Synthesis and Biological Evaluation of New 5-Fluorouracil-Substituted Ampelopsin Derivatives

Abstract
:
1. Introduction
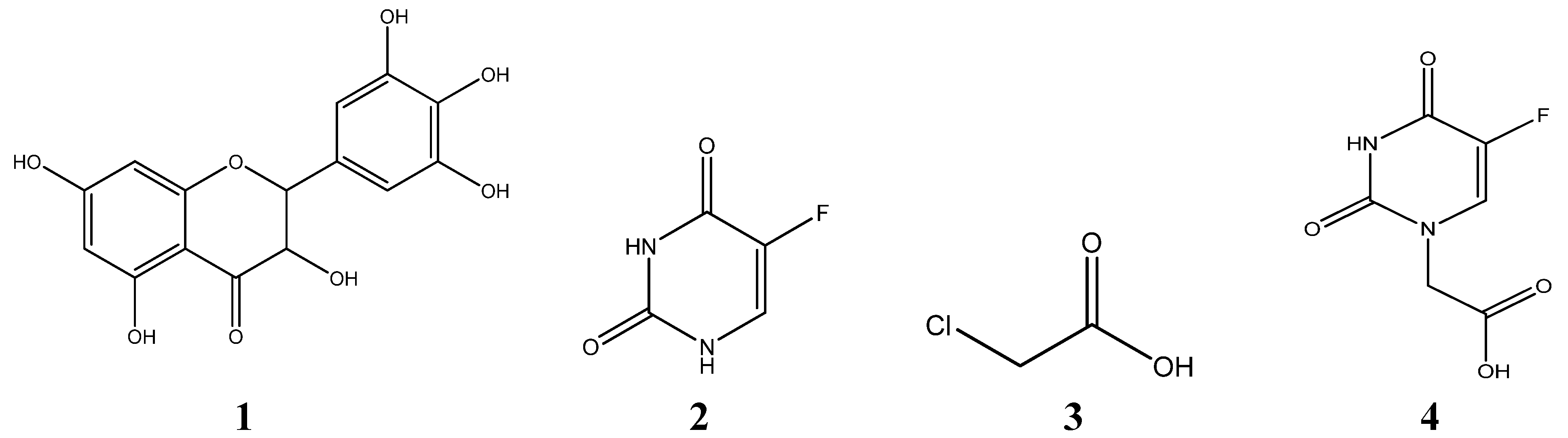
2. Results and Discussion
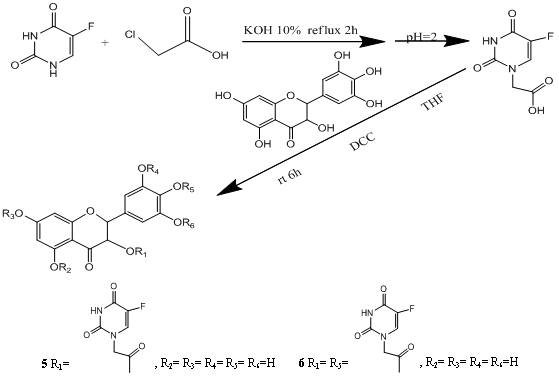
2.1. Synthesis and Post-Treatment
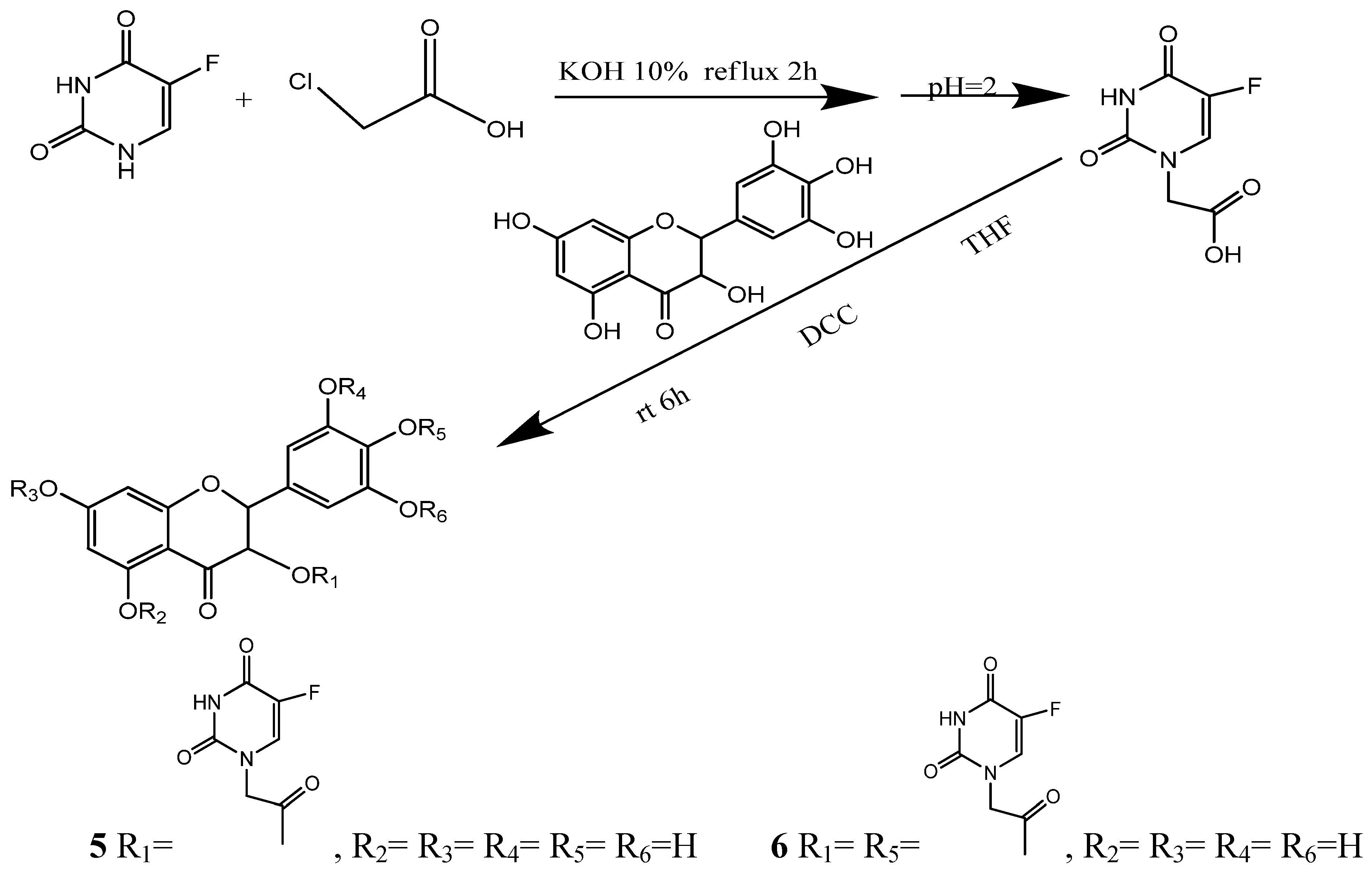
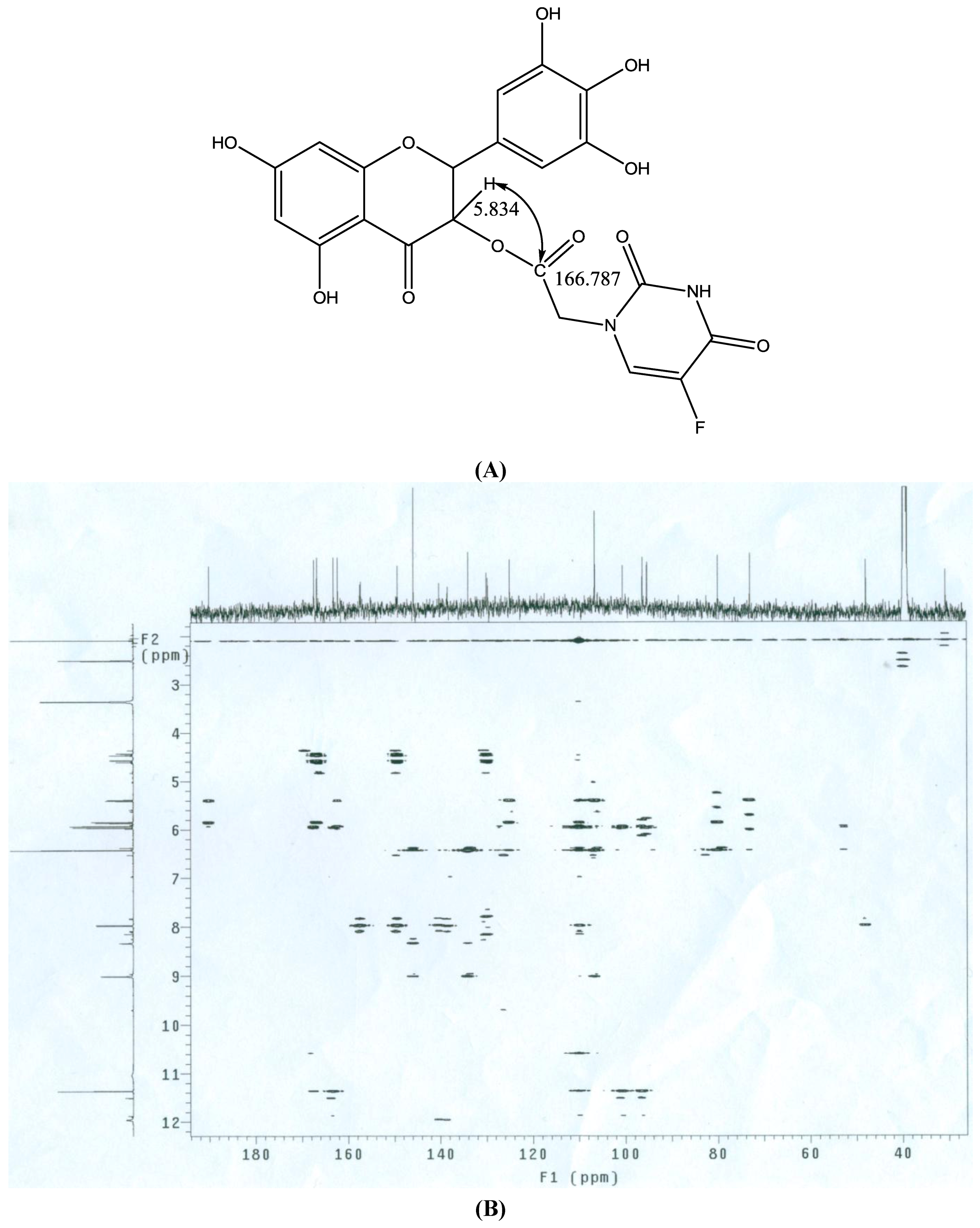
2.2. Chemical Characterization

2.3. Biological Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | K562 | K562/ADR 48 h IC50 (μmol/L) | ||
|---|---|---|---|---|
| 24 h IC50 (μmol/L) | 48 h IC50 (μmol/L) | 72 h IC50 (μmol/L) | ||
| AMP | 20.55 ± 1.10 | 13.78 ± 2.14 | 10.25 ± 0.33 | 38.89 ± 4.77 |
| 5 | 22.50 ± 2.65 | 11.62 ± 2.20 | 12.54 ± 1.02 | 7.19 ± 0.51 |
| 6 | 21.51 ± 1.84 | 10.34 ± 0.60 | 11.11 ± 1.48 | 11.26 ± 1.29 |
| AMP+5-Fu /1:1 | -- | 11.29 ± 0.56 | -- | 32.63 ± 2.67 |
| AMP+5-Fu /1:2 | -- | 12.68 ± 1.33 | -- | 29.20 ± 2.29 |
| ADR | -- | 1.26 ± 0.15 | -- | 50.52 ± 4.03 |
| Group | Concentration (μmol/L) | IC50 (μmol/L) | Fold-reversal |
|---|---|---|---|
| Control | -- | 50.52 ± 4.03 | 1.00 |
| Verapamil | 0.625 | 20.72 ± 1.62 ** | 2.44 |
| 1.25 | 12.00 ± 1.26 ** | 4.21 | |
| 5 | 0.625 | 17.94 ± 1.09 ** | 2.82 |
| 1.25 | 14.45 ± 1.52 ** | 3.50 | |
| 6 | 0.625 | 48.88 ± 8.30 | 1.03 |
| 1.25 | 36.16 ± 4.49 ** | 1.40 | |
| AMP | 0.625 | 38.34 ± 4.20 * | 1.32 |
| 1.25 | 20.19 ± 4.29 ** | 2.50 | |
| 5-Fu | 0.625 | 47.03 ± 2.79 | 1.07 |
| 1.25 | 41.12 ± 2.13 | 1.23 | |
| AMP+5-Fu /1:1 | 0.625 | 39.78 ± 2.84 * | 1.27 |
| 1.25 | 27.04 ± 4.51 ** | 1.87 |
3. Experimental
3.1. General
3.2. Synthesis of 5-fluorouracil-1-carboxylic acid (3)
3.3. Synthesis of 5,7-dihydroxy-4-oxo-2-(3,4,5-trihydroxyphenyl)chroman-3-yl 2-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetate (5) and 2-(4-(2-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetoxy)-3,5-dihydroxyphenyl)-5,7-dihydroxy-4-oxochroman-3-yl-2-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetate (6)
3.4. Biological Evaluation
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References
- Liu, D.Y.; Ye, J.T.; Yang, W.H.; Yan, J.; Zeng, C.H.; Zeng, S. Ampelopsin, a small molecule inhibitor of HIV-1 infection targeting HIV entry. Biomed. Environ. Sci. 2004, 17, 153–164. [Google Scholar]
- Liu, D.Y.; Luo, M.; Xie, B.F.; Feng, G.K.; Zhu, X.F.; Liu, Z.C. Antitumor effects of ampelopsin,in Chinese. Chin. J. Cancer. 2001, 20, 1372–1375. [Google Scholar]
- Ye, J.T.; Guan, Y.Y.; Zeng, S.; Liu, D.Y. Ampelopsin prevents apoptosis induced by H2O2 in MT-4 lymphocytes. Planta Med. 2008, 74, 252–257. [Google Scholar] [CrossRef]
- Luo, G.Q.; Zeng, S.; Liu, D.Y. Inhibitory effects of ampelopsin on angiogenesis, in Chinese. Zhong Yao Cai. 2006, 29, 146–150. [Google Scholar]
- Ruan, L.P.; Yu, B.Y.; Fu, G.M.; Zhu, D.N. Improving the solubility of ampelopsin by solid dispersions and inclusion complexes. J. Pharm. Biomed. Anal. 2005, 38, 457–464. [Google Scholar] [CrossRef]
- Murakami, T.; Zhou, Y.Y. Novel dihydroflavonol compound useful for preventing inflammation, allergy, aging, wrinkle, liver spot-freckles and drying of skin. Jpn Kokai Tokkyo KohoJP 2008007449-A 2008. Derwent Primary Accession Number: 2008, E37987. [Google Scholar]
- Ji, M.; Shannon, H.J.; Sidney, M.H. A dihydroflavonol glucoside from Commiphora africana that mediates DNA strand scission. J. Nat. Prod. 2005, 68, 115–117. [Google Scholar] [CrossRef]
- Liao, S.Y.; Chen, J.C.; Qian, L. QSAR, action mechanism and molecular design of flavone and isoflavone derivatives with cytotoxicity against Hela. Eur. J. Med. Chem. 2008, 43, 2159–2170. [Google Scholar] [CrossRef]
- Tsoukala, E.; Agelis, G.; Dolinsek, J.; Botic, T.; Cencic, A.; Komiotis, D. An efficient synthesis of 3-fluoro-5-thio-xylofuranosyl nucleosides of thymine, uracil, and 5-fluorouracil as potential antitumor or/and antiviral agents. Bioorg. Med. Chem. 2007, 15, 3241–3247. [Google Scholar] [CrossRef]
- Saniger, E.; Campos, J.M.; Entrena, A.; Marchal, J.A.; Suárez, I.; Aránega, A.; Choquesillo, D.; Niclós, J.; Gallo, M.Á.; Espinosa, A. Medium benzene-fused oxacycles with the 5-fluorouracil moiety: Synthesis, antiproliferative activities and apoptosis induction in breast cancer cells. Tetrahedron 2003, 59, 5457–5467. [Google Scholar]
- Griffon, J.F.; Mathé, C.; Faraj, A.; Aubertin, A.M.; De Clercq, E.; Balzarini, J.; Sommadossi, J.P.; Gosselin, G. Stereospecific synthesis and biological evaluations of β-L-pentofuranonucleoside derivatives of 5-fluorouracil and 5-fluorocytosine. Eur. J. Med. Chem. 2001, 36, 447–460. [Google Scholar] [CrossRef]
- Domınguez, J.F.; Marchal, J.A.; Correa, A.; Carrillo, E.; Boulaiz, H.; Aranega, A.; Gallo, M.A.; Espinosa, A. Synthesis and evaluation of new 5-fluorouracil antitumor cell differentiating derivatives. Bioorg. Med. Chem. 2003, 11, 315–323. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N. K.; Danneberg, P.; Mooren, D.; Griesbach, L. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Zhang, F.M.; Yao, X.J.; Tian, X.; Tu, Y.Q. Synthesis and biological evaluation of new 4β-5-Fu-substituted 4'-demethylepipodophyllotoxin derivatives. Molecules 2006, 11, 849–857. [Google Scholar] [CrossRef]
- Cai, T.B.; Tang, X.; Nagorski, J.; Brauschweiger, P.G.; Wang, P.G. Synthesis and cytotoxicity of 5-fluorouracil/diazeniumdiolate conjugates. Bioorg. Med. Chem. 2003, 11, 4971–4975. [Google Scholar] [CrossRef]
- Bounous, G.; Pageau, R.; Regoli, D. Enhanced 5-fluorouracil mortality in rats eating defined formula diets. Int. J. Clin. Pharmacol. Biopharm. 1978, 16, 265–267. [Google Scholar]
- Contino, C.; Maurizis, J.C.; Pucci, B. Synthesis and preliminary biological assessments of a new class of amphiphilic telomers bearing 5-fluorouracil moieties. Macromol. Chem. Phys. 1999, 200, 1351–1355. [Google Scholar] [CrossRef]
- Nichifor, M.; Schacht, E.H.; Seymour, L.W. Polymeric prodrugs of 5-fluorouracil. J. Control. Release 1997, 48, 165–178. [Google Scholar] [CrossRef]
- Garcia, O.; Blanco, M.D.; Martin, J.A.; Teijon, J.M. 5-Fluorouracil trapping in poly(2-hydroxyethyl methacrylate-co-acrylamide) hydrogels: in vitro drug delivery studies. Eur. Polym. J. 2000, 36, 111–122. [Google Scholar] [CrossRef]
- Liu, Z.F.; Rimmer, S. Synthesis and release of 5-flourouracil from poly (N-vinylpyrrolidinone) bearing 5-fluorouracil derivatives. J. Control. Release 2002, 81, 91–99. [Google Scholar] [CrossRef]
- Tian, Z.Y.; Du, G.J.; Xie, S.Q.; Zhao, J.; Gao, W.Y.; Wang, C.J. Synthesis and bioevaluation of 5-fluorouracil derivatives. Molecules 2007, 12, 2450–2457. [Google Scholar] [CrossRef]
- Alam, A.; Tsuboi, S. Total synthesis of 3,3′,4-tri-O-methylellagic acid from gallic acid. Tetrahedron 2007, 63, 10454–10465. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Y.Q.; Han, S.T. An improved synthesis metheod for 5-Fluorouracil 1-yl acetic acid,in Chinese. Huaxue Shiji 2005, 27, 500–509. [Google Scholar]
- Biasutto, L.; Marotta, E.; De Marchi, U.; Zoratti, M.; Paradisi, C. Ester-based precursors to increase the bioavailability of quercetin. J. Med. Chem. 2007, 50, 241–253. [Google Scholar] [CrossRef]
- Shaikh, H.A.; Sönnichsen, F.D.; Lindhorst, T.K. Synthesis of glycocluster peptides. Carbohyd. Res. 2008, 343, 1665–1674. [Google Scholar] [CrossRef]
- Ghatnekar, J.; Hägerlöf, M.; Oredsson, S.; Alm, K.; Elmroth, S.K.; Persson, T. Construction of polyamine-modified uridine and adenosine derivatives - evaluation of DNA binding capacity and cytotoxicity in vitro. Bioorg. Med.Chem. 2007, 15, 7426–7433. [Google Scholar] [CrossRef]
- Masao, T. Antieoplastic agents The preparatiom of 5-fluorouracil-1-acetic acid derivatives. B Chem. Soc. Jpn. 1975, 48, 3427–3428. [Google Scholar] [CrossRef]
- Ye, J.T.; Zheng, Y.L.; Liu, D.Y. Reversal effect and its mechanism of ampelopsin on multidrug resistance in K562/ADR cells, in Chinese. Zhongguo Zhong Yao Za Zhi 2009, 34, 761–765. [Google Scholar]
- Ding, Z.S.; Jiang, F.S.; Chen, N.P. Isolation and Identification of an Anti-tumor Component from Leaves of Impatiens balsamina. Molecules 2008, 13, 220–229. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, W.-M.; He, R.-R.; Ye, J.-T.; Zhang, N.; Liu, D.-Y. Synthesis and Biological Evaluation of New 5-Fluorouracil-Substituted Ampelopsin Derivatives. Molecules 2010, 15, 2114-2123. https://doi.org/10.3390/molecules15042114
Zhou W-M, He R-R, Ye J-T, Zhang N, Liu D-Y. Synthesis and Biological Evaluation of New 5-Fluorouracil-Substituted Ampelopsin Derivatives. Molecules. 2010; 15(4):2114-2123. https://doi.org/10.3390/molecules15042114
Chicago/Turabian StyleZhou, Wei-Ming, Rong-Rong He, Jian-Tao Ye, Na Zhang, and De-Yu Liu. 2010. "Synthesis and Biological Evaluation of New 5-Fluorouracil-Substituted Ampelopsin Derivatives" Molecules 15, no. 4: 2114-2123. https://doi.org/10.3390/molecules15042114
APA StyleZhou, W.-M., He, R.-R., Ye, J.-T., Zhang, N., & Liu, D.-Y. (2010). Synthesis and Biological Evaluation of New 5-Fluorouracil-Substituted Ampelopsin Derivatives. Molecules, 15(4), 2114-2123. https://doi.org/10.3390/molecules15042114