Structural and Molecular Characterization of meso-Substituted Zinc Porphyrins: A DFT Supported Study

Abstract
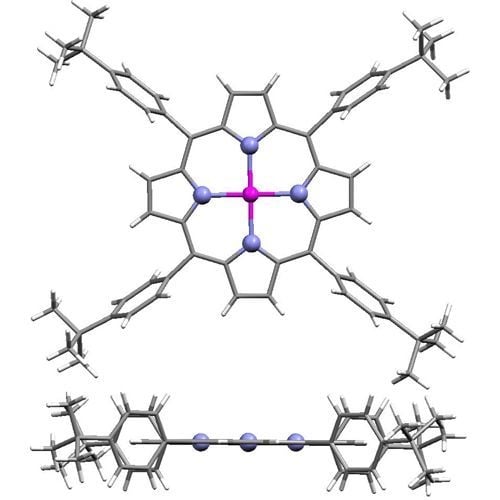
:
1. Introduction
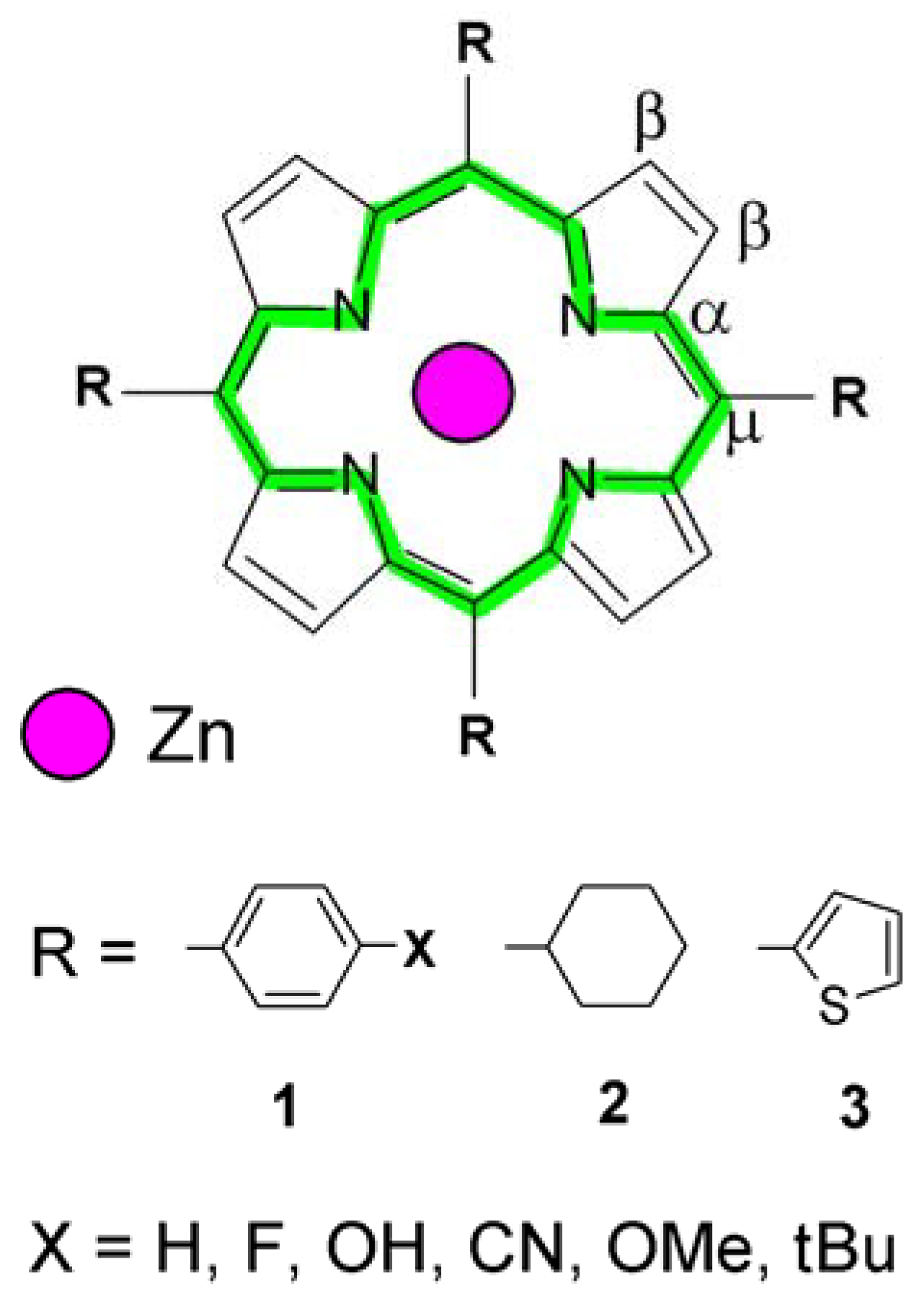
2. Results and Discussion
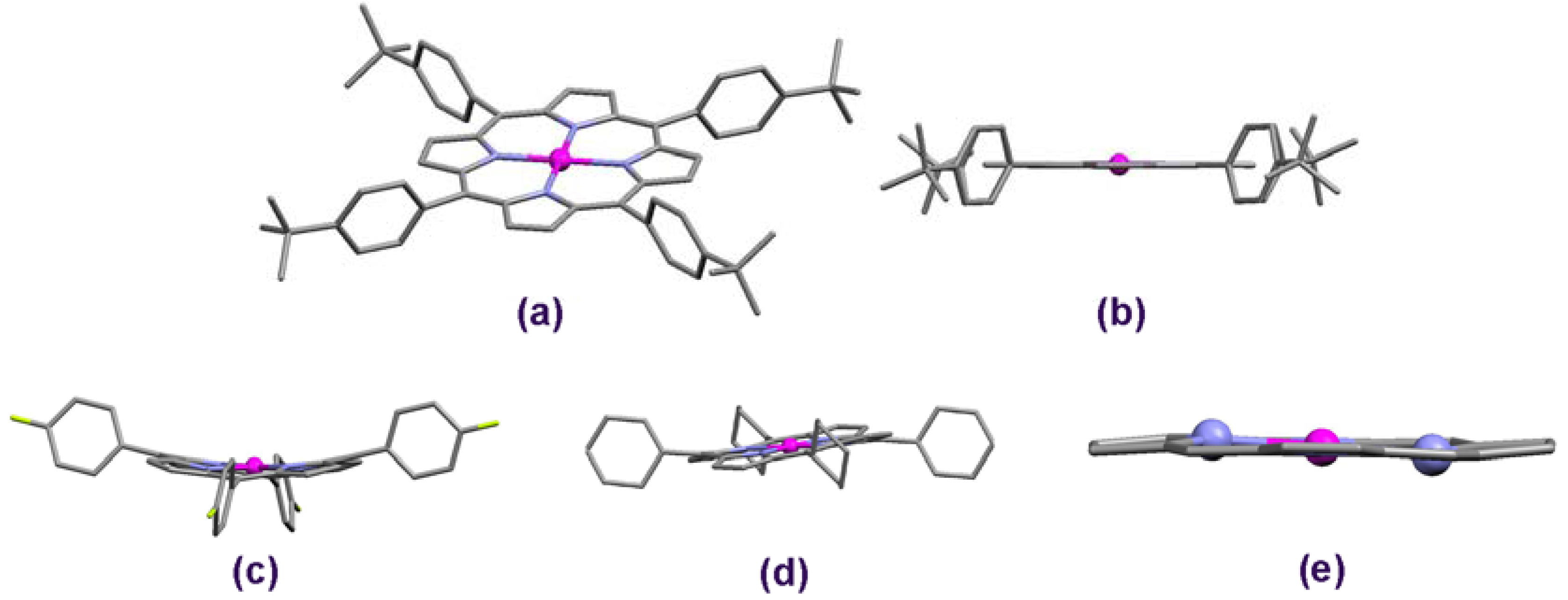
2.1. Molecular Effects in Real Crystals and DFT-Derived Zn-Porphyrins

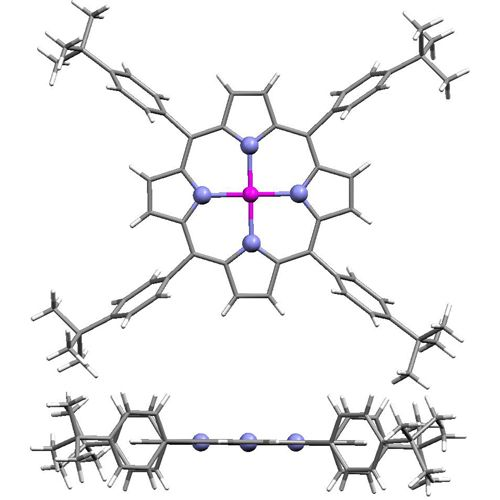{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porphyrin type | Zn-N | Cα-N | Cα-Cβ | Cβ-Cβ | Cα-Cμ | |
|---|---|---|---|---|---|---|
| ZnTBPP (crystal) | [7] | 2.04 | 1.38 | 1.44 | 1.36 | 1.40 |
| ZnTBPP (DFT) | 2.07 | 1.40 | 1.46 | 1.37 | 1.42 | |
| R = 1 (crystal) | [16,17] | 2.04 | 1.38 | 1.44 | 1.35 | 1.40 |
| R = 2 (crystal) | [10,21] | 2.03 | 1.38 | 1.44 | 1.35 | 1.40 |
| R = 3 (crystal) | [11] | 2.02 | 1.36 | 1.43 | 1.33 | 1.38 |
| Porphyrin type | References | Core perimeter (exp.) | Core perimeter (DFT) | Cμ-R distance (exp.) | Cμ-R distance (DFT) |
|---|---|---|---|---|---|
| R = H | -1) | 22.37 | -1) | 1.09 | |
| R = 1, X = H | [22] | 22.22 | 22.50 | 1.50 | 1.50 |
| R = 1, X = F | [23] | 22.14 | 22.49 | 1.48 | 1.50 |
| R = 1, X = OH | [24] | 22.24 | 22.50 | 1.50 | 1.50 |
| R = 1, X = CN | [9] | 22.24 | 22.49 | 1.50 | 1.50 |
| R = 1, X = OMe | [25] | 22.28 | 22.50 | 1.51 | 1.50 |
| 2) R = 1, X = tBu | [7] | 22.24 | 22.50 | 1.50 | 1.50 |
| R = 2 | [21] | 22.29 | 22.57 | 1.53 | 1.54 |
| R = 3 | [11] | 21.99 | 22.49 | 1.47 | 1.49 |
2.2. Crystal Surface Structure

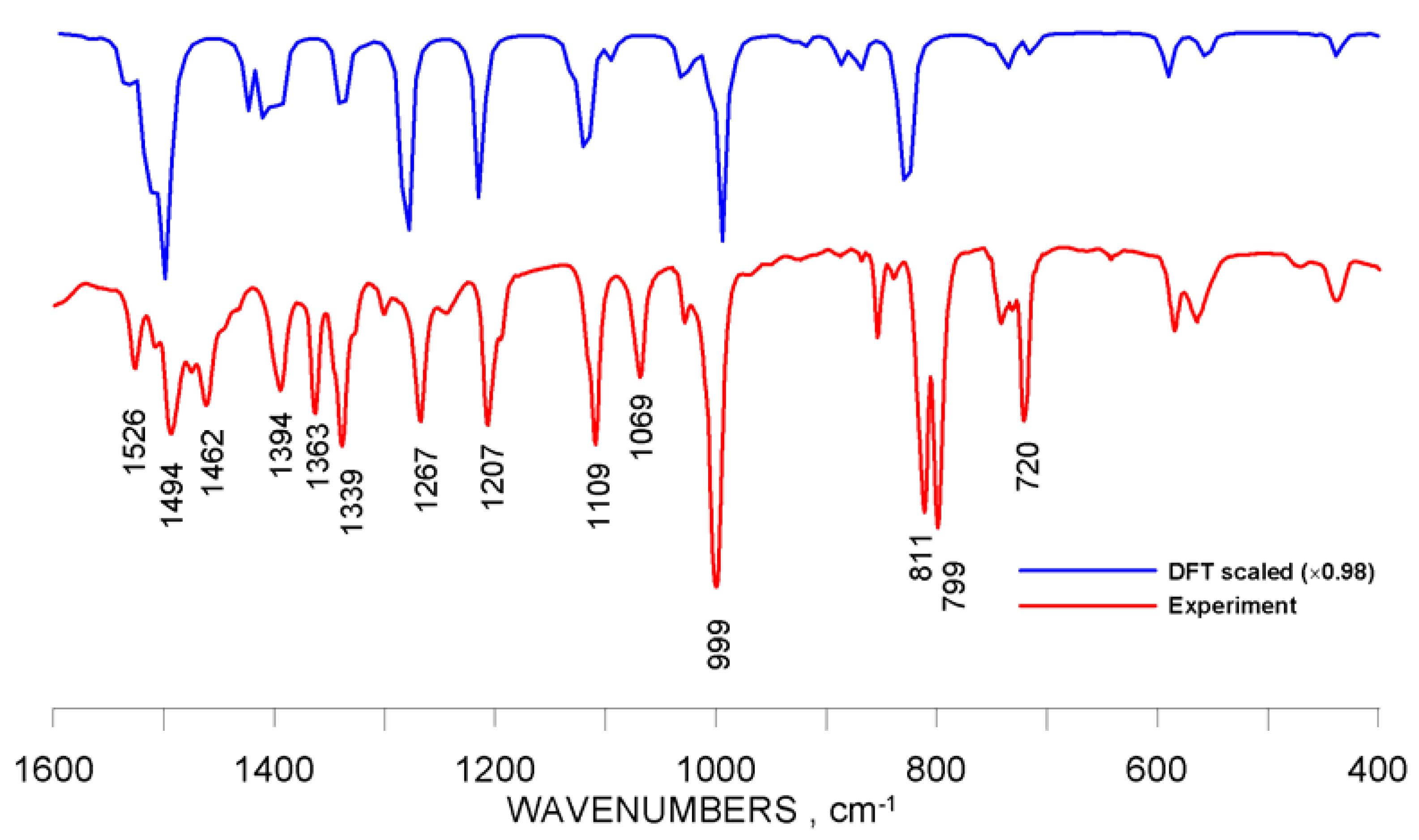
2.3. Infrared Spectra and Structure-Related Effects Based on DFT Calculations
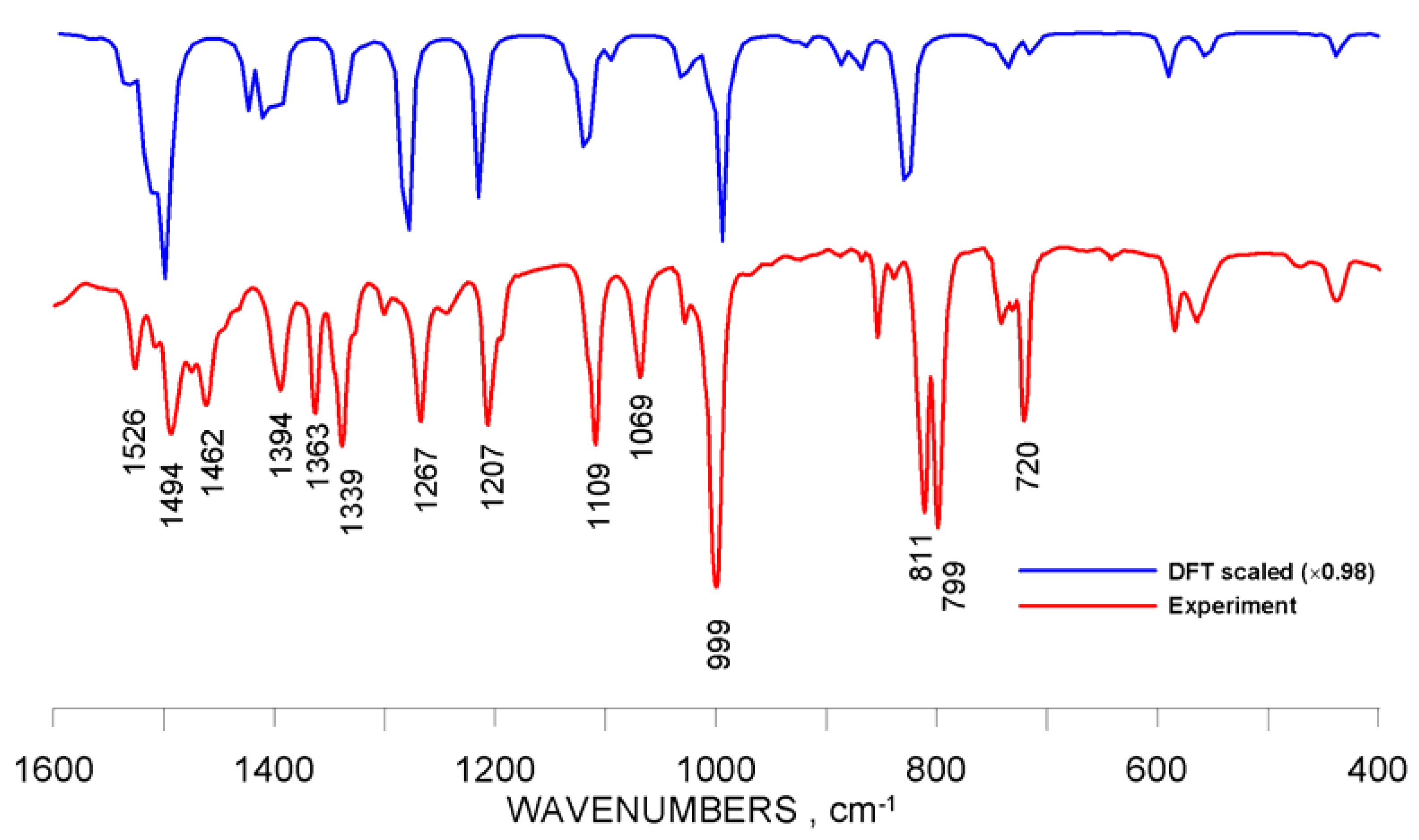
| ν, cm−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Oscillation mode | R = H | R = 1 and X as below | R = 2 | R = 3 | |||||
| H | F | CN | OH | OMe | tBu | ||||
| Cα-Cμ | 1557 | 1565 | 1566 | 1568 | 1565 | 1565 | 1565 | 1575 | 1562 |
| 1539 | 1544 | 1546 | 1547 | 1546 | 1549 | 1533 | 1538 | ||
| 1597 | 1520 | 1527 | 1531 | 1529 | 1529 | 1531 | |||
| Cμ-R | 3204 | 1648 | 1649 | 1655 | 1661 | 1658 | 1658 | 1414 | 1603 |
| 1254 | 1253 | 1254 | 1254 | 1254 | 1258 | 1179 | 1191 | ||

3. Experimental
3.1. Materials
3.2. Computational Methods
3.3. Instrumental Methods
3.3.1. X-ray Powder Diffractometry
3.3.2. Scanning Electron Microscopy (SEM)
3.3.3. Electronic Absorption Spectroscopy
3.3.4. Electronic Emission Spectroscopy (Fluorescence)
3.3.5. FTIR Spectroscopy
3.3.6. NMR Spectroscopy
3.3.7. Thermal Analysis (DSC, DTG)
4. Conclusions
Supplementary Material
Acknowledgements
References and Notes
- Kadish, K.M.; Smith, K.M.; Guilard, R. Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010-2011.
- Mele, G.; Del Sole, R.; Vasapollo, G.; García-López, E.; Palmisano, L.; Li, J.; Słota, R.; Dyrda, G. TiO2-based photocatalysts impregnated with metallo-porphyrins employed for degradation of 4-nitrophenol in aqueous solutions: Role of metal and macrocycle. Res. Chem. Intermed. 2007, 33, 433–448. [Google Scholar] [CrossRef]
- Wei, X.; Du, X.; Chen, D.; Chen, Z. Thermal analysis study of 5,10,15,20-tetrakis (methoxyphenyl) porphyrins and their nickel complexes. Thermochim. Acta 2006, 440, 181–187. [Google Scholar] [CrossRef]
- Lebedeva, N.S.; Yakubov, S.P.; Vyugin, A.I.; Parfenyuk, E.V. Thermodynamics of complex formation of natural iron(III)porphyrins with neutral ligands. Thermochim. Acta 2003, 404, 19–24. [Google Scholar] [CrossRef]
- Mele, G.; Del Sole, R.; Vasapollo, G.; García-López, E.; Palmisano, L.; Schiavello, M.J. Photocatalytic degradation of 4-nitrophenol in aqueous suspension by using polycrystalline TiO2 impregnated with functionalized Cu(II)-porphyrin or Cu(II)-phthalocyanine. J. Catal. 2003, 217, 334–342. [Google Scholar]
- Słota, R.; Dyrda, G.; Szczegot, K.; Mele, G.; Pio, I. Photocatalytic activity of nano and micro crystalline TiO2 hybrid systems involving phthalocyanine or porphyrin sensitizers. Photochem. Photobiol. Sci. 2011, 10, 361–366. [Google Scholar] [CrossRef]
- Słota, R.; Mele, G.; Ejsmont, K.; Domański, A.A.; Del Sole, R. . [5,10,15,20-Tetrakis(4-tert-butylphenyl)porphyrinato-ĸ4N]zinc(II) toluene solvate. Acta Cryst. E 2007, 63, m2582. [Google Scholar]
- Man, D.; Słota, R.; Broda, M.A.; Mele, G.; Li, J. Metalloporphyrin intercalation in liposome membranes: ESR study. J. Biol. Inorg. Chem. 2011, 16, 173–181. [Google Scholar] [CrossRef]
- Kumar, R.K.; Balasubramanian, S.; Goldberg, I. Porphyrin sponges: Structural systematics of the host lattice. Inorg. Chem. 1998, 37, 541–552. [Google Scholar] [CrossRef]
- Veyrat, M.; Ramasseul, R.; Turowska-Tyrk, I.; Scheidt, W.R.; Autret, M.; Kadish, K.M.; Marchon, J.-C. Nickel(II) and zinc(II) meso-tetracyclohexylporphyrins. Structural and electronic effects induced by meso-cyclohexyl substitution in metalloporphyrins. Inorg. Chem. 1999, 38, 1772–1779. [Google Scholar]
- Purushothaman, B.; Varghese, B.; Bhyrappa, P. [5,10,15,20-Tetrakis(2-thienyl)-porphyrinato] zinc(II)]. Acta Cryst. C 2001, 57, 252–253. [Google Scholar] [CrossRef]
- Yao, P.; Han, S.; Zhang, Y.; Zhang, X.; Jiang, J. Structures and spectroscopic properties of meso-tetrasubstituted porphyrin complexes: meso-Substitutional and central metallic effect study based on density functional theory calculations. Vib. Spectrosc. 2009, 50, 169–177. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Zhao, W.; Jiang, P.; Zhang, L.-J.; Zhang, T.; Wang, J. Structural parameters and vibrational spectra of a series of zinc meso-phenylporphyrins: A DFT and experimental study. Spectrochim. Acta A 2010, 75, 880–890. [Google Scholar] [CrossRef]
- Oliveira de, V.E.; Cimini Correa, C.; Pinheiro, C.B.; Diniz, R.; Oliveira de, L.F.C. Structural and spectroscopy studies of the zinc complex of p-hydroxyphenylporphyrin. J. Mol. Struct. 2011, 995, 125–129. [Google Scholar] [CrossRef]
- Wang, C.; Yang, G.; Li, J.; Mele, G.; Słota, R.; Broda, M.A.; Duan, M.; Vasapollo, G.; Zhang, X.; Zhang, F.X. Novel meso-substituted porphyrins: Synthesis, characterization and photocatalytic activity of their TiO2-based composites. Dyes Pigments 2009, 80, 321–328. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Cryst. B 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Van de Streek, J. Searching the Cambridge Structural Database for the best representative of each unique polymorph. Acta Cryst. B 2006, 62, 567–579. [Google Scholar] [CrossRef]
- Orpen, A.G. Applications of the Cambridge Structural Database to molecular inorganic chemistry. Acta Cryst. B 2002, 58, 398–406. [Google Scholar] [CrossRef]
- Allen, F.H.; Motherwell, W.D.S. Applications of the Cambridge Structural Database in organic chemistry and crystal chemistry. Acta Cryst. B 2002, 58, 407–422. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Veyrat, M.; Ramasseul, R.; Marchon, J.-C.; Turowska-Tyrk, I.; Scheidt, W.R. Nickel(II) and zinc(II) complexes of meso-tetrakis(cyclohexyl)porphyrin: Distinct types of porphyrin distortion in response to steric crowding. New J. Chem. 1995, 19, 1199–1202. [Google Scholar]
- Byrn, M.P.; Curtis, C.J.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Terzis, A.; Strouse, C.E. Porphyrin sponges: Conservation of host structure in over 200 porphyrin-based lattice clathrates. J. Am. Chem. Soc. 1993, 115, 9480–9497. [Google Scholar] [CrossRef]
- Krupitsky, H.; Stein, Z.; Goldberg, I. Structural patterns in clathrates and crystalline complexes of zinc-tetra(4-chlorophenyl)porphyrin and zinc-tetra(4-fluorophenyl)porphyrin. J. Incl. Phenom. Macro. Chem. 1995, 20, 211–232. [Google Scholar] [CrossRef]
- Goldberg, I.; Krupitsky, H.; Stein, Z.; Hsiou, Y.; Strouse, C.E. Supramolecular architectures of functionalized tetraphenylmetalloporphyrins in crystalline solids. Studies of the 4-methoxyphenyl, 4-hydroxyphenyl and 4-chlorophenyl derivatives. Supramol. Chem. 1994, 4, 203–221. [Google Scholar]
- Byrn, M.P.; Curtis, C.J.; Goldberg, I.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Strouse, C.E. Porphyrin sponges: Structural systematics of the host lattice. J. Am. Chem. Soc. 1991, 113, 6549–6557. [Google Scholar] [CrossRef]
- Dastidar, P.; Krupitsky, H.; Stein, Z.; Goldberg, I. Crystal inclusion chemistry of zinc-tetra(4-bromophenyl) porphyrin. J. Incl. Phenom. Macro. Chem. 1996, 24, 241–262. [Google Scholar] [CrossRef]
- Chiaroni, A.; Riche, C.; Bied-Charreton, C.; Dubois, J.C. Structure de la tetra(p-n-octylphenyl)-5,10,15,20 porphine de zinc(II). Acta Cryst. C 1988, 44, 429–432. [Google Scholar] [CrossRef]
- Golder, A.J.; Nolan, K.B.; Povey, D.C.; Milgrom, L.R. The structure of the palladium(II) and zinc(II) complexes of α,β,γ,δ-tetrakis(3,5-di-tert-butyl-4-hydroxyphenyl)porphyrin. Acta Cryst. C 1988, 44, 1916–1921. [Google Scholar] [CrossRef]
- Słota, R.; Dyrda, G.; Hnatejko, Z.; Karolczak, J.; Stryła, Z. Effect of air absorbed oxygen and moisture on the chemical stability of photoexcited phthalocyanines in dimethylformamide. J. Porphyr. Phthalocya. 2006, 10, 43–54. [Google Scholar] [CrossRef]
- Jarzęcki, A.A.; Kozłowski, P.M.; Pulay, P.; Ye, B.-H.; Li, X.-Y. Scaled quantum mechanical and experimental vibrational spectra of magnesium and zinc porphyrins. Spectrochim. Acta A 1997, 53, 1195–1209. [Google Scholar] [CrossRef]
- Andersson, L.A.; Loehr, T.M.; Thompson, R.G.; Strauss, S.H. Influence of symmetry on the vibrational spectra of Zn(TPP), Zn(TPC), and Zn(TPiBC). Inorg. Chem. 1990, 29, 2142–2147. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Bingham, J.R.; Jarzecki, A.A. Theoretical analysis of core size effect in metalloporphyrins. J. Phys. Chem. A 2008, 112, 12781–12788. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Singleton, D.A.; Wang, Z.H. Isotope effects and the nature of enantioselectivity in the shi epoxidation. The Importance of asynchronicity. J. Am. Chem. Soc. 2005, 127, 6679–6685. [Google Scholar] [CrossRef]
- Schneebeli, S.T.; Hall, M.L.; Breslow, R.; Friesner, R. Quantitative DFT modeling of the enantiomeric excess for dioxirane-catalyzed epoxidations. J. Am. Chem. Soc. 2009, 131, 3965–3973. [Google Scholar] [CrossRef]
- Samples Availability: Samples of ZnTBPP are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Słota, R.; Broda, M.A.; Dyrda, G.; Ejsmont, K.; Mele, G. Structural and Molecular Characterization of meso-Substituted Zinc Porphyrins: A DFT Supported Study. Molecules 2011, 16, 9957-9971. https://doi.org/10.3390/molecules16129957
Słota R, Broda MA, Dyrda G, Ejsmont K, Mele G. Structural and Molecular Characterization of meso-Substituted Zinc Porphyrins: A DFT Supported Study. Molecules. 2011; 16(12):9957-9971. https://doi.org/10.3390/molecules16129957
Chicago/Turabian StyleSłota, Rudolf, Małgorzata A. Broda, Gabriela Dyrda, Krzysztof Ejsmont, and Giuseppe Mele. 2011. "Structural and Molecular Characterization of meso-Substituted Zinc Porphyrins: A DFT Supported Study" Molecules 16, no. 12: 9957-9971. https://doi.org/10.3390/molecules16129957
APA StyleSłota, R., Broda, M. A., Dyrda, G., Ejsmont, K., & Mele, G. (2011). Structural and Molecular Characterization of meso-Substituted Zinc Porphyrins: A DFT Supported Study. Molecules, 16(12), 9957-9971. https://doi.org/10.3390/molecules16129957