Abstract
The anticipated worldwide increase in biodiesel production will result in an accumulation of glycerol for which there are insufficient conventional uses. The surplus of this by-product has increased rapidly during the last decade, prompting a search for new glycerol applications. We describe here the synthesis of dissymmetric chlorohydrin esters from symmetric 1,3-dichloro-2-propyl esters obtained from glycerol. We studied the influence of two solvents: 1,4-dioxane and 1-butanol and two bases: sodium carbonate and 1-butylimidazole, on the synthesis of dissymmetric chlorohydrin esters. In addition, we studied the influence of other bases (potassium and lithium carbonates) in the reaction using 1,4-dioxane as the solvent. The highest yield was obtained using 1,4-dioxane and sodium carbonate.
1. Introduction
Glycerol (1,2,3-propanetriol), is one of the most versatile and valuable chemicals known to man. Glycerol has over 1,500 known end uses, including applications as an ingredient or processing aid in cosmetics, toiletries, personal care products, pharmaceutical formulations and food [1]. Traditionally, it is obtained as a by-product in four different processes: soap manufacture (saponification), fatty acid production (hydrolysis), fatty ester production (transesterification) and microbial fermentation. It can also be synthesized from propylene oxide [2].
Production of glycerol as a by-product has increased rapidly during the last decade as a consequence of biodiesel production [3,4]. Biodiesel is a common term for the different alkyl fatty esters formed by transesterification of vegetable oils or fats with an alcohol, usually methanol, with or without a catalyst. It has been implemented as an alternative to transportation fuels in Europe, the United States, Brazil and other countries as a result of increased interest in using renewable energy sources to reduce greenhouse gas emissions and to alleviate the depletion of fossil fuel reserves [5,6].
In the biodiesel process, glycerol is usually generated at the rate of one mole of glycerol for every three moles of synthesized methyl esters; approximately 10% of the total product by mass. Thus, the increase in biodiesel production will result in an accumulation of glycerol for which there is currently insufficient conventional uses. This situation has created a glut in the glycerol market and a drop in its price.
Crude glycerol generated during the transesterification process leading to biodiesel contains impurities such as methanol, water, inorganic salts (catalyst residue), free fatty acids, unreacted mono-, di- and triglycerides, methyl esters and a variety of other organic materials, depending on the biodiesel process [7,8], hence it must be purified before subsequent use in conventional applications.
All of the factors described above have prompted a search for new glycerol applications. Recent reviews have reported the utilization of glycerol to produce different value-added chemicals [2,3,4,9,10]. As a way to revalorize glycerol, our group has adopted new approaches based on the transformation of this polyol into halohydrin esters [11,12,13]. These products can be used as building blocks for the synthesis of a range of biologically active natural and synthetic products [14,15,16,17,18,19,20,21,22,23,24,25,26,27].
In this paper, we report the results obtained when two solvents (1,4-dioxane and 1-butanol) and different bases are used to prepare dissymmetric chlorohydrin esters from symmetric 1,3-dichloro-2-propyl esters obtained from glycerol.
2. Results and Discussion
Initially, we studied the influence of combining two solvents and two bases in the synthesis of dissymmetric chlorohydrin esters. Thus, several 1,3-dichloro-2-propyl esters 1a–d were treated with water and an organic (1-butylimidazole) or inorganic (sodium carbonate) base in two different solvents: 1,4-dioxane or 1-butanol (Table 1). Most of the esters used were α,α-dimethyl-substituted (versatic) esters. Yields of the reactions carried out with sodium carbonate and 1,4-dioxane were usually higher than the corresponding reactions with sodium carbonate and 1-butanol, except in the reaction with 2-chloro-1-(chloromethyl)ethyl 2,2-dimethylpropionate (1b), where the yield was the same. A similar tendency was observed using 1-butylimidazole as the base, but this time the exception was in the reaction with 2-chloro-1-(chloromethyl)ethyl 2-methyl-2-phenylpropionate (1d).

Table 1.
Synthesis of 3-chloro-2-hydroxy-1-propyl esters using various solvents and bases.
In the reactions with 2-chloro-1-(chloromethyl)ethyl hexadecanoate (1a), the best solvent was 1,4-dioxane. There was no difference between the use of either sodium carbonate or 1-butylimidazole. The product, 2a, was not observed when 1-butanol was used. Probably, because this not an α,α-dimethyl-substituted ester, and it is not stable in such a solvent.
In the reactions with 2-chloro-1-(chloromethyl)ethyl 2,2-dimethylpropionate (1b), both solvents (1,4-dioxane and 1-butanol) with sodium carbonate and 1,4-dioxane with 1-butylimidazole produced the highest yields.
Finally, the combination 1,4-dioxane/sodium carbonate and 1-butanol/1-butylimidazole gave the highest yields with the other compounds (2c–d) when the bases were compared in the same solvent. These results can be explained by considering the high stability of the hydrolytic processes of the α,α-dimethyl-substituted esters.
We propose the mechanism shown in Scheme 1 for the formation of 3-chloro-2-hydroxy-1-propyl esters. The transposition process observed here has already been described by our group in the preparation of glycidyl palmitate [13]. This mechanism is compatible with the experimental results. Similar behaviour was described by Leggetter and Brown studying the influence of substituents on the opening of substituted 1,3-dioxolanes [28,29]. Therefore, the preferred cleavage of C2-O3 bond might be explained by electron-withdrawing substituents, which should produce on C4 a higher positive charge density than on C5. This will provoke the formation of the indicated chlorohydrin ester instead of the other putative regioisomer.
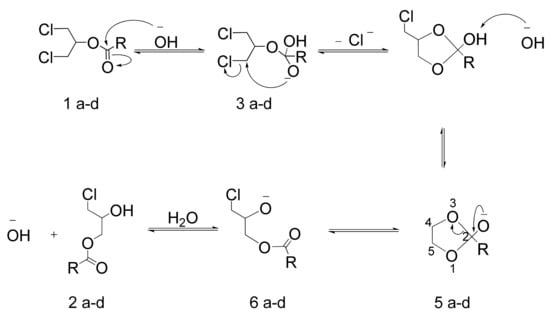
Scheme 1.
Mechanism proposed to explain the formation of 3-chloro-2-hydroxy-1-propyl esters using water and base as reagent.
In addition, we studied the influence of other bases using 1,4-dioxane as the solvent because it had been the best solvent considering all the tested compounds. Our aim was to improve the obtained yields. Two esters were chosen as model compounds. One was the palmitic ester because it does not possess α,α-dimethyl-substitution. The other was α,α-dimethylphenyl acetate. Considering that there was a small difference in the yield of compounds 2a and 2d when organic or inorganic bases were used in 1,4-dioxane, the effect of sodium, potassium and lithium carbonates on 3-chloro-2-hydroxy-1-propyl ester formation was checked.
Satisfactory yields were obtained using a cheap base such as sodium carbonate (Table 2), independent of the acyl compound used. When the effect of each base on the transformation of dichloro esters 1a and 1d was studied, the percentages of 3-chloro-2-hydroxy-1-propyl esters 2a and 2d were always higher in the transformation of 2-chloro-1-(chloromethyl)ethyl hexadecanoate (entry a) than in the transformation of phenyl acetate derivatives, irrespective of the base used.
Table 2.
Influence of different inorganic bases on 3-chloro-2-hydroxy-1-propyl ester formation in 1,4-dioxane.
The yield differences with the inorganic bases may be explained by differences in water solubility of these salts. Sodium and potassium carbonate solubility increases with increasing temperature and therefore the availability of hydroxyl groups which act as nucleophile. On the other hand, potassium cations are more basic than sodium cations. Yields in the reactions with potassium carbonate are lower because 3-chloro-2-hydroxy-1-propyl ester was transformed into an epoxide (result not shown). These compounds were not observed when sodium carbonate was used. Lithium carbonate has behaves differently because its solubility decreases with increasing temperature.
Finally, a multigram-scale synthesis of 3-chloro-2-hydroxy-1-propyl 2,2-dimethylpropanoate 2b was carried out, considering that the best yields were always obtained using the 2,2-dimethylpropanoate ester 1b. 1,4-Dioxane and sodium carbonate were used to transform 5.05 g of 1b into compound 2b. A yield of 2.9 g of 2b (62% yield) was obtained after purification by distillation, showing that the method is fully applicable to obtain gram quantities of dissymmetric chlorohydrins from the corresponding symmetric esters.
3. Experimental
3.1. General
NMR (400/100 MHz) spectra were recorded on a Varian 400 spectrometer using CDCl3 as solvent, as indicated. Chemical shifts are reported in ppm (δ) relative to the TMS signal. High-resolution mass spectral (HRMS) data were obtained by direct infusion on Agilent G6510AA Q-TOF MS using electrospray ionization source (ESI). IR spectra were recorded on a Magna IR 560 Nicolet FTIR spectrophotometer in the range 4000–600 cm−1 with KBr pellets. Spectra dates are reported in reciprocal centimetres (cm−1). Palmitic acid (98%), 2,2-dimethylbutyric acid (96%), trimethylacetic acid (99%) and butylimidazole (98%) were purchased from Aldrich. Potassium carbonate (99%) and lithium carbonate (98%) were obtained from Fluka. α,α-Dimethylphenylacetic acid was purchased from Alfa Aesar. Sodium carbonate (98%) was obtained from Panreac. Solvents and reagents were dried using conventional methods prior to use.
3.2. Procedure for the syntheses of 2-chloro-1-(chloromethyl)ethyl esters 1
Carboxylic acid (1 mmol), glycerol (184 mg, 2 mmol) and chlorotrimethylsilane (540 mg, 5 mmol) were added to a reaction vial fitted with a PTFE-lined cap. The mixture was heated at 80 °C for 48 h. After cooling, an organic solvent was added, and the mixture was washed three times with water. The organic layer was dried over anhydrous MgSO4, and the solvent was evaporated under vacuum. The residue was purified by dry flash column chromatography on silica gel.
2-Chloro-1-(chloromethyl)ethyl hexadecanoate (1a; CAS: 72165-62-9). 1H-NMR δ: 0.88 (t, J = 6.4 Hz, 3 H), 1.20–1.40 (m, 24 H) 1.60–1.75 (m, 2 H), 2.37 (t, J = 7.5 Hz, 2 H) 3.72 (dd, J = 11.6, 5.1 Hz, 2 H) 3.77 (dd, J = 11.6, 5.3 Hz, 2 H) 5.18, (tt, J = 5.1, 5.3 Hz, 1 H). 13C-NMR δ: 14.1, 22.7, 24.8, 29.0, 29.2, 29.3, 29.4, 29.6, 29.7, 31.9, 34.1, 42.4, 71.4,172.7. MS m/z: 368 (M+1)+, 366 (M-1) +, 239, 43 [12,30].
2-Chloro-1-(chloromethyl)ethyl 2,2-dimethylpropanoate (1b; CAS: 220499-01-4). 1H-NMR δ: 5.21 (m, 1H, CH), 3.83 (m, 4H, CH2-Cl), 1.24 (s, 9H, 3CH3). 13C-NMR δ: 177.3 (C=O), 43.0 (CH2-Cl), 39.0 ((CH3)2-C), 27.0 (3CH3) [12,31].
2-Chloro-1-(chloromethyl)ethyl 2,2-dimethylbutanoate (1c). 1H-NMR δ: 5.14 (quin, J = 5.2 Hz, 2H, O-CH2), 3.72 (dd, J1 = 5.2 Hz, J2 = 2.3 Hz, 4H, 2 CH2-Cl), 1.59 (q, J = 7.5 Hz, 2H, CH2-CH3), 1.17 (s, 6H, (CH3)2-C), 0.85 (t, J = 7.5 Hz, 3H, CH2-CH3). 13C-NMR δ: 176.8 (C=O), 71.4 (O-CH), 42.9 ((CH3)2-C), 42.5 (CH2-Cl), 33.1 (CH2-CH3), 24.5 ((CH3)2-C), 9.2 (CH2-CH3). IR (KBr) νmax: 2967.4, 2937.2, 2876.8, 1729.0, 1237.1, 1137.9 cm−1. HRMS (ESI+) [M+Na]+ C9H16Cl2O2Na calculated: 249.0425, found: 249.0518.
2-Chloro-1-(chloromethyl)ethyl-2-methyl-2-phenylpropanoate (1d). 1H-NMR δ: 7.46–7.21, m, 5H, CHar), 5.16 (quin, J = 5.3 Hz, 2H, O-CH2), 3.73–3.59 (m, 4H, 2 CH2-Cl), 1.63 (s, 6H, (CH3)2-C). 13C- NMR δ: 175.6 (C=O), 143.8 (Car), 128.4, 126.9, 125.7 (CHar), 72.0 (O-CH), 46.7 ((CH3)2-C), 42.3 (CH2-Cl), 26.3 ((CH3)2-C). IR (KBr) νmax: 3083.9, 3053.7, 3023.5, 2976.1, 2928.6, 2872.5, 1729.0, 1241.4, 1133.5 cm−1. HRMS (ESI+) [M+Na]+ C13H16Cl2O2Na calculated: 297.0425, found: 297.0531.
3.3. Procedure for the syntheses of 3-chloro-2-hydroxy-1-propyl esters 2
A solution of 2-chloro-1-(chloromethyl)ethyl ester (1 mmol), water (29 μL, 1.6 mmol) and either sodium carbonate (106 mg, 1 mmol) or 1-butylimidazole (124 mg, 1 mmol) in dried solvent (1,4-dioxane or 1-butanol (3 mL)) was heated at 115 °C for 48 h in a capped reaction vial. After cooling, dichloromethane was added, and the mixture was washed three times with water. The organic layer was dried over anhydrous MgSO4, and the solvent was evaporated. The crude compound was analyzed by 1H-NMR to determine the yield of the corresponding product using N,N-dimethylformamide (DMF) as an internal standard. Further purification by dry flash column chromatography on silica gel (hexane/ethyl acetate) gave the desired product.
3-Chloro-2-hydroxy-1-propyl hexadecanoate (2a; CAS: 30557-04-1). 1H-NMR δ: 0.90 (t, J = 6.3 Hz, 3H, CH3), 1.20–1.40 (m, 24H, (CH2)12), 1.66 (m, 2H, CH2-CH2COO), 2.30 (t, J = 7.5 Hz, 2H, CH2-COO), 3.59 and 3.65 (dd, J = 8.5 Hz, J = 5.2 Hz, 2H, CH2-Cl), 4.00 (quin, J = 5.2 Hz, 1H, CH-OH), 4.15 (dd, J = 5.2 Hz, J = 1.7 Hz, 2H, O-CH2-CH). 13C-NMR δ: 177 (C=O), 70 (CH-OH), 66 (O-CH2), 45 (CH2-Cl), 34 (CH2-COO), 32–22 (13C, CH2), 17 (CH3). GC-MS m/z: 348 [M]+, 299 [M-CH2Cl]+, 269 [M-C2H2ClO]+, 255 [M-C3H6ClO]+, 239 [M-C3H6ClO2]+, 152 [C5H9O3Cl]+ (McLafferty rearrangement) [13].
3-Chloro-2-hydroxy-1-propyl 2,2-dimethylpropanoate (2b; CAS: 52562-21-7). 1H-NMR δ: 4.05 (dd, J1 = 5.68 Hz, J2 = 1.26 Hz, 2H, O-CH2), 3.90 (m, 1H, CH-OH), 3.45 (m, 2H, CH2-Cl), 1.04 (s, 9H, 3CH3). 13C-NMR δ: 179.5 (C=O), 70 (CH-OH), 65.5 (O-CH2), 46 (CH2-Cl), 39 ((CH3)2-C), 27.3 (3CH3). IR ATR νmax.: 3437.5, 2953, 1718, 1500, 1296, 1156, 1046, 765, 703, 640 cm−1. HRMS (ESI+) [M+Na]+ C8H15ClO3Na calculated: 217.0607, found: 217.0701.
3-Chloro-2-hydroxy-1-propyl 2,2-dimethylbutanoate (2c). 1H-NMR δ: 4.20 (dd, J1 = 5.9 Hz, J2 = 1.2 Hz, 2H, O-CH2), 4.05 (quin, J = 5.3 Hz, 1H, CH-OH), 3.65–3.54 (m, 2H, CH2-Cl), 1.56 (q, J = 7.5 Hz, 2H, CH2-CH3), 1.16 (s, 6H, (CH3)2-C), 0.83 (t, J = 7.5 Hz, 3H, CH2-CH3). 13C-NMR δ: 178.1 (C=O), 69.8 (CH-OH), 65.0 (O-CH2), 45.9 (CH2-Cl), 42.8 ((CH3)2-C), 33.3 (CH2-CH3), 24.6 ((CH3)2-C), 9.2 (CH2-CH3). IR (KBr) νmax: 3480.9, 2967.4, 2937.2, 2876.8, 1724.7, 1241.4, 1150.8 cm−1. HRMS (ESI+) [M+Na]+ C9H17ClO3Na calculated: 231.0764, found: 231.0860.
3-Chloro-2-hydroxy-1-propyl 2-methyl-2-phenylpropanoate (2d). 1H-NMR δ: 7.39–7.23 (m, 5H, CHar), 4.21 (dd, J1 = 5.2 Hz, J2 = 2.0 Hz, 2H, O-CH2), 3.97 (quin, J = 5.3 Hz, 1H, CH-OH), 3.46–3.36 (m, 2H, CH2-Cl), 1.62 (s, 6H, (CH3)2-C). 13C-NMR δ: 176.8 (C=O) 144.2 (Car), 128.5, 126.9, 125.5 (CHar), 69.5 (CH-OH), 65.2 (O-CH2), 46.6 ((CH3)2-C), 45.7 (CH2-Cl), 26.3 ((CH3)2-C). IR (KBr) νmax: 3480.9, 3083.9, 3053.7, 3019.2, 2971.7, 2924.3, 1729.0, 1245.7, 1142.2 cm−1. HRMS (ESI+) [M+Na]+ C13H17ClO3Na calculated: 279.0764, found: 279.0873.
3.4. Procedure for the syntheses of 3-chloro-2-hydroxy-1-propyl esters 2a and 2d using different inorganic bases
A solution of 2-chloro-1-(chloromethyl)ethyl ester (1 mmol), water (29 μL, 1.6 mmol) and a carbonate (1 mmol) in dried 1,4-dioxane (3 mL) was heated at 115 °C for 48 h in a capped reaction vial. After cooling, dichloromethane was added and the mixture was washed three times with water. The organic layer was dried over anhydrous MgSO4, and the solvent was distilled. The crude compound was analyzed by 1H-NMR to determine the yield of the corresponding product using DMF as an internal standard. Further purification by dry flash column chromatography on silica gel (hexane/ethyl acetate) gave the desired product.
3.5. Procedure for the multigram-scale syntheses of 3-chloro-2-hydroxy-1-propyl 2,2-dimethylpropanoate (2b)
A solution of 2-chloro-1-(chloromethyl)ethyl 2,2-dimethylpropanoate (5.08 g, 24 mmol), water (0.7 mL, 38.4 mmol) and sodium carbonate (2.54 g, 24 mmol) in dried 1,4-dioxane (72 mL) was heated at 115 °C for 48 h in a capped reactor. After cooling, dichloromethane was added and the mixture was washed three times with water. The organic layer was dried over anhydrous MgSO4, and the mixture was distilled obtaining 2.9 g of the desired compound (62% yield) b.p.: 70 °C/ 40 Pa. The compound was identified by 1H-NMR and 13C-NMR.
4. Conclusions
1,3-Dichloro-2-propyl esters prepared from glycerol can be transformed into the corresponding 3-chloro-2-hydroxy-1-propyl esters. These dissymmetric chlorohydrin esters are potentially valuable compounds and can be used as building blocks. With the methodology described above, different 3-chloro-2-hydroxy-1-propyl esters were synthesized with all studied compounds; better reaction conditions were obtained when using sodium carbonate as base and 1,4-dioxane as solvent. In addition, when different inorganic carbonates were used, sodium carbonate gave the best results. The best procedure developed allows to prepare 3-chloro-2-hydroxy-1-propyl 2,2-dimethylpropanoate on a multigram-scale.
Acknowledgements
This work was supported by a Grant-in-Aid from the Secretaría de Estado de Política Cientifíca y Tecnológica of the Spanish Ministry of Education and Culture (contract Grant No.: CTQ2009-14699-C02-01). The authors are grateful to the Comissionat per a Universitats i Recerca del Departament d’Innovació, Universitats i Empresa de la Generalitat de Catalunya and to the European social Fund (EFS) for the FI grant of Carmen Solarte Orozco.
References and Notes
- Pagliaro, M.; Rossi, M. The Future of Glycerol: New Uses of a versatile Raw Material; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Zhou, C.H.; Beltramini, J.N.; Fan, Y.X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- Johnson, D.T.; Taconi, K.A. The glycerin glut: Options for the value-added conversion of crude glycerol resulting from biodiesel production. Environ. Prog. 2007, 26, 338–348. [Google Scholar] [CrossRef]
- Meher, L.C.; Vidya Sagar, D.; Naik, S.N. Technical aspects of biodiesel production by transesterification--a review. Renew. Sustain. Energy Rev. 2006, 10, 248–268. [Google Scholar] [CrossRef]
- Janaun, J.; Ellis, N. Perspectives on biodiesel as a sustainable fuel. Renew. Sustain. Energy Rev. 2010, 14, 1312–1320. [Google Scholar] [CrossRef]
- Chiu, C.-W.; Goff, M.J.; Suppes, G.J. Distribution of methanol and catalysts between biodiesel and glycerin phases. AIChE J. 2005, 51, 1274–1278. [Google Scholar] [CrossRef]
- Yori, J.C.; D’Ippolito, S.A.; Pieck, C.L.; Vera, C.R. Deglycerolization of Biodiesel Streams by Adsorption Over Silica Beds. Energ. Fuel. 2006, 21, 347–353. [Google Scholar] [CrossRef]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. From Glycerol to Value-Added Products. Angew. Chem. Inter. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Pina, C.D. Recent advances in the conversion of bioglycerol into value-added products. Eur. J. Lipid Sci. Technol. 2009, 111, 788–799. [Google Scholar] [CrossRef]
- Villorbina, G.; Tomàs, A.; Escribà, M.; Oromí-Farrús, M.; Eras, J.; Balcells, M.; Canela, R. Combining AlCl3·6H2O and an ionic liquid to prepare chlorohydrin esters from glycerol. Tetrahedron Lett. 2009, 50, 2828–2830. [Google Scholar] [CrossRef]
- Escribà, M.; Eras, J.; Duran, M.; Simon, S.; Butchosa, C.; Villorbina, G.; Balcells, M.; Canela, R. From glycerol to chlorohydrin esters using a solvent-free system. Microwave irradiation versus conventional heating. Tetrahedron 2009, 65, 10370–10376. [Google Scholar] [CrossRef]
- Eras, J.; Balcells, M.; Canela, R. Glycerol as a starting material to prepare palmitate derivatives. Afinidad 2007, 64, 203–206. [Google Scholar]
- Beger, J.; Jacobi, R.; Rehbeil, U.; Knoll, E. Mehrfunktionelle N-Tenside. XIV: Synthesen und kritische Mizellbildungskonzentrationen von quartären Ammoniumsalzen mit Carbonsäurederivatfunktionen (Multifunctional N-surfactants. XIV: Synthesis and micellar critical concentration of quaternary ammonium salts with carboxylic acid derivatives functions). Tenside, Surfactants, Deterg. 1992, 29, 328–332. [Google Scholar]
- Hamaguchi, S.; Ohashi, T.; Watanabe, K. Lipase-catalyzed stereoselective hydrolysis of 2-acyloxy-3-chloropropyl-p-toluenesulfonate. Agric. Biol. Chem. 1986, 50, 375–380. [Google Scholar]
- Junk, T.; Pappalardo, G.C.; Irgolic, K.J. Synthesis and characterization of rac-1,2-bis(palmitoyloxy)-3-propyl (2-trimethylarsonioethyl)phosphonate, an arsenic-containing phosphonolipid. Appl. Organometal. Chem. 1990, 4, 103–109. [Google Scholar] [CrossRef]
- Kapoor, M.; Anand, N.; Ahmad, K.; Koul, S.; Chimni, S.S.; Taneja, S.C.; Qazi, G.N. Synthesis of β-adrenergic blockers (R)-(-)-nifenalol and (S)-(+)-sotalol via a highly efficient resolution of a bromohydrin precursor. Tetrahedron Asymmetry 2005, 16, 717–725. [Google Scholar] [CrossRef]
- Kolb, H.C.; Bennani, Y.L.; Sharpless, K.B. Short and practical synthess of (R)-(-)-carnitine and (R)-(-)-γ-amino-β-hydroxybutyric acid (GABOB). Tetrahedron Asymmetry 1993, 4, 133–141. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. A simplified procedure for the stereospecific transformation of 1,2-diols into epoxides. Tetrahedron 1992, 48, 10515–10530. [Google Scholar] [CrossRef]
- Marzi, M.; Minetti, P.; Moretti, G.; Tinti, M.O.; De Angelis, F. Efficient enantioselective synthesis of (R)-(-)-carnitine from glycerol. J. Org. Chem. 2000, 65, 6766–6769. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, K. Positive-charging electrophotographic toner using bisphenol A-free linear polyester binder. JP2005326606, 24 November 2005. [Google Scholar]
- Moschidis, M.C. Synthesis of 1,2-diacyloxypropyl-3-(1′,2′-diacyl-sn-glycero)-phosphonate. Chem. Phys. Lipids 1985, 36, 297–302. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Papahatjis, D.P.; Claremon, D.A.; Magolda, R.L.; Dolle, R.E. Total synthesis of ionophore antibiotic X-14547A. J. Org. Chem. 1985, 50, 1440–1456. [Google Scholar] [CrossRef]
- Schmitt, J.D.; Nixon, A.B.; Emilsson, A.; Daniel, L.W.; Wykle, R.L. A facile synthesis of 1-O-alkyl-2-(R)-hydroxypropane-3-phosphonocholine (lyso-phosphono-platelet activating factor). Chem. Phys. Lipids 1992, 62, 263–268. [Google Scholar] [CrossRef]
- Watson, H.G.; Fung, Y.M.; Gredley, M.; Bird, G.J.; Jackson, W.R.; Gountzos, H.; Matthews, B.R. Asymmetric syntheses of (+)-diltiazem hydrochloride. J. Chem. Soc. Ser. Chem. Commun. 1990, 1018–1019. [Google Scholar] [CrossRef]
- Wilen, S.H.; Delguzzo, L.; Saferstein, R. Experimental evidence for AcO-7 neighboring group participation. Tetrahedron 1987, 43, 5089–5094. [Google Scholar] [CrossRef]
- Williams, J.R.; Boehm, J.C. The syntheses of 3[beta]-steroidal diacylglyceryl sulfides, sulfoxides, and sulfones. Steroids 1995, 60, 321–323. [Google Scholar] [CrossRef]
- Leggetter, B.E.; Brown, R.K. The influence of substituents on the ease and direction of ring opening in the LiAlH4-AlCl3 reductive cleavage of substituted 1,3-dioxolanes. Can. J. Chem. 1964, 42, 990–1004. [Google Scholar] [CrossRef]
- Leggetter, B.E.; Brown, R.K. Anomalous ring cleavage during lithium aluminum hydride - aluminum chloride reduction of 2,2,4,4-tetraalkylated-1,3-dioxolanes. Can. J. Chem. 1964, 42, 1005–1008. [Google Scholar] [CrossRef]
- Eras, J.; Mendez, J.J.; Balcells, M.; Canela, R. Chlorotrimethylsilane: A Suitable Reagent for the Synthesis of Chlorohydrin Esters. J. Org. Chem. 2002, 67, 8631–8634. [Google Scholar] [CrossRef] [PubMed]
- Mormann, W.; Demeter, J.; Wagner, T. Selectivity in the acylation of partially silylated hydroxy polymers - a study with trimethylsilyl cellulose and model compounds. Acta Polym. 1999, 50, 20–27. [Google Scholar] [CrossRef]
Sample Availability: Samples are available from the authors. |


© 2011 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).