3.1. General
All reactions were performed under an argon atmosphere and all glassware was flame dried prior to use. CH2Cl2 and THF were dried by passing through a column of activated alumina. Reactions carried out at −78 °C employed a CO2/acetone bath. Reactions were monitored by TLC analysis (pre-coated silica gel 60 F254 plates, 250 μm layer thickness) and visualization was accomplished with a 254 nm UV light and by staining with a KMnO4 solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of a 0.1% NaOH solution), CAM solution (5 g of cerium sulfate, 25 g of ammonium molybdate, 50 mL of conc. H2SO4 and 450 mL of H2O, p-anisaldehyde solution (2.5 mL of p-anisaldehyde, 2 mL of AcOH, and 3.5 mL of conc. H2SO4 in 100 mL of 95% EtOH). Purifications by chromatography were performed using SiO2 (SiliaFlash® F60, Silicycle). 1H-NMR spectra were recorded on Bruker Avance 300/400/600 MHz instruments in CDCl3. Chemical shifts were reported in parts per million with the residual solvent peak used as an internal standard. Chemical shifts are tabulated as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, dd = doublet of doublet, dt = doublet of triplet, dq = doublet of quartet, sp = septet), coupling constant(s), and integration. 13C NMR spectra were run at 75 or 100 MHz using a proton-decoupled pulse sequence with a d1 of 3 sec, and are tabulated by observed peak. Mass spectra were obtained on a Micromass Autospec double focusing instrument. IR spectra were obtained on a Identify IR-ATR spectrometer.
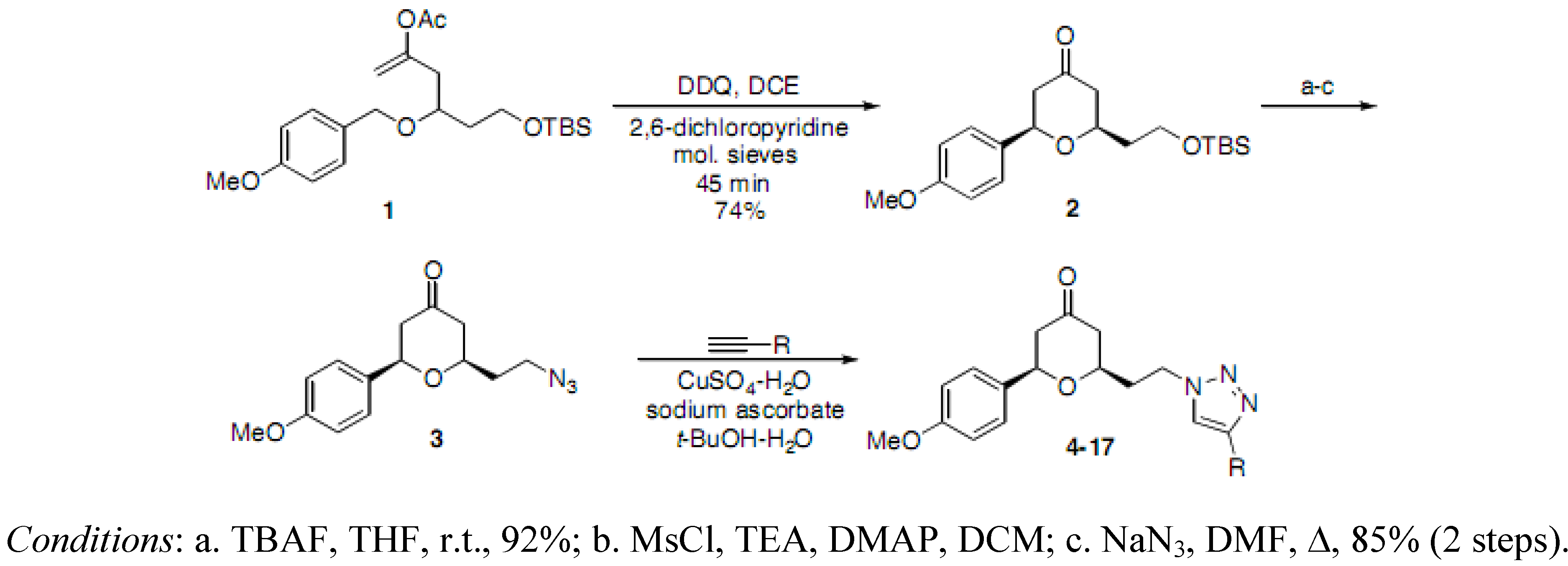
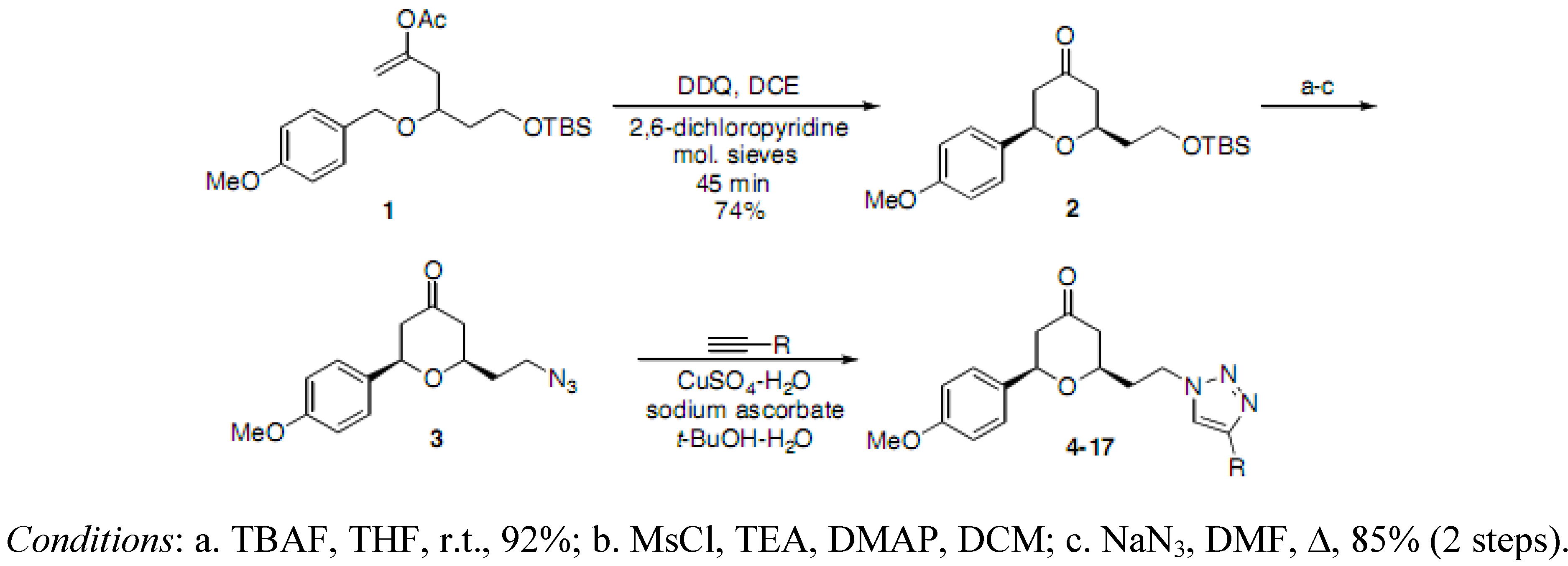
(2RS,6RS)-2-(2-Hydroxyethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one. To a solution of (2
RS,6
RS)-2-(2-((
tert-butyldimethylsilyl)oxy)ethyl)-6-(4-methoxyphenyl)dihydro-2
H-pyran-4(3
H)-one
2 [21] (0.104 g, 0.285 mmol) in THF (3.5 mL) was added TBAF (0.429 mL, 1 M solution in THF) dropwise at 0 °C over 10 min. The reaction mixture was allowed to warm to r.t. and stirred for 1.5 h, quenched with water (5 mL), extracted with DCM, dried (Na
2SO
4), filtered, concentrated under reduced pressure, and purified by chromatography (SiO
2) eluting with hexane-ethyl acetate (3:7) to afford the primary alcohol (0.065 g, 92%) as a colorless oil: IR (neat) 3419, 2954, 2915, 1711, 1610, 1513, 1461, 1302, 1245, 1174, 1149, 1031, 826 cm
−1;
1H-NMR (300 MHz, CDCl
3) δ 7.29 (d,
J = 8.4 Hz, 2H), 6.92 (d,
J = 8.7 Hz, 2H), 4.65 (t,
J = 7.2 Hz, 1H), 4.07–4.03 (m, 1H), 3.86 (q,
J = 6.0 Hz, 2H), 3.82 (s, 3H), 2.61 (d,
J = 7.8 Hz, 2H), 2.48 (d,
J = 7.8 Hz, 2H), 2.21 (t,
J = 5.1 Hz, 1H), 2.07–1.97 (m, 1H), 1.95–1.85 (m, 1H);
13C-NMR (75 MHz, CDCl
3) δ 206.5, 159.7, 132.8, 127.2, 114.3, 78.9, 60.5, 55.6, 55.5, 49.6, 47.9, 38.7; HRMS (ESI)
m/z calcd for C
14H
18O
4Na (M+Na) 273.1103, found 273.1118.
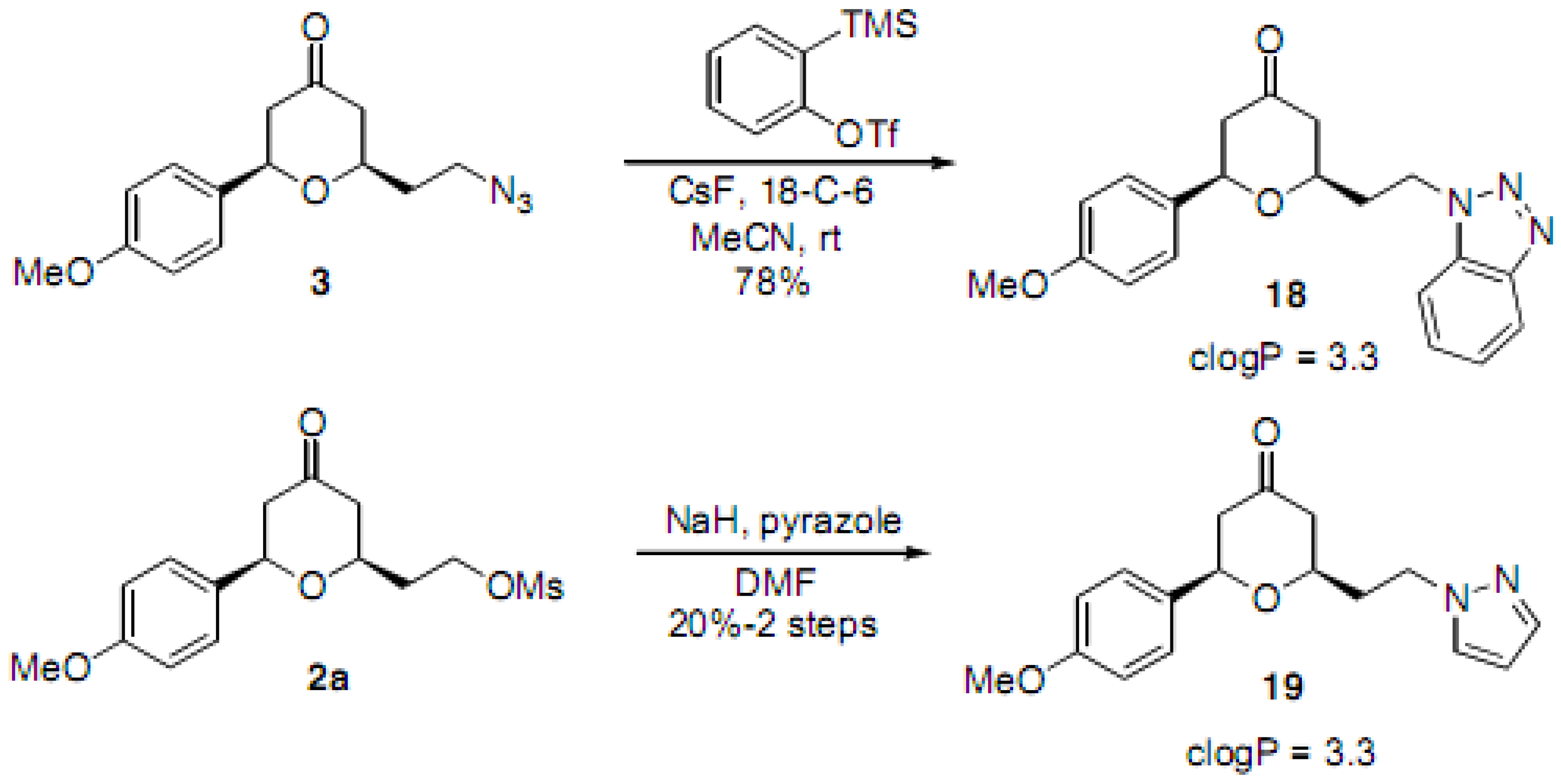
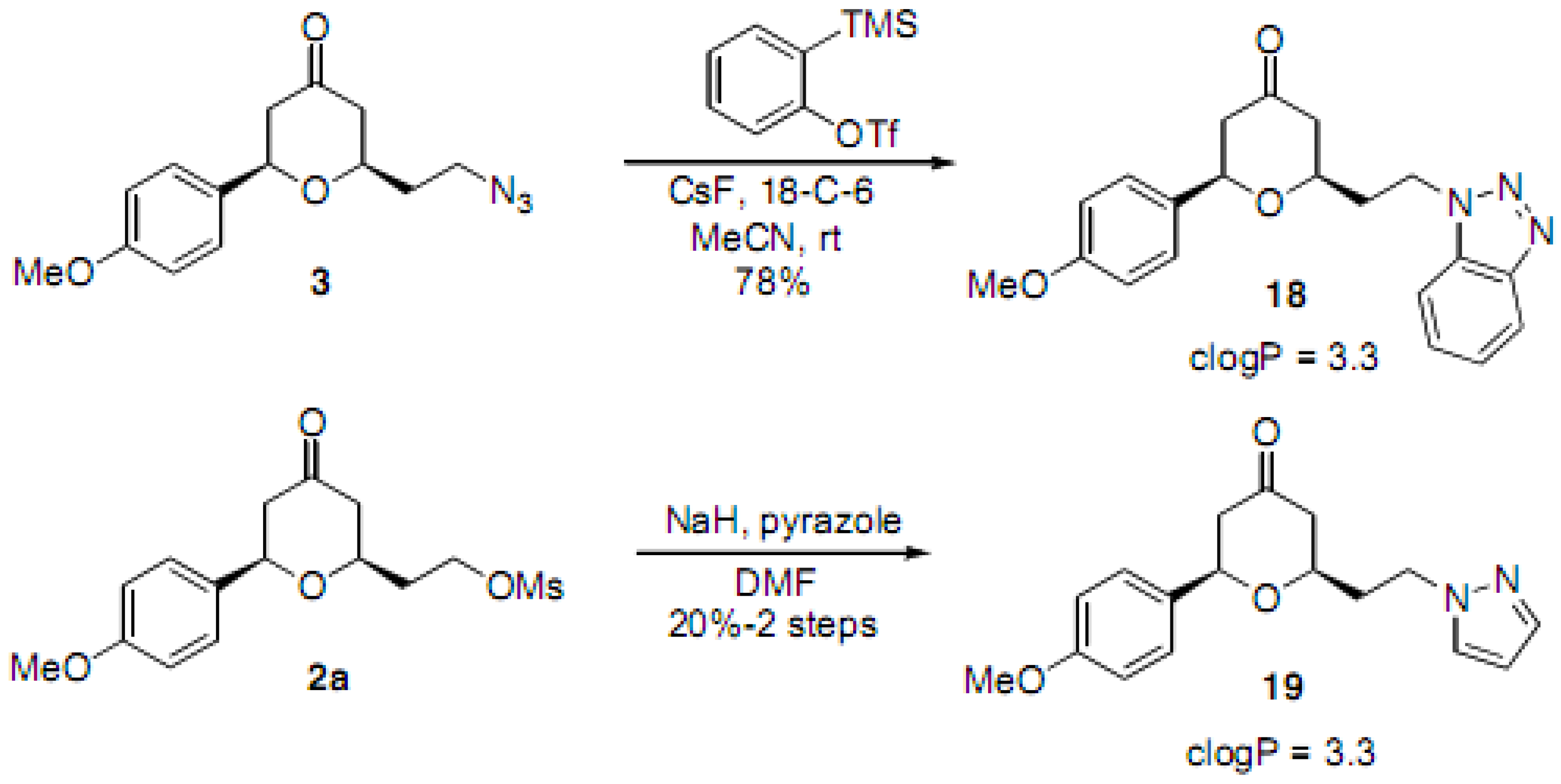
(2RS,6RS)-2-(2-Azidoethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (3). To a solution of the alcohol intermediate (0.122 g, 0.487 mmol), triethylamine (0.103 mL, 0.731 mmol) and DMAP (0.003 g, 0.025 mmol) in DCM (2.5 mL) was added methanesulfonyl chloride (0.045 mL, 0.576 mmol) dropwise at 0 °C. The resulting solution was stirred at r.t. for 15 min. The reaction mixture was diluted with HCl (0.5 M) and extracted with DCM. The aqueous layer was back extracted with DCM. The combined organic layers were washed with saturated NaHCO3, brine, dried (Na2SO4), filtered and concentrated under reduced pressure to obtain the crude 2-((2RS,6RS)-6-(4-methoxyphenyl)-4-oxotetrahydro-2H-pyran-2-yl)ethyl methanesulfonate 2a (0.171 g) as a yellow oil which was used without further purification: 1H-NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H,), 4.63 (dd, J = 4.4, 9.6 Hz, 1H), 4.51–4.45 (m, 1H), 4.42–4.38 (m, 1H), 4.01–3.95 (m, 1H), 3.83 (s, 3H), 2.98 (s, 3H), 2.63-2.59 (m, 2H), 2.52-2.38 (m, 2H), 2.12-2.07 (m, 2H). To a solution of 2a (0.171 g, 0.520 mmol) in DMF (1 mL) was added sodium azide (0.063 g, 0.969 mmol) at r.t. The reaction mixture was stirred at r.t. for 13.5 h. TLC analysis indicated remaining starting material. The reaction mixture was then heated at 40 °C for 6 h and at 50 °C for 1 h, diluted with water, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated under reduced pressure and purified by chromatography on SiO2 (hexane/EtOAc, 3:7) to afford the azide 3 (0.114 g, 85% over two steps) as a colorless oil: IR (neat) 3424, 2098, 1717, 1614, 1515, 1461, 1355, 1303, 1247, 1177, 1032 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 6.92 (d, J = 8.7 Hz, 2H), 4.62 (dd, J = 4.8, 9.6 Hz, 1H), 3.94–3.86 (m, 1H), 3.81 (s, 3H), 3.55–3.46 (m, 2H), 2.65–2.58 (m, 2H), 2.50–2.35 (m, 2H), 1.99–1.85 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 206.2, 159.5, 132.7, 127.1, 114.0, 78.3, 74.1, 55.3, 49.2, 47.6, 47.5, 35.6; MS (EI) m/z 275 (M+, 25%), 135 (100); HRMS (EI) m/z calcd for C14H17N3O3 275.1269, found 275.1265.
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (4). To a suspension of 3 (0.114 g, 0.414 mmol) and phenylacetylene (0.045 mL, 0.410 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.7 mL) was added sodium ascorbate (0.032 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 33 h, recharged with phenylacetylene (0.090 mL, 0.820 mmol) and stirred at r.t. for 21 h. The reaction mixture was diluted with water (7 mL), cooled in ice bath, and filtered. The precipitate was dissolved in ethyl acetate and chloroform, dried (Na2SO4), filtered, and concentrated to afford the corresponding triazole 4 (0.110 g, 71%) as an off-white solid: m.p. 151–152 °C; IR (neat) 3089, 2964, 1714, 1611, 1515, 1470, 1355, 1242, 1174, 1036, 816, 769 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.77 (d, J = 6.9 Hz, 2H), 7.72 (s, 1H), 7.42 (t, J = 6.9 Hz, 2H), 7.35–7.32 (m, 1H), 7.29 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 4.62–4.54 (m, 3H), 3.81 (s, 3H), 3.79–3.72 (m, 1H), 2.59 (d, J = 7.5 Hz, 2H), 2.45–2.42 (m, 2H), 2.32–2.25 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 205.7, 159.7, 147.8, 132.6, 130.7, 128.9, 128.3, 127.3, 125.8, 120.3, 114.3, 78.7, 74.2, 55.5, 49.3, 47.4, 46.9, 36.6; HRMS (ESI) m/z calcd for C22H23N3O3Na (M+Na) 400.1637, found 400.1642.
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-(trimethylsilyl)-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (5). To a suspension of 3 (0.072 g, 0.262 mmol) and trimethylsilylacetylene (0.075 mL, 0.529 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.1 mL) was added sodium ascorbate (0.025 mL, 1 M solution in water) followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 14 h, recharged with trimethylsilylacetylene (0.075 mL, 0.529 mmol) and stirred at r.t. for an additional 6 h. The reaction mixture was diluted with water, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 5 (0.053 g, 54%) as a colorless oil: IR (neat) 2958, 1717, 1614, 1515, 1463, 1351, 1248, 1177, 1035, 839, 752 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.46 (s, 1H), 7.31 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 4.61–4.51 (m, 3H), 3.83 (s, 3H), 3.75–3.65 (m, 1H), 2.61–2.58 (d, J= 7.2 Hz, 2H), 2.43–2.39 (m, 2H), 2.32–2.19 (m, 2H), 0.30 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ 205.9, 159.8, 146.7, 132.7, 129.5, 127.3, 114.3, 78.7, 74.0, 55.5, 49.4, 47.5, 46.1, 36.8, −0.9; HRMS (ESI) m/z calcd for C19H27N3O3Si Na (M+Na) 396.1719, found 396.1698.
(2RS,6RS)-2-(2-(1H-1,2,3-Triazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (6). To a solution of 5 (0.050 g, 0.134 mmol) in THF (1.5 mL) was added TBAF (0.402 ml, 1 M solution in THF) dropwise at 0 °C followed by the addition of acetic acid (0.008 mL, 0.140 mmol). The reaction mixture was stirred at r.t. for 14 h, recharged with acetic acid (0.016 mL, 0.280 mmol) and stirred at r.t. for 4 h, concentrated, and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (3:7) to afford 6 (0.021 g, 53%) as a colorless oil. IR (neat) 2958, 1713, 1611, 1585, 1514, 1446, 1303, 1247, 1176, 1029, 813 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.51 (s, 1H), 7.30 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 4.62–4.54 (m, 3H), 3.83 (s, 3H), 3.72–3.65 (m, 1H), 2.61 (d, J= 8.8 Hz, 2H), 2.45–2.37 (m, 2H), 2.31–2.21 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.7, 159.8, 133.9, 132.6, 127.3, 123.9, 114.3, 78.7, 73.9, 55.5, 49.4, 47.4, 46.6, 36.7, 29.9; HRMS (ESI) m/z calcd for C16H20N3O3 (M+H+) 302.1505, found 302.1531.
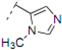
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-methyl-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (7). Propyne (1 mL, 17.7 mmol) was condensed into a 10 mL flask using a dry-ice/acetone condenser. A suspension of 3 (0.060 g, 0.218 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.7 mL) was added to the flask, followed by the addition of sodium ascorbate (0.02 mL, 1 M solution in water), and copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The reaction mixture was stirred at r.t. for 19 h, diluted with water, extracted with ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (2:3) to obtain the triazole 7 (0.038 g, 55%) as a colorless oil: IR (neat) 3584, 2956, 1715, 1612, 1514, 1303, 1247, 1176, 1049, 1029, 813 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 7.22 (s, 1H), 6.93 (d, J = 8.7 Hz, 2H), 4.59–4.49 (m, 3H), 3.83 (s, 3H), 3.74–3.66 (m, 1H), 2.60–2.57 (m, 2H), 2.42–2.39 (m, 2H), 2.32 (s, 3H), 2.27–2.22 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.9, 159.7, 143.5, 132.7, 127.3, 121.7, 114.2, 78.6, 73.9, 55.5, 49.4, 47.4, 46.5, 36.8, 10.9; MS (EI) m/z 338 (M+Na, 100%), 316 (M+H, 18), 298 (18), 233 (50), 215 (75); HRMS (ESI) m/z calcd for C17H21N3O3Na (M+Na) 338.1481, found 338.1487.
(2RS,6RS)-2-(2-(4-Isopropyl-1H-1,2,3-triazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (8). To a suspension of 3 (0.088 g, 0.319 mmol) and 3-methyl-1-butyne (0.065 mL, 0.635 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.1 mL) was added sodium ascorbate (0.03 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 18 h, diluted with water, extracted with ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 8 (0.050 g, 46%) as an off-white solid: m.p. 66–67 °C; IR (neat) 3629, 2960, 1715, 1514, 1446, 1245, 1176, 1045, 813 cm-1; 1H-NMR (300 MHz, CDCl3) δ 7.30 (d, J = 8.7 Hz, 2H), 7.21 (s, 1H), 6.93 (d, J = 8.7 Hz, 2H), 4.58–4.49 (m, 3H), 3.82 (s, 3H), 3.76–3.65 (m, 1H), 3.06 (sp, J = 6.9 Hz, 1H), 2.59 (d, J= 7.5 Hz, 2H), 2.42–2.39 (m, 2H), 2.28–2.19 (m, 2H), 1.27 (dd, J = 1.8, 6.9 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 205.9, 159.7, 154.7, 132.7, 127.3, 119.8, 114.2, 78.7, 74.0, 55.5, 49.4, 47.4, 46.5, 36.7, 25.9, 22.7; MS (EI) m/z 366 (M+Na, 100%), 344 (M+H, 55), 326 (40), 233 (50), 215 (60); HRMS (ESI) m/z calcd for C19H26N3O3 344.1974 (M+H), found 344.1960.
(2RS,6RS)-2-(2-(4-(4-Fluorophenyl)-1H-1,2,3-triazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (9). To a suspension of 3 (0.052 g, 0.189 mmol) and 1-ethynyl-4-fluorobenzene (0.043 mL, 0.37 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.1 mL) was added sodium ascorbate (0.02 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 12 h, diluted with water and filtered. The solid was dissolved in ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 9 (0.054 g, 72%) as a white solid: m.p. 158–159 °C; IR (neat) 3559, 2957, 1715, 1613, 1514, 1497, 1247, 1176, 1027, 1027, 841, 813 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.74–7.69 (m, 2H), 7.67 (s, 1H), 7.29 (d, J = 8.7 Hz, 2H), 7.10 (app t, J= 8.4 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 4.67–4.54 (m, 3H), 3.82 (s, 3H), 3.79–3.74 (m, 1H), 2.59 (d, J= 7.2 Hz, 2H), 2.49–2.42 (m, 2H), 2.34–2.26 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 205.7, 164.5, 161.2, 159.8, 147.0, 132.5, 127.6, 127.5, 127.3, 1267.0, 126.9; 120.1, 116.1, 115.8, 114.3, 78.8, 74.3, 55.5, 49.4, 47.4, 47.0, 36.6; HRMS (ESI) m/z calcd for C22H22FN3O3Na (M+Na) 418.1543, found 418.1570.
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (10). To a suspension of 3 (0.075 g, 0.272 mmol) and 1-ethynyl-3-methoxybenzene (0.069 mL, 0.543 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.5 mL) was added sodium ascorbate (0.025 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 12 h, diluted with water, extracted with ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 10 (0.102 g, 92%) as an off-white solid: m.p. 111–112 °C; IR (neat) 3633, 2957, 1713, 1611, 1585, 1245, 1174, 1040, 813 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.71 (s, 1H), 7.42–7.41 (m, 1H), 7.34–7.26 (m, 4H), 6.90 (d, J = 8.7 Hz, 2H), 6.88–6.86 (m, 1H), 4.68–4.53 (m, 3H), 3.86 (s, 3H), 3.81 (s, 3H), 3.77–3.69 (m, 1H), 2.59–2.57 (d, J = 7.5 Hz, 2H), 2.47–2.41 (m, 2H), 2.33–2.24 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 205.7, 160.2, 159.7, 147.7, 132.5, 1312.0, 130.0, 127.3, 120.5, 118.2, 114.4, 114.2, 110.8, 78.7, 74.1, 55.5, 55.4, 49.3, 47.4, 46.9, 36.6; MS (EI) m/z 430 (M+Na, 90%), 408 (M+H, 60), 365 (70), 233 (15), 215 (20); HRMS (ESI)m/z calcd for C23H25N3O4Na (M+Na) 430.1743, found 430.1727.
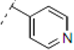
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (11). To a suspension of 3 (0.080 g, 0.291 mmol) and 2-ethynylpyridine (0.294 mL, 2.91 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.6 mL) was added sodium ascorbate (0.029 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 42 h, diluted with water, extracted with ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (3:7) to obtain the triazole 11 (0.095 g, 86%) as a brown oil: IR (neat) 3286, 2960, 1713, 1605, 1514, 1245, 1174, 1038, 719 cm−1; 1H-NMR (400 MHz, CDCl3) δ 8.56 (app dq, J = 0.8, 0.8, 0.8, 4.8 Hz, 1H), 8.14 (app dt, J = 1.2, 1.2, 8.0 Hz, 1H), 8.11 (s, 1H), 7.76 (app dt, J = 1.6, 7.6, 7.6 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), 7.23 (ddd, J = 1.2, 4.8, 7.6 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 4.69–4.54 (m, 3H), 3.81 (s, 1H), 3.79–3.72 (m, 1H), 2.59 (d, J = 7.6 Hz, 2H), 2.48–2.41 (m, 2H), 2.32–2.27 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.8, 159.7, 150.4, 149.6, 148.6, 137.1, 132.6, 127.4, 123.1, 122.6, 120.4, 114.2, 78.6, 73.9, 55.5, 49.3, 47.4, 46.9, 36.7; HRMS (ESI) m/z calcd for C21H22N4O3Na (M+Na) 401.1590, found 401.1559.
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-(pyridin-3-yl)-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (12). To a suspension of 3 (0.087 g, 0.316 mmol) and 3-ethynylpyridine (0.065 g, 0.630 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.7 mL) was added sodium ascorbate (0.032 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 14 h, diluted with water and filtered. The solid was dissolved in ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate-triethylamine (1:9:0.1) to obtain the triazole 12 (0.099 g, 83%) as a brown solid: m.p. 146–147 °C; IR (neat) 2960, 1708, 1611, 1514, 1446, 1247, 1174, 1049, 1025, 813, 707 cm−1; 1H-NMR (300 MHz, CDCl3) δ 8.92 (dd, J = 0.6, 2.0 Hz, 1H), 8.57 (dd, J = 1.5, 4.8 Hz, 1H), 8.14 (app dt, J = 1.8, 1.8, 8.3 Hz, 1H) , 7.80 (s, 1H), 7.36 (ddd, J = 0.6, 4.8, 8.1 Hz, 1H), 7.28 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 4.73–4.54 (m, 3H), 3.82 (s, 3H), 3.79–3.73 (m, 1H), 2.59 (d, J= 7.5 Hz, 2H), 2.50–2.43 (m, 2H), 2.39–2.25 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.6, 159.8, 149.4, 147.2, 144.8, 133.1, 132.5, 127.3, 126.9, 123.9, 120.7, 114.3, 78.9, 74.2, 55.5, 49.4, 47.4, 47.2, 36.6; HRMS (ESI) m/z calcd for C21H23N4O3 (M+H) 379.1770, found 379.1757.
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-(pyridin-4-yl)-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (13). To a suspension of 3 (0.087 g, 0.316 mmol) and 4-ethynylpyridine hydrochloride (0.088 g, 0.630 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.7 mL) was added sodium ascorbate (0.032 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 12 h, diluted with water, triethylamine (1 mL) was added, extracted with ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate-triethylamine (1:9:0.1). The residue was washed with saturated sodium bicarbonate, extracted with ethyl acetate, dried (Na2SO4), filtered and concentrated to obtain the triazole 13 (0.034 g, 28%) as an off-white solid: m.p. 144–145 °C; IR (neat) 3038, 2959, 1715, 1610, 1247, 1213, 746, 719 cm−1; 1H-NMR (400 MHz, CDCl3) δ 8.65 (bs, 2H), 7.85 (s, 1H), 7.63 (bs, 2H), 7.27 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 4.73–4.52 (m, 3H), 3.81 (s, 3H), 3.79–3.74 (m, 1H), 2.59 (d, J = 7.6 Hz, 2H), 2.49–2.40 (m, 2H), 2.35–2.28 (m, 2H); 13C- NMR (100 MHz, CDCl3) δ 205.5, 159.8, 150.6, 145.3, 138.1, 132.4, 127.3, 121.9, 114.3, 78.9, 74.4, 55.5, 49.4, 47.4, 47.3, 36.5, 29.9; HRMS (ESI) m/z calcd for C21H23N4O3 (M+H) 379.1770, found 379.1754.
(2RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-(1-methyl-1H-imidazol-5-yl)-1H-1,2,3-triazol-1-yl)ethyl)-dihydro-2H-pyran-4(3H)-one (14). To a suspension of 3 (0.075 g, 0.272 mmol) and 5-ethynyl-1-methyl-1H-imidazole (0.056 mL, 0.551 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.5 mL) was added sodium ascorbate (0.025 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 12 h, diluted with water, extracted with ethyl acetate, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with chloroform-methanol (20:1) to obtain the triazole 14 (0.090 g, 87%) as a colorless oil: IR (neat) 3003, 1713, 1610, 1514, 1245, 1174, 1027, 747 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.63 (s, 1H), 7.48 (bs, 1H), 7.27 (d, J = 8.7 Hz, 2H), 7.15 (bs, 1H), 6.89 (d, J = 8.7 Hz, 2H), 4.68–4.52 (m, 3H), 3.88 (s, 3H), 3.81 (s, 3H), 3.78–3.67 (m, 1H), 2.58 (d, J = 7.2 Hz, 2H), 2.48–2.36 (m, 2H), 2.33–2.22 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.6, 159.7, 139.6, 138.7, 132.4, 128.6, 127.2, 123.5, 121.5, 114.2, 78.7, 74.0, 55.5, 49.3, 47.4, 46.9, 36.5, 33.8; MS (EI) m/z 405 (M+Na, 20%), 382 (M+H, 60), 365 (100); HRMS (ESI) m/z calcd for C20H24N5O3 (M+H) 382.1879, found 382.1854.
(2RS,6RS)-2-(2-(4-(Cyclopentylmethyl)-1H-1,2,3-triazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (15). To a suspension of 3 (0.072 g, 0.262 mmol) and 3-cyclopentyl-1-propyne (0.068 mL, 0.520 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.1 mL) was added sodium ascorbate (0.025 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 17 h, diluted with water, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 15 (0.086 g, 86%) as a colorless oil: IR (neat) 2945, 1715, 1613, 1514, 1446, 1247, 1174, 1045, 828, 813 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 7.22 (s, 1H), 6.92 (d, J = 8.7 Hz, 2H), 4.57–4.49 (m, 3H), 3.82 (s, 3H), 3.73–3.64 (m, 1H), 2.59 (d, J = 6.0 Hz, 2H), 2.42–2.38 (m, 2H), 2.29–2.06 (m, 3H), 1.78–1.68 (m, 2H), 1.63–1.51 (m, 4H), 1.25–1.17 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.9, 159.7, 148.0, 132.6, 127.2, 121.4, 114.2, 78.6, 73.9, 55.5, 49.4, 47.4, 46.4, 40.1, 36.7, 32.6, 31.8, 25.2; HRMS (ESI) m/z calcd for C22H29N3O3Na (M+Na) 406.2107, found 406.2069.
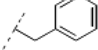
(2RS,6RS)-2-(2-(4-Benzyl-1H-1,2,3-triazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (16). To a suspension of 3 (0.072 g, 0.262 mmol) and 3-phenyl-1-propyne (0.065 mL, 0.522 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.1 mL) was added sodium ascorbate (0.025 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 17 h, diluted with water, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 16 (0.086 g, 84%) as a colorless oil: IR (neat) 2953, 1713, 1611, 1514, 1446, 1245, 1174, 1046, 813, 725 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.33–7.19 (m, 7H), 7.09 (s, 1H), 6.90 (d, J = 8.7 Hz, 2H), 4.50–4.43 (m, 3H), 4.06 (bs, 2H), 3.82 (s, 3H), 3.68–3.58 (m, 1H), 2.56 (d, J = 7.5 Hz, 2H), 2.43–2.36 (m, 2H), 2.27–2.13 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 205.8, 159.7, 147.9, 139.3, 132.5, 128.9, 128.8, 127.3, 126.7, 122.1, 114.2, 78.6, 73.8, 55.5, 49.2, 47.4, 46.5, 36.6, 32.4; HRMS (ESI) m/z calcd for C23H25N3O3Na (M+Na) 414.1794, found 414.1768.
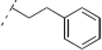
(2R,6R)-2-(4-Methoxyphenyl)-6-(2-(4-phenethyl-1H-1,2,3-triazol-1-yl)ethyl)dihydro-2H-pyran-4(3H)-one (17). To a suspension of 3 (0.072 g, 0.262 mmol) and 4-phenyl-1-butyne (0.074 mL, 0.526 mmol) in a mixture of water and tert-butyl alcohol (1:1, 1.1 mL) was added sodium ascorbate (0.025 mL, 1 M solution in water), followed by copper(II) sulfate pentahydrate (0.001 g, 0.004 mmol). The heterogeneous mixture was stirred at r.t. for 14 h, diluted with water, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to obtain the triazole 17 (0.087 g, 82%) as a colorless oil: IR (neat) 2932, 1717, 1613, 1515, 1454, 1248, 1177, 1048, 831, 749 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.29–7.14 (m, 7H), 7.01 (s, 1H), 6.93 (d, J = 8.7 Hz, 2H), 4.48 (dd, J = 6.2, 7.2 Hz, 2H), 4.38 (dd, J = 5.9, 8.3 Hz, 1H), 3.83 (s, 3H), 3.52 (m, 1H), 3.09–2.93 (m, 4H), 2.57 (d, J = 5.7 Hz, 2H), 2.39–2.36 (m, 2H), 2.23–2.11 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 205.8, 159.7, 147.2, 141.3, 132.6, 128.7, 128.6, 127.3, 126.3, 121.7, 114.2, 78.4, 73.6, 55.5, 49.3, 47.4, 46.4, 36.6, 35.6, 27.5; HRMS (ESI) m/z calcd for C24H27N3O3Na (M+Na) 428.1950, found 428.1982.
(2RS,6RS)-2-(2-(1H-Benzo[d][1,2,3]triazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (18). To a solution of 3 (0.080 g, 0.291 mmol) in acetonitrile (1.5 mL) was added 18-crown-6 (0.173 g, 0.654 mmol) and cesium fluoride (0.121 g, 0.797 mmol). The reaction mixture was stirred at r.t. for 15 min and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (0.194 mL, 0.798 mmol) in acetonitrile (1 mL) was added. The reaction mixture was stirred at r.t. for 2 h, quenched with saturated sodium bicarbonate solution, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (3:2) to obtain the triazole 18 (0.080 g, 78%) as a white solid: m.p. 137–138 °C; IR (neat) 2954, 1717, 1612, 1514, 1446, 1303, 1247, 1174, 1049, 1027, 813, 747 cm−1; 1H-NMR (300 MHz, CDCl3) δ 8.05 (dd, J = 1.8, 6.9 Hz, 1H), 7.44–7.27 (m, 3H), 7.31 (d, J = 8.7 Hz, 2H), 6.96 (d, J = 8.7 Hz, 2H), 4.89–4.77 (m, 2H), 4.43 (dd, J = 4.9, 9.4 Hz, 1H), 3.85 (s, 3H), 3.61–3.50 (m, 1H), 2.58–2.55 (m, 2H), 2.41–2.32 (m, 4H); 13C- NMR (100 MHz, CDCl3) δ 205.7, 159.7, 145.9, 133.5, 132.8, 127.5, 127.3, 124.1, 120.2, 114.3, 109.5, 78.7, 73.7, 55.5, 49.6, 47.5, 44.1, 36.5; MS (EI) m/z 374 (M+Na, 40%), 365 (100); HRMS (ESI) m/z calcd for C20H21N3O3Na (M+Na) 374.1481, found 374.1450.
(2RS,6RS)-2-(2-(1H-Pyrazol-1-yl)ethyl)-6-(4-methoxyphenyl)dihydro-2H-pyran-4(3H)-one (19). To a solution of alcohol intermediate (0.091 g, 0.364 mmol), triethylamine (0.076 mL, 0.546 mmol) and DMAP (0.002 g, 0.016 mmol) in DCM (2 mL) was added methane sulfonyl chloride (0.034 mL, 0.437 mmol) dropwise at 0 °C. The resulting solution was stirred for 15 min, diluted with HCl (0.5 M) and extracted with DCM. The aqueous layer was back extracted with DCM, and the combined organic layers were washed with saturated sodium bicarbonate and brine. The organic layer was dried (Na2SO4), filtered and concentrated to obtain crude 2-((2R,6R)-6-(4-methoxyphenyl)-4-oxotetrahydro-2H-pyran-2-yl)ethyl methanesulfonate 2a (0.120 g, quant.) as a yellow oil which was used without further purification. Pyrazole (0.029 g, 0.426 mmol) in DMF (1 mL) was added to a slurry of sodium hydride (0.013 g, 0.542 mmol) in DMF (3 mL) and stirred for 2 h. The resulting solution was added dropwise to a solution of the mesylate 2a (0.120 g, 0.365 mmol) in DMF (0.5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 5 h, diluted with water, extracted with diethyl ether, dried (Na2SO4), filtered, concentrated, and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (7:3) to afford 19 (0.022 g, 20% over two steps) as a colorless oil: IR (neat) 2956, 1713, 1514, 1247, 1027, 906, 725 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.52 (d, J = 1.5 Hz, 1H), 7.34–7.31 (m, 3H), 6.95 (d, J = 8.7 Hz, 2H), 6.22 (t, J = 1.8 Hz, 1H), 4.54 (dd, J = 5.4, 8.7 Hz, 1H), 4.34 (dd, J = 5.7, 7.8 Hz, 1H), 3.84 (s, 3H), 3.64–3.55 (m, 1H), 2.63–2.52 (m, 2H), 2.44–2.37 (m, 2H), 2.33–2.13 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 206.4, 159.7, 139.8, 132.9, 129.7, 127.2, 114.2, 105.5, 78.5, 74.1, 55.5, 49.5, 48.2, 47.6, 36.8; HRMS (ESI) m/z calcd for C17H20N2O3 300.1474, found 300.1468.
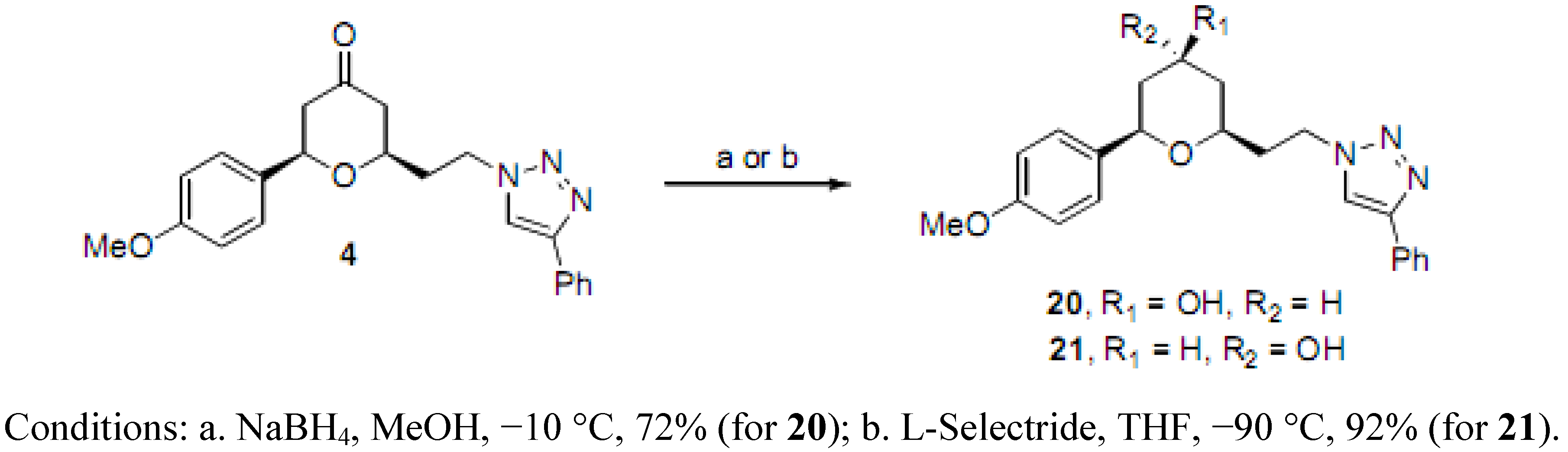
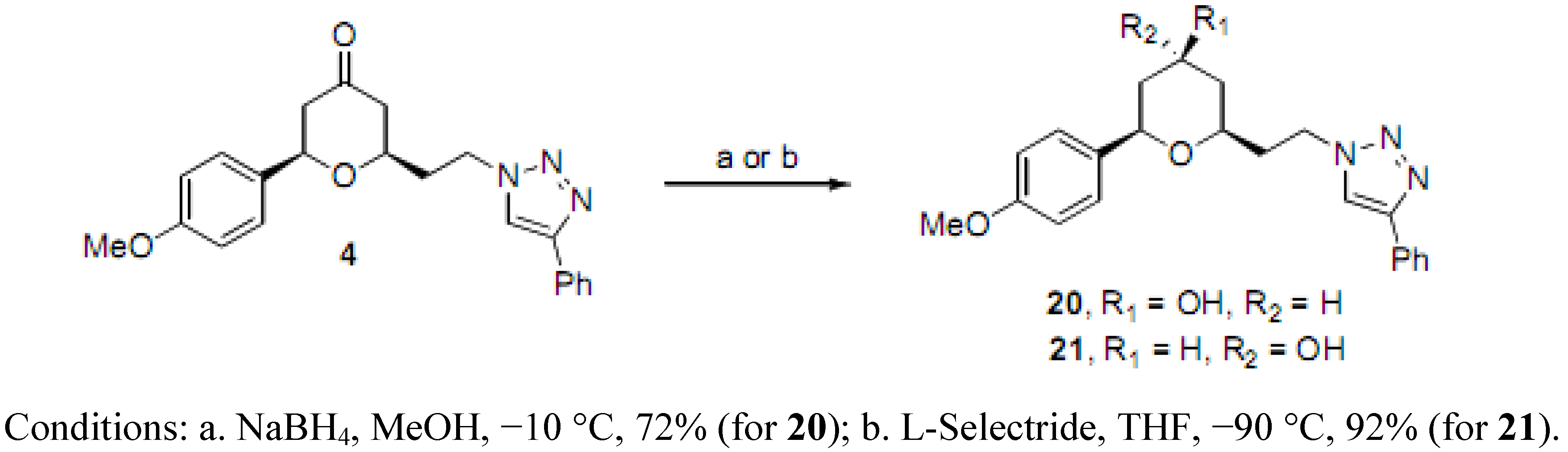
(2RS,4SR,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethyl)tetrahydro-2H-pyran-4-ol (20). To a solution of 4 (0.040 g, 0.106 mmol) in MeOH (5 mL) at −10 °C was added NaBH4 (0.002 g, 0.053 mmol) in one portion. The reaction mixture was stirred at −10 °C for 4 h, quenched with water (2 drops), concentrated, purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to afford 20 (0.023 g, 72%-based on recovered 4, 0.008 g) as an off-white solid: m.p. 139–140 °C; IR (neat) 3430, 2917, 1612, 1514, 1446, 1174, 1075, 1027, 701 cm−1; 1H-NMR (600 MHz, CDCl3) δ 7.78 (d, J = 7.2 Hz, 2H), 7.72 (s, 1H), 7.42 (t, J = 7.2 Hz, 2H), 7.33 (t, J = 7.2 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 4.62–4.52 (m, 2H), 4.28 (dd, J = 1.8, 11.4 Hz, 1H), 3.96–3.91 (m, 1H), 3.82 (s, 3H), 3.45–3.42 (m, 1H), 2.28-2.23 (m, 1H), 2.19–2.13 (m, 2H), 2.02–1.99 (m, 1H), 1.88 (bs, 1H), 1.53 (ap q, J = 10.8 Hz, 1H), 1.35 (ap q, J = 11.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 159.4, 147.7, 133.9, 130.9, 129.0, 128.3, 127.5, 125.9, 120.5, 114.1, 72.7, 68.3, 55.5, 47.1, 42.7, 40.9, 36.4; HRMS (ESI) m/z calcd for C22H25N3O3Na (M+Na) 402.1794, found 402.1766.
(2RS,4RS,6RS)-2-(4-Methoxyphenyl)-6-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethyl)tetrahydro-2H-pyran-4-ol (21). To a solution of 4 (0.050 g, 0.132 mmol) in THF (5.5 mL) at −90 °C was added L-Selectride (0.199 mL, 1 M in THF) dropwise over 10 min. The reaction mixture was stirred at −90 °C for 15 min, and quenched with saturated potassium sodium tartrate (10 mL). The mixture was diluted with diethyl ether (10 mL), stirred for 1 h, extracted with diethyl ether, concentrated, and purified by chromatography (SiO2) eluting with hexane-ethyl acetate (1:1) to give 21 (0.046 g, 92%) as an off-white solid: m.p. 127–128 °C; IR (neat) 3485, 2952, 1724, 1609, 1512, 1463, 1297, 1236, 1174, 1081, 1046, 826 cm−1; 1H-NMR (600 MHz, CDCl3) δ 7.76 (d, J = 7.2 Hz, 2H), 7.73 (s, 1H), 7.41 (t, J = 7.8 Hz, 2H), 7.34–7.31 (m, 1H), 7.29 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 4.80 (dd, J = 1.8, 12.0 Hz, 1H), 4.57–4.55 (m, 2H), 4.35–4.34 (m, 1H), 3.99–3.95 (m, 1H), 3.81 (s, 3H), 2.18 (bs, 1H), 2.21–2.15 (m, 1H), 2.11–2.05 (m, 1H), 1.91–1.89 (m, 1H), 1.78 (ddd, J = 2.6, 11.9, 14.2 Hz, 1H), 1.73 (dd, J = 2.3, 13.9 Hz, 1H), 1.62 (ddd, J = 3.0, 3.0, 12.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 159.2, 147.7, 134.9, 130.9, 128.9, 128.2, 127.5, 125.9, 120.6, 114.0, 73.6, 69.3, 64.8, 55.5, 47.3, 40.4, 38.4, 36.6; HRMS (ESI) m/z calcd for C22H25N3O3Na (M+Na) 402.1794, found 402.1798.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}