1. Introduction
Nitrone spin traps such as the most commonly used 5,5-dimethylpyrroline
N-oxide (DMPO) are important reagents for the detection of free radicals by means of ESR spin trapping [
1]. For certain mass spectrometry experiments or to investigate the fidelity of spin trapping, it is helpful to use isotopically labeled spin traps.
The di(trideuteromethyl) analog of DMPO with its mass shift of +6 compared to unlabeled DMPO can be used for dual spin-trapping mass spectrometry experiments. Here, spin traps labeled with stable isotopes (
2H,
15N or
13C) are utilized to simplify the interpretation of mass spectrometry experiments [
2,
3]. With equal amounts of the labeled and unlabeled spin trap present, adducts of the trapped radicals will appear as ion pairs in the mass spectrum (with the mass differences depending on the isotopes incorporated in the spin trap and on the charge state of the ion). This facilitates and clarifies the identification of radical-derived analytes.
The
15N analog can be used to unambiguously determine the susceptibility of a particular spin-trapping experiment to the Forrester-Hepburn artifact in an ESR experiment [
4]. A Forrester-Hepburn artifact is the result of nucleophilic attack of the radical precursor on the spin trap with subsequent oxidation to the identical nitroxide radical as derived from genuine spin trapping. It is difficult to distinguish between nucleophilic attack and free-radical trapping, with normal chemical and biological control experiments being of no use. Timmins
et al. reported a method based on spin traps with different isotopes at the α- or β-positions to the nitrogen of the spin trap [
4]. The substrate is preincubated with a spin trap (first isotope), and then the spin trap labeled with the second isotope is added simultaneously with the initiation of free radical formation. Because the ESR signals are different, the origin of the signal can be determined as artifact or genuine signal. Employing that technique, we were able to identify the DMPO/
•CN radical, supposedly generated by horseradish peroxidase and hydrogen peroxide, as an artifact (unpublished data).
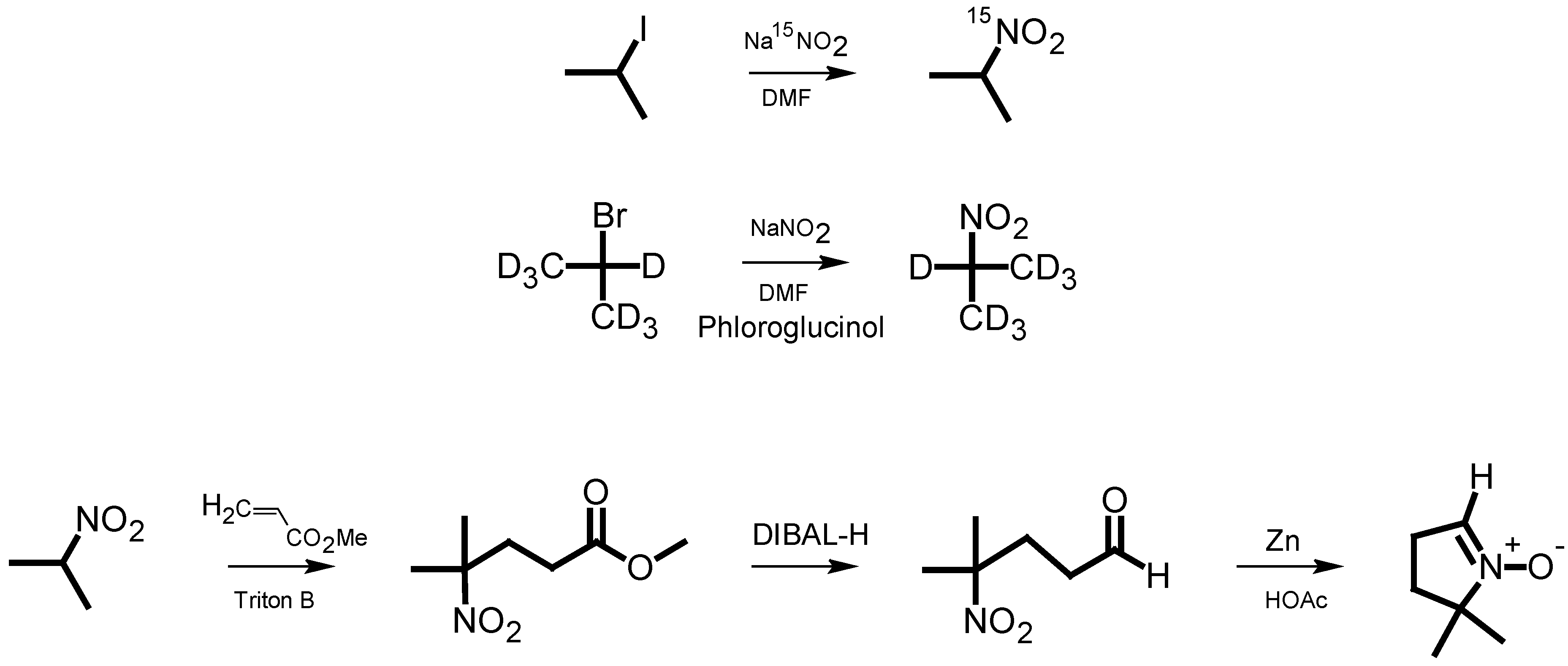
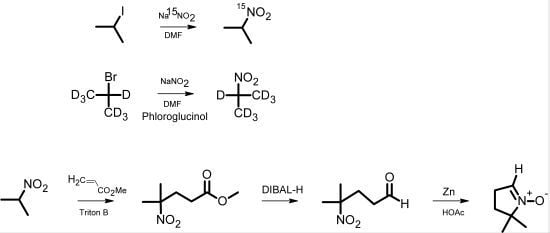
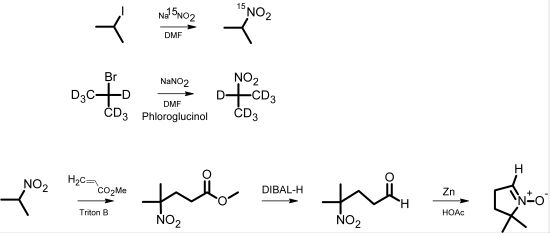
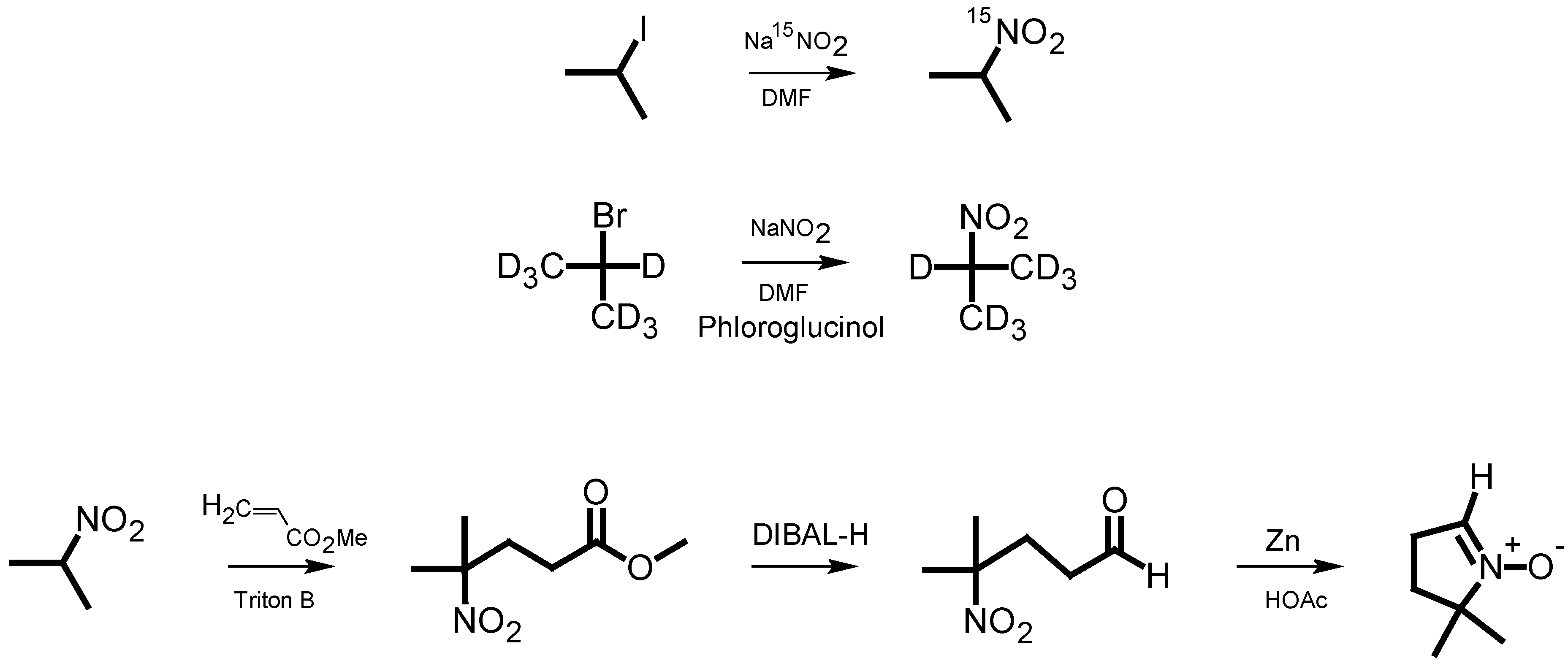
The classical synthesis of DMPO, as reported by Janzen
et al. [
5], is based on the synthesis of pyrrolines [
6]. Later, Le
et al. [
7] published a synthesis of 2-
14C-DMPO that avoided the direct Michael reaction of nitropropane with methyl acrylate, thereby improving the low yield and eliminating the difficult purification of this step. For the synthesis of isotopically labeled DMPO, Pou
et al. [
8] published an effective method starting from
15N-hydroxylamine, which involved the use of hydrogen gas in an autoclave as well as a reaction with ozone derived from an ozone generator. We have developed a more facile synthetic pathway for the synthesis of DMPO based on
15N-sodium nitrite or 2-bromopropane (D7) as the isotopically labeled starting material. The intermediate 2-nitropropane was prepared in a one-step reaction according to the reaction principle described by Kornblum
et al. [
9]. This principle has been used for nitrone spin trap synthesis [
10]. Nitropropane was then used for a DMPO synthesis similar to that of Le
et al. [
7] (
Scheme 1).
Scheme 1.
Pathway of DMPO analog synthesis.
Scheme 1.
Pathway of DMPO analog synthesis.
3. Experimental
15N-NaNO2 and 2-bromopropane (D7) were purchased from Cambridge Isotope Laboratories (Andover, MA, USA). Phloroglucinol was purchased from Alfa Aesar (Ward Hill, MA, USA). 14N-DMPO was purchased from Dojindo (Rockville, MD, USA). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Where dry solvents were required, the glassware was oven-dried and a dry argon atmosphere was used. These solvents were purchased as ‘anhydrous’ grade and handled under air exclusion. ESR spectra were recorded with a Bruker ElexSys E-500 spectrometer with ER 4122SHQ cavity, flow injection electrospray ionization (ESI/MS) analyses were performed with a Waters Q-TOF Premier mass spectrometer. 1H NMR spectra were acquired at 25 °C on a Varian INOVA 600 spectrometer operating at 1H frequency of 599.763 MHz.
3.1. Synthesis of 15N-5,5-Dimethyl-1-pyrroline N-oxide
15N-2-Nitropropane. In a 500 mL three-neck flask equipped with a magnetic stir bar, septum, Ar supply and a gas bubbler, urea (17 g, 0.28 mol) was dissolved in dry N,N-dimethylformamide (250 mL) under an Ar atmosphere. Then, 15N-NaNO2 (10 g, 145 mmol) was added and allowed to dissolve. 2-Iodopropane (18 mL; 180 mmol) was added and the solution was stirred for 5 h at room temperature. The reaction was quenched by pouring the reaction mixture into ice water (300 mL) with diethyl ether (100 mL). The organic layer was separated and retained, and the aqueous layer was extracted four times with diethyl ether. The combined organic layers were washed twice with sodium thiosulfate solution (10 wt%, 25 mL) followed by washing with saturated sodium bicarbonate solution (2 × 100 mL). The organic layer was dried over anhydrous magnesium sulfate, filtered, and the solvent removed with a rotary evaporator to obtain the crude product. The product was purified by vacuum distillation (−80 kPa, 75 °C) to give 4.63 g (51 mmol, 35%) of 15N-2-nitropropane. 1H-NMR (CDCl3): 1.51 (m, 6H), 4.62 (m, 1H).
15N-4-Methyl-4-nitropentanoic acid methyl ester. To a 100 mL three-neck flask with magnetic stir bar, condenser and addition funnel, 15N-2-nitropropane (4.63 g, 51 mmol), 1,4-dioxane (5 mL) and an aqueous solution of benzyltrimethylammonium hydroxide (Triton B, 40%, 1 mL) were added. The contents were heated to 70 °C (water bath) with stirring, and methyl acrylate (4.5 mL, 52 mmol) was added dropwise over 15 min. The temperature was raised and the solution stirred at 85 °C for 3 h. After cooling to room temperature, the solution was diluted (100 mL of H2O) and acidified with diluted hydrochloric acid (1 M). The reaction mixture was then extracted five times with diethyl ether (100 mL each). The combined organic layers were washed with water, saturated sodium bicarbonate solution and water again (100 mL each), then dried over anhydrous NaSO4. The solvent was removed on a rotary evaporator to give the crude product, which was purified by column chromatography on silica gel (hexane:diethyl ether = 1:1) to give 4.62 g (26.4 mmol, 51%) of 15N-4-methyl-4-nitropentanoic acid methyl ester. An rf of 0.33 was found. The product was stored under an argon atmosphere. 1H-NMR (CDCl3): 1.57 (s, 3H), 1.78 (s, 3H), 2.26 (m, 2H), 2.32 (m, 2H), 3.67 (s, 3H).
15N-4-Nitro-4-methyl-1-pentanal. A 500 mL three-neck flask equipped with a magnetic stir bar, septum, Ar supply and gas bubbler was filled with dry dichloromethane (125 mL) with the exclusion of air. 15N-4-methyl-4-nitropentanoic acid methyl ester (4.62 g; 26.4 mmol) was added, and the solution was cooled down to −95 °C with a methanol bath using dry ice initially, then liquid nitrogen for temperature adjustment. Next, 1 M diisobutylaluminum hydride solution in dichloromethane (29 mL) was added dropwise over 5 min, keeping the temperature below −90 °C. After 40 min of stirring at −95 °C, the solution was quenched by the addition of wet silica gel (20 g of silica gel well-mixed with 10 mL of H2O) while maintaining the reaction mixture at −95 °C. After 10 min of stirring at −95 °C, the cold bath was removed and the mixture was allowed to equilibrate to room temperature. The silica gel was separated by filtration and extracted three times with dichloromethane (100 mL each portion). The combined organic layers were washed with dilute hydrochloric acid, saturated sodium bicarbonate solution and water (100 mL each) and dried over anhydrous sodium sulfate. The removal of dichloromethane with a rotary evaporator gave the raw product, which was purified by silica gel flash chromatography (ethyl acetate:hexane = 1:5) to give 2.85 g (19.2 mmol, 74% yield) of 15N-4-nitro-4-methyl-1-pentanal. The rf value was determined to be 0.29. The product was stored under argon at −20 °C. 1H-NMR (CDCl3): 1.58 (m, 6H), 2.22 (bt, 2H), 2.49 (t, J = 7.2 Hz, 2H), 9.75 (s, 1H).
15N-5,5-Dimethyl-1-pyrroline N-oxide. In a 500 mL three-neck flask with an addition funnel, ethanol (95%, 60 mL) was cooled to 2 °C. Then 15N-4-nitro-4-methyl-1-pentanal (2.85 g, 19.2 mmol) and zinc dust (2.68 g, 0.41 mmol, 2.1 equivalents) were added. Under brisk magnetic stirring, glacial acetic acid (4.2 mL) was added dropwise while the temperature was kept below 10 °C. After stirring 1 h, the apparatus was put in a refrigerator suitable for flammable materials. Any hydrogen gas which may have formed was allowed to dissipate. After an additional 30 h of stirring at 10 °C, the solution was filtered to remove zinc and any undissolved zinc acetate. The filtrate was extracted with cold ethanol (3 × 100 mL). From the combined solutions, the ethanol was removed with a rotational evaporator. The residue was diluted with dichloromethane (200 mL) and washed twice with saturated sodium bicarbonate solution and water (50 mL each). The organic layer was dried with magnesium sulfate, filtered and the dichloromethane removed with a rotary evaporator to give a reddish-brown crude product. The product was purified by two consecutive sublimations (0.1 torr) from room temperature to 0 °C, and 305 mg (2.7 mmol, 15%) of 15N-DMPO was obtained. 1H-NMR (CDCl3): 1.42 (m, 6H), 2.12 (td, 1.5 Hz, 7.6 Hz, 2H), 2.56 (m, 2H), 6.80 (t, J = 2.6 Hz, 1H).
3.2. Synthesis of 5,5-Di-(trideuteromethyl)-1-pyrroline N-oxide
2-Nitropropane (D7). Urea (26.5 g, 0.44 mol), and phloroglucinol (21 g, 0.17 mol) were dissolved in dry N,N-dimethylformamide (600 mL) in a 1000 mL three-neck flask equipped with a magnetic stir bar, septum, Ar supply and bubbler, under an Ar atmosphere. Then, NaNO2 (24.74 g, 0.36 mol) was added and dissolved. Heptadeutero-2-bromopropane (20 g, 154 mmol) was added, and the solution was stirred for 48 h at room temperature. The reaction was quenched with ice water (1000 mL) covered with diethyl ether (200 mL). The organic layer was separated and collected, and the aqueous layer was extracted four times with diethyl ether (100 mL each). The combined organic layers were washed with saturated sodium bicarbonate solution (100 mL) and water (2 × 100 mL). The organic layer was dried over anhydrous magnesium sulfate and filtered. The solvent was removed with a rotational evaporator to obtain the crude product. The product was purified by vacuum distillation (−80 kPa, 75 °C) to give 4.02 g (42 mmol, 27%) of 2-nitropropane (D7).
4,4,4-Trideuteromethyl-4-nitro-5,5,5-trideuteropentanoic acid methyl ester. The ester was synthesized from 2-nitropropane (D7) (4.02 g, 42 mmol), dioxane (5 mL), Triton B (1 mL) and methyl acrylate (4.1 mL) as described earlier for 15N-4-methyl-4-nitropentanoic acid methyl ester. The yield was 6.11 g (33.72 mmol, 80%) after column chromatography (hexane:diethyl ether = 4:1) and the removal of the solvent. 1H-NMR (CDCl3): 2.26 (m, 2H), 2.32 (m, 2H), 3.67 (s, 3H).
4-Nitro-4,4,4-trideuteromethyl-5,5,5-trideutero-1-pentanal. The aldehyde was synthesized as described for 15N-4-nitro-4-methyl-1-pentanal with dry dichloromethane (200 mL), the deuterated nitromethylpentanoic acid methyl ester (6.11 g, 33.72 mmol) and a 1 M stock solution of diisobutylaluminum hydride (37.1 mL) using water-free chemicals under an argon atmosphere. After flash chromatography (hexane:ethyl acetate = 5:1) and removal of the solvent, 2.86 g (19.67 mmol, 58%) of the pure product was obtained. 1H-NMR (CDCl3): 2.14 (t, J = 7.4 Hz, 2H), 2.42 (t, 7.4 Hz, 2H), 9.67 (s, 1H).
5,5-Di(trideuteromethyl)-1-pyrroline N-oxide. The DMPO analog was synthesized similarly to 15N-5,5-dimethyl-1-pyrroline N-oxide, using ethanol (100 mL), the aldehyde intermediate (4.08 g; 19.67 mmol), zinc dust (3.69 g, 56.43 mmol, 2.9 eq) and acetic acid (6.75 g, 112.4 mmol). Purification gave 357 mg (2.75 mmol) of colorless DMPO crystals (14% yield). 1H-NMR (CDCl3): 2.12 (td, 1.5 Hz, 7.6 Hz, 2H), 2.56 (td, 2.5 Hz, 7.4 Hz, 2H), 6.82 (t, J = 2.5 Hz, 1H).
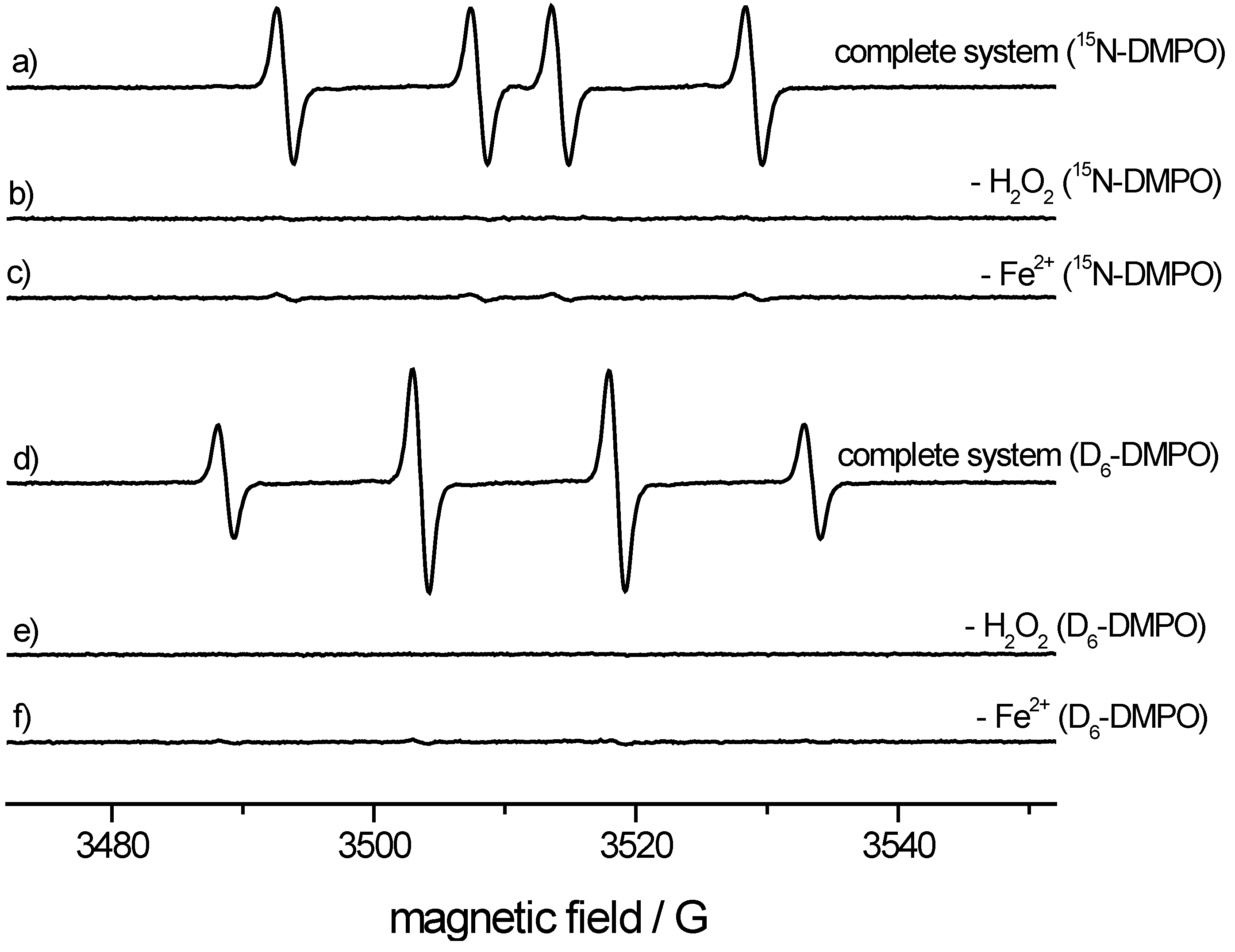
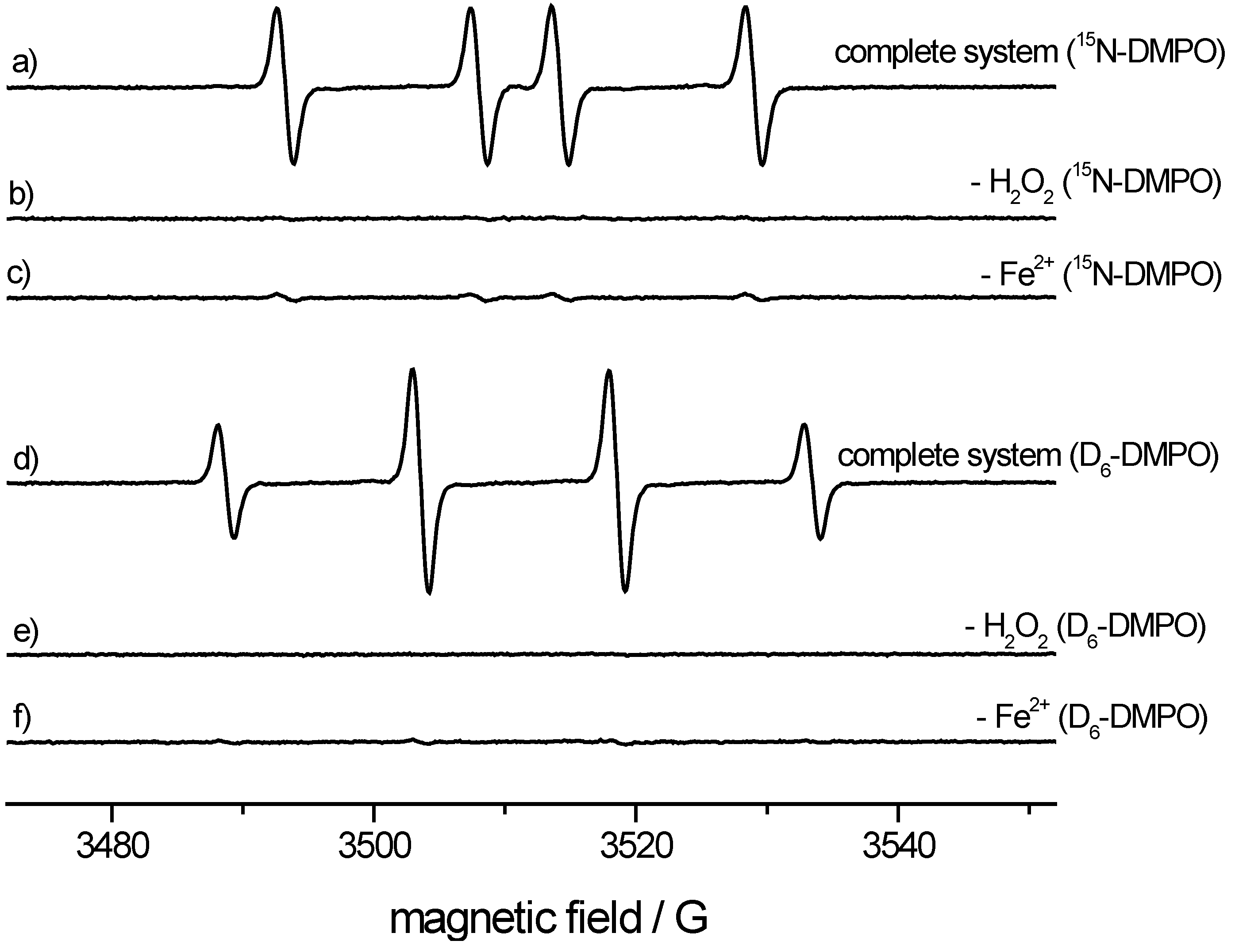
ESR spectroscopy. We used a microwave frequency of 9.78 GHz (X-Band), a microwave power of 20 mW and 1 × 104 receiver gain, a modulation frequency of 100 kHz and modulation amplitude of 1.0 G. Time constant and conversion time were 40.96 ms. The field range was 80 G with a center field of 3512 G. For the spin-trapping experiment, 100 mM of the 15N- or di(trideuteromethyl)DMPO analog was mixed with 100 μM FeSO4, 200 μM diethylenetriaminepentaacetic acid and 200 μM hydrogen peroxide. The phosphate buffer (100 mM, pH = 7.4) was treated with Chelex-100 to reduce metal contamination. In control experiments, the iron or H2O2 was omitted for comparison. The experiment was repeated with regular 14N-DMPO.
Mass Spectrometry. Samples of the DMPO analogs were initially prepared at 100 mM in acetonitrile, diluted 1000-fold just prior to analysis with a solution of water with 0.1% formic acid and infused into the mass spectrometer at 500 nL/min using a syringe pump. The instrumental parameters for the MS analyses were as follows: capillary voltage, 3.5 kV; cone voltage, 30 V; collision energy, 4 eV and source temperature, 80 ºC. MS/MS data of the DMPO analogs were acquired using collision energies of 15–25 eV. For calibration, a solution of glu-fibrinopeptide B (500 fmol/μL) in water/acetonitrile 80:20 (v/v) with 0.1% formic acid and a mass of 785.8496 (2+) was used. Data analysis was accomplished using MassLynx software supplied by the manufacturer.

{kind=link}
{kind=link}
{kind=link}