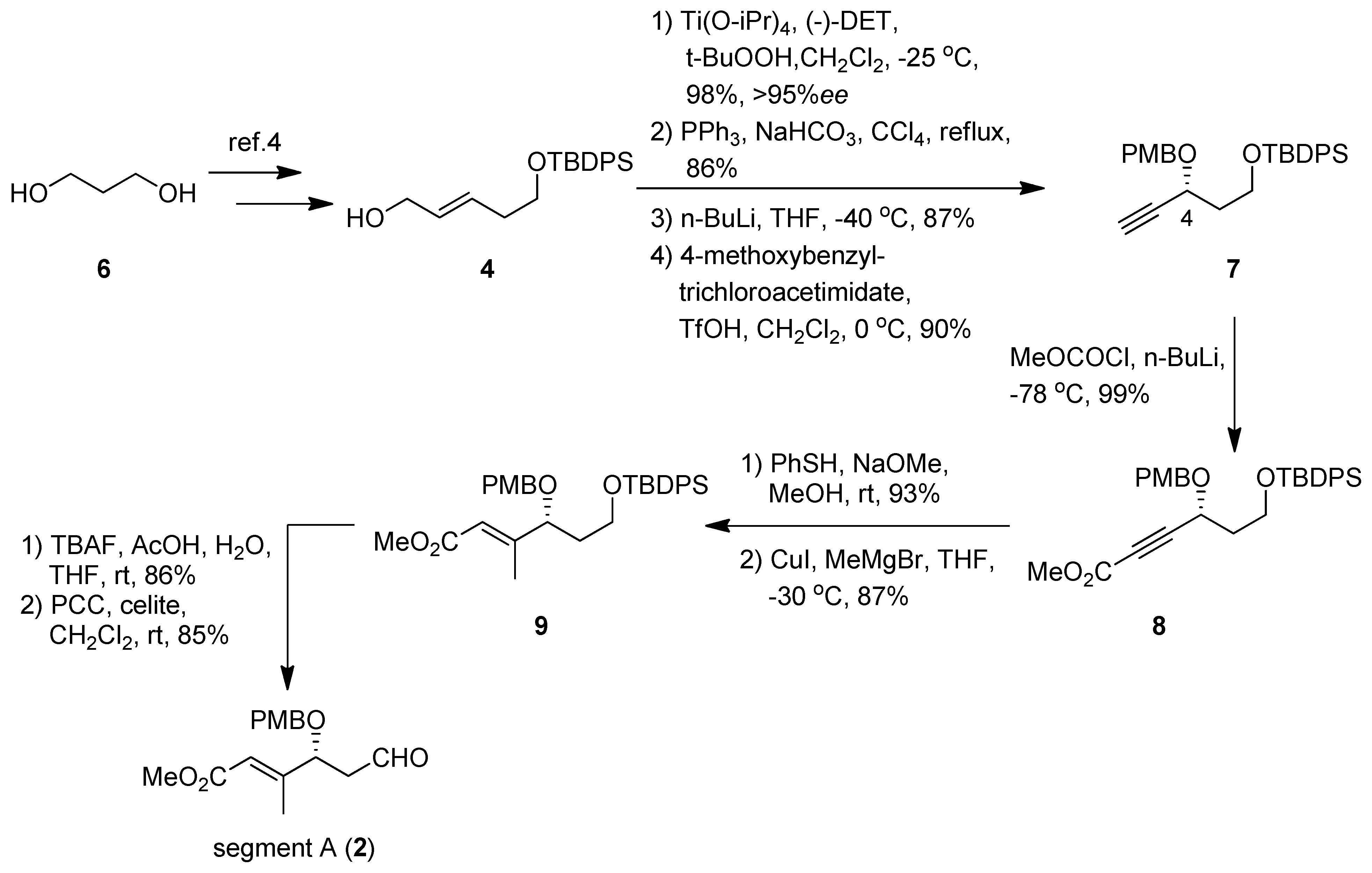
(R)-tert-Butyl(3-(4-methoxybenzyloxy)pent-4-ynyloxy)diphenylsilane (7). To a suspension of Ti(OiPr)4 (4.28 mL, 14.6 mmol) and MS4A (4.9 g) in CH2Cl2 (100 mL) was added (-)-DET (3.32 mL, 19.5 mmol) at −25 °C. To the mixture were added 4 (3.31 g, 9.73 mmol) and t-BuOOH (3.17 mL, 29.2 mmol); the solution was stirred overnight. The reaction was quenched by L-(+)-tartaric acid (12.4 g, 82.6 mmol) aqueous and iron (II) sulfate heptahydrate (12.2 g, 43.9 mmol). The mixture was extracted with Et2O, and the organic extracts were dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel column chromatography using hexane-ethyl acetate (3:1) to give an alcohol (3.38 g, 98%, 95% ee) as a colorless oil: [α]23D +14.7 (c 1.00, CHCl3); IR (film) 3437, 3048, 1428, 1110, 936, 822 cm−1; 1H-NMR (400 MHz) δ 1.06 (9H, s), 1.81 (2H, td, J = 12.1, 6.0 Hz), 2.98 (1H, td, J = 4.7. 2.5 Hz), 3.13 (1H, td, J = 6.0, 2.5 Hz), 3.63 (1H, m), 3.80 (2H, m), 3.91 (1H, m), 7.40 (6H, m), 7.66 (4H, m); 13C-NMR (100 MHz) δ 19.2, 26.8, 34.8, 53.7, 58.5, 60.7, 61.6, 127.7, 129.7, 133.6, 135.5. ESI-MS: calcd for C21H29O3Si 357.1886 (M+H)+, found, m/z 357.1897.
A stirred mixture of the alcohol (3.38 g, 9.49 mmol), PPh3 (7.47 g, 28.5 mmol), and NaHCO3 (0.80 g, 9.5 mmol) in CCl4 (100 mL) was refluxed under argon atmosphere overnight. After completion of the reaction, CCl4 was removed under reduced pressure, and the residue was purified by silica gel column chromatography (hexane-ethyl acetate 40:1) to furnish an epoxy chloride (3.04 g, 86%) as a colorless oil: [α]23D +12.9 (c 1.00, CHCl3); IR (film) 3049, 1428, 1111, 939, 822 cm−1; 1H-NMR (400 MHz) δ 1.06 (9H, s), 1.81 (2H, td, J = 11.2, 5.6 Hz), 3.07 (2H, m), 3.56 (2H, m), 3.80 (2H, m), 7.41 (6H, m), 7.66 (4H, m); 13C-NMR (100 MHz) δ 19.2, 26.8, 34.7, 44.7, 56.7, 57.3, 60.5, 127.7, 129.7, 133.5, 135.5. ESI-MS: calcd for C21H28O2SiCl 375.1547 (M+H)+, found, m/z 375.1539.
To a stirred solution of the epoxy chloride (0.23 g, 0.63 mmol) in dry THF (7 mL) was added n-BuLi (2.4 mL, 1.6 M solution in n-hexane, 3.79 mmol) dropwise at −40 °C under argon atmosphere; and the mixture was stirred for an additional 2 h. The mixture was quenched with saturated aqueous NH4Cl, and the mixture was extracted with ethyl acetate. The organic extracts were dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 7:1) to give an alcohol (0.18 g, 87%) as a colorless oil: [α]23D +3.67 (c 1.00, CHCl3); IR (film) 3422, 3304, 3071, 1428, 1111, 823 cm−1; 1H-NMR (400 MHz) δ 1.06 (9H, s), 1.92 (1H, tdd, J = 14.0, 6.3, 3.6 Hz), 2.05 (1H, tdd, J = 14.0, 8.0, 4.3 Hz), 2.48 (1H, d, J = 5.8 Hz), 3.34 (1H, d, J = 5.8 Hz), 3.85 (1H, ddd, J = 12.0, 6.3, 4.3 Hz), 4.07 (1H, ddd, J = 12.0, 8.0, 3.6 Hz), 4.71 (1H, m), 7.42 (6H, m), 7.69 (4H, m); 13C-NMR (100 MHz) δ 19.0, 26.8, 38.5, 61.5, 61.7, 73.0, 84.4, 127.8, 129.9, 132.8, 135.6. ESI-MS: calcd for C21H27O2Si 339.1780 (M+H)+, found, m/z 339.1775.
To a solution of the alcohol (0.10 g, 0.30 mmol) in anhydrous CH2Cl2 (7 mL) were added 4-methoxybenzyl trichloroacetimidate (0.493 g, 1.74 mmol) and TfOH (cat.) at 0 ºC, and the mixture was stirred overnight. The reaction was quenched with saturated aqueous NH4Cl, and extracted with ethyl acetate. The organic extracts were dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 7:1) to give 7 (0.12 g, 90%) as a colorless oil: [α]23D +45.1 (c 1.02, CHCl3); IR (film) 3286, 1514, 1249, 1111, 823 cm−1; 1H-NMR (400 MHz) δ 0.94 (9H, s), 1.91 (2H, m), 2.36 (1H, d, J = 2.1 Hz), 3.72 (5H, m), 4.34 (2H, m), 4.66 (1H, d, J = 11.2 Hz), 6.77 (2H, m), 7.20 (2H, m), 7.31 (6H, m), 7.56 (4H, m); 13C-NMR (100 MHz) δ 19.2, 26.8, 38.7, 55.2, 59.7, 65.0, 70.3, 73.8, 83.0, 113.8, 127.6, 129.6, 133.7, 135.5, 159.2. ESI-MS: calcd for C29H34O3SiK 497.1914 (M+K)+, found, m/z 497.1911.
(R)-Metyl-6-(tert-Butyldiphenylsilyloxy)-4-(4-methoxybenzyloxy)hex-2-ynoate (8). To a solution of 7 (50.8 mg, 0.11 mmol) in THF (1.5 mL) was added dropwise n-BuLi (0.3 mL, 1.6 M solution in hexane, 0.48 mmol) at −78 °C, and the mixture was stirred at the same temperature for 1 h. Methyl chloroformate (0.09 mL, 1.11 mmol) was added dropwise to the mixture; the resulting mixture was stirred at 0 °C for 2.5 h. After being quenched with saturated aqueous NH4Cl, the mixture extracted with ethyl acetate. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 9:1) to give 8 (56.4 mg, 99%) as a colorless oil: [α]23D +59.8 (c 0.99, CHCl3); IR (film) 3071, 2233, 1719, 1249, 1111 cm−1; 1H-NMR (400 MHz) δ 1.00 (9H, s), 2.01 (2H, ddd, J = 12.7, 7.6, 6.0 Hz), 3.79 (8H, m), 4.43 (1H, d, J = 11.2 Hz), 4.55 (1H, d, J = 7.6, 6.0 Hz), 4.74 (1H, d, J = 11.2 Hz), 6.86 (2H, m), 7.26 (2H, m), 7.37 (6H, m), 7.60 (4H, m); 13C-NMR (100 MHz) δ 19.1, 26.7, 38.0, 52.8, 55.2, 59.3, 64.7, 71.0, 86.9, 113.8, 127.7, 129.2, 129.6, 129.7, 133.5, 135.5, 153.7, 159.4. ESI-MS: calcd for C31H36O5SiK 555.1969 (M+K)+, found, m/z 555.1970.
(R,E)-Methyl 6-(tert-butyldiphenylsilyloxy)-4-(4-methoxybenzyloxy)-3-methylhex-2-enoate (9). To a solution of 8 (90.8 mg, 0.18 mmol) in MeOH was added PhSH (36 μL, 0.35 mmol) and NaOMe (0.18 mL, 0.02 mmol) at room temperature. After being stirred at the same temperature for 3 h, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane- ethyl acetate 9:1) to give a methyl ester (102.1 mg, 93%) as a colorless oil: [α]23D -91.3 (c 1.0, CHCl3); IR (film) 1705, 1513, 1248, 1110 cm−1; 1H-NMR (400 MHz) δ 0.93 (9H, s), 1.63 (1H, m), 2.02 (1H, m), 3.46 (1H, m), 3.50 (1H, m), 3.77 (3H, s), 3.79 (3H, s), 3.96 (1H, dd, J = 2.5, 9.9 Hz), 4.04 (1H, d, J = 10.8 Hz), 4.46 (1H, d, J = 10.8 Hz), 6.35 (1H, s), 6.79 (2H, m), 7.07 (2H, m), 7.21 (3H, m), 7.34 (4H, m), 7.43 (4H, m), 7.53 (4H, m); 13C-NMR (100 MHz) δ 19.1, 26.8, 39.8, 51.4, 55.3, 60.2, 70.7, 75.3, 113.8, 111.6, 113.7, 127.5, 129.2, 129.3, 129.5, 135.3, 135.5, 159.2, 160.5, 166.8. ESI-MS: calcd for C37H42O5SiSNa 649.2420 (M+Na)+, found, m/z 649.2440.
To a suspension of CuI (0.62 g, 3.2 mmol) in THF was added MeMgBr (6.7 mL, 0.95 M in THF, 6.3 mmol) at −78 °C; the solution was warmed to −30 °C and stirred for 1.5 h. The solution was cooled to −78 °C, and the methyl ester (0.10 g, 0.16 mmol) was added. The reaction mixture was stirred at −30 °C for 1 h, then quenched with saturated aqueous NH4Cl and NH4OH. The reaction mixture was extracted with ethyl acetate, and the organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography (benzene-hexane 7:1) to give 9 (75 mg, 87%) as a colorless oil: [α]23D +64.0 (c 0.50, CHCl3); IR (film) 1720, 1428, 1249, 1111, 822 cm−1; 1H-NMR (400 MHz) δ 0.96 (9H, s), 1.69 (2H, m), 2.05 (3H, d, J = 1.0 Hz), 3.64 (8H, m), 3.99 (1H, t, J = 6.6 Hz), 4.08 (1H, d, J = 11.2 Hz), 4.34 (1H, d, J = 11.2 Hz), 5.86 (1H, s), 6.77 (2H, m), 7.12 (2H, m), 7.31 (6H, m), 7.56 (4H, m); 13C-NMR (100 MHz) δ 14.3, 19.2, 26.8, 37.2, 51.0, 55.2, 59.9, 70.6, 80.2, 113.8, 116.4, 127.6, 129.4, 129.6, 130.1, 133.7, 135.5, 158.9, 159.2, 167.0. ESI-MS: calcd for C32H40O5SiNa 555.2543 (M+Na)+, found, m/z 555.2530.
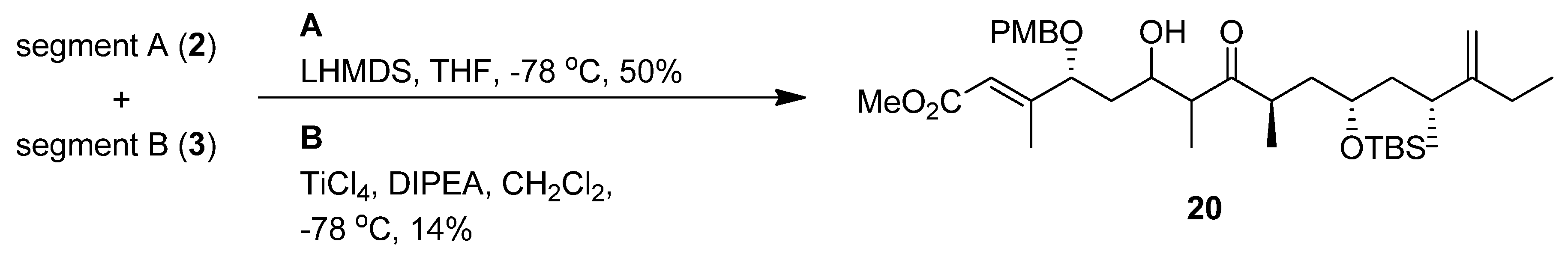
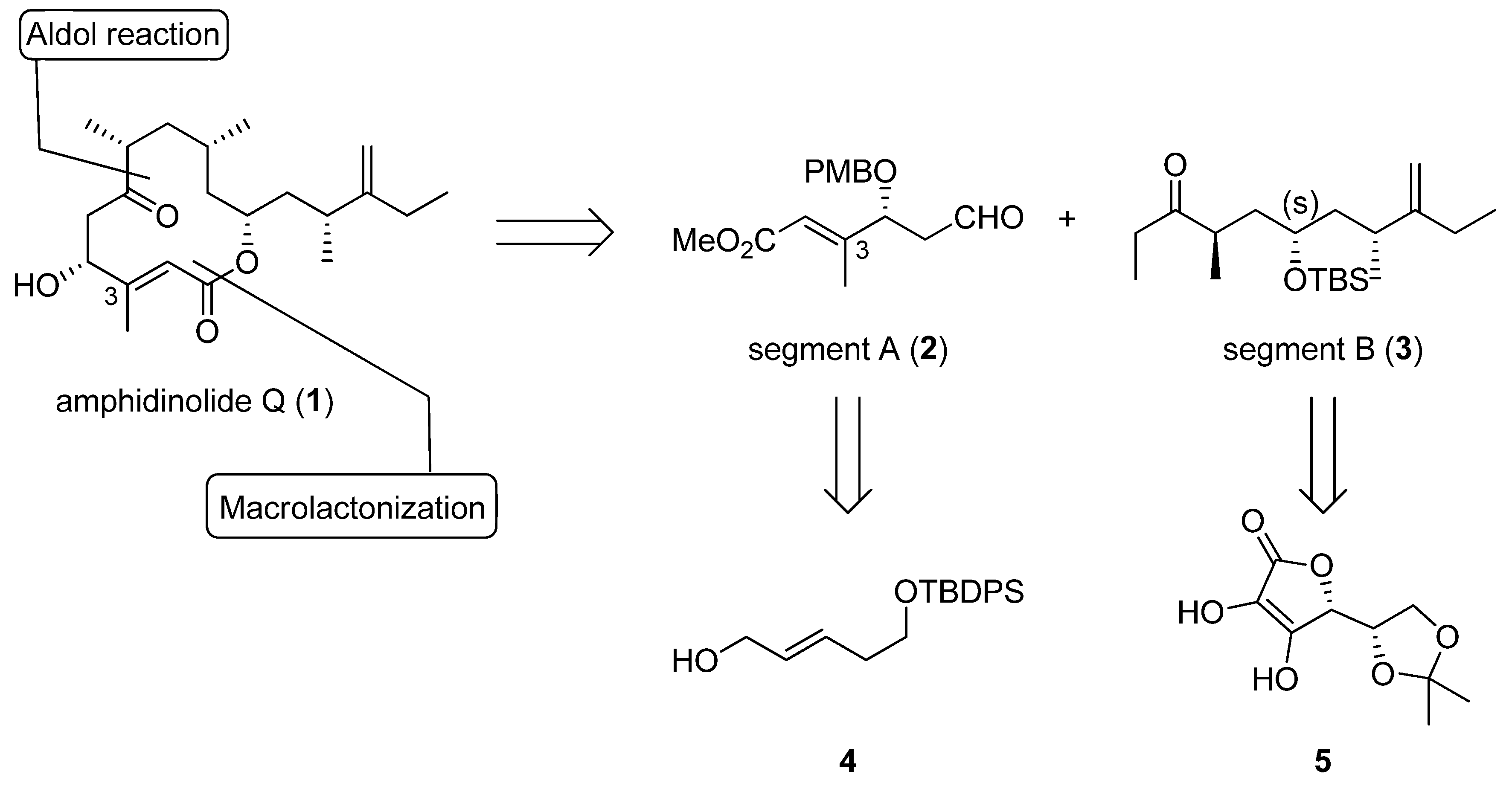
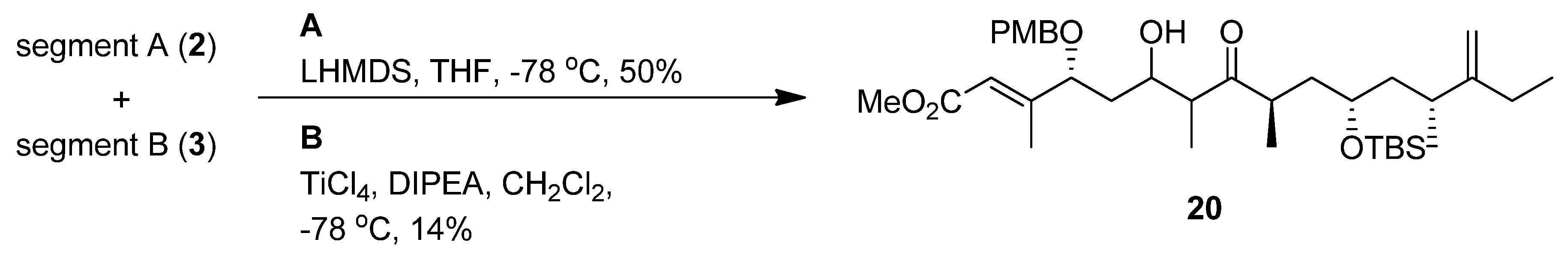
(R,E)-Methyl 4-(4-methoxybenzyloxy)-3-methyl-6-oxohex-2-enoate (segment A, 2). To a solution of 9 (0.33 g, 0.62 mmol) in THF (6 mL) was added reagent (TBAF-AcOH-H2O= 1:1:5 0.1 M in THF, 0.3 mmol) at room temperature; the mixture was stirred at the same temperature overnight. After the addition of cold water, the mixture was extracted with Et2O. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 1:1) to give an alcohol (0.16 g, 88%) as a colorless oil: [α]23D +68.7 (c 0.99, CHCl3); IR (film) 3429, 1718, 1654, 1613, 1513, 1156, 1035 cm−1; 1H-NMR (270 MHz) δ 1.84 (2H, m), 2.15 (3H, d, J = 1.3 Hz), 3.73 (5H, m), 3.81 (3H, s), 3.98 (1H, dd, J = 9.1, 3.8 Hz), 4.20 (1H, d, J = 11.2 Hz), 4.48 (1H, d, J = 11.2 Hz), 5.95 (1H, s), 6.89 (2H, m), 7.23 (2H, m); 13C-NMR (67.5 MHz) δ 14.4, 29.7, 36.3, 51.1, 55.2, 60.4, 70.6, 82.03, 113.9, 116.6, 128.3, 129.5, 129.6, 157.7, 159.4, 166.8. ESI-MS: calcd for C16H22O5Na 317.1365 (M+Na)+, found, m/z 317.1363.
To a mixture of PCC (35 mg, 0.16 mmol) and Celite in CH2Cl2 (2 mL) was added the alcohol (31.7 mg, 0.11 mmol) at 0 °C; the mixture was stirred at room temperature for 30 min. After concentration of the reaction mixture, the residue was purified by silica gel column chromatography (Et2O-hexane 2:1) to give segment A (26.7 mg, 85%) as a colorless oil: [α]23D +28.9 (c 0.91, CHCl3); IR (film) 1720, 1655, 1612, 1513, 1248, 1034 cm−1; 1H-NMR (270 MHz, C6D6) δ 1.90 (1H, ddd, J = 1.1, 3.6, 16.6 Hz), 2.14 (3H, m), 2.34 (1H, ddd, J = 2.5, 9.2, 16.6 Hz), 3.38 (3H, s), 3.53 (3H, s), 4.05 (1H, m), 4.07 (1H, d, J = 11.2 Hz), 4.32 (1H, d, J = 11.2 Hz), 6.09 (1H, m), 6.84 (2H, m), 7.18 (2H, m), 9.38 (1H, dd, J = 1.1, 2.5 Hz); 13C-NMR (67.5 MHz, C6D6) δ 14.3, 47.5, 50.7, 54.7, 70.7, 77.6, 78.2, 114.1, 117.4, 129.7, 130.0, 156.6, 166.3, 198.3. ESI-MS: calcd for 293.1389 (M+H)+, found, m/z 293.1402.
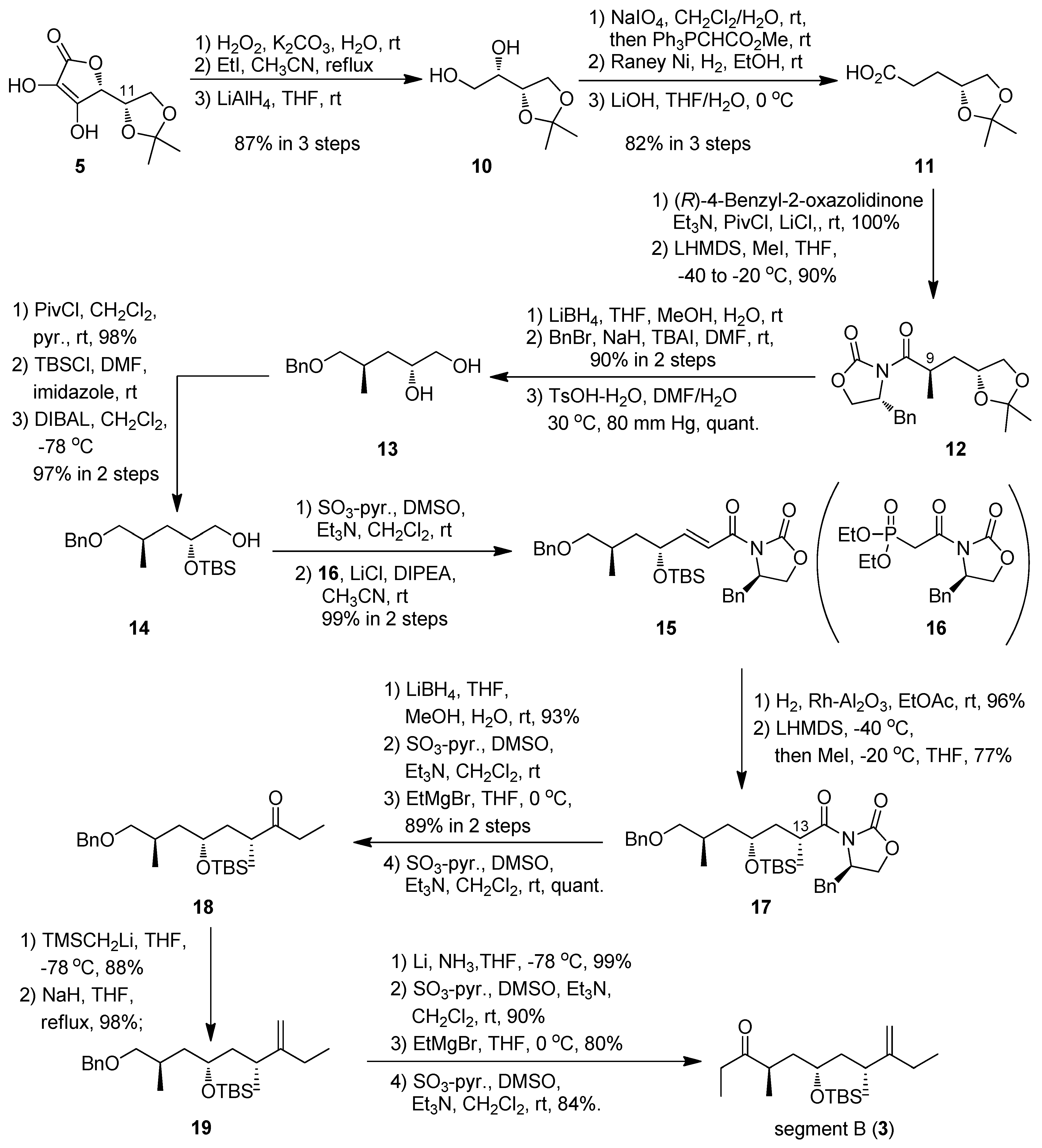
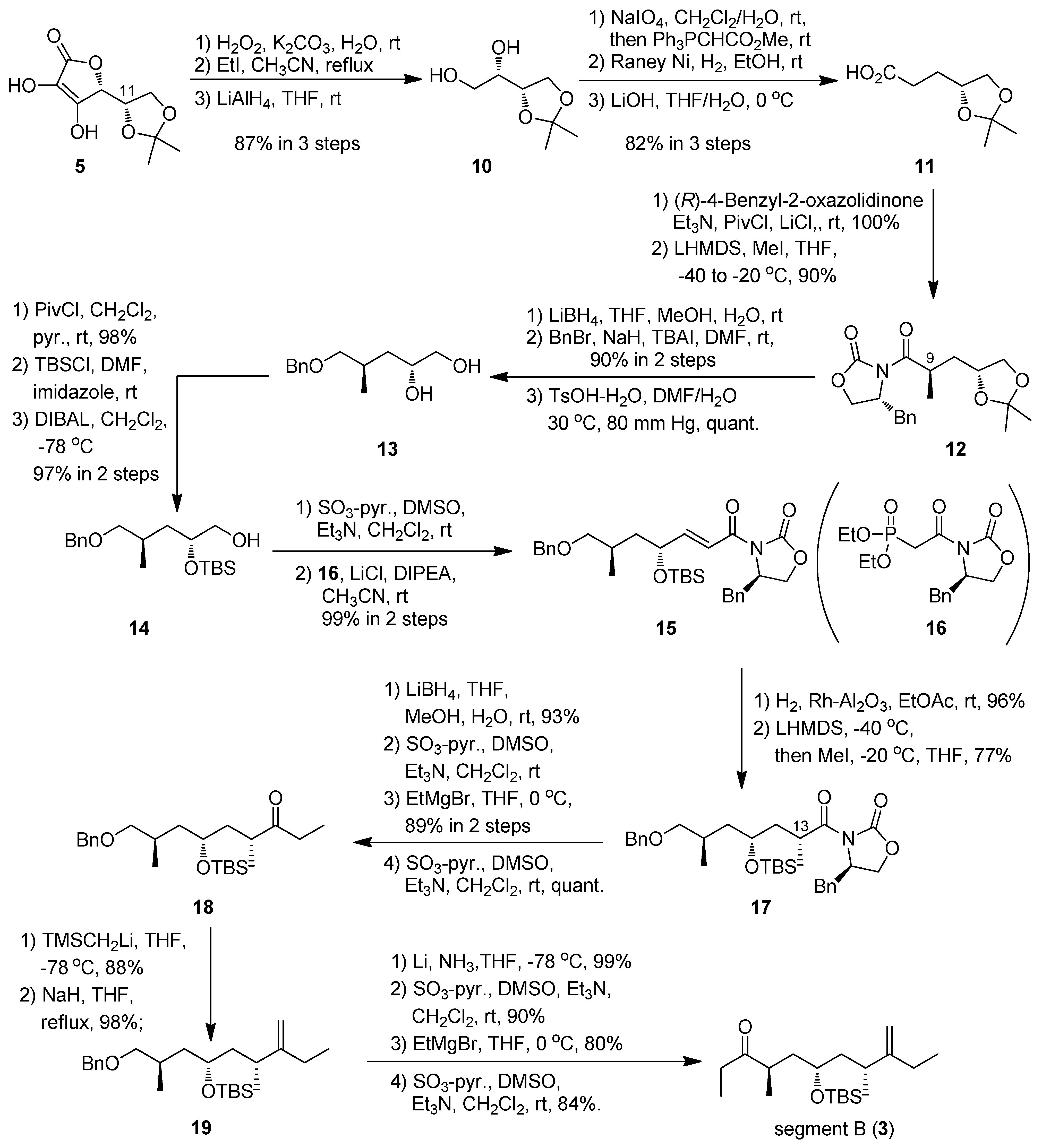
(R)-3-(2,2-Dimethyl-1,3-dioxolan-4-yl)propanoic acid (11). To a solution of NaIO4 (3.78 g, 17.7 mmol) in CH2Cl2 (60 mL) and H2O (30 mL) was added 10 (1.91 g, 11.8 mmol) at 0 °C; the mixture was stirred at room temperature for 1.5 h. After the addition of Ph3PCHCO2Me (7.89 g, 23.6 mmol) at 0 °C, the mixture was stirred at room temperature overnight. The organic layer was separated and the aqueous layer was extracted with CHCl3 three times. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 3:1) to give an α,β-unsaturated ester (1.84 g, 84%, E:Z = 1:3) as a colorless oil.
The ester (1.02 g, 5.48 mmol) was dissolved in EtOH (55 mL) in the presence of Raney Ni W-4. The mixture was stirred at room temperature under hydrogen atmosphere overnight. After filtration, the filtrate was concentrated at 110 °C to afford the corresponding ester as a crude oil. To a solution of the ester in THF (17 mL) was added 1.5 M LiOH aqueous (17 mL) at 0 °C. The mixture was stirred at 0 °C for 3 h. The mixture was acidified to pH 4 with 10% aqueous citric acid and extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo to give 11 (0.935 g, 98%) as a colorless oil: [α]23D +1.2 (c 1.00, CHCl3); IR (film) 2987, 1712, 1215, 1154, 1073 cm−1; 1H-NMR (270 MHz) δ 1.35 (3H, s), 1.42 (3H, s), 1.89 (2H, m), 2.51 (2H, m), 3.57 (1H, dd, J = 6.5, 7.8 Hz), 4.06 (1H, dd, J = 5.9, 7.8 Hz), 4.14 (1H, m); 13C- NMR (67.5 MHz) δ 25.6, 26.9, 28.5, 30.2, 68.9, 74.7, 109.1, 178.9. ESI-MS: calcd for C8H15O4 175.0970 (M+H)+, found, m/z 175.0968.
(R)-4-Benzyl-3-((R)-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2-methylpropanoyl)oxazolidin-2-one (12). To a solution of 11 (6.10 g, 35.0 mmol) in THF (325 mL) were added Et3N (15.0 mL, 0.190 mol) and PivCl (6.40 mL, 52.5 mmol) at 0 °C. After 2 h, LiCl (7.42 g, 175 mmol) and (R)-4-benzyl-2-oxazolidinone (9.30 g, 52.5 mmol) were added to the reaction mixture, and then the mixture was stirred at room temperature overnight. The reaction was quenched by the addition of saturated aqueous NH4Cl at 0 °C, then the resulting slurry was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 3:1) to give an amide (10.8 g, 92%) as a colorless oil: [α]24D -42.4 (c 1.01, CHCl3); IR (film) 2984, 1782, 1690, 1382, 1212, 1054 cm−1; 1H-NMR (400 MHz) δ 1.35 (3H, s), 1.42 (3H, s), 1.96 (2H, m), 2.78 (1H, dd, J = 9.8, 13.2 Hz), 3.07 (2H, t, J = 7.8 Hz), 3.30 (1H, dd, J = 3.4, 13.2 Hz), 3.60 (1H, dd, J = 6.8, 7.8 Hz), 4.08 (1H, dd, J = 5.9, 7.8 Hz), 4.64 (3H, m), 4.67 (1H, m), 7.27 (5H, m); 13C-NMR (100 MHz) δ 25.7, 27.0, 28.2, 32.0, 37.9, 55.1, 66.2, 69.2, 74.9, 108.9, 127.2, 128.8, 129.3, 135.1, 153.3, 172.5, 180.1. ESI-MS: calcd for C18H23NO5Na 356.1474 (M+Na)+, found, m/z 356.1489.
To a solution of LHMDS (73.0 mL, 1.0 M solution in THF, 73.0 mmol) was added the amide (12.2 g, 36.5 mmol) in THF (400 mL) at −40 °C. After 1.5 h, MeI (22.7 mL, 0.365 mol) was added to the reaction mixture. The mixture was stirred at −20 °C for 4 h. The reaction was quenched by the addition of saturated aqueous NH4Cl at 0 °C, then the resulting slurry was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 3:1) to give 12 (10.8 g, 85%) as a white needles: m.p. 93–93.5 °C (ethyl acetate); [α]24D -65.4 (c 1.00, CHCl3); IR (film) 2983, 1780, 1697, 1381, 1213, 1159, 1085, 1053 cm−1; 1H-NMR (400 MHz) δ 1.26 (3H, d, J = 7.3 Hz), 1.30 (3H, s), 1.36 (3H, s), 1.66 (1H, ddd, J = 4.4, 5.4, 13.7 Hz), 2.05 (1H, ddd, J = 8.8, 8.3, 13.7 Hz), 2.79 (1H, dd, J = 9.8, 13.2 Hz), 3.26 (1H, dd, J = 3.4, 13.2 Hz), 3.50 (1H, dd, J = 7.3, 7.8 Hz), 4.01 (1H, m), 4.03 (1H, dd, J = 5.4, 7.8 Hz), 4.17 (3H, m), 4.67 (1H, m), 7.28 (5H, m); 13C- NMR (100 MHz) δ 18.2, 25.7, 26.9, 34.7, 38.0, 38.1, 55.4, 66.0, 69.5, 74.2, 108.9, 127.2, 128.8, 129.3, 153.2, 176.9, 180.1. ESI-MS: calcd for C19H26NO5Na (M+Na)+ 348.1811, found, m/z 348.1805.
(2R,4R)-5-(Benzyloxy)-4-methylpentane-1,2-diol (13). To a solution of 12 (7.44 g, 21.4 mmol) in THF-MeOH-H2O (4:4:1), (220 mL) was added LiBH4 (2.1 g, 86 mmol) at 0 °C. The mixture was stirred at room temperature for 2.5 h. The reaction was quenched by the addition of saturated aqueous NH4Cl at 0 °C, then the resulting slurry was extracted with Et2O three times. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane-ethyl acetate (1:1)) to give an alcohol. To a solution of the alcohol in DMF (220 mL) was added BnBr (8 mL, 64.3 mmol), TBAI (3.2 g, 8.57 mmol) and NaH (3.1 g, 64.3 mmol) at 0 °C. The mixture was stirred at room temperature overnight. The reaction was quenched by the addition of saturated aqueous NH4Cl at 0 °C, then the resulting slurry was extracted with Et2O three times. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 10:1) to give an acetonide (5.09 g, 90%) as a colorless oil: [α]24D -11.5 (c 1.03, CHCl3); IR (film) 2984, 1454, 1368, 1213, 1098, 1061 cm−1; 1H-NMR (400 MHz) δ 0.91 (3H, d, J = 6.8 Hz), 1.29 (7H, m), 1.71 (1H, ddd, J = 5.8, 8.3, 13.7 Hz), 1.89 (1H, m), 3.24 (2H, m), 3.42 (1H, t, J = 7.8 Hz), 3.97 (1H, dd, J = 7.8, 5.8 Hz), 4.12 (1H, tt, J = 7.8, 5.8 Hz), 4.43 (2H, s), 7.26 (5H, m); 13C-NMR (100 MHz) δ 17.1, 25.8, 27.1, 30.7, 37.8, 69.9, 72.9, 74.2, 108.5, 127.4, 127.5, 128.3, 138.6. ESI-MS: calcd for C16H25O3 265.1804 (M+H)+, found, m/z 265.1803.
To a solution of the acetonide (0.73 g, 2.8 mmol) in DMF (30 mL) were added TsOH·H2O (1.6 g, 8.3 mmol) and H2O (10 mL) at 0 °C; the mixture was stirred at 30 ºC under 80 mmHg for 3 h. After the addition of saturated aqueous NH4Cl, the mixture was extracted with Et2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 1:1) to give 13 (0.66 g, quant.) as a colorless oil: [α]24D +16.0 (c 0.99, CHCl3); IR (film) 3375, 1454, 1363, 1074 cm−1; 1H-NMR (400 MHz) δ 0.87 (3H, d, J = 6.8 Hz), 1.39 (2H,m), 1.93 (2H, m), 3.22 (1H, m), 3.37 (2H, m), 3.52 (2H, m), 3.70 (1H, m), 4.47 (2H, s), 7.24 (5H, m); 13C- NMR (100 MHz) δ 18.2, 31.8, 39.6, 67.4, 71.0, 73.3, 76.6, 127.7, 127.8, 128.5, 137.6. ESI-MS: calcd for C13H20O3Na 247.1310 (M+Na)+, found, m/z 247.1309.
(2R,4R)-5-(Benzyloxy)-2-(tert-butyldimethylsilyloxy)-4-methylpentan-1-ol (14). To a solution of 13 (3.39 g, 15.1 mmol) in CH2Cl2 (75 mL) and pyridine (75 mL) was added PivCl (2.8 mL, 22.7 mmol) at 0 °C; the mixture was stirred at room temperature for 1.5 h. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with Et2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 4:1) to give an alcohol (4.57 g, 98%) as a colorless oil: [α]24D +10.7 (c 1.01, CHCl3); IR (film) 3442, 1730, 1455, 1285, 1164, 1097 cm−1; 1H-NMR (400 MHz) δ 0.88 (3H, d, J = 7.3 Hz), 1.15 (9H, s), 1.33 (1H, ddd, J = 14.4, 6.8, 2.9 Hz), 1.46 (1H, m), 1.95 (1H, m), 2.98 (1H, br), 3.22 (1H, dd, J = 9.8, 6.3 Hz), 3.32 (1H, dd, J = 9.8, 5.4 Hz), 3.84 (1H, m), 3.96 (2H, m), 4.46 (2H, s), 7.26 (5H, m); 13C-NMR (100 MHz) δ 17.7, 27.2, 31.2, 38.8, 39.0, 68.8, 73.1, 76.2, 127.7, 128.3, 128.4, 138.0, 178.6. ESI-MS: calcd for C18H28O4Na 331.1885 (M+Na)+, found, m/z 331.1880.
To a solution of the alcohol (4.57 g, 14.8 mmol) in DMF (150 mL) were added imidazole (3.03 g, 44.5 mmol) and TBSCl (6.7 g, 44.5 mmol) at 0 °C; the mixture was stirred at room temperature overnight. After the addition of saturated aqueous NH4Cl, the mixture was extracted with Et2O. The organic layer dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 7:1) to give a silyl ether (6.31 g, quant.) as a colorless oil: [α]24D +10.6 (c 1.02, CHCl3); IR (film) 1733, 1160, 836 cm−1; 1H-NMR (400 MHz) δ 0.09 (6H, s), 0.88 (9H, s), 0.94 (3H, m), 1.20 (9H, s), 1.28 (1H, m), 1.63 (1H, ddd, J = 4.5, 7.9, 13.7 Hz), 1.97 (1H, m), 3.27 (1H, dd, J = 6.1, 9.2 Hz), 3.33 (1H, dd, J = 6.1, 9.2 Hz), 3.97 (3H, m), 4.50 (2H, s), 7.34 (5H, m); 13C- NMR (100 MHz) δ –4.7, –4.4, 17.1, 18.0, 25.5, 25.8, 27.1, 27.2, 29.5, 38.7, 38.8, 68.1, 68.4, 76.2, 127.3, 127.4, 128.3, 138.7, 178.5. ESI-MS: calcd for C24H42O4SiNa 445.2750 (M+Na)+, found, m/z 445.2747.
To a solution of the silyl ether (11.7 mg, 27.7 μmol) in CH2Cl2 (0.3 mL) was added DIBAL-H (0.08 mL, 1.03 M in n-hexane, 83.1 μmol) at −78 °C. After being stirred for 1 h then the reaction was quenched by potassium-sodium tartrate aqueous. The mixture was extracted with CHCl3, dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel column chromatography using hexane-ethyl acetate (5:1) to give 14 (9.1 mg, 97%) as a colorless oil: [α]24D -2.3 (c 0.97, CHCl3); IR (film) 3437, 2857, 836 cm−1; 1H-NMR (400 MHz) δ 0.08 (6H, s), 0.90 (9H, s), 0.95 (3H, m), 1.29 (2H, m), 1.66 (1H, m), 1.90 (1H, m), 3.29 (2H, m), 3.45 (1H, dd, J = 4.9, 11.0 Hz), 3.58 (1H, dd, J = 4.9, 11.0 Hz), 3.88 (1H, m), 4.49 (2H, s), 7.33 (5H, m); 13C-NMR (100 MHz) δ –4.5, –4.4, 17.7, 18.1, 25.8, 29.6, 38.2, 66.6, 70.8, 73.0, 76.1, 127.4, 127.5, 128.3, 138.5. ESI-MS: calcd for C19H34O3SiNa 361.2175 (M+Na)+, found, m/z 361.2168.
(R)-4-Benzyl-3-((4R,6R,E)-7-(benzyloxy)-6-methyl-4-(tert-butyldimethylsilyloxy)hept-2-enoyl)oxazol-idin-2-one (15). To a solution of 14 (4.39 g, 13 mmol) in CH2Cl2 (65 mL) and DMSO (65 mL) were added Et3N (5.4 mL, 39 mmol) and SO3-pyridine (6.2 g, 39 mmol) complex at 0 °C. After 2 h, the reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned between Et2O and H2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 6:1) to give an aldehyde (4.3 g, 99%) as a colorless oil.
To a suspension of LiCl (0.09 g, 2.2 mmol) in anhydrous CH3CN (8 mL) were added 16 (0.29 g, 0.81 mmol), DIPEA (0.3 mL, 1.65 mmol), and the aldehyde (0.18 g, 0.55 mmol) at room temperature; the mixture was stirred at room temperature for overnight. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned between Et2O and H2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 6:1) to give 15 (0.33 g, quant.) as a colorless oil: [α]24D -23.1 (c 1.02, CHCl3); IR (film) 1782, 1683, 1639, 1352, 1207, 1100, 836 cm−1; 1H-NMR (400 MHz) δ 0.07 (6H, s), 0.93 (9H, s), 0.99 (3H, d, J = 6.8 Hz), 1.35 (1H, ddd, J = 4.9, 8.8, 13.7 Hz), 1.75 (1H, ddd, J = 4.9, 8.8, 13.7 Hz), 2.02 (1H, m), 2.81 (1H, dd, J = 9.8, 13.7 Hz), 3.32 (3H, m), 4.19 (2H, m), 4.50 (3H, m), 4.73 (1H, ddd, J = 3.4, 6.8, 12.7 Hz), 7.37 (12H, m); 13C-NMR (100 MHz) δ –5.0, –4.3, 17.3, 18.1, 25.8, 29.7, 37.9, 41.6, 55.3, 66.1, 70.2, 72.9, 76.0, 77.3, 118.7, 127.3, 127.4, 127.5, 128.3, 128.9, 129.5, 135.3, 138.6, 153.2, 153.6, 165.0. ESI-MS: calcd for C31H43NO5SiNa 560.2808 (M+Na)+, found, m/z 560.2808.
(R)-4-Benzyl-3-((2R,4R,6R)-7-(benzyloxy)-2,6-dimethyl-4-(tert-butyldimethylsilyloxy)heptanoyl)oxazol-idin-2-one (17). To a solution of 15 (6.21 g, 11.6 mmol) in ethyl acetate (120 mL) was added Rh-Al2O3 (cat.) at room temperature under hydrogen atmosphere; the mixture was stirred for 2 h. The reaction mixture was filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 6:1) to give an amide (5.99 g, 96%) as a colorless oil: [α]24D -23.6 (c 0.99, CHCl3); IR (film) 1784, 1701, 1210, 835 cm−1; 1H-NMR (400 MHz,) δ 0.07 (6H, d, J = 4.9 Hz), 0.90 (9H, s), 0.97 (3H, d, J = 6.5 Hz), 1.26 (1H, ddd, J = 5.4, 8.1, 13.7 Hz), 1.62 (1H, ddd, J = 5.4, 7.2, 13.7 Hz), 1.88 (3H, m), 2.76 (1H, dd, J = 9.6, 13.7 Hz), 2.92 (1H, ddd, J = 5.4, 9.6, 17.3 Hz), 3.05 (1H, ddd, J = 5.4, 9.6, 17.3 Hz), 3.31 (3H, m), 3.89 (1H, tt, J = 5.4, 11.7 Hz), 4.17 (2H, m), 4.50 (2H, s), 4.66 (1H, ddd, J = 3.4, 7.2, 13.0 Hz), 7.34 (10H, m); 13C-NMR (100 MHz) δ –4.5, –4.4, 17.6, 18.0, 25.9, 29.8, 31.5, 31.7, 37.9, 41.3, 55.1, 66.1, 69.2, 72.8, 76.1, 127.4, 127.5, 128.2, 128.3, 128.9, 129.4, 135.3, 153.4, 173.4. ESI-MS: calcd for C31H45NO5SiNa 562.2965 (M+Na)+, found, m/z 562.2962.
To a solution of LHMDS (47 mL, 1.0 M in THF, 47 mmol) in THF (100 mL) was added the amide (5.07 g, 9.4 mmol) at −40 °C. After being stirred at same temperature for 2 h, MeI (12 mL, 0.19 mol) was added and the resultant mixture stirred at −20 °C for 0.5 h. After the addition of saturated aqueous NH4Cl, the mixture was extracted with ethyl acetate. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 9:1) to give 17 (4.03 g, 77%) as a colorless oil: [α]24D -41.5 (c 1.00, CHCl3); IR (film) 1783, 1697, 1455, 1386, 1209, 836 cm−1; 1H-NMR (400 MHz) δ 0.05 (6H, d, J = 14.6 Hz), 0.90 (9H, s), 1.02 (3H, d, J = 6.1 Hz), 1.32 (4H, m), 1.58 (2H, m), 1.98 (1H, ddd, J = 6.7, 13.2, 19.7 Hz), 2.11 (1H, m), 2.80 (1H, dd, J = 10.1, 13.2 Hz), 3.34 (3H, m), 3.84 (2H, m), 4.18 (2H, m), 4.55 (2H, s), 4.67 (1H, m), 7.33 (10H, m); 13C-NMR (100 MHz) δ –4.7, –4.3, 17.9, 18.0, 18.6, 25.8, 29.9, 34.3, 37.9, 40.7, 41.6, 55.3, 65.9, 68.7, 72.9, 76.0, 127.3, 127.5, 128.2, 128.9, 129.4, 135.3, 138.8, 152.8, 176.8. ESI-MS: calcd for C32H48NO5Si 554.3302 (M+H)+, found, m/z 554.3292.
(4R,6R,8R)-9-(Benzyloxy)-4,8-dimethyl-6-(tert-butyldimethylsilyloxy)nonan-3-one (18). To a solution of 17 (4.7 g, 8.49 mmol) in THF-MeOH-H2O (4:4:1, 90 mL) was added LiBH4 (0.82 g, 34 mmol) at 0 °C; the mixture was stirred at room temperature for 1 h. After the addition of saturated aqueous NH4Cl, the mixture was extracted with Et2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 5:1) to give an alcohol (3.02 g, 93%) as a yellow oil: [α]24D +9.1 (c 1.00, CHCl3); IR (film) 1459, 1255, 1049, 835 cm−1; 1H-NMR (400 MHz) δ 0.08 (6H, d, J = 3.6 Hz), 0.87 (3H, d, J = 7.2 Hz), 0.90 (9H, s), 0.95 (3H, d, J = 7.2 Hz), 1.38 (1H, ddd, J = 6.3, 7.2, 13.7 Hz), 1.55 (4H, m), 1.86 (2H, m), 3.26 (2H, m), 3.33 (1H, m), 3.44 (1H, m), 3.98 (1H, tt, J = 4.5, 11.4 Hz), 4.49 (2H, s), 7.33 (5H, m); 13C-NMR (100 MHz) δ –4.6, –4.4, 17.8, 18.0, 18.3, 25.8, 30.0, 31.8, 40.6, 42.6, 69.3, 73.0, 76.2, 127.4, 127.5, 128.3, 138.6. ESI-MS: calcd for C22H41O3Si 381.2825 (M+H)+, found, m/z 381.2821.
To a solution of the alcohol (2.5 mg, 6.6 μmol) in CH2Cl2 (0.05 mL) and DMSO (0.05 mL) were added Et3N (3 μL, 19.7 μmol) and SO3-pyridine (3 mg, 19.7 μmol) complex at 0 °C. After 1.5 h, the reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned between Et2O and H2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by PLC (hexane-ethyl acetate 3:1) to give an aldehyde as a colorless oil.
To a solution of the aldehyde in THF (0.1 mL) was added EtMgBr (0.02 mL, 1.0 M in THF, 19.7 μmol) at 0 °C; the mixture was stirred at the same temperature for 15 min. The reaction mixture was quenched with saturated aqueous NH4Cl, and extracted with CH2Cl2. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by PLC (hexane-ethyl acetate (5:1)) to give an alcohol as a colorless oil.
To a solution of the alcohol in CH2Cl2 (0.05 mL) and DMSO (0.05 mL) were added Et3N (3 μL, 19.7 μmol) and SO3-pyridine (3 mg, 19.7 μmol) complex at 0 °C. After 4 h, the reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned between Et2O and H2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by PLC (hexane-ethyl acetate (5:1) to give 18 (2.5 mg 89% in 3 steps) as a colorless oil: [α]24D +1.1 (c 0.98, CHCl3); IR (film) 1714, 1459, 1255, 1099, 835 cm−1; 1H-NMR (400 MHz) δ 0.02 (6H, d, J = 6.7 Hz), 0.87 (9H, s), 0.95 (3H, d, J = 7.2 Hz), 1.05 (6H, m), 1.22 (1H, m), 1.35 (1H, m), 1.57 (1H, m), 1.93 (1H, m), 2.46 (2H, m), 2.66 (qt, 1H, J = 7.2, 13.7 Hz), 3.26 (1H, dd, J = 6.1, 8.8 Hz), 3.33 (1H, dd, J = 6.1, 8.8 Hz), 3.75 (1H, tt, J = 5.8, 12.1 Hz), 4.50 (2H, s), 7.33 (5H, m); 13C-NMR (100 MHz) δ –4.34, –4.30, 7.8, 17.5, 17.7, 18.1, 25.9, 29.8, 34.2, 40.6, 41.4, 42.3, 68.8, 75.9, 127.4, 127.5, 128.3, 138.8, 214.7. ESI-MS: calcd for C24H43O3Si 407.2981 (M+H)+, found, m/z 407.2982.
((2R,4S,6R)-1-(Benzyloxy)-2,6-dimethyl-7-methylenenonan-4-yloxy)(tert-butyl)dimethylsilane (19). To a solution of ethyl ketone 18 (60.0 mg, 0.12 mmol) in THF (1 mL) was added (trimethylsilylmethyl) lithium (0.5 mL, 1.0 M in pentane, 0.5 mmol) at −78 °C; the mixture was stirred at the same temperature for 20 min. The reaction mixture was quenched with saturated aqueous NH4Cl, and extracted with ethyl acetate. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by PLC (hexane-ethyl acetate 9:1) to give an alcohol as a colorless oil.
To a solution of the alcohol in THF (2 mL) was added NaH (60% dispersion in mineral oil, 50 mg, 0.1 mmol) at room temperature; the mixture was stirred at reflux temperature overnight. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with ethyl acetate. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by PLC (hexane-ethyl acetate 9:1) to give 19 as a colorless oil (48 mg, 86% in 2 steps): [α]24D +17.8 (c 1.01, CHCl3); IR (film) 2958, 1642, 1460, 1254, 1097, 835 cm−1; 1H-NMR (400 MHz) δ 0.05 (6H, s), 0.89 (9H, s), 0.95 (3H, d, J = 6.5 Hz), 1.02 (6H, m), 1.24 (1H, m), 1.46 (2H, m), 1.61 (1H, m), 1.99 (3H, m), 2.19 (1H, m), 3.23 (1H, dd, J = 6.5, 9.1 Hz), 3.34 (1H, dd, J = 6.5, 9.1 Hz), 3.77 (1H, m), 4.50 (2H, s), 4.72 (2H, dd, J = 1.5, 11.9 Hz), 7.33 (5H, m); 13C-NMR (100 MHz) δ –4.2, –4.1, 12.4, 17.4, 18.1, 20.3, 26.0, 26.2, 29.7, 36.8, 41.3, 44.2, 69.0, 72.8, 76.3, 106.4, 127.4, 127.5, 128.3, 138.8, 156.3. ESI-MS: calcd for C25H45O2Si 405.3189 (M+H)+, found, m/z 405.3181.
(4R,6S,8R)-6-(tert-Butyldimethylsilyloxy)-4,8-dimethyl-9-methyleneundecan-3-one (segment B, 3). To a solution of lithium (12.4 mg, 1.9 mmol) in liquid NH3 (10 mL) was added 19 (46.4 mg, 0.11 mmol) in dry THF (3 mL) at −78 °C. The reaction mixture was stirred for 15 min at the same temperature; and quenched with solid NH4Cl. The mixture was extracted with ethyl acetate, and the organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 5:1) to give an alcohol (35.6 mg, 99%) as a colorless oil: [α]24D +36.4 (c 0.98, CHCl3); IR (film) 3346, 2959, 1643, 1461, 1254, 1046, 835 cm−1; 1H-NMR (400 MHz) δ 0.06 (6H, d, J = 1.6 Hz), 0.89 (9H, s), 0.93 (3H, d, J = 6.3 Hz), 1.02 (6H, m), 1.27 (1H, ddd, J = 3.7, 8.5, 13.9 Hz), 1.46 (2H, m), 1.59 (1H, m), 1.81 (1H, ttd, J = 2.2, 6.3, 13.0 Hz), 2.00 (2H, m), 2.16 (1H, qt, J = 6.3, 7.0 Hz), 3.45 (2H, dd, J = 4.5, 6.3 Hz), 3.76 (1H, m), 4.72 (2H, ddd, J = 1.6, 2.9, 11.9 Hz); 13C-NMR (100 MHz) δ –4.2, –4.0, 12.4, 17.0, 18.1, 20.1, 25.8, 25.9, 26.3, 32.3, 36.9, 40.8, 44.4, 68.8, 69.3, 106.4, 156.3. ESI-MS: calcd for C18H39O2Si 315.2719 (M+H)+, found, m/z 315.2717.
To a solution of the alcohol (0.35 g, 1.10 mmol) in CH2Cl2 (6 mL) and DMSO (6 mL) were added Et3N (0.5 mL, 3.29 mmol) and SO3-pyridine (0.52 g, 3.29 mmol) complex at 0 °C. After 1 h, the reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned between Et2O and H2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 7:1) to give an aldehyde as a colorless oil.
To a solution of the aldehyde in THF (10 mL) was added EtMgBr (3 mL, 1.0 M in THF, 2.95 mmol) at 0 °C; the mixture was stirred at the same temperature for 30 min. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with CH2Cl2. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate (9:1)) to give an alcohol as a colorless oil.
To a solution of the alcohol in CH2Cl2 (4 mL) and DMSO (4 mL) were added Et3N (0.32 mL, 2.34 mmol) and SO3-pyridine (0.4 g, 2.34 mmol) complex at 0 °C. After 40 min, the reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned between Et2O and H2O. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane-ethyl acetate 11:1) to give segment B (0.22 g 60% in 3 steps) as a colorless oil.: [α]24D +11.7 (c 0.99, CHCl3); IR (film) 2960, 1716, 1643, 1461, 1255, 1101, 835 cm−1; 1H-NMR (400 MHz) δ 0.06 (6H, d, J = 3.8 Hz), 0.89 (9H, s), 1.02 (12H, m), 1.38 (2H, m), 1.55 (1H, ddd, J = 6.7, 7.0, 13.5 Hz), 1.74 (1H, ddd, J = 5.2, 7.0, 13.5 Hz), 1.97 (2H, m), 2.16 (1H, dd, J = 7.0, 14.0 Hz), 2.47 (2H, q, J = 7.0 Hz), 2.73 (1H, qt, J = 7.0, 14.0 Hz), 3.66 (1H, m), 4.70 (2H, m); 13C- NMR (100 MHz) δ –4.3, –4.1, 7.9, 12.3, 16.4, 18.1, 20.3, 25.9, 26.2, 34.2, 36.8, 40.1, 41.8, 44.0, 68.7, 77.3, 106.6, 156.0, 215.2. ESI-MS: calcd for C20H39O2Si 339.2719 (M+H)+, found, m/z 339.2689.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
