Functionality Pattern Matching as an Efficient Complementary Structure/Reaction Search Tool: an Open-Source Approach


Abstract
:1. Introduction
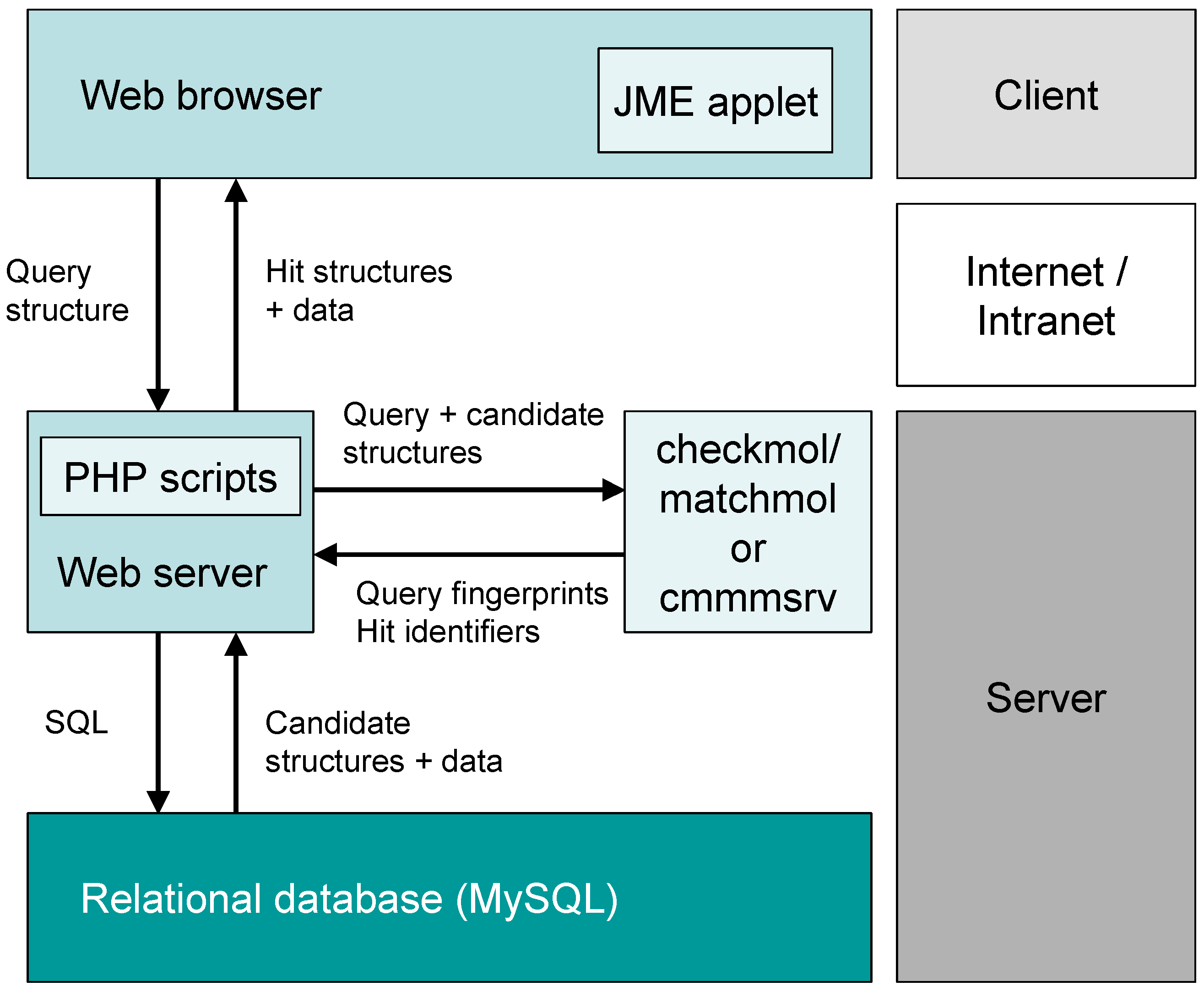
2. System Description
2.1. General Concept
- text search in structure and reaction data collections
- structure/substructure/similarity search in structure data collections
- structure/substructure search in reaction data collections
- functional group search in structure and reaction data collections
- administration front-end for entering/editing structures or reactions and associated data
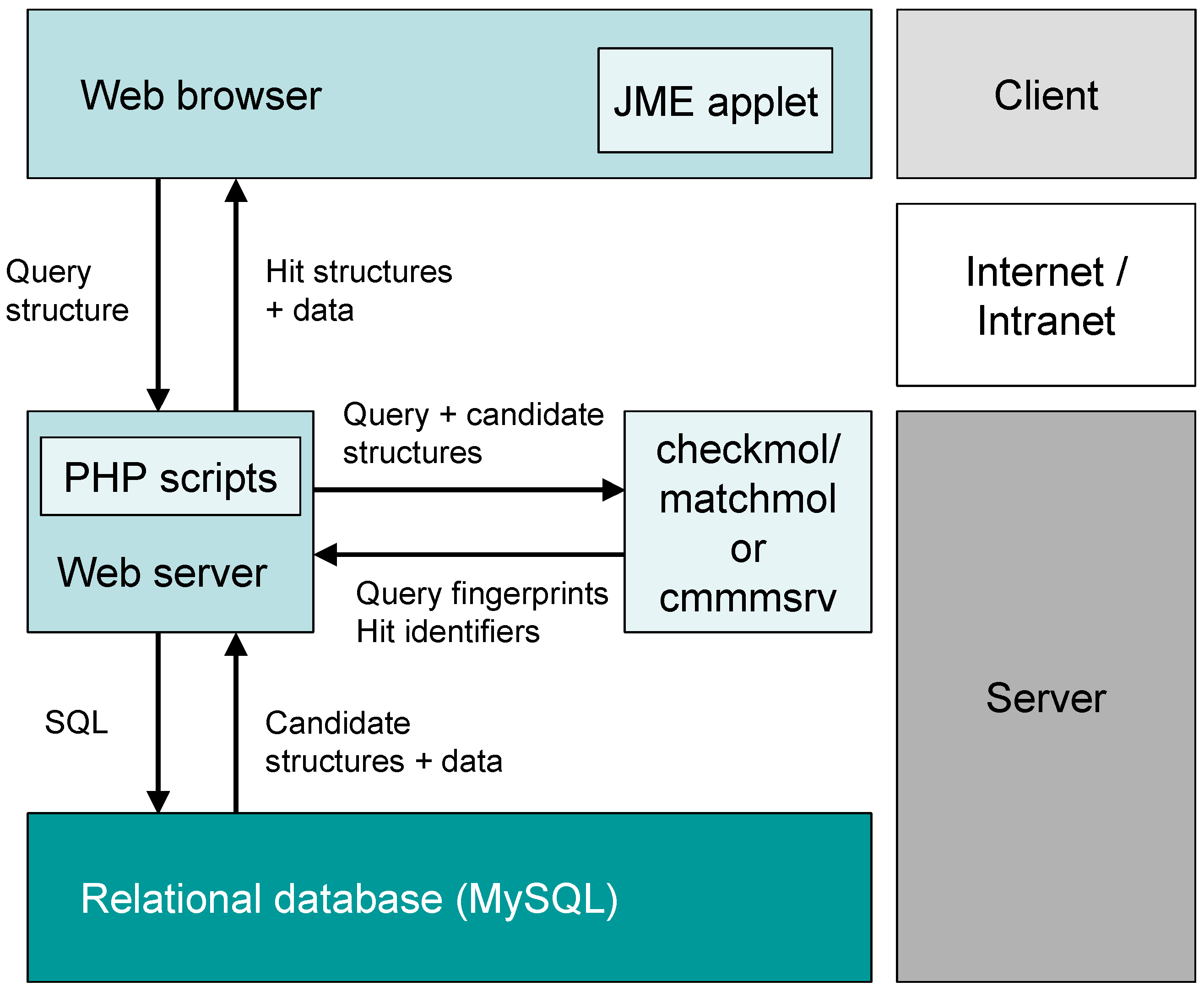
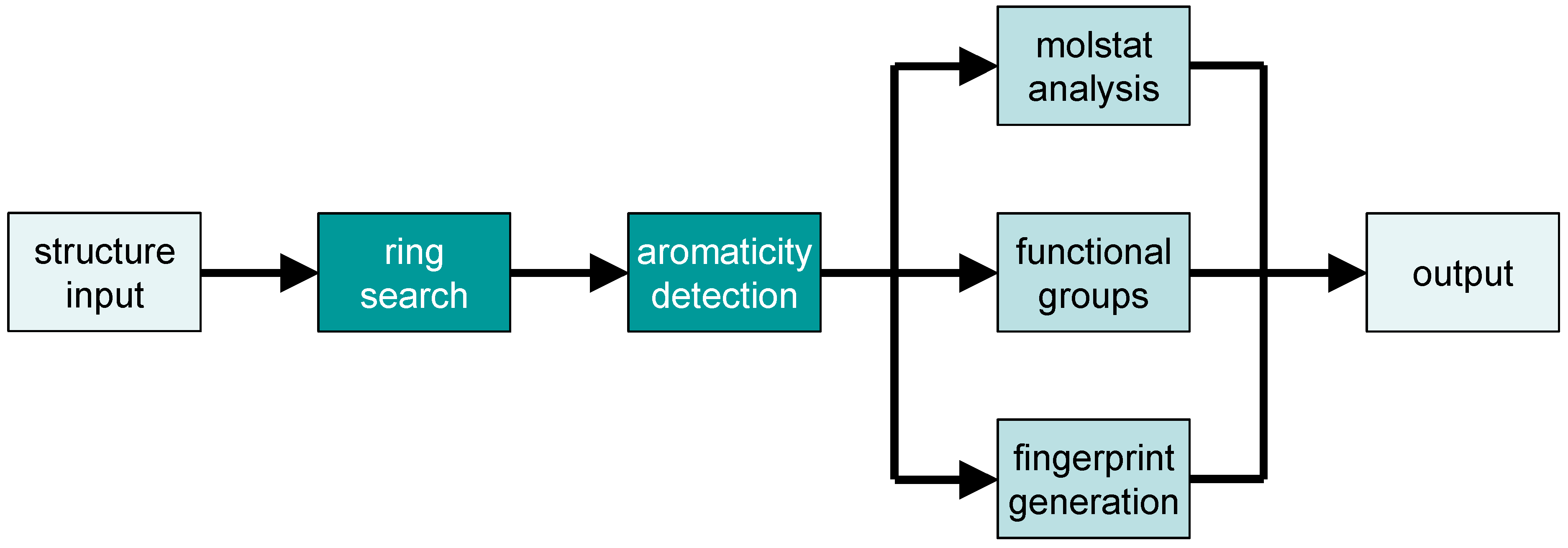
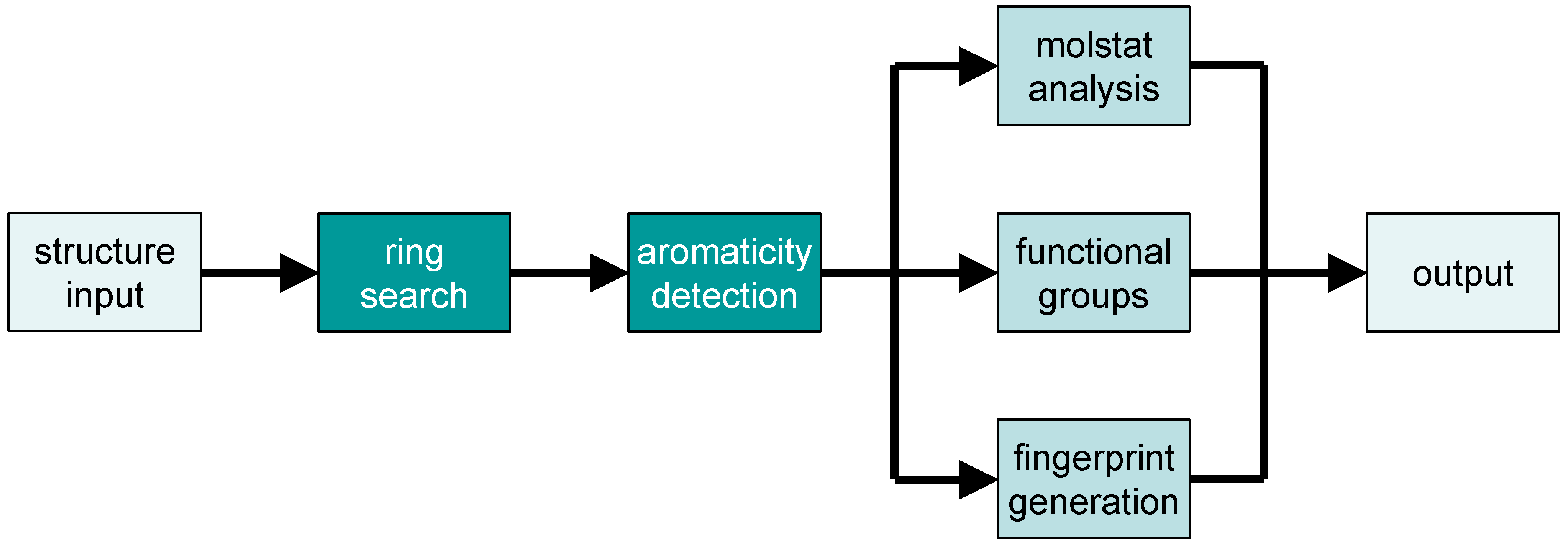
2.2. Analysis of Chemical Structure and Functionality with Checkmol
- a list of all detected functional groups in clear text (English or German)
- the same list as a bitstring (in 32-bit segments, each represented as unsigned integer number)
- the same bitstring in ASCII representation (i.e., as a string composed of “0” and “1” symbols); in both representations, each position in the bitstring represents the presence or absence of a particular functional group in the molecule
- a list (in one of two possible output formats) of discrete structural descriptors (“molecular statistics” = “molstat”), useful for pre-selection in a structure database
- a list (in one of two possible output formats) of hash-based fingerprints, useful for pre-selection in a structure database
- the chemical structure in MDL molfile format with additional encoding for aromaticity
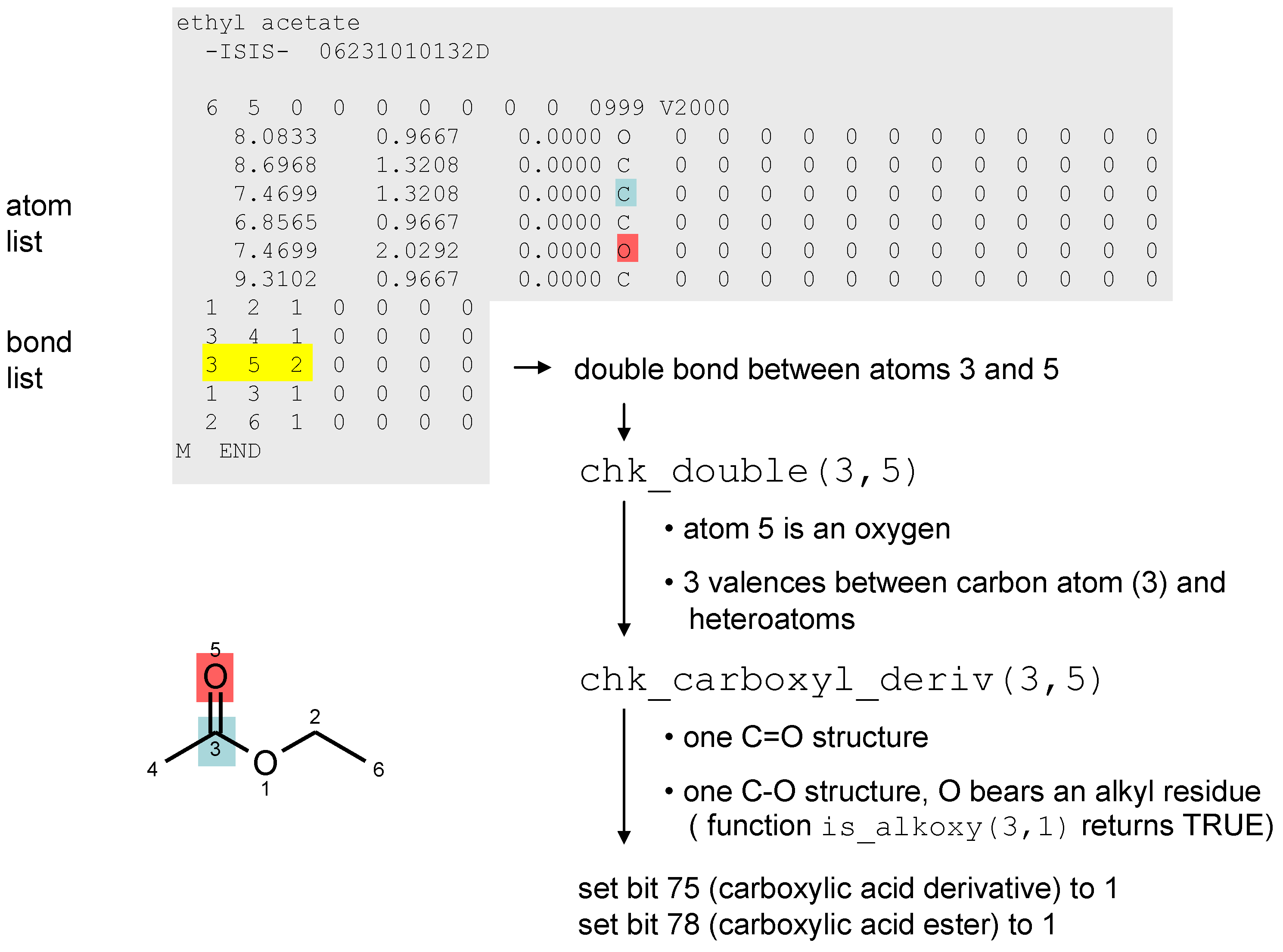
2.3. Structure Matching with Matchmol
2.4. Structure Search Operation: Typical Procedure
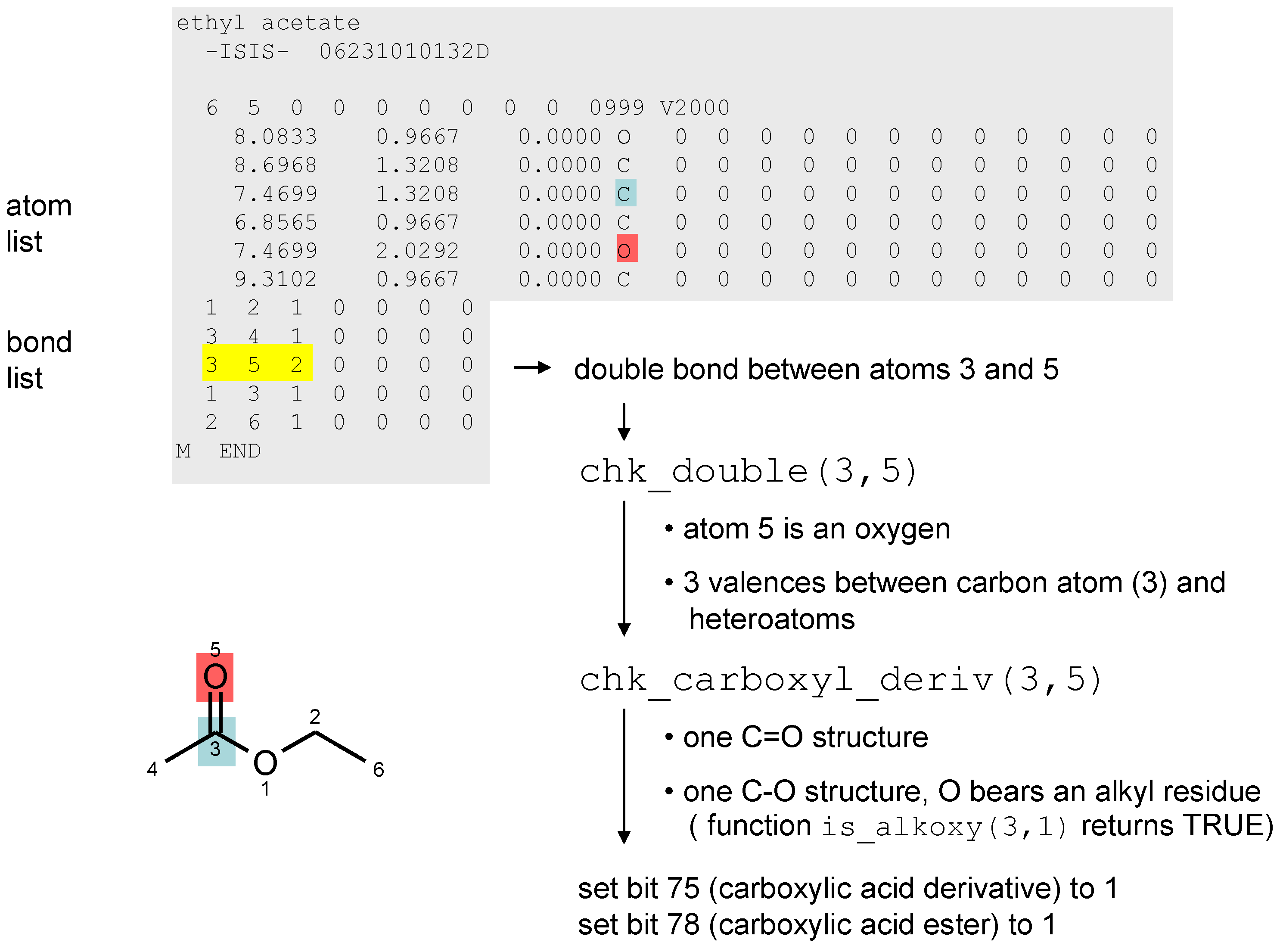
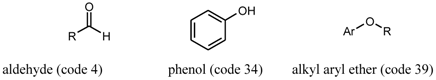
3. Chemical Functionality as a Search Criterion
- 1)
- 00010000000000000000000000000000 → bit number 3 is set, decimal value: 23 = 8
- 2)
- 01000010000000000000000000000000 → bits 1 and 6 are set, decimal value: 21 + 26 = 66
4. Results and Discussion
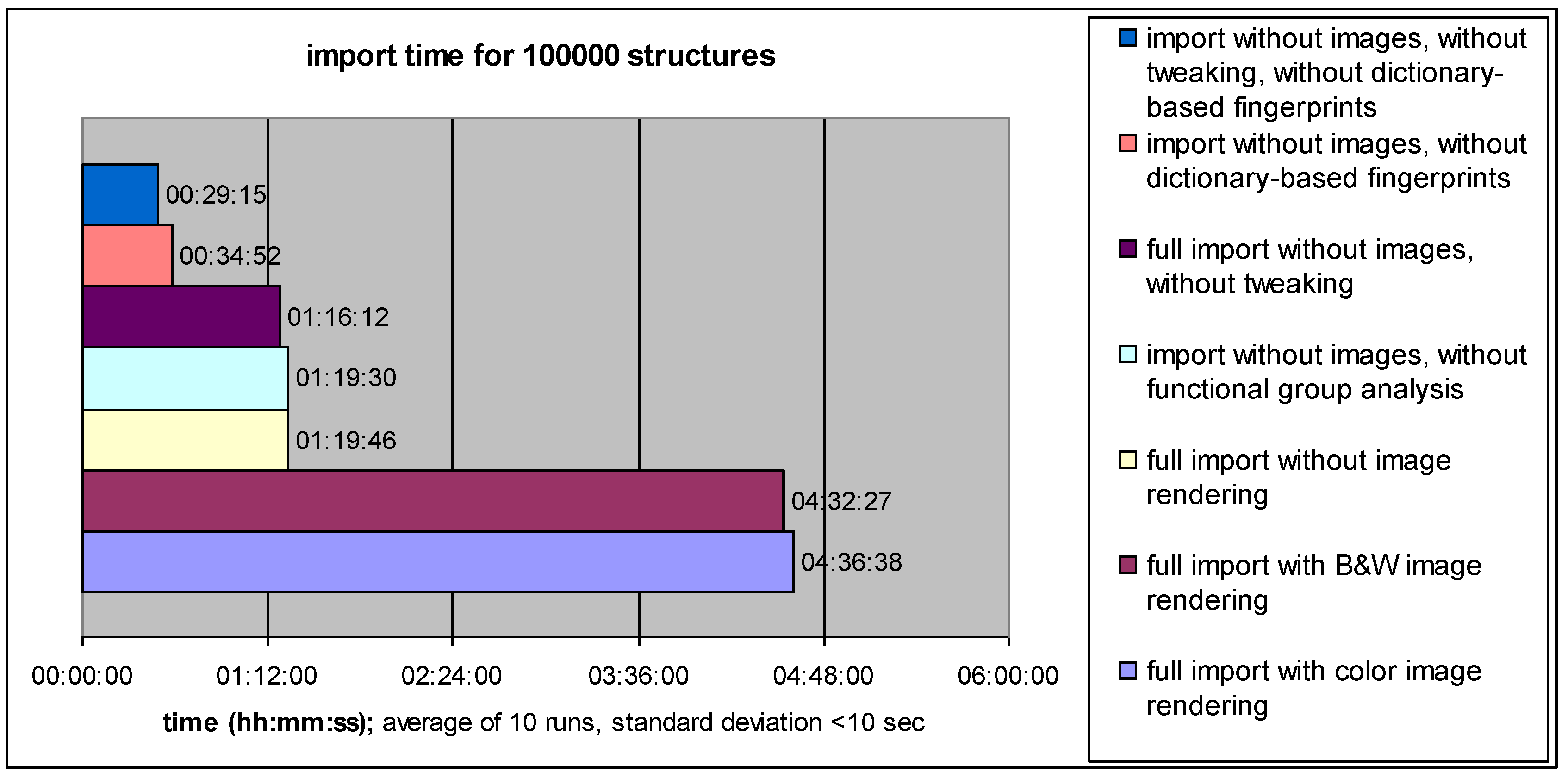
4.1. Data Import Performance: Speed versus Information Content
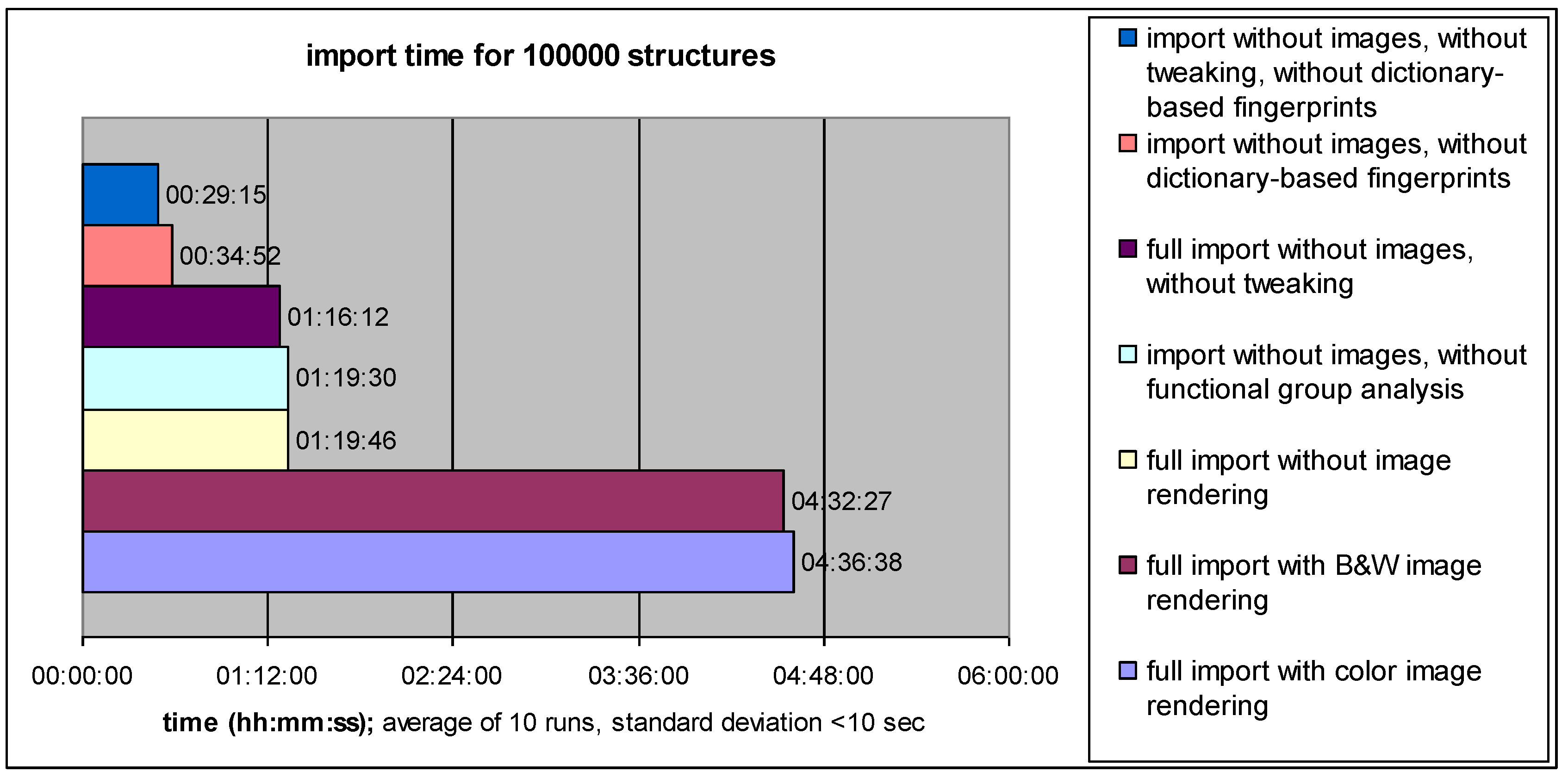
4.2. Search Performance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Search type | Query | Remarks | Hits | Time (sec) |
|---|---|---|---|---|---|
| 1 | substructure | 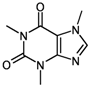 caffeine caffeine | with untweaked molfiles | 3518 (of 3613 candidates) | 11.20 (± 0.029) |
| 2 | – | 3518 (of 3613 candidates) | 6.56 (± 0.136) | ||
| 3 | substructure |  ellipticine ellipticine | with untweaked molfiles | 131 (of 216 candidates) | 2.30 (± 0.007) |
| 4 | – | 131 (of 216 candidates) | 0.86 (± 0.010) | ||
| 5 | substructure |  | – | 905 (of 8520 candidates) | 13.46 (± 0.799) |
| 6 | functional groups | “primary aromatic amine” + “sulfonamide” | groups taken from entry 5 | 1912 | 0.16 (± 0.015) |
| 7 | substructure |  | – | 452 (of 591 candidates) | 1.81 (± 0.029) |
| 8 | functional groups | “heterocycle” + “acetal” + “secondary carboxamide" + “primary alcohol” + “secondary alcohol” | groups taken from entry 7 | 688 | 0.06 (± 0.001) |
| 9 | substructure |  diazepam diazepam | – | 127 (of 155 candidates) | 0.48 (± 0.004) |
| 10 | functional groups | “aromatic” + “heterocycle" + “imine” + “tert. carboxamide” + “lactam” + “aryl chloride” | groups taken from entry 9 | 273 | 0.02 (± 0.000) |
| Entry | Search type | Query | Remarks | Hits | Time (sec) |
|---|---|---|---|---|---|
| 1 | reaction substructure |  | – | 38 (of 56 candidates) | 7.01 (± 0.004) |
| 2 | functional groups | “imine” created during reaction | group taken from entry 1 | 37 | 0.019 (± 0.000) |
| 3 | reaction substructure |  | – | 8 (of 20 candidates) | 2.68 (± 0.002) |
| 4 | functional groups | “carboxylic acid hydrazide”created during reaction | group taken from entry 3 | 10 | 0.004 (± 0.000) |
| 5 | reaction substructure |  (half reaction) (half reaction) | – | 10 (of 16 candidates) | 0.73 (± 0.002) |
| 6 | functional groups | “isocyanate” lost during reaction | group taken from entry 5 | 10 | 0.005 (± 0.000) |
5. Conclusions
References and Notes
- The PubChem Project. Available online: http://pubchem.ncbi.nlm.nih.gov/ (accessed on 29 June 2010).
- OpenEye Scientific Software-OEChem TK. Available online: http://www.eyesopen.com/products/toolkits/ oechem.html/ (accessed on 29 June 2010).
- Xemistry Chemoinformatics. Available online: http://www.xemistry.com/ (accessed on 16 June 2010).
- Ihlenfeldt, W.D.; Takahashi, Y.; Abe, H.; Sasaki, S. Computation and Management of Chemical Properties in CACTVS: An extensible Networked Approach toward Modularity and Flexibility. J. Chem. Inf. Comput. Sci. 1994, 34, 109–116. [Google Scholar] [CrossRef]
- ChemAxon - Toolkits and Desktop Applications for Cheminformatics. Available online: http://www.chemaxon.com/ (accessed on 16 June 2010).
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar]
- MySQL Website. Available online: http://www.mysql.com/ (accessed on 29 June 2010).
- PostgreSQL Website. Available online: http://www.postgresql.org/ (accessed on 29 June 2010).
- Oracle Database Website. Available online: http://www.oracle.com/us/products/database/ (accessed on 29 June 2010).
- Open Babel: The Open Source Chemistry Toolbox. Available online: http://openbabel.org/wiki/Main_Page/ (accessed on 29 June 2010).
- PgFoundry: pgchem::tigress: project info. Available online: http://pgfoundry.org/projects/pgchem/ (accessed on 29 June 2010).
- ChemiSQL Wiki website. Available online: http://chemdb.sourceforge.net/wiki/index.php/Main_Page/ (accessed on 29 June 2010).
- The MolDB5 homepage. Available online: http://merian.pch.univie.ac.at/~nhaider/cheminf/moldb5.html (accessed on 29 June 2010).
- Apache HTTP server. Available online: http://projects.apache.org/projects/http_server.html/ (accessed on 29 June 2010).
- PHP Hypertext Preprocessor. Available online: http://php.net/index.php/ (accessed on 29 June 2010).
- The checkmol/matchmol homepage. Available online: http://merian.pch.univie.ac.at/~nhaider/cheminf/cmmm.html/ (accessed on 29 June 2010).
- Symyx CTfile formats. Available online: http://www.symyx.com/downloads/public/ctfile/ctfile.jsp/ (accessed on 29 June 2010).
- JME Molecular Editor Applet. Available online: http://www.molinspiration.com/jme/ (accessed on 29 June 2010).
- Ertl, P.; Jacob, O. WWW-based chemical information system. Theochem 1997, 419, 113–120. [Google Scholar]
- Hanser, T.; Jauffret, P.; Kaufmann, G. A new algorithm for exhaustive ring perception in a molecular graph. J. Chem. Inf. Comput. Sci. 1996, 36, 1146–1152. [Google Scholar]
- A complete list of these (more than 50) molstat descriptors can be obtained by invoking the checkmol program with the -l option (“checkmol -l”).
- Gasteiger, J.; Engel, T. Chemoinformatics: A Textbook; Wiley-VCH: Weinheim, Germany, 2003; pp. 71–75. [Google Scholar]
- Leach, A.R.; Gillet, V.J. An Introduction to Chemoinformatics; Kluwer Academic Publishers: Dordrecht, Boston, London, 2003; pp. 11–13. [Google Scholar]
- Daylight Chemical Information Systems, Inc.: Fingerprints – Screening and Similarity. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.finger.html/ (accessed on 29 June 2009).
- General Purpose Hash Function Algorithms. Available online: http://www.partow.net/programming/hashfunctions/ (accessed on 17 June 2010).
- Chemical Hashed Fingerprints - ChemAxon toolkits and desktop applications for cheminformatics. Available online: http://www.chemaxon.com/jchem/doc/user/fingerprint.html (accessed on 17 June 2010).
- List of functional groups recognized by checkmol. Available online: http://merian.pch.univie.ac.at/ ~nhaider/cheminf/fgtable.pdf/ (accessed on 17 June 2010).
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures - A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- The mol2ps homepage. Available online: http://merian.pch.univie.ac.at/~nhaider/cheminf/mol2ps.html/ (accessed on 17 June 2010).
- Ghostscript, Ghostview and GSview. Available online: http://pages.cs.wisc.edu/~ghost/ (accessed on 17 June 2010).
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- SciFinder Strategies-Functional Group Searching. Available online: http://www.cas.org/support/scifi/strategies/ wreact3.html/ (accessed on 26 July 2010).
- Korea Institute of Science and Technology Information. ChemDB. Available online: http://chemdb.kisti.re.kr/help/ 07_tutorial.html/ (accessed on 21 June 2010).
- University of Erlangen-Nürnberg. NCI Screening Data 3D Miner. Available online: http://www2.chemie.uni-erlangen.de/services/nciscreen/index.html/ (accessed on 21 June 2010).
- Feldman, H.; Dumontier, M.; Ling, S.; Haider, N.; Hogue, C.W.V. CO: a chemical ontology for identification of functional groups and semantic comparison of small molecules. FEBS Lett. 2005, 579, 4685–4691. [Google Scholar] [CrossRef]
- Hardware used: HP xw4600 workstation with 3.1 GHz Intel Core 2 Duo CPU, 4GB RAM, 2x250 GB SATA disks; operating system: Ubuntu Linux 10.04 workstation edition with kernel 2.6.32; application software: Apache 2.2.14, PHP 5.3.2, MySQL 5.1.41, Perl 5.10.1, JME v2008.01, Ghostscript 8.71, mol2ps v0.1f, checkmol/matchmol/cmmmsrv v0.4c (compiled with Free Pascal 2.4.0).
- Molbank. An Open Access Journal from MDPI. Available online: https://www.mdpi.com/journal/molbank/ (accessed on 17 June 2010).
- This untypical candidate/hit ratio reflects the fact that hydrogen atoms are ignored in the hash-based fingerprinting algorithm, whereas the query structure contained explicit hydrogens at the (frequently substituted) amino nitrogen.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Haider, N. Functionality Pattern Matching as an Efficient Complementary Structure/Reaction Search Tool: an Open-Source Approach. Molecules 2010, 15, 5079-5092. https://doi.org/10.3390/molecules15085079
Haider N. Functionality Pattern Matching as an Efficient Complementary Structure/Reaction Search Tool: an Open-Source Approach. Molecules. 2010; 15(8):5079-5092. https://doi.org/10.3390/molecules15085079
Chicago/Turabian StyleHaider, Norbert. 2010. "Functionality Pattern Matching as an Efficient Complementary Structure/Reaction Search Tool: an Open-Source Approach" Molecules 15, no. 8: 5079-5092. https://doi.org/10.3390/molecules15085079
APA StyleHaider, N. (2010). Functionality Pattern Matching as an Efficient Complementary Structure/Reaction Search Tool: an Open-Source Approach. Molecules, 15(8), 5079-5092. https://doi.org/10.3390/molecules15085079