N-Cyanomethylnorboldine: A New Aporphine Isolated from Alseodaphne perakensis (Lauraceae)

Abstract
:1. Introduction
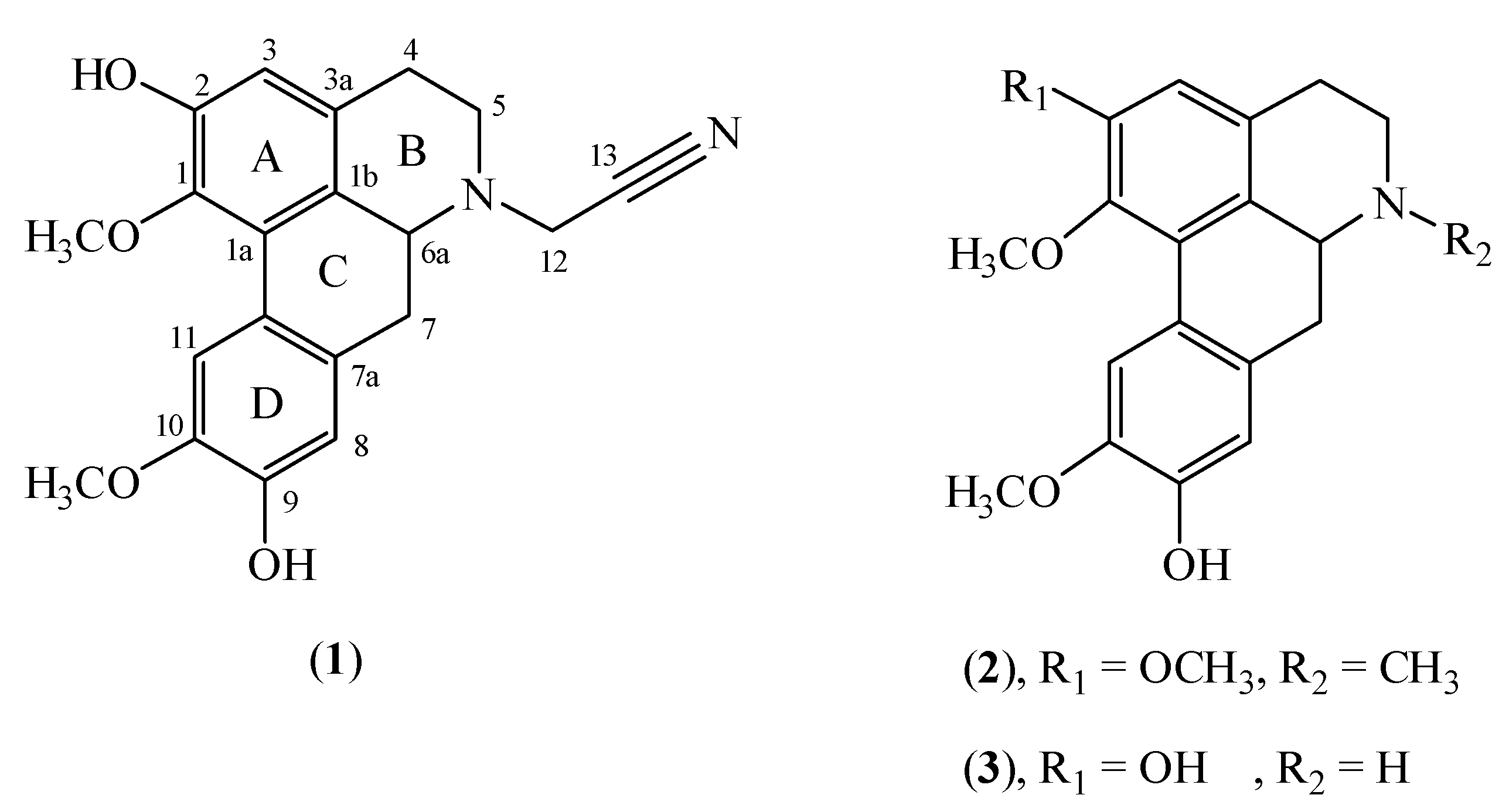
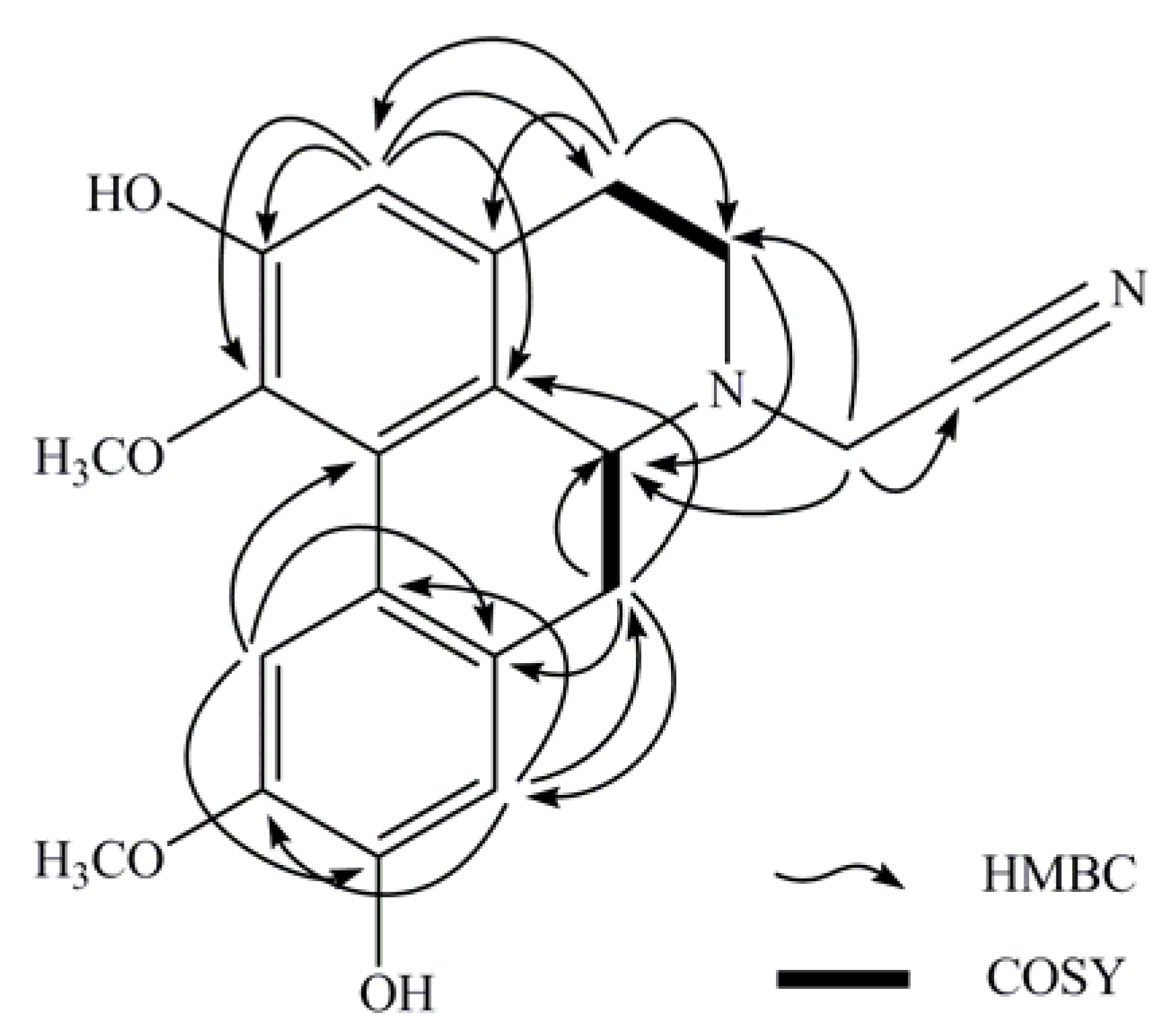
2. Results and Discussion



{kind=link}
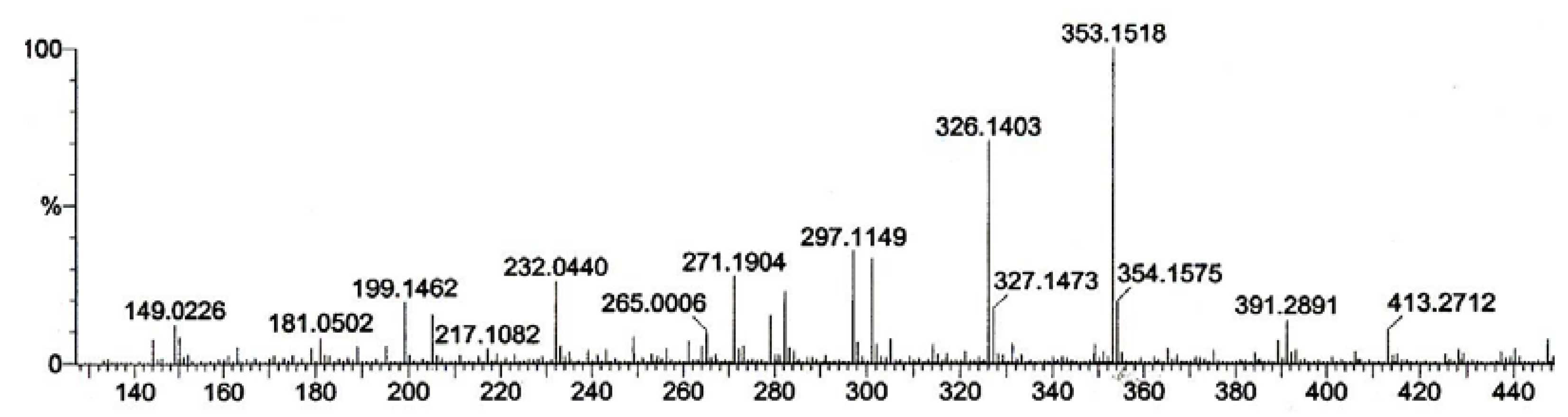{kind=link}
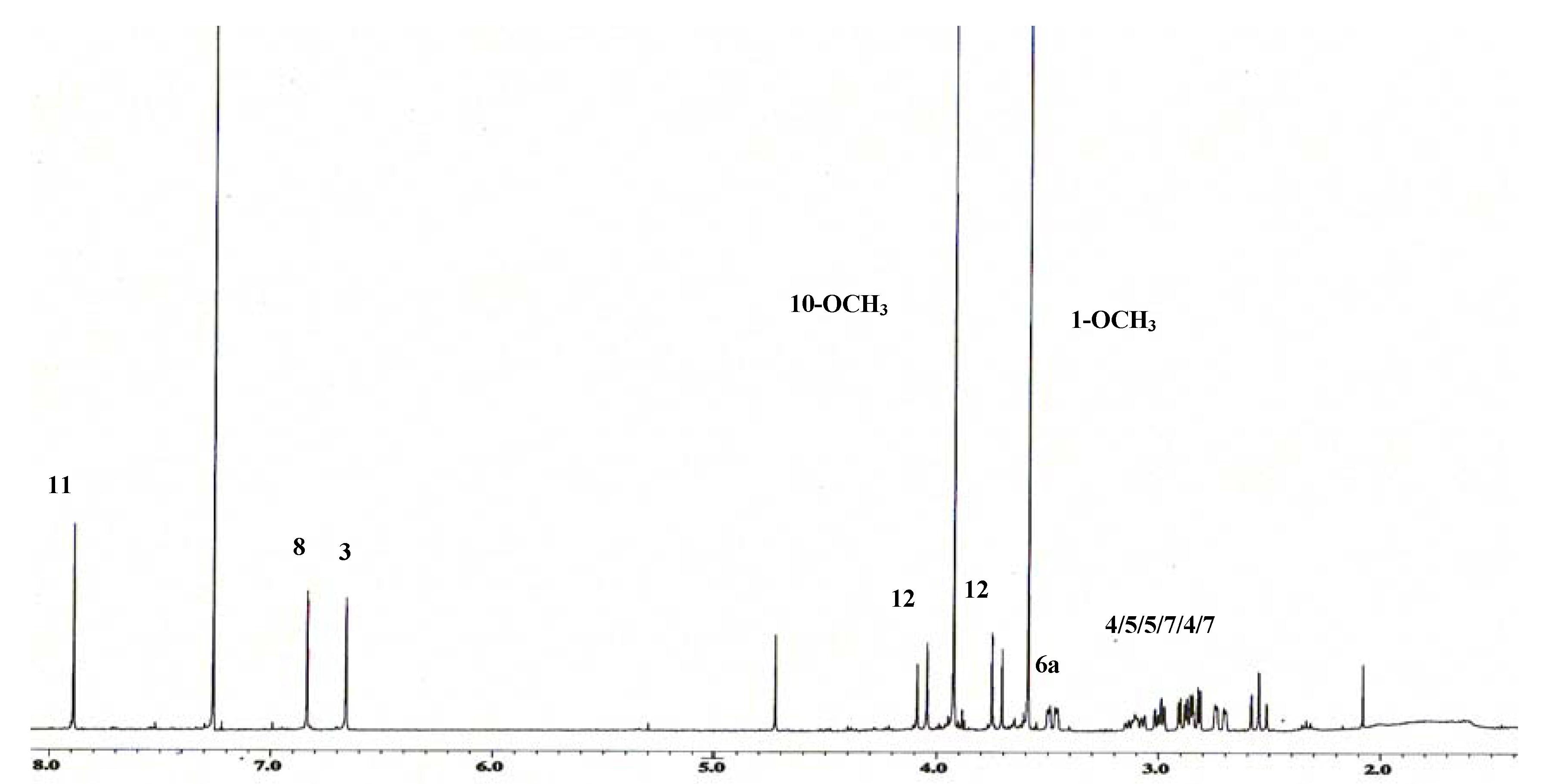{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
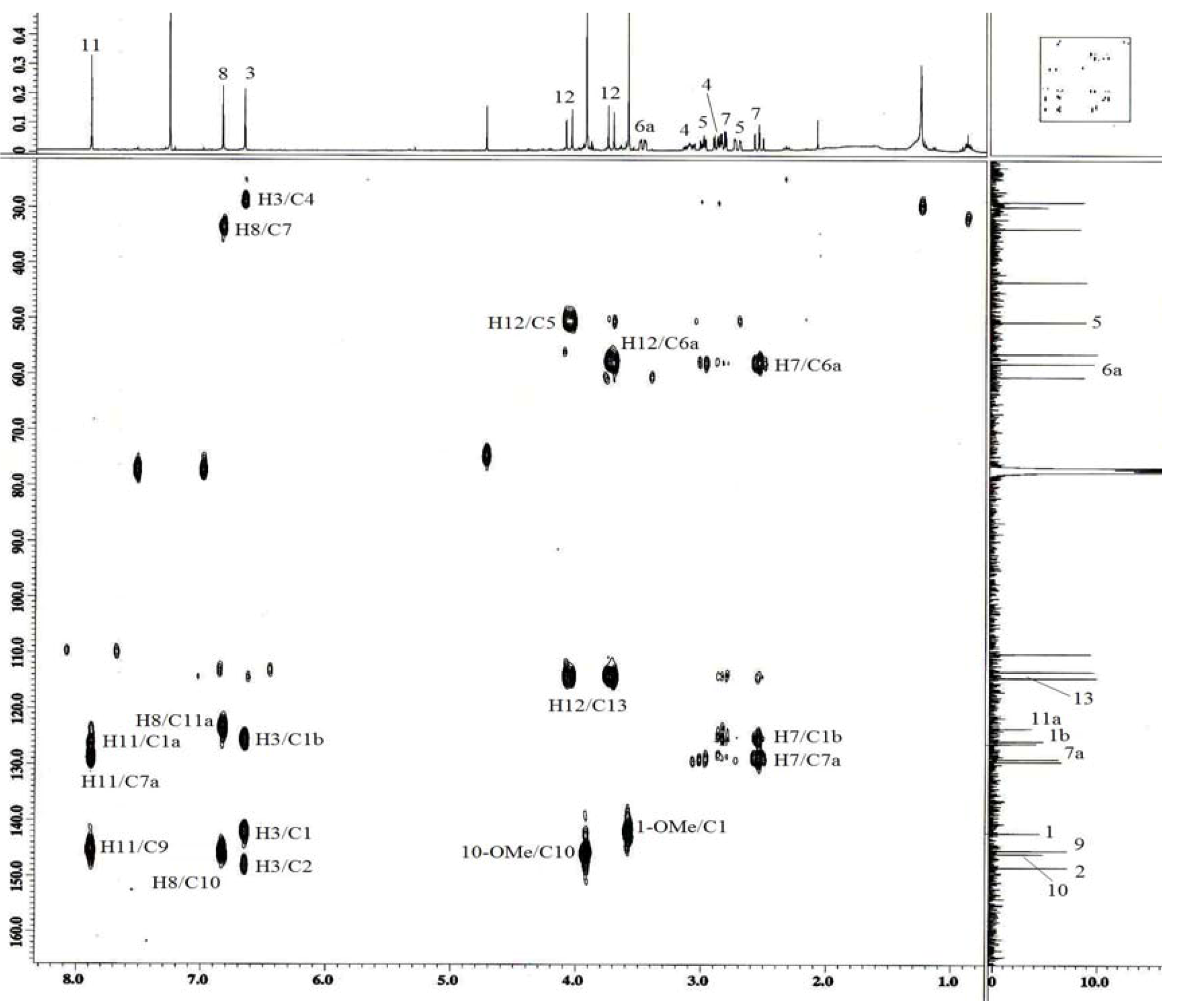
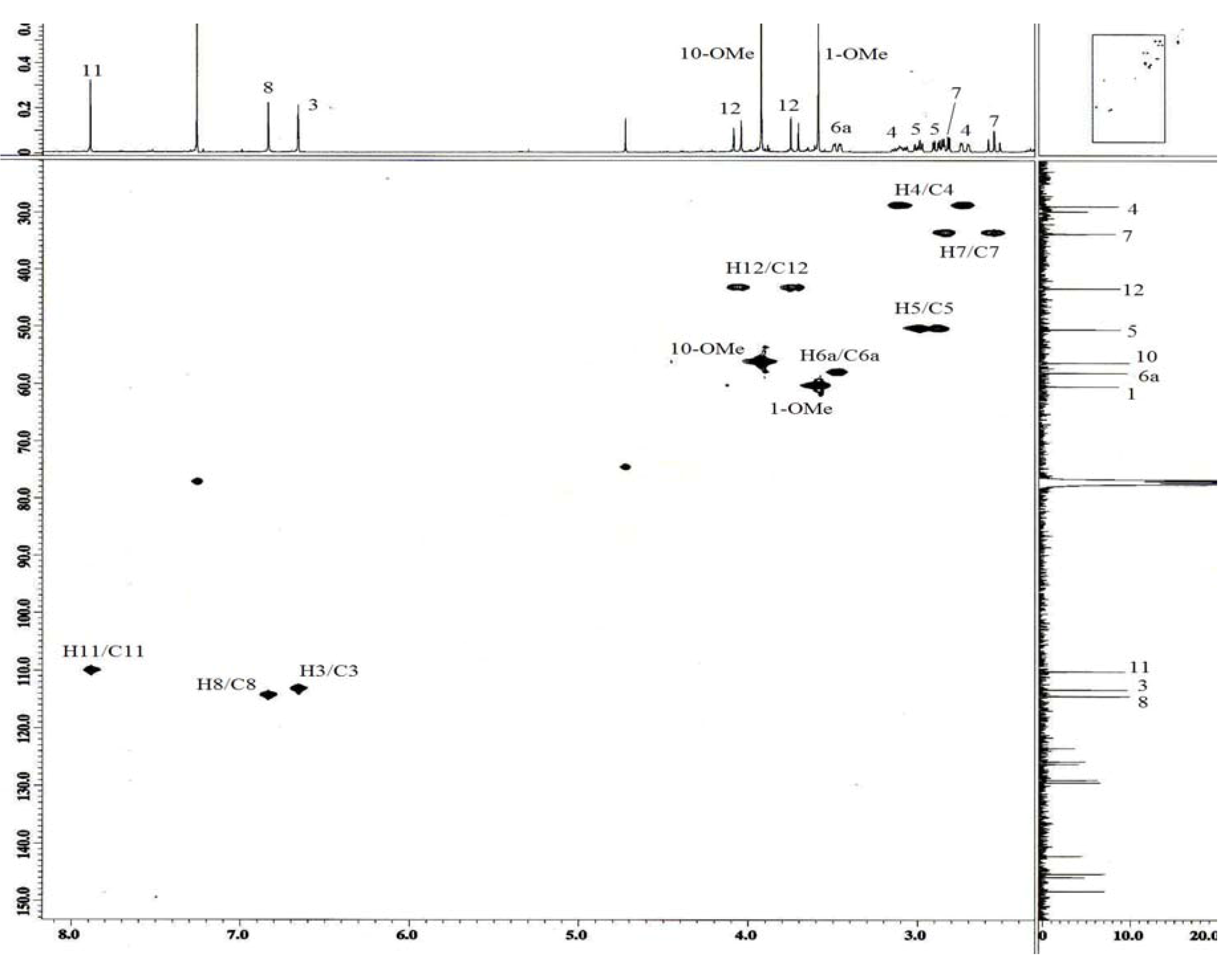
| Position | δ 1H ( J, Hz) | δ 13C | HMBC (2J, 3J) | COSY |
|---|---|---|---|---|
| 1 | 142.1 | |||
| 1-OCH3 | 3.58 ( s) | 60.4 | ||
| 1a | 126.1 | |||
| 1b | 125.7 | |||
| 2 | 148.3 | |||
| 3 | 6.66 ( s) | 113.2 | 1,1b,2,4 | |
| 3a | 129.3 | |||
| 4 | 3.11 ( m) | 28.8 | 3a,5 | H5 |
| 2.72 ( dd,16.4,3.2) | ||||
| 5 | 2.89 ( dd,12.3,3.6) | 50.4 | 3a,6a | H4 |
| 3.00 ( dd,11.4,5.9) | ||||
| 6a | 3.48 ( dd,13.7,4.5) | 58.0 | H7 | |
| 7 | 2.54 ( ddd,13.7,0.9) | 33.7 | 1b,6a,7a,8 | H6a |
| 2.83 ( dd,13.7,4.1) | ||||
| 7a | 128.9 | |||
| 8 | 6.83 ( s) | 114.4 | 7,10,11a | |
| 9 | 145.2 | |||
| 10 | 145.8 | |||
| 10-OCH3 | 3.92 ( s) | 56.2 | ||
| 11 | 7.89 ( s) | 110.1 | 7a,9,1b | |
| 11a | 123.4 | |||
| 12 | 3.73 ( d,17.8) | 43.3 | 5,6a,13 | |
| 4.06 ( d,17.8) | ||||
| 13 | 114.3 |

3. Experimental
3.1. General
3.2. Extraction and Isolation of the Alkaloids
3.3. Vasorelaxant Activity
4. Conclusions
Acknowledgments
References
- Mukhtar, M.R.; Zahari, A.; Nafiah, M.A.; A. Hadi, A.H.; Thomas, N.F.; Hiroko, A.; Morita, H.; Litaudon, M.; Ahmad, K. 3',4'-Dihydrostephasubine, A New Bisbenzylisoquinoline from the Bark of Alseodaphne corneri. Heterocycles 2009, 78, 2571–2578. [Google Scholar] [CrossRef]
- Mukhtar, M.R.; Nafiah, M.A.; Ahmad, K.; Thomas, N.F.; Kazumasa, Z., Morita; Litaudon, M.; A. Hadi, A.H. α′-Oxoperakensimine A-C, New Bisbenzylisoquinoline Alkaloids From Alseodaphne perakensis (Gamble) Kosterm. Heterocycles 2009, 78, 2085–2092. [Google Scholar]
- Rachmatiah, T.; Mukhtar, M.R.; Nafiah, M.A.; Hanafi, M.; Kosela, S.; Morita, H.; Litaudon, M.; Awang, K.; Omar, H.; A. Hadi, A.H. (+)-N-(2-Hydrxypropyl)lindcarpine: A New Cytotoxic Aporphine Isolated from Actinodaphne pruinosa Ness. Molecules 2009, 14, 2850–2856. [Google Scholar]
- Morita, H.; Iizuka, T.; Choo, C.Y.; Chan, K.L.; Takeya, K.; Kobayashi, J. Vasorelaxant Activity of Cyclic Peptide, Cyclosquamosin B, from Annona squamosa. Bioorg. Med. Chem. Lett. 2006, 16, 4609–4611. [Google Scholar] [CrossRef]
- Wu, W.N.; Beal, J.L.; Doskotch, R.W. Alkaloids of Thalictrum XXXV. Northalicarpine, A New Aporphine-Benzylisoquinoline Dimer, N-methyllaurotetanine and Thalflavidine From The Roots of Thalictrum revolutum. J. Nat. Prod. 1980, 43, 567–570. [Google Scholar] [CrossRef]
- Tewari, S.; Bhakuni, D.S.; Dhar, M.M. The Aporphine Alkaloids of Litsea glutenosa. Phytochemistry 1972, 11, 1149–1152. [Google Scholar] [CrossRef]
- Guinaudeau, H.; Lebeouf, M.; Cave, A. Aporphine alkaloids. Lloydia 1975, 38, 275–338. [Google Scholar]
- Bhakuni, D.S.; Gupta, S. Alkaloids of Litsea wightiana1. Planta Med. 1983, 48, 52–54. [Google Scholar] [CrossRef]
- Kozuka, M.; Miyazawa, S.; Yokoyama, K.; Odani, T.; Kubo, M. Alkaloids from Lindera umbellata, Lindera sericea, and Their Varieties. J. Nat. Prod. 1985, 48, 160–161. [Google Scholar]
- Ross, S.A.; Minard, R.D.; Shamma, M.; Fagbule, M.O.; Olatunji, G.; Gbile, Z. Thaliporphinemethine: A New Phenanthrene Alkaloid from Illigera pentaphylla. J. Nat. Prod. 1985, 48, 835–836. [Google Scholar]
- Weber, J.F.; Bruneton, J.; Pusset, J. Plants of New Caledonia Part 103.1 Alkaloids of Litsea lecardii. Planta Med. 1986, 52, 74. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nafiah, M.A.; Mukhtar, M.R.; Omar, H.; Ahmad, K.; Morita, H.; Litaudon, M.; Awang, K.; Hadi, A.H.A. N-Cyanomethylnorboldine: A New Aporphine Isolated from Alseodaphne perakensis (Lauraceae). Molecules 2011, 16, 3402-3409. https://doi.org/10.3390/molecules16043402
Nafiah MA, Mukhtar MR, Omar H, Ahmad K, Morita H, Litaudon M, Awang K, Hadi AHA. N-Cyanomethylnorboldine: A New Aporphine Isolated from Alseodaphne perakensis (Lauraceae). Molecules. 2011; 16(4):3402-3409. https://doi.org/10.3390/molecules16043402
Chicago/Turabian StyleNafiah, Mohd Azlan, Mat Ropi Mukhtar, Hanita Omar, Kartini Ahmad, Hiroshi Morita, Marc Litaudon, Khalijah Awang, and A. Hamid A. Hadi. 2011. "N-Cyanomethylnorboldine: A New Aporphine Isolated from Alseodaphne perakensis (Lauraceae)" Molecules 16, no. 4: 3402-3409. https://doi.org/10.3390/molecules16043402
APA StyleNafiah, M. A., Mukhtar, M. R., Omar, H., Ahmad, K., Morita, H., Litaudon, M., Awang, K., & Hadi, A. H. A. (2011). N-Cyanomethylnorboldine: A New Aporphine Isolated from Alseodaphne perakensis (Lauraceae). Molecules, 16(4), 3402-3409. https://doi.org/10.3390/molecules16043402