Cranberry Proanthocyanidins Mediate Growth Arrest of Lung Cancer Cells through Modulation of Gene Expression and Rapid Induction of Apoptosis

Abstract
:1. Introduction
2. Results and Discussion
2.1. Modulation of global gene expression patterns by PAC

{kind=link}
{kind=link}
{kind=link}
| Up-Regulated Biological Processes (n = 30/221) | % | P-Value | Down-Regulated Biological Processes (n = 30/207) | % | P-Value |
|---|---|---|---|---|---|
| Protein transport | 5.89 | 7.2E-11 | RNA metabolic process | 7.46 | 9.5E-21 |
| RNA metabolic process | 6.81 | 3.8E-09 | Cellular protein metabolic process | 15.79 | 1.4E-19 |
| Cellular protein metabolic process | 15.17 | 7.2E-09 | RNA processing | 4.61 | 8.9E-16 |
| RNA processing | 4.24 | 3.5E-08 | M phase of mitotic cell cycle | 2.21 | 1.1E-12 |
| Intracellular protein transport | 3.02 | 3.9E-07 | Mitosis | 2.16 | 2.1E-12 |
| Golgi vesicle transport | 1.27 | 4.3E-06 | M phase | 2.91 | 4.0E-12 |
| RNA splicing | 2.29 | 1.2E-05 | DNA repair | 2.59 | 6.5E-12 |
| Cellular protein complex assembly | 1.44 | 2.7E-05 | Regulation of gene expression | 17.66 | 1.3E-11 |
| Protein modification process | 9.24 | 5.2E-05 | Regulation of macromolecule biosynthetic process | 17.51 | 1.4E-11 |
| Regulation of gene expression, epigenetic | 0.80 | 7.0E-05 | mRNA metabolic process | 3.13 | 3.8E-11 |
| Regulation of apoptosis | 5.37 | 1.2E-04 | DNA metabolic process | 3.96 | 1.7E-10 |
| ncRNA processing | 1.56 | 1.2E-04 | Modification-dependent macromolecule catabolic process | 4.37 | 3.1E-10 |
| mRNA metabolic process | 2.73 | 1.2E-04 | mRNA processing | 2.74 | 3.1E-10 |
| phospholipid biosynthetic process | 0.97 | 1.4E-04 | Cellular protein catabolic process | 4.54 | 3.9E-10 |
| Regulation of programmed cell death | 5.40 | 1.7E-04 | Regulation of cellular biosynthetic process | 17.91 | 4.3E-10 |
| mRNA processing | 2.40 | 1.9E-04 | Regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 17.14 | 8.0E-10 |
| Positive regulation of cell death | 3.09 | 3.0E-04 | Protein catabolic process | 4.61 | 1.6E-09 |
| Negative regulation of macromolecule metabolic process | 4.88 | 3.5E-04 | RNA splicing | 2.44 | 2.0E-09 |
| Positive regulation of programmed cell death | 3.06 | 3.7E-04 | Regulation of transcription | 15.79 | 1.1E-08 |
| Positive regulation of apoptosis | 3.04 | 4.0E-04 | Protein transport | 5.25 | 1.5E-07 |
| Apoptotic mitochondrial changes | 0.40 | 4.3E-04 | Protein modification process | 9.15 | 5.5E-07 |
| Glycerophospholipid biosynthetic process | 0.68 | 5.3E-04 | Cell cycle checkpoint | 0.94 | 1.1E-06 |
| Negative regulation of cellular metabolic process | 4.76 | 5.6E-04 | Regulation of cell cycle process | 1.07 | 6.2E-06 |
| Chromatin modification | 2.05 | 5.9E-04 | Negative regulation of macromolecule metabolic process | 4.89 | 7.3E-06 |
| ncRNA metabolic process | 1.77 | 6.3E-04 | DNA replication | 1.56 | 1.3E-05 |
| Actin filament organization | 0.71 | 6.6E-04 | Negative regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 3.51 | 2.7E-05 |
| Induction of programmed cell death | 2.31 | 1.1E-03 | RNA biosynthetic process | 2.21 | 2.9E-05 |
| Heme metabolic process | 0.31 | 1.2E-03 | Translation | 2.42 | 3.0E-05 |
| Negative regulation of gene expression | 3.42 | 1.2E-03 | DNA damage response, signal transduction | 0.79 | 3.1E-05 |
| DNA metabolic process | 3.42 | 1.4E-03 | Negative regulation of gene expression | 3.43 | 5.7E-05 |
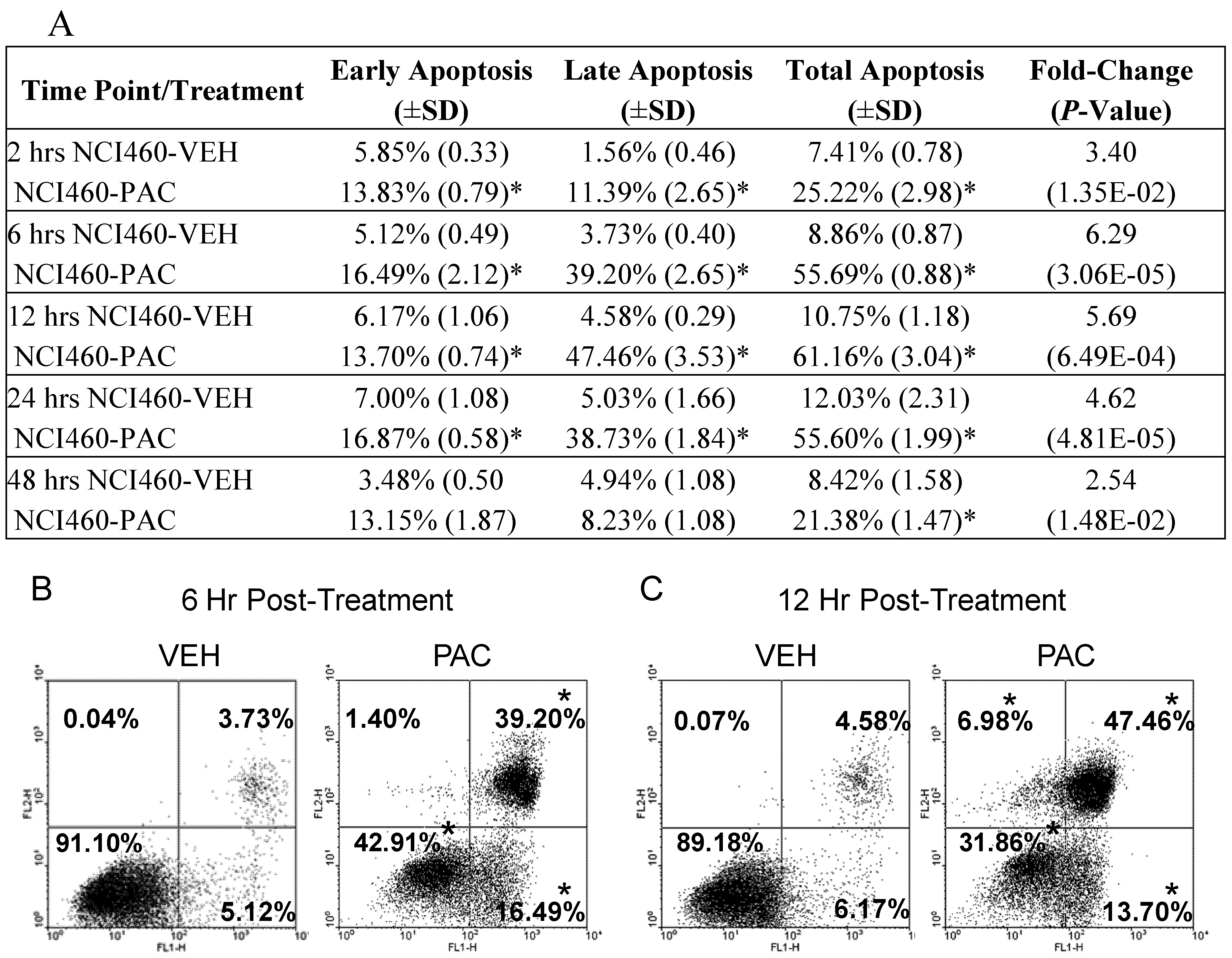
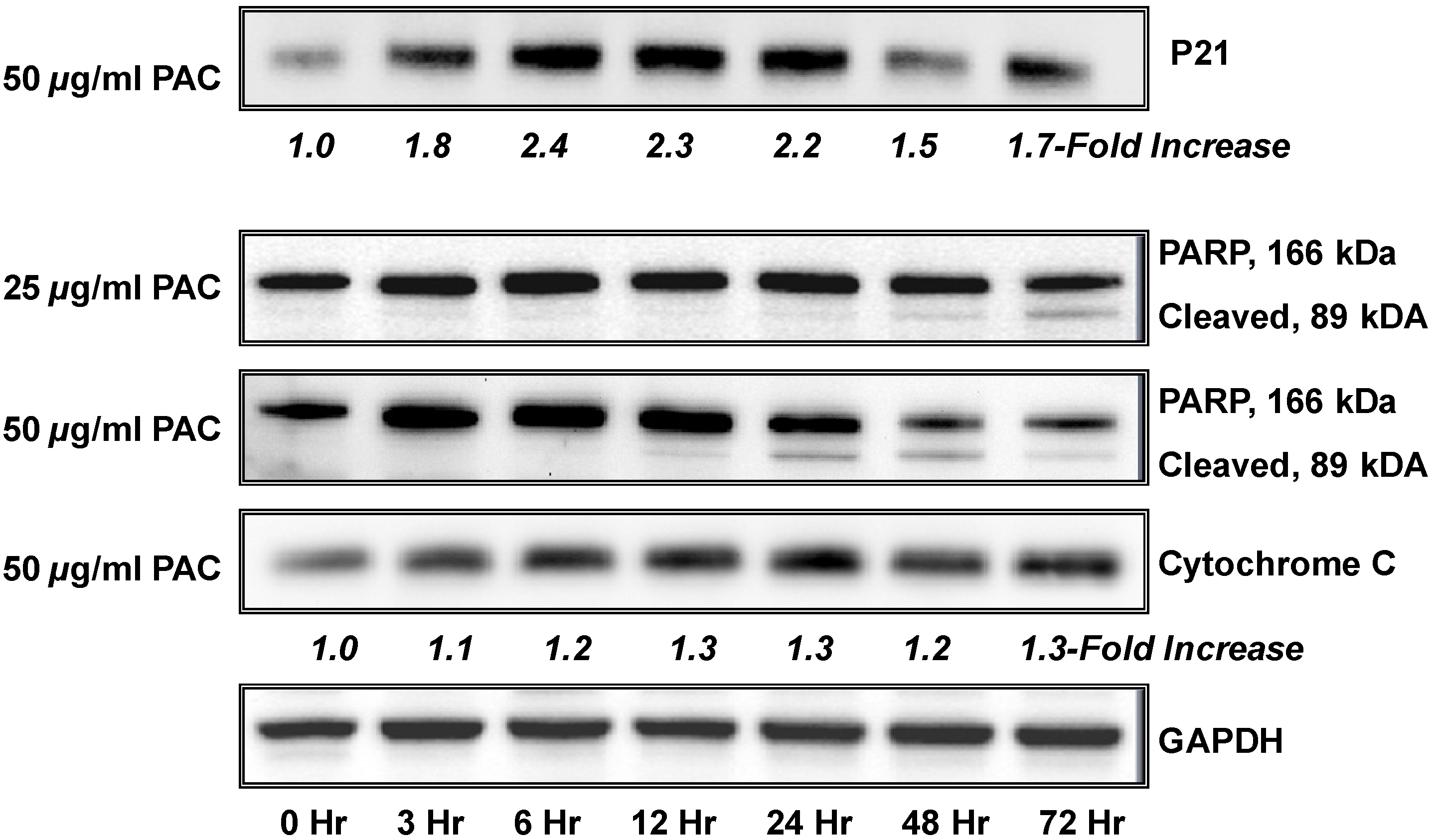
2.2. Pac induces rapid and significant apoptosis in lung cancer cells
| Up-Regulated by PAC Treatment (>2.0 fold) | |||
|---|---|---|---|
| Gene | Name | Fold-Change | Function |
| BCL2L10 | BCL2-like 10 (apoptosis facilitator) | +3.25 | Anti-apoptotic member of the Bcl-2 family that blocks apoptosis in the mitochondrial death pathway, but not in the death receptor pathway [42]. |
| BID | BH3 interacting domain death agonist | +3.25 | Pro-apoptotic member of Bcl-2 proteins and encodes a death agonist that heterodimerizes with either agonist BAX or antagonist BCL2. |
| DFFA | DNA fragmentation factor | +2.00 | A substrate for caspase-3 and triggers DNA fragmentation during apoptosis [43]. |
| MCL1 | myeloid cell leukemia sequence 1 (BCL2-related) | +2.00 | Involved in the regulation of apoptosis versus cell survival and maintenance of viability, but not of proliferation. Two isoforms have been identified, isoform 1 inhibits apoptosis and isoform 2 promotes apoptosis [44]. |
| TNF | tumor necrosis factor | +9.19 | Cytokine that binds to TNFRSF1A/TNFR1 and TNFRSF1B/TNFBR, involved in the regulation cell proliferation, differentiation, apoptosis, lipid metabolism, and coagulation. Induces cell death of certain tumor cell lines. |
| TNFRSF10A | tumor necrosis factor receptor superfamily, member 10a | +3.03 | Transduces cell death signal and induces cell apoptosis via activation by tumor necrosis factor-related apoptosis inducing ligand (TNFSF10/TRAIL) [45]. |
| TNFRSF25 | tumor necrosis factor receptor superfamily, member 25 | +14.93 | TNFSF12/APO3L/TWEAK receptor, interacts directly with the TRADD, mediates activation of NF-kappa-B and induces apoptosis [46)]. |
| TNFSF7 | tumor necrosis factor receptor superfamily, member 7 | +2.83 | Cytokine that binds to CD27 and involved in T-cell activation. Induces proliferation of co-stimulated T-cells and enhances the generation of cytolytic T-cells [47]. |
| TP73 | tumor protein P73 | +22.63 | Postulated tumor suppressor protein and p53 family member. Family members include p53, p63, and p73 and have high sequence similarity, which allows p63 and p73 to transactivate p53-responsive genes causing cell cycle arrest and apoptosis. [33]. |
| TRADD | TNFRSF1A-associated via death domain | +12.13 | Adaptor molecule that interacts with TNFRSF1A/TNFR1 and mediates programmed cell death signaling and NF-kappaB activation. This protein reduces recruitment of inhibitor-of-apoptosis proteins (IAPs) by TRAF2 [48]. |
| TRAF3 | TNF receptor-associated factor 3 | +2.43 | Adapter protein and signal transducer that links members of the tumor necrosis factor receptor family to signaling pathways. Involved in the activation of NF-kappa-B and JNK and in apoptosis [49]. |
| BAG4 | BCL2-associated athanogene 4 | -2.46 | Member of the BAG1 anti-apoptotic protein family [31,32]. |
| XIAP | baculoviral IAP repeat-containing protein 4 | -6.96 | Apoptotic suppressor through binding to tumor necrosis factor receptor-associated factors TRAF1 and TRAF2 [28,29]. |
| BFAR | bifunctional apoptosis regulator | -3.25 | Apoptosis regulator with bifunctional anti-apoptotic activity for apoptosis triggered by death-receptors and mitochondrial factors [50]. |
| BNIP2 | BCL2/adenovirus E1B 19kDa interacting protein 2 | -6.50 | Member of the BCL2/adenovirus E1B 19 kD-interacting protein family. Its specific function is unknown; however, it interacts with the E1B 19 kD protein which is responsible for the protection of virally-induced cell death, as well as E1B 19 kD-like sequences of BCL2, an apoptotic protector [51]. |
| BNIP3L | BCL2/adenovirus E1B19kDa interacting protein 3-like | -3.25 | Same as BNIP2. May also function as a tumor suppressor and inhibits apoptosis induced by BNIP3 [51,52]. |
| CARD8 | caspase recruitment domain family, member 8 | -3.73 | Postulated to be a component of the inflammasome, a protein complex that plays a role in the activation of proinflammatory caspases. Also, acts as an adaptor molecule negatively regulating NFKB activation, CASP1-dependent IL1B secretion, and apoptosis [53]. |
| CASP3 | caspase 3, apoptosis-related cysteine peptidase | -8.00 | Involved in the activation cascade of caspases responsible for apoptosis execution. An effector caspases, responsible for cleaving downstream substrates [36]. |
| CASP4 | caspase 4, apoptosis-related cysteine peptidase | -13.00 | An initiator caspase able to cleave and activate its own precursor protein, as well as caspase 1 precursor. |
| CRADD | CASP2 and RIPK1 domain containing adaptor with death domain | -3.03 | Apoptotic adaptor molecule specific for caspase-2 and FASL/TNF receptor-interacting protein RIP [54]. |
| LTBR | lymphotoxin beta receptor, TNFR superfamily member 3 | -2.83 | Receptor for the heterotrimeric lymphotoxin containing LTA, LTB, and TNFS14/LIGHT. Pro-apoptotic via TRAF3 and TRAF5 [55,56]. |
| TNFS8 | tumor necrosis factor (ligand) superfamily, member 8 | -3.73 | A cytokine that belongs to the tumor necrosis factor (TNF) ligand family and has been reported to induce cell proliferation [57]. |
| TP53BP2 | tumor protein p53 binding protein | -5.66 | Regulates apoptosis and cell growth through interactions with other p53 regulatory molecules. Inhibits the ability of APPBP1 to conjugate NEDD8 to CUL1 decreasing apoptosis induction by APPBP1. Impedes cell cycle progression at G2/M checkpoint [58,59]. |
3. Experimental
3.1. Cell cultures
3.2. Cranberry proanthocyanidins
3.3. Flow cytometry analysis of cellular apoptosis
3.4. Western blot analysis
3.5. Isolation of RNA and synthesis of cDNA
3.6. Microarray studies
3.7. Validation by Real Time PCR
3.8. Statistical and microarray analysis
4. Conclusions
References
- American Cancer Society. Cancer Facts and Figures 2010; American Cancer Society: Atlanta, GA, USA, 2010. [Google Scholar]
- Blot, W.J.; Fraumeni, J.F., Jr. Cancers of the lung and pleura. In Cancer Epidemiology and Prevention; Schottenfeld, D., Fraumeni, J., Jr., Eds.; Oxford University Press: New York, NY, USA, 1996. [Google Scholar]
- International Agency for Research on Cancer. Overall evaluation of carcinogenicity: An updating of IARC Monographs Volumes 1 to 42. IARC Monogr. Eval. Carcinog. Risks Hum. 1987. No. 1-42, supplement 7. [Google Scholar]
- World Cancer Research Fund, American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; American Institute for Cancer Research: Washington, DC, USA, 2007. [Google Scholar]
- Su, X.; Howell, A.B.; D'Souza, D.H. Antiviral effects of cranberry juice and cranberry proanthocyanidins on foodborne viral surrogates--a time dependence study in vitro. Food Microbiol. 2010, 8, 985–991. [Google Scholar]
- Lipson, S.M.; Sethi, L.; Cohen, P.; Gordon, R.E.; Tan, I.P.; Burdowski, A.; Stotzky, G. Antiviral effects on bacteriophages and rotavirus by cranberry juice. Phytomedicine 2007, 1, 23–30. [Google Scholar]
- Avorn, J.; Monane, M.; Gurwitz, J.H.; Glynn, R.J.; Choodnovskiy, I.; Lipsitz, L.A. Reduction of bacteriuria and pyuria after ingestion of cranberry juice. JAMA 1994, 271, 751–754. [Google Scholar]
- Foo, L.Y.; Lu, Y.; Howell, A.B.; Vorsa, N. The structure of cranberry proanthocyanidins which inhibit adherence of uropathogenic P-fimbriated Escherichia coli in vitro. Phytochemistry 2000, 54, 173–181. [Google Scholar]
- Foo, L.; Lu, Y.; Howell, A.B.; Vorsa, N. A-type proanthocyanidin trimers from cranberry that inhibit adherence of uropathogenic P-fimbriated Escherichia coli. J. Nat. Prod. 2000, 63, 1225–1228. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, J.; Pan, K.; Go, V.L.; You, W.C. Efficacy of cranberry juice on Helicobacter pylori infection: A double-blind, randomized placebo-controlled trial. Helicobacter 2005, 2, 139–145. [Google Scholar]
- Howell, A.B.; Vorsa, N.; Der Marderosian, A.; Foo, L.Y. Inhibition of the adherence of P-fimbriated Escherichia coli to uroepithelial-cell surfaces by proanthocyanidin extracts from cranberries. N. Engl. J. Med. 1998, 339, 1085–1086. [Google Scholar] [CrossRef]
- Liu, M.; Lin, L.Q.; Song, B.B.; Wang, L.F.; Zhang, C.P.; Zhao, J.L.; Liu, J.R. Cranberry phytochemical extract inhibits SGC-7901 cell growth and human tumor xenografts in Balb/c nu/nu mice. J. Agric. Food Chem. 2009, 57, 762–768. [Google Scholar] [CrossRef]
- Evans, S.; Dizeyi, N.; Abrahamsson, P.-A.; Persson, J. The effect of a novel botanical agent TBS-101 on invasive prostate cancer in animal models. Anticancer Res. 2009, 29, 3917–3924. [Google Scholar]
- Ferguson, P.J.; Kurowska, E.M.; Freeman, D.J. In vivo inhibition of growth of human tumor lines by flavonoid fractions from cranberry extract. Nutr. Cancer. 2006, 56, 86–94. [Google Scholar] [CrossRef]
- Hochman, N.; Houri-Haddad, Y.; Koblinski, J.; Wahl, L.; Roniger, M.; Bar-Sinai, A.; Weiss, E.I.; Hochman, J. Cranberry juice constituents impair lymphoma growth and augment the generation of antilymphoma antibodies in syngeneic mice. Nutr. Cancer 2008, 60, 511–517. [Google Scholar] [CrossRef]
- Prasain, J.K.; Jones, K.; Moore, R.; Barnes, S.; Leahy, M.; Roderick, R.; Juliana, M.M.; Grubbs, C.J. Effect of cranberry juice concentrate on chemically-induced urinary bladder cancers. Oncol. Rep. 2008, 19, 1565–1570. [Google Scholar] [Green Version]
- Kresty, L.A.; Howell, A.B.; Baird, M. Cranberry proanthocyanidins induce apoptosis and inhibit acid-induced proliferation of human esophageal adenocarcinoma cells. J. Agric. Food Chem. 2008, 56, 676–80. [Google Scholar] [CrossRef]
- Slatore, C.G.; Littman, A.J.; Hu, D.H.; Satia, J.A.; White, E. Long-term use of supplemental multivitamins, vitamin C, vitamin E, and folate does not reduce the risk of lung cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 524–530. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.; Simonetti, R.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar]
- The Alpha Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Omenn, G.; Goodman, G.; Thornquist, M.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.I.; Valanis, B.; Williams, J.H.; Barnhart, S.; Hammer, S. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar]
- Hoque, M.O.; Brait, M.; Rosenbaum, E.; Poeta, M.L.; Pal, P.; Begum, S.; Dasgupta, S.; Carvalho, A.L.; Ahrendt, S.A.; Westra, W.H.; Sidransky, D. Genetic and epigenetic analysis of erbB signaling pathway genes in lung cancer. J. Thorac. Oncol. 2010, 12, 1887–1893. [Google Scholar]
- Memmott, R.M.; Dennis, P.A. The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis. Clin. Cancer Res. 2010, 16, 4–10. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; and Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucl. Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Lai, S.-L.; Pemg, R.-P.; Hwang, J. p53 gene status modulates the chemosensitivity of non-small cell lung cancer cells. J. Biomed. Sci. 2000, 7, 64–70. [Google Scholar] [CrossRef]
- Yang, L.; Mashima, T.; Sato, S. Predominate suppression of apoptosome by inhibitor of apoptosis protein in non-small cell ling cancer H460 cells: Therapeutic effect of a novel polyarginine conjugated Smac peptide. Cancer Res. 2003, 63, 831–837. [Google Scholar]
- Chen, Z.; Naito, M.; Hori, S.; Mashima, T.; Yamori, T.; Tsuruo, T. A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem. Biophys. Res. Commun. 1999, 264, 847–854. [Google Scholar] [CrossRef]
- Straub, C.S. Targeting IAPs as an Approach to Anti-cancer Therapy. Curr. Top. Med. Chem. 2011, 11, 291–316. [Google Scholar] [CrossRef]
- Pore, M.M.; Hiltermann, T.J.; Kruyt, F.A. Targeting apoptosis pathways in lung cancer. Cancer Lett. 2010, in press. [Google Scholar]
- Jiang, Y.; Woronicz, J.D.; Liu, W.; Goeddel, D.V. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science 1999, 283, 543–546. [Google Scholar] [CrossRef]
- Wang, K.; Yin, X.-M.; Chao, D.T.; Milliman, C.L.; Korsmeyer, S.J. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The emerging p53 gene family. J. Natl. Cancer Inst. 1999, 91, 594–598. [Google Scholar] [CrossRef]
- Bitomsky, N.; Hofmann, T.G. Apoptosis and autophagy: Regulation of apoptosis by DNA damage signalling - roles of p53, p73 and HIPK2. FEBS J. 2009, 276, 6074–6083. [Google Scholar] [CrossRef]
- Kim, K.W.; Hwang, M.; Moretti, L.; Jaboin, J.J.; Cha, Y.I.; Lu, B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 2008, 4, 659–668. [Google Scholar]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; Munday, N.A.; Raju, S.M.; Smulson, M.E.; Yamin, T.-T.; Li, V.L.; Miller, D.K. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar]
- Chipuk, J.E.; Fisher, J.C.; Dillon, C.P.; Kriwacki, R.W.; Kuwana, T.; Green, D.R. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 20327–20332. [Google Scholar]
- Dasgupta, P.; Kinkade, R.; Joshi, B.; Decook, C.; Haura, E.; Chellappan, S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc. Natl. Acad. Sci. USA 2003, 103, 6332–6337. [Google Scholar]
- Jin, Z.; Gao, F.; Flagg, T.; Deng, X. Nicotine induces multi-site phosphorylation of Bad in association with suppression of apoptosis. J. Biol. Chem. 2004, 279, 23837–23844. [Google Scholar]
- Xin, M.; Deng, X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J. Biol. Chem. 2005, 280, 10781–10789. [Google Scholar]
- Jin, Z.; Gao, F.; Flagg, T.; Deng, X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 2004, 279, 40209–40219. [Google Scholar] [CrossRef]
- Zhang, H.; Holzgreve, W.; De Geyter, C. Bcl2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway. Hum. Mol. Genet. 2001, 10, 2329–2339. [Google Scholar] [CrossRef]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar] [CrossRef]
- Bingle, C.D.; Craig, R.W.; Swales, B.M.; Singleton, V.; Zhou, P.; Whyte, M.K.B. Exon skipping in Mcl-1 results in a Bcl-2 homology domain 3 only gene product that promotes cell death. J. Biol. Chem. 2000, 275, 22136–22146. [Google Scholar]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Kitson, J.; Raven, T.; Jiang, Y.-P.; Goeddel, D.V.; Giles, K.M.; Pun, K.-T.; Grinham, C.J.; Brown, R.; Farrow, S.N. A death-domain-containing receptor that mediates apoptosis. Nature 1996, 384, 372–375. [Google Scholar]
- Bowman, M.R.; Crimmins, M.A.; Yetz-Aldape, J.; Kriz, R.; Kelleher, K.; Herrmann, S. The cloning of CD70 and its identification as the ligand for CD27. J. Immunol. 1994, 152, 1756–1761. [Google Scholar]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- He, L.; Grammer, A.C.; Wu, X.; Lipsky, P.E. TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-{kappa}B activation. J. Biol. Chem. 2004, 279, 55855–55865. [Google Scholar] [CrossRef]
- Roth, W.; Kermer, P.; Krajewska, M.; Welsh, K.; Davis, S.; Krajewski, S.; Reed, J.C. Bifunctional apoptosis inhibitor (BAR) protects neurons from diverse cell death pathways. Cell Death Differ. 2003, 10, 1178–1187. [Google Scholar] [CrossRef]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.K.; Schaeper, U.; Elangovan, B.; D'Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Ohi, N.; Tokunaga, A.; Tsunoda, H.; Nakano, K.; Haraguchi, K.; Oda, K.; Motoyama, N.; Nakajima, T. A novel adenovirus E1B19K-binding protein B5 inhibits apoptosis induced by Nip3 by forming a heterodimer through the C-terminal hydrophobic region. Cell Death Differ. 1999, 6, 314–325. [Google Scholar]
- Razmara, M.; Srinivasula, S.M.; Wang, L.; Poyet, J.-L.; Geddes, B.J.; DiStefano, P.S.; Bertin, J.; Alnemri, E.S. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J. Biol. Chem. 2002, 277, 13952–13958. [Google Scholar]
- Ahmad, M.; Srinivasula, S.M.; Wang, L.; Talanian, R.V.; Litwack, G.; Fernandes-Alnemri, T.; Alnemri, E.S. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. 1997, 57, 615–619. [Google Scholar]
- Crowe, P.D.; VanArsdale, T.L.; Walter, B.N.; Ware, C.F.; Hession, C.; Ehrenfels, B.; Browning, J.L.; Din, W.S.; Goodwin, R.G.; Smith, C.A. A lymphotoxin-beta-specific receptor. Science 1994, 264, 707–710. [Google Scholar]
- Rooney, I.A.; Butrovich, K.D.; Glass, A.A.; Borboroglu, S.; Benedict, C.A.; Whitbeck, J.C.; Cohen, G.H.; Eisenberg, R.J.; Ware, C.F. The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J. Biol. Chem. 2002, 275, 14307–14315. [Google Scholar]
- Cerutti, A.; Schaffer, A.; Goodwin, R.G.; Shah, S.; Zan, H.; Ely, S.; Casali, P. Engagement of CD153 (CD30 ligand) by CD30-positive T cells inhibits class switch DNA recombination and antibody production in human IgD-positive IgM-positive B cells. J. Immun. 2000, 165, 786–794. [Google Scholar]
- Naumovski, L.; Cleary, M.L. The p53-binding protein 53BP2 also interacts with Bcl2 and impedes cell cycle progression at G2/M. Mol. Cell. Biol. 2002, 16, 3884–3892. [Google Scholar]
- Samuels-Lev, Y.; O'Connor, D.J.; Bergamaschi, D.; Trigiante, G.; Hsieh, J.-K.; Zhong, S.; Campargue, I.; Naumovski, L.; Crook, T.; Lu, X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 2001, 8, 781–794. [Google Scholar] [CrossRef]
- Bendjennat, M.; Boulaire, J.; Jascur, T.; Brickner, H.; Barbier, V.; Sarasin, H.; Fotedar, A.; Fotedar, R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003, 114, 599–610. [Google Scholar] [CrossRef]
- Wei, J.; Zhao, J.; Long, M.; Han, Y.; Wang, X.; Lin, F.; Ren, J.; He, T.; Zhang, H. p21WAF1/CIP1 gene transcriptional activation exerts cell growth inhibition and enhances chemosensitivity to cisplatin in lung carcinoma cell. BMC Cancer 2010, 10, 632–639. [Google Scholar] [CrossRef]
- American Type Culture Collection. NCI-H460, ATCC HTB-177TM. http://www.atcc.org/ (accessed on 23 January 2011).
- Howell, A.B.; Reed, J.D.; Krueger, C.G.; Winterbottom, R.; Cunningham, D.G.; Leahy, M. A-type proanthocyanidins and uropathogenic bacterial anti-adhesion activity. Phytochemistry 2005, 66, 2281–2291. [Google Scholar]
- Foo, L.Y.; Lu, Y.; Howell, A.B.; Vorsa, N. The structure of cranberry proanthocyanidins which inhibit adherence of uropathogenic P-fimbriated Escherichia coliin vitro. Phytochemistry 2000, 54, 173–181. [Google Scholar]
- Foo, L.Y.; Lu, Y.; Howell, A.B.; Vorsa, N. A-type proanthocyanidin trimers from cranberry that inhibit adherence of uropathogenic P-fimbriated Escherichia coli. J. Nat. Prod. 2000, 63, 1225–1228. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bruno, S.; Del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992, 13, 795–808. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, 3. [Google Scholar] [CrossRef]
- Auer, H.; Lyianarachchi, S.; Newsom, D.; Klisovic, M.I.; Marcucci, G.; Kornacker, K. Chipping away at the chip bias: RNA degradation in microarray analysis. Nat. Genet. 2003, 35, 292–293. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix Gene Chip probe level data. Nucl. Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Raponi, M.; Zhang, Y.; Yu, J.; Chen, G.; Lee, G.; Taylor, J.M.; Macdonald, J.; Thomas, D.; Moskaluk, C.; Wang, Y.; Beer, D.G. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66, 7466–7472. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucl. Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef]
- Sample Availability: Not Avaialble.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kresty, L.A.; Howell, A.B.; Baird, M. Cranberry Proanthocyanidins Mediate Growth Arrest of Lung Cancer Cells through Modulation of Gene Expression and Rapid Induction of Apoptosis. Molecules 2011, 16, 2375-2390. https://doi.org/10.3390/molecules16032375
Kresty LA, Howell AB, Baird M. Cranberry Proanthocyanidins Mediate Growth Arrest of Lung Cancer Cells through Modulation of Gene Expression and Rapid Induction of Apoptosis. Molecules. 2011; 16(3):2375-2390. https://doi.org/10.3390/molecules16032375
Chicago/Turabian StyleKresty, Laura A., Amy B. Howell, and Maureen Baird. 2011. "Cranberry Proanthocyanidins Mediate Growth Arrest of Lung Cancer Cells through Modulation of Gene Expression and Rapid Induction of Apoptosis" Molecules 16, no. 3: 2375-2390. https://doi.org/10.3390/molecules16032375
APA StyleKresty, L. A., Howell, A. B., & Baird, M. (2011). Cranberry Proanthocyanidins Mediate Growth Arrest of Lung Cancer Cells through Modulation of Gene Expression and Rapid Induction of Apoptosis. Molecules, 16(3), 2375-2390. https://doi.org/10.3390/molecules16032375