Conversion of Aryl Iodides into Aryliodine(III) Dichlorides by an Oxidative Halogenation Strategy Using 30% Aqueous Hydrogen Peroxide in Fluorinated Alcohol

Abstract
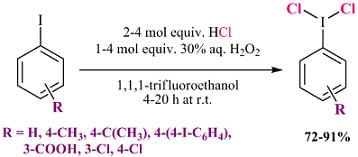
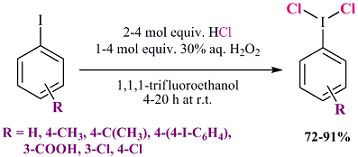
:
1. Introduction
2. Results and Discussion
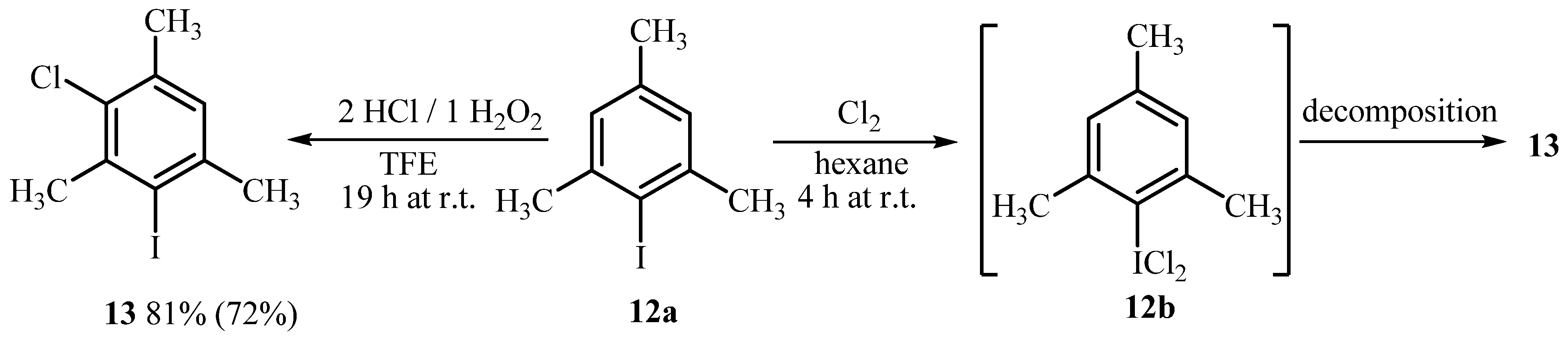
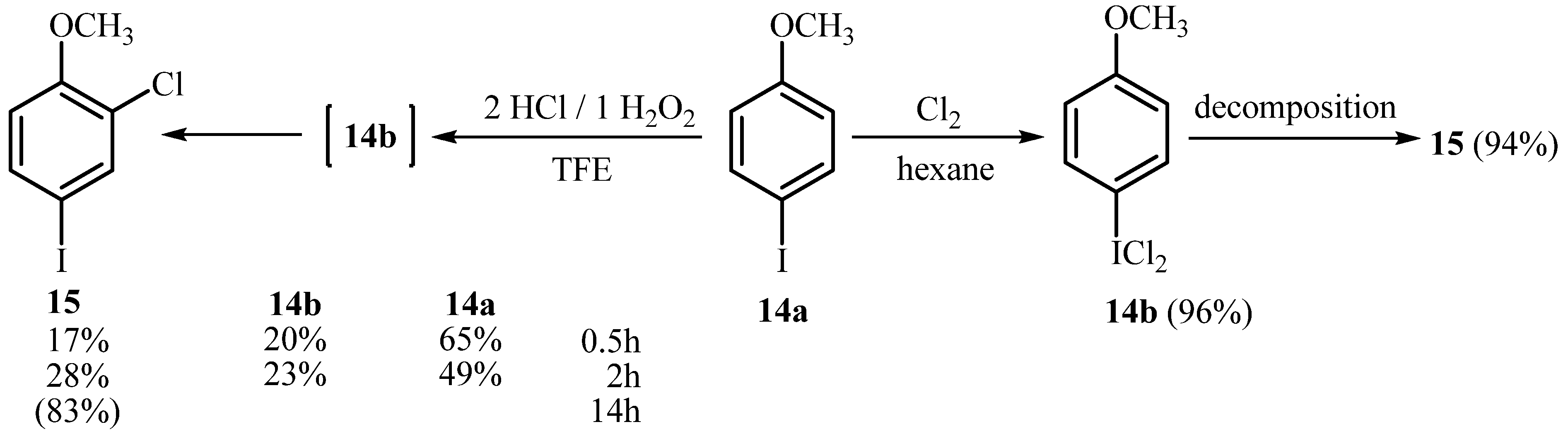
2.1. Synthesis of Aryl Iodine (III) Dichlorides
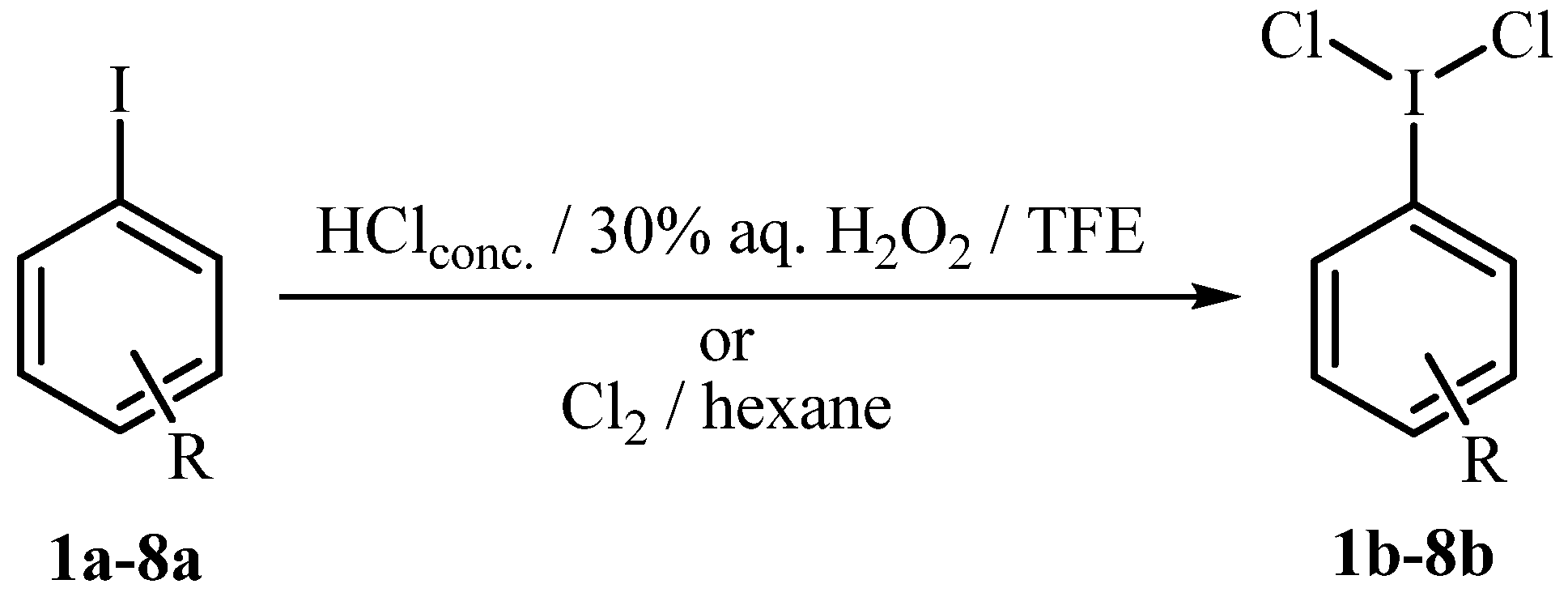

{kind=link}
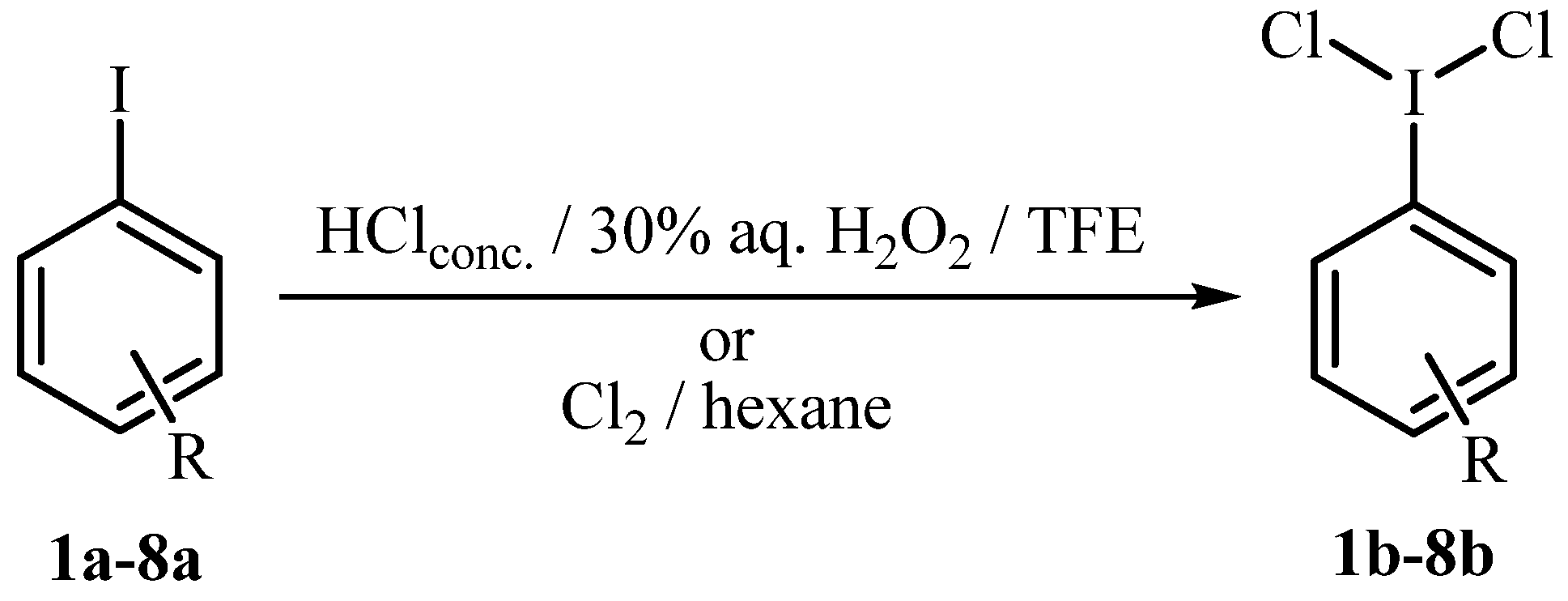{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
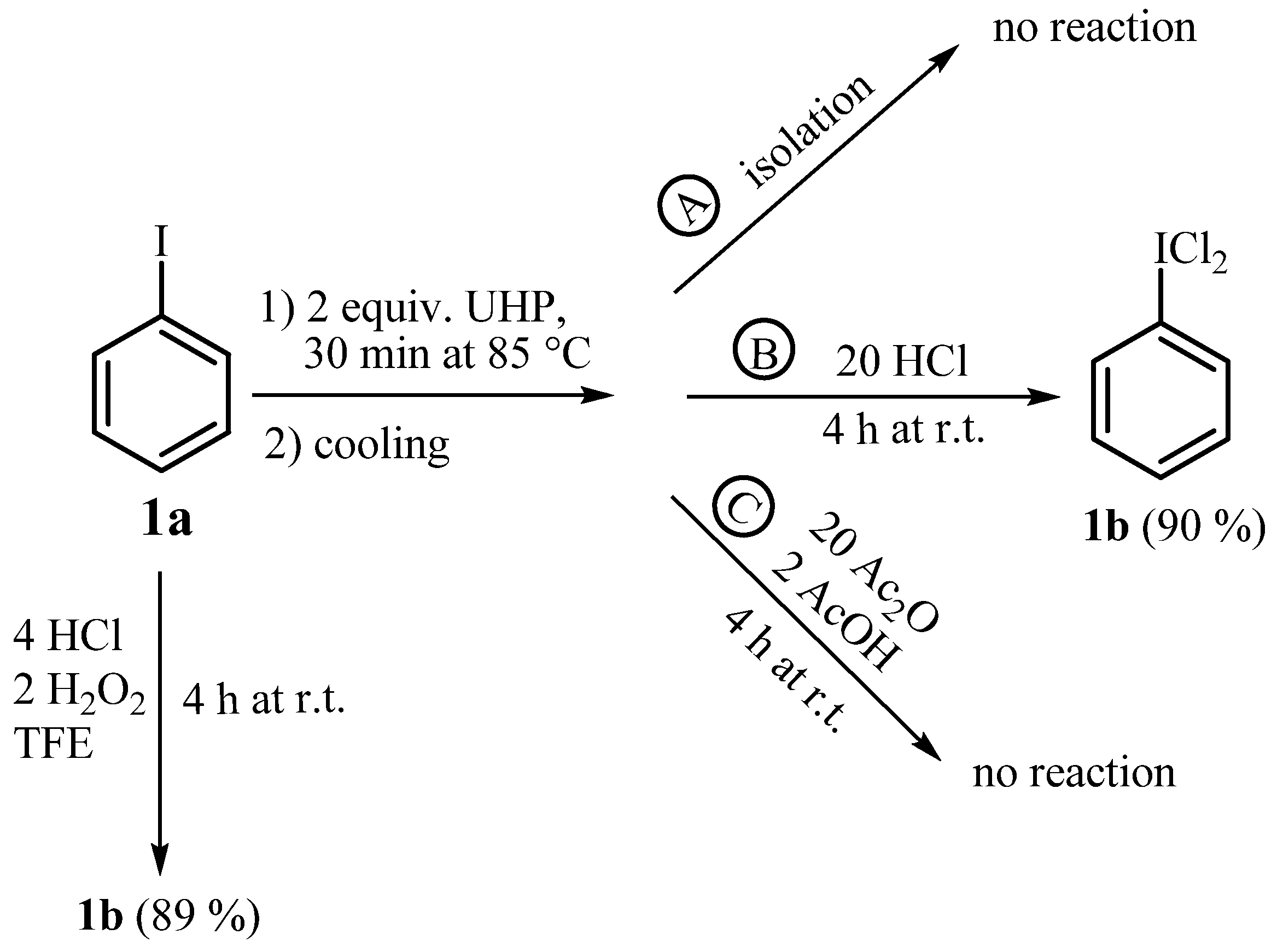{kind=link}
| Entry | Substrate | Method a | Time (h) | Yield b (%) |
|---|---|---|---|---|
| 1 | 1a, R=H | A | 4 | 1b, 80 |
| B | 4 | 89 | ||
| D | 3 | 96 | ||
| 2 | 2a, R=4-CH3 | A | 16 | 2b, 66 |
| B | 16 | 90 | ||
| D | 3 | 92 | ||
| 3 | 3a, R=4-C(CH3)3 | A | 19 | 3b, 55 |
| C | 19 | 80 | ||
| D | 19 | 85 | ||
| 4 | 4a, R=4-(4-I-C6H4) | B | 5 | 4b, 85 |
| C | 19 | 89 | ||
| D | 10 | 96 | ||
| 5 | 5a, R=3-COOH | A | 5 | 5b, 40 |
| B | 20 | 78 | ||
| D | 10 | 82 | ||
| 6 | 6a, R=3-NO2 | B | 14 | 6b, 20 |
| C | 20 | 72 | ||
| D | 19 | 76 | ||
| 7 | 7a, R=3-Cl | B | 19 | 7b, 59 |
| C | 20 | 85 | ||
| D | 19 | 90 | ||
| 8 | 8a, R=4-Cl | B | 14 | 8b, 50 |
| C | 19 | 85 | ||
| D | 19 | 92 |
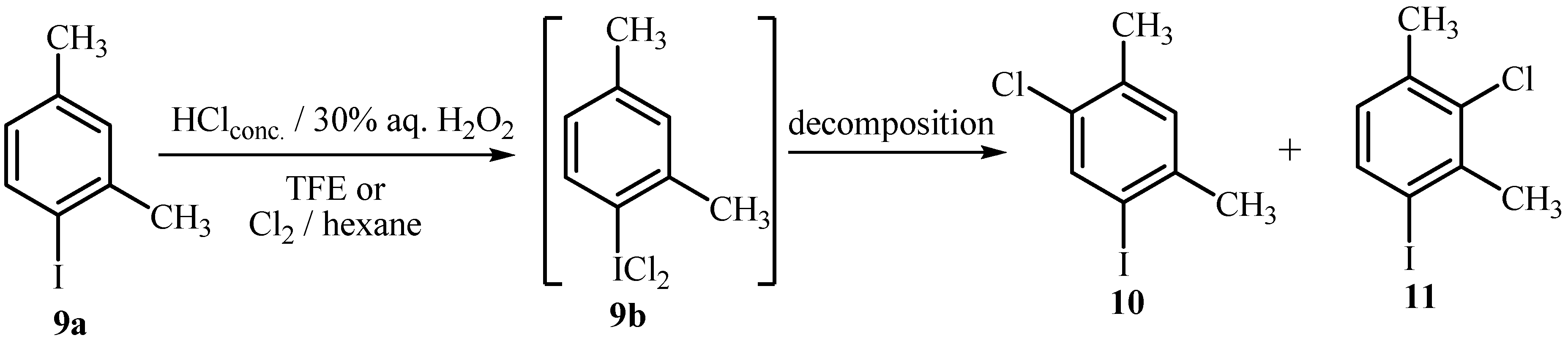
2.2. Chlorination of Activated Aryliodides with HCl/H2O2/TFE and with Molecular Chlorine
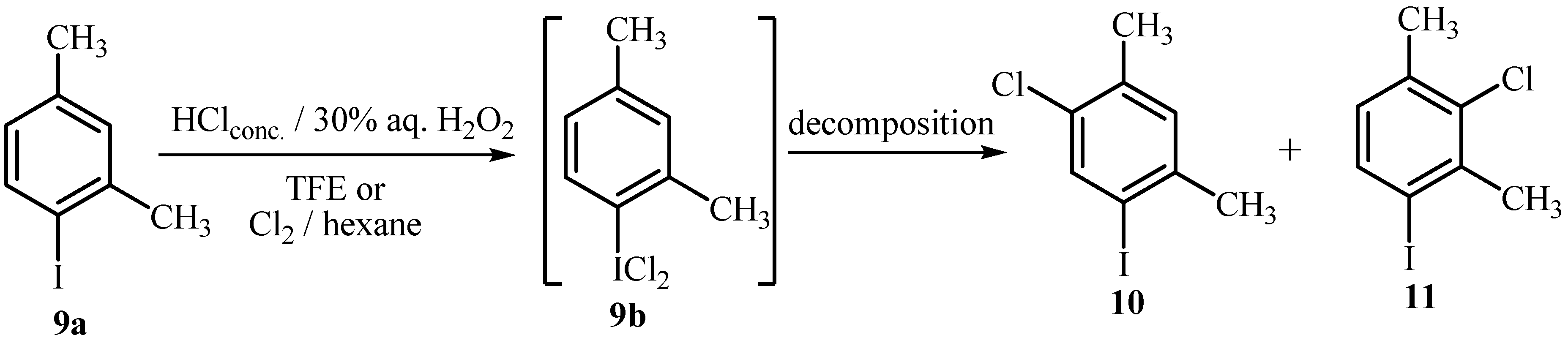
| Distributionb (%) | |||
|---|---|---|---|
| Entry | Methoda | Time (h) | 9a : 9bc : 10 : 11 |
| 1 | A | 0.5 | 83 : 17 : / : / |
| 2 | A | 2 | 54 : 46 : trace : trace |
| 3 | A | 20 | 33 : / : 30 : 37 |
| 4 | B | 20 | / : / : 47 : 53 |
| 5 | D | 6 | 19 : 22 : 28 : 31 |
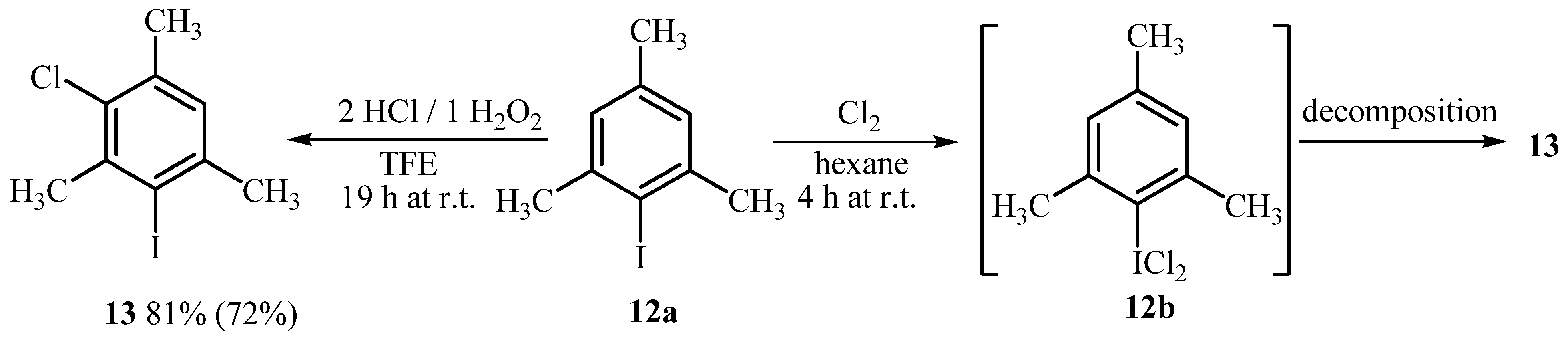
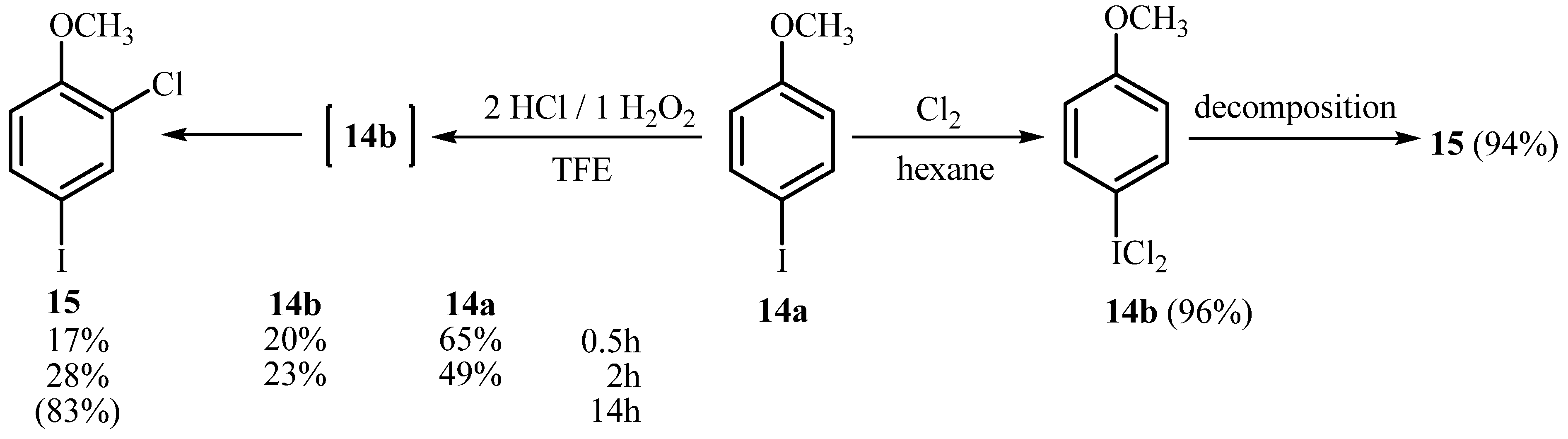
2.3. Investigation of the Nature of Reactive Chlorinating Species
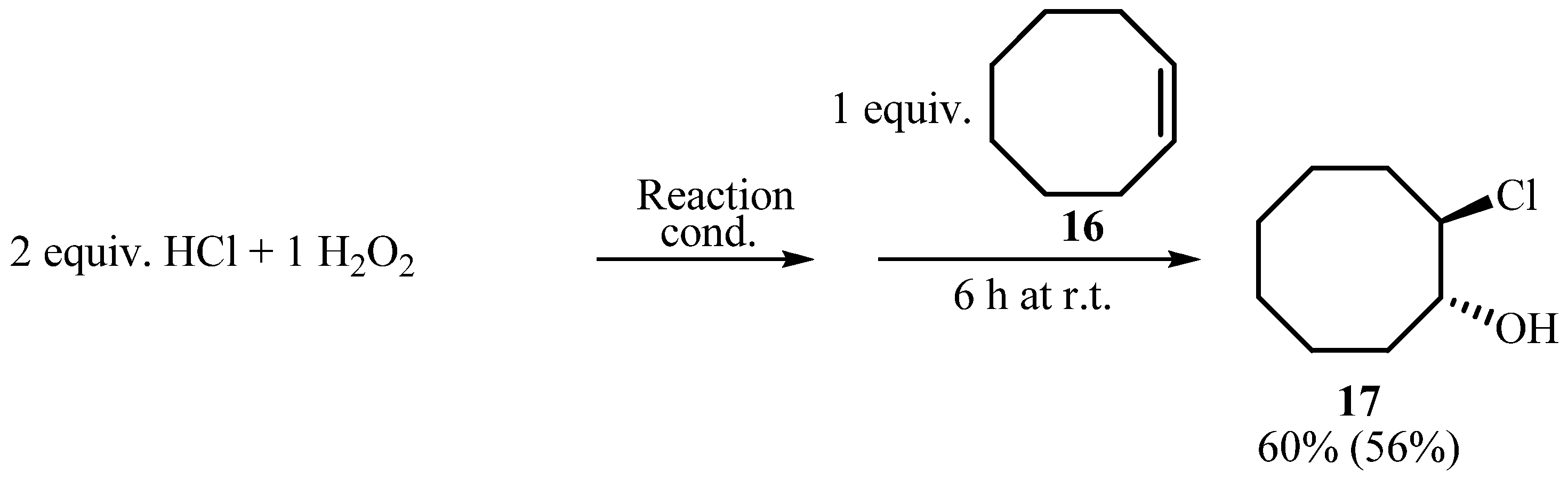
| Entry | Oxidant | React. cond.a | Conv.b (%) |
|---|---|---|---|
| 1 | H2O2 | TFEc, 2 h at r.t. | 60 |
| 2 | H2O2 | 2 h at 85°C | 62 |
| 3 | UHP | 2 h at r.t. | 5 |
| 4 | UHP | 2 h at 85°C | 17 |
| 5 | UHPd | 2 h at r.t. | 38 |
| 6 | UHP | TFEc, 2 h at r.t. | 84 |
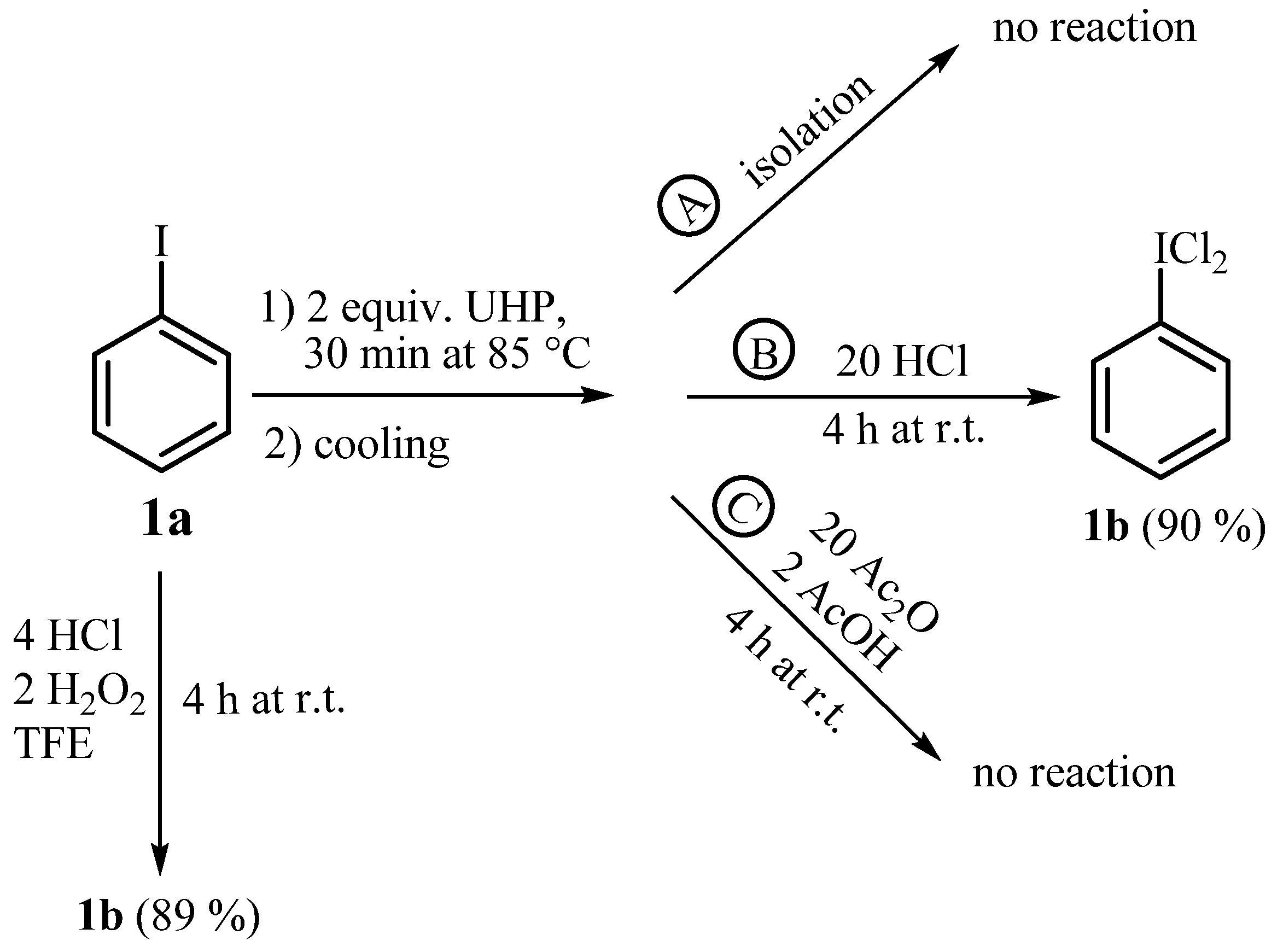
3. Experimental Section
3.1. General Reaction Procedure for the Synthesis of Aryliodine(III) Dichlorides 1b-8b with HCl H2O2 (Methods A, B and C, Table 1)
3.2. General Reaction Procedure for the Synthesis of Aryliodine(III) Dichlorides 1b-8b with Cl2 in Hexane (Method D, Table 1)
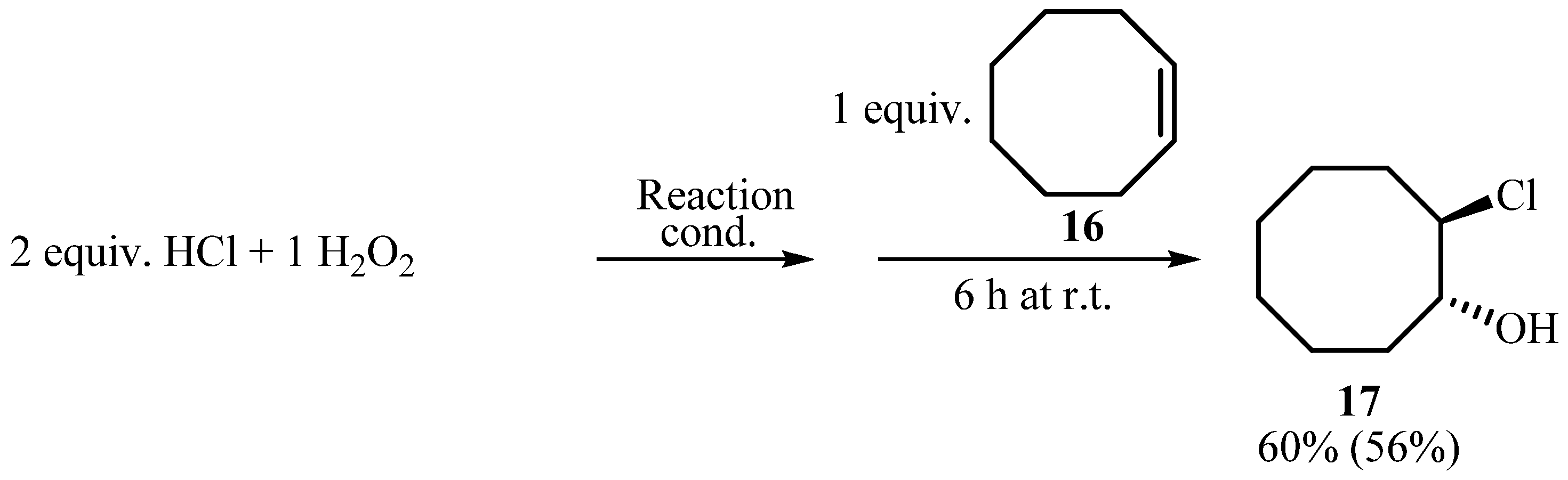
3.3. Oxidative chlorination of cyclooctene (16, Scheme 6)
3.4. Oxidative transformations of iodobenzene (1a, Scheme 6)
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds 1b-8b are available from the authors.
References and Notes
- Varvoglis, A. The Organic Chemistry of Polycoordinated Iodine; VCH Publishers, Inc.: New York, NY, USA, 1992. [Google Scholar]
- Zhdankin, V. V.; Stang, P. J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Tohma, H.; Kita, Y. Hypervalent iodine reagents for the oxidation of alcohols and their application to complex molecule synthesis. Adv. Synth. Catal. 2004, 346, 111–124. [Google Scholar] [CrossRef]
- Kita, Y.; Takada, T.; Ibaraki, M.; Gyoten, M.; Mihara, S.; Fujita, S.; Tohma, H. An Intramolecular Cyclization of Phenol Derivatives Bearing Aminoquinones Using a Hypervalent Iodine Reagents. J. Org. Chem. 1996, 61, 223–227. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef]
- Richardson, R. D.; Wirth, T. Hypervalent Iodine Goes Catalytic. Angew. Chem., Int. Ed. 2006, 45, 4402–4404. [Google Scholar] [CrossRef]
- ket, B.; Zupan, M.; Zupet, P. Role of the Polymer Backbone on the Reactivity of Polymer- Supported (Dichloroiodo)Benzene. Tetrahedron 1984, 40, 1603–1606. [Google Scholar] [CrossRef]
- Yusubov, M. S.; Drygunova, L. A.; Zhdankin, V. V. 4,4 '-Bis(Dichloroiodo)Biphenyl and 3-(Dichloroiodo)Benzoic Acid: New Recyclable Hypervalent Iodine Reagents for Vicinal Halomethoxylation of Unsaturated Compounds. Synthesis 2004, 2289–2292. [Google Scholar]
- Podgoršek, A.; Jurisch, M.; Stavber, S.; Zupan, M.; Iskra, J.; Gladysz, J. A. Synthesis and Reactivity of Fluorous and Nonfluorous Aryl and Alkyl Iodine(III) Dichlorides: New Chlorinating Reagents that are Easily Recycled Using Biphasic Protocols. J. Org. Chem. 2009, 74, 3133–3140. [Google Scholar] [CrossRef]
- Willgerodt, O. Über Einige Aromatische Jodidchloride. J. Prakt. Chem. 1886, 33, 154–160. [Google Scholar] [CrossRef]
- Bravo, P.; Montanari, V.; Resnati, G.; DesMarteau, D. D. 1-(Dihloroiodo)-1H,1H-Perfluoroalkanes. J. Org. Chem. 1994, 59, 6093–6094. [Google Scholar]
- Taylor, R. T.; Stevenson, T. A. Mercury Mediated Synthesis of Bis(Carboxy)Iodobenzenes. Tetrahedron Lett. 1988, 29, 2033–2036. [Google Scholar] [CrossRef]
- Zanka, A.; Takeuchi, H.; Kubota, A. Large-Scale Preparation of Iodobenzene Dichloride and Efficient Monochlorination of 4-Aminoacetophenone. Org. Proc. Res. Dev. 1998, 2, 270–273. [Google Scholar]
- Skulski, L. Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland. Molecules 2000, 5, 1331–1371. [Google Scholar] [CrossRef]
- Podgoršek, A.; Zupan, M.; Iskra, J. Oxidative Halogenation with "Green" Oxidants: Oxygen and Hydrogen Peroxide. Angew. Chem., Int. Ed. 2009, 48, 8424–8450. [Google Scholar] [CrossRef]
- Obeid, N.; Skulski, L. Novel Oxidative, Liquid-Phase Chlorination Procedures for the Preparation of (Dichloroidoarenes) from Iodoarenes. Pol. J. Chem. 2000, 74, 1609–1615. [Google Scholar]
- Krassowska-Swiebocka, B.; Prokopienko, G.; Skulski, L. Biphasic Chlorination of Iodoarenes to (Dichloroiodo)Arenes. Synlett 1999, 1409–1410. [Google Scholar]
- Baranowski, A.; Plachta, D.; Skulski, L.; Klimaszewska, M. Liquid-Phase and Biphasic Chlorination of some Iodoarenes to form (Dichloroiodo)Arenes with Sodium Peroxodisulfate as the Oxidant. J. Chem. Res.-S 2000, 435–437. [Google Scholar]
- Kazmierczak, P.; Skulski, L.; Obeid, N. Oxidative Chlorination of Various Iodoarenes to (Dichloroiodo)Arenes With Chromium(VI) Oxide as the Oxidant. J. Chem. Res.-S 1999, 64-65. [Google Scholar]
- Zhao, X. F.; Zhang, C. Iodobenzene Dichloride as a Stoichiometric Oxidant for the Conversion of Alcohols into Carbonyl Compounds; Two Facile Methods for its Preparation. Synthesis 2007, 551–557. [Google Scholar]
- Koyuncu, D.; Mckillop, A.; Mclaren, L. A Simple and Inexpensive Procedure for the Preparation of (Dichloroiodo)Arenes. J. Chem. Res. (S) 1990, 21. [Google Scholar]
- Zielinska, A.; Skulski, L. A Solvent-Free Synthesis of (Dichloroiodo)Arenes from Iodoarenes. Tetrahedron Lett. 2004, 45, 1087–1089. [Google Scholar] [CrossRef]
- Obeid, N.; Skulski, L. One-Pot Procedures for Preparing (Dichloroiodo)Arenes from Arenes and Diiodine, With Chromium(VI) Oxide as the Oxidant. Molecules 2001, 6, 869–874. [Google Scholar] [CrossRef]
- Lulinski, P.; Obeid, N.; Skulski, L. One-Pot Preparations of (Dichloroiodo)Arenes from some Arenes. Bull. Chem. Soc. Jpn. 2001, 74, 2433–2434. [Google Scholar] [CrossRef]
- Iskra, J.; Stavber, S.; Zupan, M. Nonmetal-Catalyzed Iodination of Arenes with Iodide and Hydrogen Peroxide. Synthesis 2004, 1869–1873. [Google Scholar]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Free Radical Bromination by the H2O2-HBr System on Water. Tetrahedron Lett. 2006, 47, 7245–7247. [Google Scholar] [CrossRef]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Bromination of Ketones with H2O2-HBr "on Water''. Green Chem. 2007, 9, 1212–1218. [Google Scholar] [CrossRef]
- Iskra, J.; Stavber, S.; Zupan, M. Aerobic Oxidative Iodination of Organic Molecules Activated by Sodium Nitrite. Tetrahedron Lett. 2008, 49, 893–895. [Google Scholar] [CrossRef]
- Podgoršek, A.; Eissen, M.; Fleckenstein, J.; Stavber, S.; Zupan, M.; Iskra, J. Selective Aerobic Oxidative Dibromination of Alkenes with Aqueous HBr and Sodium Nitrite as a Catalyst. Green Chem. 2009, 11, 120–126. [Google Scholar] [CrossRef]
- Ben Daniel, R.; de Visser, S. P.; Shaik, S.; Neumann, R. Electrophilic Aromatic Chlorination and Haloperoxidation of Chloride Catalyzed by Polyfluorinated Alcohols: A New Manifestation of Template Catalysis. J. Am. Chem. Soc. 2003, 125, 12116–12117. [Google Scholar] [CrossRef]
- Zielinska, A.; Skulski, L. Easy Preparation of (Diacetoxyiodo)Arenes from Iodoarenes with Sodium Percarbonate as the Oxidant. Molecules 2002, 7, 806–809. [Google Scholar] [CrossRef]
- Willgerodt, C. Die Organischen Verbindungen Mit Mehrvertigem Jod; Enke Verlag: Stuttgart, Germany, 1914. [Google Scholar]
- Ranganathan, S.; Ranganathan, D.; Ramachandran, P. V. Iodoxybenzene - A Remarkably Close Ozone Equivalent. Tetrahedron 1984, 40, 3145–3151. [Google Scholar] [CrossRef]
- Keefer, R. M.; Andrews, L. J. The Kinetics of Dissociation of Derivatives of Iodobenzene Dichloride in Acetic Acid. J. Am. Chem. Soc. 1958, 80, 277–281. [Google Scholar] [CrossRef]
- Varma, P. S.; Raman, V. Halogenation. Part X. Preparation of Mixed Halogen Derivatives of Xylenes. J. Indian Chem. Soc. 1935, 12, 245–247. [Google Scholar]
- Carey, J. V.; Chaloner, P. A.; Hitchcock, P. B.; Neugebauer, T.; Seddon, K. R. Synthesis and Decomposition of Dichloroiodoarenes. An Improved Low Temperature X-Ray Structure of Dichloroiodobenzene and the Structure of 1-Chloro-2,3,5,6-Tetrakis(Chloromethyl)-4-Methylbenzene. J. Chem. Res. (S) 1996, 8, 2031–2054. [Google Scholar]
- Wilgerodt, C.; Roggatz, H. Über Jodoso-, Jodo- Ind Jodiniumverbindungen, die sich von Jod- und Chlorjodmesitylen Ableiten. J. Prakt. Chem. 1900, 61, 423–430. [Google Scholar]
- Xu, G.; Hartman, T. L.; Wargo, H.; Turpin, J. A.; Buckheit, R. W.; Cushman, M. Synthesis of Alkenyldiarylmethane (ADAM) Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors with Non-Identical Aromatic Rings. Bioorg. Med. Chem. Lett. 2002, 10, 283–290. [Google Scholar] [CrossRef]
- Reymond, S.; Legrand, O.; Brunel, J. M.; Buono, G. Chiral O-Methoxyaryldiazaphosphonamides - A New Class of Efficient Lewis Bases in the Catalytic Asymmetric Ring Opening of Cyclooctene Oxide with Silicon Tetrachloride. Eur. J. Org. Chem. 2001, 2819–2823. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Podgoršek, A.; Iskra, J. Conversion of Aryl Iodides into Aryliodine(III) Dichlorides by an Oxidative Halogenation Strategy Using 30% Aqueous Hydrogen Peroxide in Fluorinated Alcohol. Molecules 2010, 15, 2857-2871. https://doi.org/10.3390/molecules15042857
Podgoršek A, Iskra J. Conversion of Aryl Iodides into Aryliodine(III) Dichlorides by an Oxidative Halogenation Strategy Using 30% Aqueous Hydrogen Peroxide in Fluorinated Alcohol. Molecules. 2010; 15(4):2857-2871. https://doi.org/10.3390/molecules15042857
Chicago/Turabian StylePodgoršek, Ajda, and Jernej Iskra. 2010. "Conversion of Aryl Iodides into Aryliodine(III) Dichlorides by an Oxidative Halogenation Strategy Using 30% Aqueous Hydrogen Peroxide in Fluorinated Alcohol" Molecules 15, no. 4: 2857-2871. https://doi.org/10.3390/molecules15042857
APA StylePodgoršek, A., & Iskra, J. (2010). Conversion of Aryl Iodides into Aryliodine(III) Dichlorides by an Oxidative Halogenation Strategy Using 30% Aqueous Hydrogen Peroxide in Fluorinated Alcohol. Molecules, 15(4), 2857-2871. https://doi.org/10.3390/molecules15042857